The Influence of Tumor Microenvironment on Immune Escape of Melanoma

 , , , and
, , , and
Abstract
:1. Introduction
2. Immune Cells Present within the Melanoma Microenvironment
2.1. Functions of Immune Cells

{kind=link}
{kind=link}
| Type of Immune Cell | Role in the Anti-Cancer Response |
|---|---|
| Natural killer cells | By binding to the tumor cells and releasing cytolytic molecules, they cause tumor cell death. They also participate in the recruitment of APCs by the secretion of cytokines [10]. |
| Macrophages, neutrophils, dendritic cells | Phagocytosis of dead melanoma cells and presentation of cancer antigens that activate secondary adaptive immune responses [10,11]. |
| Th (helper) cells | Binding to the APCs via MHC class II protein complex and secretion of cytokines, eventually leading to tumor cell death [15]. |
| Teff/Tc (effector/cytotoxic) cells | Recognition of antigens presented by immune cells via MHC class I molecules and induction of a cytotoxic effect in tumor cells [10,13]. |
| Treg (regulatory) cells | Secretion of cytokines and chemokines with immunosuppressive activity [16,17]. |
2.2. T Lymphocytes and T Cell-Related Immunotherapy
2.2.1. CTLA-4 and PD-1/PD-L1
| Drug Name | Therapeutic Target and Effect |
|---|---|
| Ipilimumab | Human monoclonal IgG1 anti-CTLA-4 antibody blocking inhibitory signaling based on CTLA-4 [27]. |
| Nivolumab | Human monoclonal IgG4 anti-PD-1 antibody binding to PD-1, thereby preventing interaction between this receptor and its ligands present in the tumor niche [28]. |
| Pembrolizumab | Fully humanized monoclonal IgG4 antibody directed against PD-1, which prevents it from interacting with PD-L1 [28]. |
| Atezolizumab | Fully humanized monoclonal IgG1 antibody, interfering with the binding of PD-L1 ligand to its receptors, PD-1 and B7.1 [29]. |
2.2.2. BRAF V600E-Mutated Melanoma
| Immunosuppression in the BRAF V600E-Positive Melanoma |
|---|
| Low T cell infiltration into the tumor [37] |
| Increase in the number of immunosuppressive cells—Tregs and MDSCs within the TME [35] |
| Inhibition of dendritic cell maturation and, thus, production of TNF-α and IL-12 [35] |
| Low expression of melanoma differentiation antigens and a decrease in the level of MHC molecules, which results in diminished recognition of melanoma cells by the immune system [38] |
2.2.3. Potential Markers for Multitargeted Immunotherapy
2.2.4. Metabolic Mechanisms Involved in Melanoma Immunosuppression
| Immunosuppressive Factor | Action |
|---|---|
| Decreased production of arginine | |
| High level of adenosine | |
| High level of indoleamine 2,3-dioxygenase (IDO) | |
| High level of kinurenine |
|
| Acidification within the tumor niche | |
| Exosomes |
|
3. B Lymphocytes
4. Tumor-Associated Macrophages
5. Myeloid-Derived Suppressor Cells
6. Dendritic Cells
7. Neutrophils
8. Natural Killer Cells
| Cell Type | Role in Melanoma Immunosuppression |
|---|---|
| T lymphocytes | |
| B lymphocytes |
|
| Tumor-associated macrophages (TAMs) | |
| Myeloid-derived suppressor cells (MDSCs) | |
| Dendritic cells (DCs) |
|
| Neutrophils | |
| Natural killer cells (NK cells) |
|
9. Other Elements of the TME Regulating Immune Response
9.1. microRNAs
| The Type of Immune Cells | The Immunosuppressive Role of miRNAs | The Name of miRNA |
|---|---|---|
| T cells | Impaired effector T cells recruitment, increased IL-10 secretion, and regulatory T lymphocytes infiltration | miR-30b/-30d [134] |
| Increased PD-L1 expression in BRAF-mutated melanoma | miR-17-5p [140,141] | |
| Macrophages | Polarization of macrophages toward the M2 type | miR-21, miR-29a, and miR-125b-5p [143,144,145] |
| Enhancement of the expression of CD80, which binds to the CTLA-4 receptor present on T cells, inhibiting their proliferation and function | miR-125b-5p [144] | |
| MDSCs | Conversion of monocytes to MDSCs | miRs: -146a, -155, -125b, -100, -125a, -146b, -99b, and let-7e [135] |
| MDSC induction in IL-1βHIGH melanoma | miR-155 [145] | |
| TGF-β1-dependent increased accumulation and activity of MDSCs | miR-494 [146] | |
| NK cells | Diminished recognition of melanoma cells by NK cells through decreased expression of NKG2D ligands | miR- 34a and miR-34c [147] |
| The Immunogenic Role of miRNAs | The Name of miRNAs | |
| T cells | Positive correlation with anti-PD-L1 therapy efficiency | miR-16-5p, miR-17-5p, and miR-20a-5p [136] |
| Decreased PD-L1 expression | miR-28 [141,142] | |
| Elevated production of IFN-γ corresponding with diminished migration rate of melanoma cells | miR-146a [137] | |
| Macrophages | Polarization of macrophages toward the M1 type | miR-155 [148] |
9.2. Exosomes
9.3. Acidification
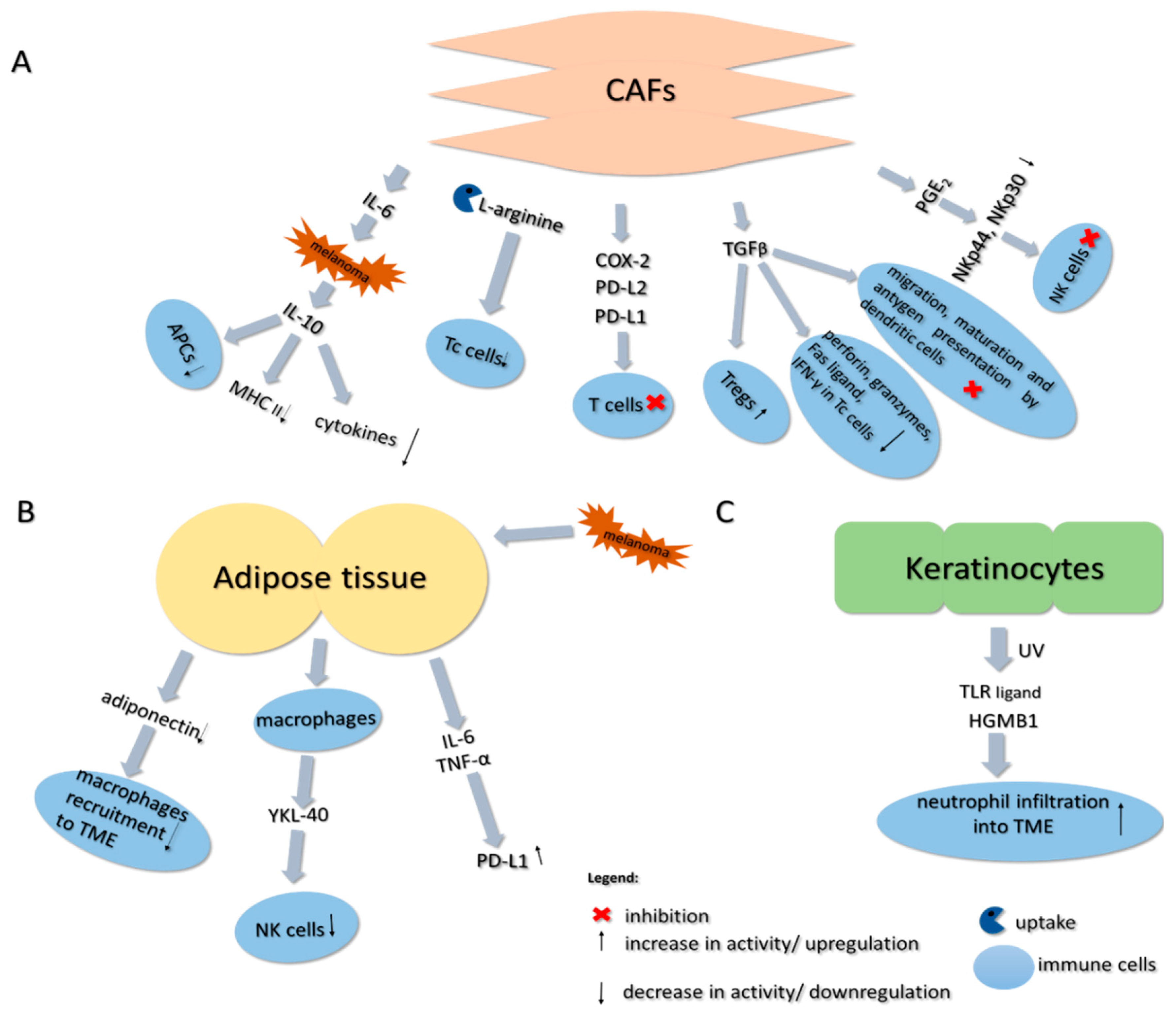
9.4. Cancer-Associated Fibroblasts
9.5. Adipose Tissue
9.6. Keratinocytes
10. Conclusions
Funding
Conflicts of Interest
References
- Miller, A.J.; Mihm, M.C. Melanoma. N. Engl. J. Med. 2006, 355, 51–65. [Google Scholar] [CrossRef] [PubMed]
- Garbe, C.; Amaral, T.; Peris, K.; Hauschild, A.; Arenberger, P.; Bastholt, L.; Bataille, V.; del Marmol, V.; Dréno, B.; Fargnoli, M.C.; et al. European consensus-based interdisciplinary guideline for melanoma. Part 2: Treatment—Update 2019. Eur. J. Cancer 2019, 126, 159–177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ekedahl, H.; Cirenajwis, H.; Harbst, K.; Carneiro, A.; Nielsen, K.; Olsson, H.; Lundgren, L.; Ingvar, C.; Jönsson, G. The clinical significance of BRAF and NRAS mutations in a clinic-based metastatic melanoma cohort. Br. J. Dermatol. 2013, 169, 1049–1055. [Google Scholar] [CrossRef] [PubMed]
- Piris, A.; Mihm, M.C.; Hoang, M.P. BAP1 and BRAFV600E expression in benign and malignant melanocytic proliferations. Hum. Pathol. 2015, 46, 239–245. [Google Scholar] [CrossRef] [PubMed]
- Long, G.V.; Menzies, A.M.; Nagrial, A.M.; Haydu, L.E.; Hamilton, A.L.; Mann, G.J.; Hughes, T.M.; Thompson, J.F.; Scolyer, R.A.; Kefford, R.F. Prognostic and clinicopathologic associations of oncogenic BRAF in metastatic melanoma. J. Clin. Oncol. 2011, 29, 1239–1246. [Google Scholar] [CrossRef] [PubMed]
- Guadarrama-Orozco, J.A.; Ortega-Gómez, A.; Ruíz-García, E.B.; Astudillo-de la Vega, H.; Meneses-García, A.; Lopez-Camarillo, C. Braf V600E mutation in melanoma: Translational current scenario. Clin. Transl. Oncol. 2016, 18, 863–871. [Google Scholar] [CrossRef]
- Hertzman Johansson, C.; Egyhazi Brage, S. BRAF inhibitors in cancer therapy. Pharmacol. Ther. 2014, 142, 176–182. [Google Scholar] [CrossRef]
- Grimaldi, A.M.; Simeone, E.; Festino, L.; Vanella, V.; Strudel, M.; Ascierto, P.A. MEK Inhibitors in the Treatment of Metastatic Melanoma and Solid Tumors. Am. J. Clin. Dermatol. 2017, 18, 745–754. [Google Scholar] [CrossRef]
- Passarelli, A.; Mannavola, F.; Stucci, L.S.; Tucci, M.; Silvestris, F. Immune system and melanoma biology: A balance between immunosurveillance and immune escape. Oncotarget 2017, 8, 106132–106142. [Google Scholar] [CrossRef]
- Marzagalli, M.; Ebelt, N.D.; Manuel, E.R. Unraveling the crosstalk between melanoma and immune cells in the tumor microenvironment. Semin. Cancer Biol. 2019, 59, 236–250. [Google Scholar] [CrossRef]
- Huang, Z.; Gan, J.; Long, Z.; Guo, G.; Shi, X.; Wang, C.; Zang, Y.; Ding, Z.; Chen, J.; Zhang, J.; et al. Targeted delivery of let-7b to reprogramme tumor-associated macrophages and tumor infiltrating dendritic cells for tumor rejection. Biomaterials 2016, 90, 72–84. [Google Scholar] [CrossRef] [PubMed]
- Straten, P.T.; Guldberg, P.; Grønbæk, K.; Hansen, M.R.; Kirkin, A.F.; Seremet, T.; Zeuthen, J.; Becker, J.C. In situ T cell responses against melanoma comprise high numbers of locally expanded T cell clonotypes. J. Immunol. 1999, 163, 443–447. [Google Scholar]
- Durgeau, A.; Virk, Y.; Corgnac, S.; Mami-Chouaib, F. Recent advances in targeting CD8 T-cell immunity for more effective cancer immunotherapy. Front. Immunol. 2018, 9, 14. [Google Scholar] [CrossRef] [PubMed]
- Serpa, J. Tumor Microenvironment; Wiley: Hoboken, NJ, USA, 2020; ISBN 9780470749968. [Google Scholar]
- Giavina-Bianchi, M.H.; Giavina-Bianchi Junior, P.F.; Festa Neto, C. Melanoma: Tumor microenvironment and new treatments. An. Bras. Dermatol. 2017, 92, 156–166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Viguier, M.; Lemaître, F.; Verola, O.; Cho, M.-S.; Gorochov, G.; Dubertret, L.; Bachelez, H.; Kourilsky, P.; Ferradini, L. Foxp3 Expressing CD4 + CD25 high Regulatory T Cells Are Overrepresented in Human Metastatic Melanoma Lymph Nodes and Inhibit the Function of Infiltrating T Cells. J. Immunol. 2004, 173, 1444–1453. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Facciabene, A.; Motz, G.T.; Coukos, G. T-Regulatory cells: Key players in tumor immune escape and angiogenesis. Cancer Res. 2012, 72, 2162–2171. [Google Scholar] [CrossRef] [Green Version]
- Marincola, F.M.; Jaffee, E.M.; Hicklin, D.J.; Ferrone, S. Escape of Human Solid Tumors from T–Cell Recognition: Molecular Mechanisms and Functional Significance. In Advances in Immunology; ScienceDirect: Cambridge, MA, USA, 1999; Volume 74, pp. 181–273. [Google Scholar]
- Mahmoud, F.; Shields, B.; Makhoul, I.; Avaritt, N.; Wong, H.K.; Hutchins, L.F.; Shalin, S.; Tackett, A.J. Immune surveillance in melanoma: From immune attack to melanoma escape and even counterattack. Cancer Biol. Ther. 2017, 18, 451–469. [Google Scholar] [CrossRef] [Green Version]
- Ahmadzadeh, M.; Johnson, L.A.; Heemskerk, B.; Wunderlich, J.R.; Dudley, M.E.; White, D.E.; Rosenberg, S.A. Tumor antigen-specific CD8 T cells infiltrating the tumor express high levels of PD-1 and are functionally impaired. Blood 2009, 114, 1537–1544. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; van der Leun, A.M.; Yofe, I.; Lubling, Y.; Gelbard-Solodkin, D.; van Akkooi, A.C.J.; van den Braber, M.; Rozeman, E.A.; Haanen, J.B.A.G.; Blank, C.U.; et al. Dysfunctional CD8 T Cells Form a Proliferative, Dynamically Regulated Compartment within Human Melanoma. Cell 2019, 176, 775–789.e18. [Google Scholar] [CrossRef]
- Marconcini, R.; Spagnolo, F.; Stucci, L.S.; Ribero, S.; Marra, E.; de Rosa, F.D.; Picasso, V.; di Guardo, L.D.; Cimminiello, C.; Cavalieri, S.; et al. Current status and perspectives in immunotherapy for metastatic melanoma. Oncotarget 2018, 9, 12452–12470. [Google Scholar] [CrossRef] [Green Version]
- Harlin, H.; Kuna, T.V.; Peterson, A.C.; Meng, Y.; Gajewski, T.F. Tumor progression despite massive influx of activated CD8+ T cells in a patient with malignant melanoma ascites. Cancer Immunol. Immunother. 2006, 55, 1185–1197. [Google Scholar] [CrossRef]
- Peggs, K.S.; Quezada, S.A.; Chambers, C.A.; Korman, A.J.; Allison, J.P. Blockade of CTLA-4 on both effector and regulatory T cell compartments contributes to the antitumor activity of anti-CTLA-4 antibodies. J. Exp. Med. 2009, 206, 1717–1725. [Google Scholar] [CrossRef] [Green Version]
- Sharma, P.; Hu-Lieskovan, S.; Wargo, J.A.; Ribas, A. Primary, Adaptive, and Acquired Resistance to Cancer Immunotherapy. Cell 2017, 168, 707–723. [Google Scholar] [CrossRef] [Green Version]
- Allison, J.P.; Hurwitz, A.A.; Leach, D.R. Manipulation of costimulatory signals to enhance antitumor T-cell responses. Curr. Opin. Immunol. 1995, 7, 682–686. [Google Scholar] [CrossRef]
- Specenier, P. Ipilimumab in melanoma. Expert Rev. Anticancer Ther. 2016, 16, 811–826. [Google Scholar] [CrossRef]
- Franklin, C.; Livingstone, E.; Roesch, A.; Schilling, B.; Schadendorf, D. Immunotherapy in melanoma: Recent advances and future directions. Eur. J. Surg. Oncol. 2017, 43, 604–611. [Google Scholar] [CrossRef] [PubMed]
- Inman, B.A.; Longo, T.A.; Ramalingam, S.; Harrison, M.R. Atezolizumab: A PD-L1-blocking antibody for bladder cancer. Clin. Cancer Res. 2017, 23, 1886–1890. [Google Scholar] [CrossRef] [Green Version]
- Larkin, J.; Chiarion-Sileni, V.; Gonzalez, R.; Grob, J.J.; Cowey, C.L.; Lao, C.D.; Schadendorf, D.; Dummer, R.; Smylie, M.; Rutkowski, P.; et al. Combined nivolumab and ipilimumab or monotherapy in untreated Melanoma. N. Engl. J. Med. 2015, 373, 23–34. [Google Scholar] [CrossRef] [Green Version]
- Mourah, S.; Louveau, B.; Dumaz, N. Mechanisms of resistance and predictive biomarkers of response to targeted therapies and immunotherapies in metastatic melanoma. Curr. Opin. Oncol. 2020, 32, 91–97. [Google Scholar] [CrossRef]
- Petrova, V.; Arkhypov, I.; Weber, R.; Groth, C.; Altevogt, P.; Utikal, J.; Umansky, V. Modern Aspects of Immunotherapy with Checkpoint Inhibitors in Melanoma. Int. J. Mol. Sci. 2020, 21, 2367. [Google Scholar] [CrossRef] [Green Version]
- Antohe, M.; Nedelcu, R.I.; Nichita, L.; Popp, C.G.; Cioplea, M.; Brinzea, A.; Hodorogea, A.; Calinescu, A.; Balaban, M.; Ion, D.A.; et al. Tumor infiltrating lymphocytes: The regulator of melanoma evolution (Review). Oncol. Lett. 2019, 17, 4155–4161. [Google Scholar] [CrossRef] [PubMed]
- Spranger, S.; Bao, R.; Gajewski, T.F. Melanoma-intrinsic β-catenin signalling prevents anti-tumour immunity. Nature 2015, 523, 231–235. [Google Scholar] [CrossRef]
- Sumimoto, H.; Imabayashi, F.; Iwata, T.; Kawakami, Y. The BRAF-MAPK signaling pathway is essential for cancer-immune evasion in human melanoma cells. J. Exp. Med. 2006, 203, 1651–1656. [Google Scholar] [CrossRef] [Green Version]
- Pelster, M.S.; Amaria, R. Combined targeted therapy and immunotherapy in melanoma: A review of the impact on the tumor microenvironment and outcomes of early clinical trials. Ther. Adv. Med. Oncol. 2019, 11, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Ho, P.C.; Meeth, K.M.; Tsui, Y.C.; Srivastava, B.; Bosenberg, M.W.; Kaech, S.M. Immune-based antitumor effects of BRAF inhibitors rely on signaling by CD40L and IFNγ. Cancer Res. 2014, 74, 3205–3217. [Google Scholar] [CrossRef] [Green Version]
- Frederick, D.T.; Piris, A.; Cogdill, A.P.; Cooper, Z.A.; Lezcano, C.; Ferrone, C.R.; Mitra, D.; Boni, A.; Newton, L.P.; Liu, C.; et al. BRAF inhibition is associated with enhanced melanoma antigen expression and a more favorable tumor microenvironment in patients with metastatic melanoma. Clin. Cancer Res. 2013, 19, 1225–1231. [Google Scholar] [CrossRef] [Green Version]
- Boni, A.; Cogdill, A.P.; Dang, P.; Udayakumar, D.; Njauw, C.N.J.; Sloss, C.M.; Ferrone, C.R.; Flaherty, K.T.; Lawrence, D.P.; Fisher, D.E.; et al. Selective BRAFV600E inhibition enhances T-cell recognition of melanoma without affecting lymphocyte function. Cancer Res. 2010, 70, 5213–5219. [Google Scholar] [CrossRef] [Green Version]
- Bradley, S.D.; Chen, Z.; Melendez, B.; Talukder, A.; Khalili, J.S.; Rodriguez-Cruz, T.; Liu, S.; Whittington, M.; Deng, W.; Li, F.; et al. BRAFV600E co-opts a conserved MHC class I internalization pathway to diminish antigen presentation and CD8+ T-cell recognition of melanoma. Cancer Immunol. Res. 2015, 3, 602–609. [Google Scholar] [CrossRef] [Green Version]
- Ott, P.A.; Henry, T.; Baranda, S.J.; Frleta, D.; Manches, O.; Bogunovic, D.; Bhardwaj, N. Inhibition of both BRAF and MEK in BRAFV600E mutant melanoma restores compromised dendritic cell (DC) function while having differential direct effects on DC properties. Cancer Immunol. Immunother. 2013, 62, 811–822. [Google Scholar] [CrossRef]
- Steinberg, S.M.; Zhang, P.; Malik, B.T.; Boni, A.; Shabaneh, T.B.; Byrne, K.T.; Mullins, D.W.; Brinckerhoff, C.E.; Ernstoff, M.S.; Bosenberg, M.W.; et al. BRAF inhibition alleviates immune suppression in murine autochthonous melanoma. Cancer Immunol. Res. 2014, 2, 1044–1050. [Google Scholar] [CrossRef] [Green Version]
- Ebert, P.J.R.; Cheung, J.; Yang, Y.; McNamara, E.; Hong, R.; Moskalenko, M.; Gould, S.E.; Maecker, H.; Irving, B.A.; Kim, J.M.; et al. MAP Kinase Inhibition Promotes T Cell and Anti-tumor Activity in Combination with PD-L1 Checkpoint Blockade. Immunity 2016, 44, 609–621. [Google Scholar] [CrossRef] [Green Version]
- Wilmott, J.S.; Haydu, L.E.; Menzies, A.M.; Lum, T.; Hyman, J.; Thompson, J.F.; Hersey, P.; Kefford, R.F.; Scolyer, R.A.; Long, G.V. Dynamics of Chemokine, Cytokine, and Growth Factor Serum Levels in BRAF-Mutant Melanoma Patients during BRAF Inhibitor Treatment. J. Immunol. 2014, 192, 2505–2513. [Google Scholar] [CrossRef] [Green Version]
- Naderi-Azad, S.; Sullivan, R. The potential of BRAF-targeted therapy combined with immunotherapy in melanoma. Expert Rev. Anticancer Ther. 2020, 20, 131–136. [Google Scholar] [CrossRef]
- Homet Moreno, B.; Mok, S.; Comin-Anduix, B.; Hu-Lieskovan, S.; Ribas, A. Combined treatment with dabrafenib and trametinib with immune-stimulating antibodies for BRAF mutant melanoma. Oncoimmunology 2016, 5, e1052212. [Google Scholar] [CrossRef] [Green Version]
- Long, G.V.; Lebbe, C.; Atkinson, V.; Mandalà, M.; Nathan, P.D.; Arance Fernandez, A.M.; Richtig, E.; Yamazaki, N.; Robert, C.; Schadendorf, D.; et al. The anti–PD-1 antibody spartalizumab (S) in combination with dabrafenib (D) and trametinib (T) in previously untreated patients (pts) with advanced BRAF V600–mutant melanoma: Updated efficacy and safety from parts 1 and 2 of COMBI-i. J. Clin. Oncol. 2019, 37, 9531. [Google Scholar] [CrossRef]
- Sullivan, R.J.; Hamid, O.; Gonzalez, R.; Infante, J.R.; Patel, M.R.; Hodi, F.S.; Lewis, K.D.; Tawbi, H.A.; Hernandez, G.; Wongchenko, M.J.; et al. Atezolizumab plus cobimetinib and vemurafenib in BRAF-mutated melanoma patients. Nat. Med. 2019, 25, 929–935. [Google Scholar] [CrossRef]
- Gutzmer, R.; Stroyakovskiy, D.; Gogas, H.; Robert, C.; Lewis, K.; Protsenko, S.; Pereira, R.P.; Eigentler, T.; Rutkowski, P.; Demidov, L.; et al. Atezolizumab, vemurafenib, and cobimetinib as first-line treatment for unresectable advanced BRAFV600 mutation-positive melanoma (IMspire150): Primary analysis of the randomised, double-blind, placebo-controlled, phase 3 trial. Lancet 2020, 395, 1835–1844. [Google Scholar] [CrossRef]
- Anderson, A.C.; Joller, N.; Kuchroo, V.K. Lag-3, Tim-3, and TIGIT: Co-inhibitory Receptors with Specialized Functions in Immune Regulation. Immunity 2016, 44, 989–1004. [Google Scholar] [CrossRef] [Green Version]
- Knee, D.A.; Hewes, B.; Brogdon, J.L. Rationale for anti-GITR cancer immunotherapy. Eur. J. Cancer 2016, 67, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Guo, Y.; Yang, L.; Lei, S.; Tan, W.; Long, J. NEDD4 negatively regulates GITR via ubiquitination in immune microenvironment of melanoma. OncoTargets Ther. 2019, 12, 10629–10637. [Google Scholar] [CrossRef] [Green Version]
- Imbert, C.; Montfort, A.; Fraisse, M.; Marcheteau, E.; Gilhodes, J.; Martin, E.; Bertrand, F.; Marcellin, M.; Burlet-Schiltz, O.; de Peredo, A.G.; et al. Resistance of melanoma to immune checkpoint inhibitors is overcome by targeting the sphingosine kinase-1. Nat. Commun. 2020, 11, 1–14. [Google Scholar] [CrossRef]
- Johnson, L.A.; Morgan, R.A.; Dudley, M.E.; Cassard, L.; Yang, J.C.; Hughes, M.S.; Kammula, U.S.; Royal, R.E.; Sherry, R.M.; Wunderlich, J.R.; et al. Gene therapy with human and mouse T-cell receptors mediates cancer regression and targets normal tissues expressing cognate antigen. Blood 2009, 114, 535–546. [Google Scholar] [CrossRef] [Green Version]
- Chodon, T.; Comin-Anduix, B.; Chmielowski, B.; Koya, R.C.; Wu, Z.; Auerbach, M.; Ng, C.; Avramis, E.; Seja, E.; Villanueva, A.; et al. Adoptive transfer of MART-1 T-cell receptor transgenic lymphocytes and dendritic cell vaccination in patients with metastatic melanoma. Clin. Cancer Res. 2014, 20, 2457–2465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Landsberg, J.; Kohlmeyer, J.; Renn, M.; Bald, T.; Rogava, M.; Cron, M.; Fatho, M.; Lennerz, V.; Wölfel, T.; Hölzel, M.; et al. Melanomas resist T-cell therapy through inflammation-induced reversible dedifferentiation. Nature 2012, 490, 412–416. [Google Scholar] [CrossRef]
- Kim, S.H.; Roszik, J.; Grimm, E.A.; Ekmekcioglu, S. Impact of l-arginine metabolism on immune response and anticancer immunotherapy. Front. Oncol. 2018, 8, 1–5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dillon, B.J.; Prieto, V.G.; Curley, S.A.; Ensor, C.M.; Holtsberg, F.W.; Bomalaski, J.S.; Clark, M.A. Incidence and Distribution of Argininosuccinate Synthetase Deficiency in Human Cancers: A Method for Identifying Cancers Sensitive to Arginine Deprivation. Cancer 2004, 100, 826–833. [Google Scholar] [CrossRef]
- Yoon, J.K.; Frankel, A.E.; Feun, L.G.; Ekmekcioglu, S.; Kim, K.B. Arginine deprivation therapy for malignant melanoma. Clin. Pharmacol. Adv. Appl. 2012, 5, 11–19. [Google Scholar]
- Geiger, R.; Rieckmann, J.C.; Wolf, T.; Basso, C.; Feng, Y.; Fuhrer, T.; Kogadeeva, M.; Picotti, P.; Meissner, F.; Mann, M.; et al. L-Arginine Modulates T Cell Metabolism and Enhances Survival and Anti-tumor Activity. Cell 2016, 167, 829–842.e13. [Google Scholar] [CrossRef] [Green Version]
- Bronte, V.; Serafini, P.; Mazzoni, A.; Segal, D.M.; Zanovello, P. L-arginine metabolism in myeloid cells controls T-lymphocyte functions. Trends Immunol. 2003, 24, 301–305. [Google Scholar] [CrossRef]
- Yarlagadda, K.; Hassani, J.; Foote, I.P.; Markowitz, J. The role of nitric oxide in melanoma. Biochim. Biophys. Acta Rev. Cancer 2017, 1868, 500–509. [Google Scholar] [CrossRef]
- Cekic, C.; Day, Y.J.; Sag, D.; Linden, J. Myeloid expression of adenosine a2A receptor suppresses T and NK cell responses in the solid tumor microenvironment. Cancer Res. 2014, 74, 7250–7259. [Google Scholar] [CrossRef] [Green Version]
- Krähenbühl, L.; Goldinger, S.M.; Mangana, J.; Kerl, K.; Chevolet, I.; Brochez, L.; Horak, C.; Levesque, M.; Dummer, R.; Cheng, P.F. A Longitudinal Analysis of IDO and PDL1 Expression during Immune- or Targeted Therapy in Advanced Melanoma. Neoplasia 2018, 20, 218–225. [Google Scholar] [CrossRef]
- Rubel, F.; Kern, J.S.; Technau-Hafsi, K.; Uhrich, S.; Thoma, K.; Häcker, G.; von Bubnoff, N.; Meiss, F.; von Bubnoff, D. Indoleamine 2,3-Dioxygenase Expression in Primary Cutaneous Melanoma Correlates with Breslow Thickness and Is of Significant Prognostic Value for Progression-Free Survival. J. Investig. Dermatol. 2018, 138, 679–687. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neagu, M.; Constantin, C.; Caruntu, C.; Dumitru, C.; Surcel, M.; Zurac, S. Inflammation: A key process in skin tumorigenesis (Review). Oncol. Lett. 2019, 17, 4068–4084. [Google Scholar] [CrossRef] [Green Version]
- Holmgaard, R.B.; Zamarin, D.; Li, Y.; Gasmi, B.; Munn, D.H.; Allison, J.P.; Merghoub, T.; Wolchok, J.D. Tumor-Expressed IDO Recruits and Activates MDSCs in a Treg-Dependent Manner. Cell Rep. 2015, 13, 412–424. [Google Scholar] [CrossRef] [Green Version]
- Chevolet, I.; Speeckaert, R.; Schreuer, M.; Neyns, B.; Krysko, O.; Bachert, C.; Hennart, B.; Allorge, D.; van Geel, N.; van Gele, M.; et al. Characterization of the in vivo immune network of IDO, tryptophan metabolism, PD-L1, and CTLA-4 in circulating immune cells in melanoma. Oncoimmunology 2015, 4, 1–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moon, Y.W.; Hajjar, J.; Hwu, P.; Naing, A. Targeting the indoleamine 2,3-dioxygenase pathway in cancer. J. Immunother. Cancer 2015, 3, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Pietra, G.; Manzini, C.; Rivara, S.; Vitale, M.; Cantoni, C.; Petretto, A.; Balsamo, M.; Conte, R.; Benelli, R.; Minghelli, S.; et al. Melanoma cells inhibit natural killer cell function by modulating the expression of activating receptors and cytolytic activity. Cancer Res. 2012, 72, 1407–1415. [Google Scholar] [CrossRef] [Green Version]
- Labadie, B.W.; Bao, R.; Luke, J.J. Reimagining IDO pathway inhibition in cancer immunotherapy via downstream focus on the tryptophan–kynurenine–aryl hydrocarbon axis. Clin. Cancer Res. 2019, 25, 1462–1471. [Google Scholar] [CrossRef] [Green Version]
- Ercolano, G.; Garcia-Garijo, A.; Salomé, B.; Gomez-Cadena, A.; Vanoni, G.; Mastelic-Gavillet, B.; Ianaro, A.; Speiser, D.E.; Romero, P.; Trabanelli, S.; et al. Immunosuppressive Mediators Impair Proinflammatory Innate Lymphoid Cell Function in Human Malignant Melanoma. Cancer Immunol. Res. 2020, 8, 556–564. [Google Scholar] [CrossRef] [PubMed]
- Spranger, S.; Koblish, H.K.; Horton, B.; Scherle, P.A.; Newton, R.; Gajewski, T.F. Mechanism of tumor rejection with doublets of CTLA-4, PD-1/PD-L1, or IDO blockade involves restored IL-2 production and proliferation of CD8+ T cells directly within the tumor microenvironment. J. Immunother. Cancer 2014, 2, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hennequart, M.; Pilotte, L.; Cane, S.; Hoffmann, D.; Stroobant, V.; De Plaen, E.; Van Den Eynde, B.J. Constitutive IDO1 expression in human tumors is driven by cyclooxygenase-2 and mediates intrinsic immune resistance. Cancer Immunol. Res. 2017, 5, 695–709. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tudor, D.V.; Bâldea, I.; Lupu, M.; Kacso, T.; Kutasi, E.; Hopârtean, A.; Stretea, R.; Filip, A.G. COX-2 as a potential biomarker and therapeutic target in melanoma. Cancer Biol. Med. 2020, 17, 20–31. [Google Scholar]
- Ferreira, M.; Krykbaeva, I.; Damsky, W.; Kluger, H.M.; Bosenberg, M. Evaluating the role of the COX2/PGE2 pathway in anti-melanoma immunity. J. Clin. Oncol. 2019, 37, e14114. [Google Scholar] [CrossRef]
- Bohn, T.; Rapp, S.; Luther, N.; Klein, M.; Bruehl, T.J.; Kojima, N.; Aranda Lopez, P.; Hahlbrock, J.; Muth, S.; Endo, S.; et al. Tumor immunoevasion via acidosis-dependent induction of regulatory tumor-associated macrophages. Nat. Immunol. 2018, 19, 1319–1329. [Google Scholar] [CrossRef] [PubMed]
- Díaz, F.E.; Dantas, E.; Geffner, J. Unravelling the Interplay between Extracellular Acidosis and Immune Cells. Mediators Inflamm. 2018, 2018, 1218297. [Google Scholar]
- Chen, G.; Huang, A.C.; Zhang, W.; Zhang, G.; Wu, M.; Xu, W.; Yu, Z.; Yang, J.; Wang, B.; Sun, H.; et al. Exosomal PD-L1 contributes to immunosuppression and is associated with anti-PD-1 response. Nature 2018, 560, 382–386. [Google Scholar] [CrossRef]
- Andreola, G.; Rivoltini, L.; Castelli, C.; Huber, V.; Perego, P.; Deho, P.; Squarcina, P.; Accornero, P.; Lozupone, F.; Lugini, L.; et al. Induction of lymphocyte apoptosis by tumor cell secretion of FasL-bearing microvesicles. J. Exp. Med. 2002, 195, 1303–1316. [Google Scholar] [CrossRef]
- Tucci, M.; Mannavola, F.; Passarelli, A.; Stucci, L.S.; Cives, M.; Silvestris, F. Exosomes in melanoma: A role in tumor progression, metastasis and impaired immune system activity. Oncotarget 2018, 9, 20826–20837. [Google Scholar] [CrossRef] [Green Version]
- Ladányi, A.; Kiss, J.; Mohos, A.; Somlai, B.; Liszkay, G.; Gilde, K.; Fejős, Z.; Gaudi, I.; Dobos, J.; Tímár, J. Prognostic impact of B-cell density in cutaneous melanoma. Cancer Immunol. Immunother. 2011, 60, 1729–1738. [Google Scholar] [CrossRef]
- Somasundaram, R.; Zhang, G.; Fukunaga-Kalabis, M.; Perego, M.; Krepler, C.; Xu, X.; Wagner, C.; Hristova, D.; Zhang, J.; Tian, T.; et al. Tumor-associated B-cells induce tumor heterogeneity and therapy resistance. Nat. Commun. 2017, 8, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Georgescu, S.R.; Tampa, M.; Mitran, C.I.; Mitran, M.I.; Caruntu, C.; Caruntu, A.; Lupu, M.; Matei, C.; Constantin, C.; Georgescu, M.N.R.; et al. Tumor Microenvironments in Organs, Tumour Microenvironment in Skin Carcinogenesis. In Tumor Microenvironments in Organs; Springer: Cham, Switzerland, 2020; pp. 123–142. ISBN 9783030362133. [Google Scholar]
- Botti, G.; Cerrone, M.; Scognamiglio, G.; Anniciello, A.; Ascierto, P.A.; Cantile, M. Microenvironment and tumor progression of melanoma: New therapeutic prospectives. J. Immunotoxicol. 2013, 10, 235–252. [Google Scholar] [CrossRef]
- Falleni, M.; Savi, F.; Tosi, D.; Agape, E.; Cerri, A.; Moneghini, L.; Bulfamante, G.P. M1 and M2 macrophages’ clinicopathological significance in cutaneous melanoma. Melanoma Res. 2017, 27, 200–210. [Google Scholar] [CrossRef]
- Chanmee, T.; Ontong, P.; Konno, K.; Itano, N. Tumor-associated macrophages as major players in the tumor microenvironment. Cancers 2014, 6, 1670–1690. [Google Scholar] [CrossRef] [Green Version]
- Nevaia, W.K.; Vachon, C.M.; Leontovich, A.A.; Scott, C.G.; Thompson, M.A.; Markovic, S.N. Evidence of systemic Th2-driven chronic inflammation in patients with metastatic melanoma. Clin. Cancer Res. 2009, 15, 1931–1939. [Google Scholar] [CrossRef] [Green Version]
- Solinas, G.; Germano, G.; Mantovani, A.; Allavena, P. Tumor-associated macrophages (TAM) as major players of the cancer-related inflammation. J. Leukoc. Biol. 2009, 86, 1065–1073. [Google Scholar] [CrossRef] [Green Version]
- Ding, L.; Linsley, P.S.; Huang, L.Y.; Germain, R.N.; Shevach, E.M. IL-10 inhibits macrophage costimulatory activity by selectively inhibiting the up-regulation of B7 expression. J. Immunol. 1993, 151, 1224–1234. [Google Scholar]
- Fiorentino, D.F.; Zlotnik, A.; Vieira, P.; Mosmann, T.R.; Howard, M.; Moore, K.W.; O’Garra, A. IL-10 acts on the antigen-presenting cell to inhibit cytokine production by Th1 cells. J. Immunol. 1991, 146, 3444–3451. [Google Scholar]
- Varney, M.L.; Olsen, K.J.; Mosley, R.L.; Singh, R.K. Paracrine Regulation of Vascular Endothelial Growth Factor-A Expression During Macrophage-Melanoma Cell Interaction: Role of Monocyte Chemotactic Protein-1 and Macrophage Colony-Stimulating Factor. J. Interf. Cytokine Res. 2005, 25, 674–683. [Google Scholar] [CrossRef]
- Varney, M.L.; Olsen, K.J.; Mosley, R.L.; Bucana, C.D.; Talmadge, J.E.; Singh, R.K. Monocyte/macrophage recruitment, activation and differentiation modulate interleukin-8 production: A paracrine role of tumor-associated macrophages in tumor angiogenesis. In Vivo (Brooklyn) 2002, 16, 471–477. [Google Scholar]
- Roda, J.M.; Sumner, L.A.; Evans, R.; Phillips, G.S.; Marsh, C.B.; Eubank, T.D. Hypoxia-Inducible Factor-2α Regulates GM-CSF–Derived Soluble Vascular Endothelial Growth Factor Receptor 1 Production from Macrophages and Inhibits Tumor Growth and Angiogenesis. J. Immunol. 2011, 187, 1970–1976. [Google Scholar] [CrossRef] [Green Version]
- Chen, P.; Huang, Y.; Bong, R.; Ding, Y.; Song, N.; Wang, X.; Song, X.; Luo, Y. Tumor-associated macrophages promote angiogenesis and melanoma growth via adrenomedullin in a paracrine and autocrine manner. Clin. Cancer Res. 2011, 17, 7230–7239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Somasundaram, R.; Herlyn, M.; Wagner, S.N. The role of tumor microenvironment in melanoma therapy resistance. Melanoma Manag. 2016, 3, 23–32. [Google Scholar] [CrossRef]
- Wang, H.; Yang, L.; Wang, D.; Zhang, Q.; Zhang, L. Pro-tumor activities of macrophages in the progression of melanoma. Hum. Vaccines Immunother. 2017, 13, 1556–1562. [Google Scholar] [CrossRef]
- Di Martile, M.; Farini, V.; Consonni, F.M.; Trisciuoglio, D.; Desideri, M.; Valentini, E.; D’aguanno, S.; Tupone, M.G.; Buglioni, S.; Ercolani, C.; et al. Melanoma-specific bcl-2 promotes a protumoral M2-like phenotype by tumor-associated macrophages. J. Immunother. Cancer 2020, 8, 1–14. [Google Scholar] [CrossRef]
- Smith, M.P.; Sanchez-laorden, B.; Brien, K.O.; Brunton, H.; Young, H.; Dhomen, N.; Flaherty, K.T.; Frederick, D.T.; Cooper, Z.A.; Wargo, J.A.; et al. The immune-microenvironment confers resistance to MAP kinase pathway inhibitors through macrophage-derived TNFα. Cancer Discov. 2014, 4, 1214–1229. [Google Scholar] [CrossRef] [Green Version]
- Ohanna, M.; Giuliano, S.; Bonet, C.; Imbert, V.; Hofman, V.; Zangari, J.; Bille, K.; Robert, C.; Bressac-de Paillerets, B.; Hofman, P.; et al. Senescent cells develop a parp-1 and nuclear factor-κB-associated secretome (PNAS). Genes Dev. 2011, 25, 1245–1261. [Google Scholar] [CrossRef] [Green Version]
- Gabrilovich, D.I.; Nagaraj, S. Myeloid-derived suppressor cells as regulators of the immune system. Nat. Rev. Immunol. 2009, 9, 162–174. [Google Scholar] [CrossRef]
- Horton, B.L.; Williams, J.B.; Cabanov, A.; Spranger, S.; Gajewski, T.F. Intratumoral CD8+ T-cell apoptosis is a major component of T-cell dysfunction and impedes antitumor immunity. Cancer Immunol. Res. 2018, 6, 14–24. [Google Scholar] [CrossRef] [Green Version]
- Blattner, C.; Fleming, V.; Weber, R.; Himmelhan, B.; Altevogt, P.; Gebhardt, C.; Schulze, T.J.; Razon, H.; Hawila, E.; Wildbaum, G.; et al. CCR5+ myeloid-derived suppressor cells are enriched and activated in melanoma lesions. Cancer Res. 2018, 78, 157–167. [Google Scholar] [CrossRef] [Green Version]
- Serafini, P.; Meckel, K.; Kelso, M.; Noonan, K.; Califano, J.; Koch, W.; Dolcetti, L.; Bronte, V.; Borrello, I. Phosphodiesterase-5 inhibition augments endogenous antitumor immunity by reducing myeloid-derived suppressor cell function. J. Exp. Med. 2006, 203, 2691–2702. [Google Scholar] [CrossRef]
- Hassel, J.C.; Jiang, H.; Bender, C.; Winkler, J.; Sevko, A.; Shevchenko, I.; Halama, N.; Dimitrakopoulou-Strauss, A.; Haefeli, W.E.; Jäger, D.; et al. Tadalafil has biologic activity in human melanoma. Results of a pilot trial with Tadalafil in patients with metastatic Melanoma (TaMe). Oncoimmunology 2017, 6, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Yang, L.; DeBusk, L.M.; Fukuda, K.; Fingleton, B.; Green-Jarvis, B.; Shyr, Y.; Matrisian, L.M.; Carbone, D.P.; Lin, P.C. Expansion of myeloid immune suppressor Gr+CD11b+ cells in tumor-bearing host directly promotes tumor angiogenesis. Cancer Cell 2004, 6, 409–421. [Google Scholar] [CrossRef] [Green Version]
- Poschke, I.; Mougiakakos, D.; Hansson, J.; Masucci, G.V.; Kiessling, R. Immature immunosuppressive CD14+HLA-DR-/low cells in melanoma patients are Stat3hi and overexpress CD80, CD83, and DC-sign. Cancer Res. 2010, 70, 4335–4345. [Google Scholar] [CrossRef] [Green Version]
- Tobin, R.P.; Jordan, K.R.; Robinson, W.A.; Davis, D.; Borges, V.F.; Gonzalez, R.; Lewis, K.D.; McCarter, M.D. Targeting myeloid-derived suppressor cells using all-trans retinoic acid in melanoma patients treated with Ipilimumab. Int. Immunopharmacol. 2018, 63, 282–291. [Google Scholar] [CrossRef]
- Entinostat Helps Thwart Immunotherapy Resistance. Cancer Discov. 2019, 9, 685–686.
- Roberts, E.W.; Broz, M.L.; Binnewies, M.; Headley, M.B.; Nelson, A.E.; Wolf, D.M.; Kaisho, T.; Bogunovic, D.; Bhardwaj, N.; Krummel, M.F. Critical Role for CD103+/CD141+ Dendritic Cells Bearing CCR7 for Tumor Antigen Trafficking and Priming of T Cell Immunity in Melanoma. Cancer Cell 2016, 30, 324–336. [Google Scholar] [CrossRef] [Green Version]
- Tucci, M.; Stucci, L.S.; Mannavola, F.; Passarelli, A.; D’Oronzo, S.; Lospalluti, L.; Giudice, G.; Silvestris, F. Defective levels of both circulating dendritic cells and T-regulatory cells correlate with risk of recurrence in cutaneous melanoma. Clin. Transl. Oncol. 2019, 21, 845–854. [Google Scholar] [CrossRef]
- Di Blasio, S.; van Wigcheren, G.F.; Becker, A.; van Duffelen, A.; Gorris, M.; Verrijp, K.; Stefanini, I.; Bakker, G.J.; Bloemendal, M.; Halilovic, A.; et al. The tumour microenvironment shapes dendritic cell plasticity in a human organotypic melanoma culture. Nat. Commun. 2020, 11, 1–17. [Google Scholar] [CrossRef]
- Steinman, R.M.; Hawiger, D.; Nussenzweig, M.C. Tolerogenic Dendritic C Ells. Annu. Rev. Immunol. 2003, 21, 685–711. [Google Scholar] [CrossRef] [Green Version]
- Ellis, L.M.; Hicklin, D.J. VEGF-targeted therapy: Mechanisms of anti-tumour activity. Nat. Rev. Cancer 2008, 8, 579–591. [Google Scholar] [CrossRef]
- Yaguchi, T.; Goto, Y.; Kido, K.; Mochimaru, H.; Sakurai, T.; Tsukamoto, N.; Kudo-Saito, C.; Fujita, T.; Sumimoto, H.; Kawakami, Y. Immune Suppression and Resistance Mediated by Constitutive Activation of Wnt/β-Catenin Signaling in Human Melanoma Cells. J. Immunol. 2012, 189, 2110–2117. [Google Scholar] [CrossRef] [Green Version]
- Dissanayake, S.K.; Olkhanud, P.B.; O’Connell, M.P.; Carter, A.; French, A.D.; Camilli, T.C.; Emeche, C.D.; Hewitt, K.J.; Rosenthal, D.T.; Leotlela, P.D.; et al. Wnt5A regulates expression of tumor-associated antigens in melanoma via changes in signal transducers and activators of transcription 3 phosphorylation. Cancer Res. 2008, 68, 10205–10214. [Google Scholar] [CrossRef] [Green Version]
- Holtzhausen, A.; Zhao, F.; Evans, K.S.; Tsutsui, M.; Orabona, C.; Tyler, D.S.; Hanks, B.A. Melanoma-derived Wnt5a promotes local dendritic-cell expression of IDO and immunotolerance: Opportunities for pharmacologic enhancement of immunotherapy. Cancer Immunol. Res. 2015, 3, 1082–1095. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, F.; Xiao, C.; Evans, K.S.; Theivanthiran, T.; DeVito, N.; Holtzhausen, A.; Liu, J.; Liu, X.; Boczkowski, D.; Nair, S.; et al. Paracrine Wnt5a-β-Catenin Signaling Triggers a Metabolic Program that Drives Dendritic Cell Tolerization. Immunity 2018, 48, 147–160.e7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Diao, J.; Mikhailova, A.; Tang, M.; Gu, H.; Zhao, J.; Cattral, M.S. Immunostimulatory conventional dendritic cells evolve into regulatory macrophage-like cells. Blood 2012, 119, 4919–4927. [Google Scholar] [CrossRef] [Green Version]
- Rajarathnam, K.; Schnoor, M.; Richardson, R.M.; Rajagopal, S. How do chemokines navigate neutrophils to the target site: Dissecting the structural mechanisms and signaling pathways. Cell. Signal. 2019, 54, 69–80. [Google Scholar] [CrossRef] [PubMed]
- Strickland, I.; Rhodes, L.E.; Flanagan, B.F.; Friedmann, P.S. TNF-α and IL-8 are upregulated in the epidermis of normal human skin after UVB exposure: Correlation with neutrophil accumulation and E-selectin expression. J. Investig. Dermatol. 1997, 108, 763–768. [Google Scholar] [CrossRef] [Green Version]
- Mangahas, C.R.; Dela Cruz, G.V.; Friedman-Jiménez, G.; Jamal, S. Endothelin-1 induces CXCL1 and CXCL8 secretion in human melanoma cells. J. Investig. Dermatol. 2005, 125, 307–311. [Google Scholar] [CrossRef]
- Mishalian, I.; Bayuh, R.; Levy, L.; Zolotarov, L.; Michaeli, J.; Fridlender, Z.G. Tumor-associated neutrophils (TAN) develop pro-tumorigenic properties during tumor progression. Cancer Immunol. Immunother. 2013, 62, 1745–1756. [Google Scholar] [CrossRef]
- Granot, Z.; Jablonska, J. Distinct Functions of Neutrophil in Cancer and Its Regulation. Mediators Inflamm. 2015, 2015, 701067. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.C.; Zou, J.M.; Luo, C.; Shu, Y.; Luo, J.; Qin, J.; Wang, Y.; Li, D.; Wang, S.S.; Chi, G.; et al. Circulating tumor cells promote the metastatic colonization of disseminated carcinoma cells by inducing systemic inflammation. Oncotarget 2017, 8, 28418–28430. [Google Scholar] [CrossRef]
- Peng, H.H.; Liang, S.; Henderson, A.J.; Dong, C. Regulation of interleukin-8 expression in melanoma-stimulated neutrophil inflammatory response. Exp. Cell Res. 2007, 313, 551–559. [Google Scholar] [CrossRef] [Green Version]
- Tazzyman, S.; Lewis, C.E.; Murdoch, C. Neutrophils: Key mediators of tumour angiogenesis. Int. J. Exp. Pathol. 2009, 90, 222–231. [Google Scholar] [CrossRef]
- Shaul, M.E.; Fridlender, Z.G. Cancer-related circulating and tumor-associated neutrophils—Subtypes, sources and function. FEBS J. 2018, 285, 4316–4342. [Google Scholar] [CrossRef]
- Cruz, J.; Reis-Filho, J.S.; Silva, P.; Lopes, J.M. Expression of c-met Tyrosine Kinase Receptor Is Biologically and Prognostically Relevant for Primary Cutaneous Malignant Melanomas. Oncology 2003, 65, 72–82. [Google Scholar] [CrossRef]
- Glodde, N.; Bald, T.; van den Boorn-Konijnenberg, D.; Nakamura, K.; O’Donnell, J.S.; Szczepanski, S.; Brandes, M.; Eickhoff, S.; Das, I.; Shridhar, N.; et al. Reactive Neutrophil Responses Dependent on the Receptor Tyrosine Kinase c-MET Limit Cancer Immunotherapy. Immunity 2017, 47, 789–802.e9. [Google Scholar] [CrossRef] [Green Version]
- Huergo-Zapico, L.; Parodi, M.; Cantoni, C.; Lavarello, C.; Fernández-Martínez, J.L.; Petretto, A.; DeAndrés-Galiana, E.J.; Balsamo, M.; López-Soto, A.; Pietra, G.; et al. NK-cell editing mediates epithelial-to-mesenchymal transition via phenotypic and proteomic changes in melanoma cell lines. Cancer Res. 2018, 78, 3913–3925. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Xiang, Y.; Li, F.; Yin, C.; Li, B.; Ke, X. WNT/β-catenin signaling pathway regulating T cell-inflammation in the tumor microenvironment. Front. Immunol. 2019, 10, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Michniewicz, A.G.; Czyz, M. Role of mirnas in melanoma metastasis. Cancers 2019, 11, 326. [Google Scholar] [CrossRef] [Green Version]
- Gaziel-Sovran, A.; Segura, M.F.; Di Micco, R.; Collins, M.K.; Hanniford, D.; Vega-Saenz de Miera, E.; Rakus, J.F.; Dankert, J.F.; Shang, S.; Kerbel, R.S.; et al. MiR-30b/30d Regulation of GalNAc Transferases Enhances Invasion and Immunosuppression during Metastasis. Cancer Cell 2011, 20, 104–118. [Google Scholar] [CrossRef] [Green Version]
- Huber, V.; Vallacchi, V.; Fleming, V.; Hu, X.; Cova, A.; Dugo, M.; Shahaj, E.; Sulsenti, R.; Vergani, E.; Filipazzi, P.; et al. Tumor-derived microRNAs induce myeloid suppressor cells and predict immunotherapy resistance in melanoma. J. Clin. Investig. 2018, 128, 5517–5530. [Google Scholar] [CrossRef] [Green Version]
- Nakahara, S.; Fukushima, S.; Okada, E.; Morinaga, J.; Kubo, Y.; Tokuzumi, A.; Matsumoto, S.; Tsuruta-Kadohisa, M.; Kimura, T.; Kuriyama, H.; et al. MicroRNAs that predict the effectiveness of anti-PD-1 therapies in patients with advanced melanoma. J. Dermatol. Sci. 2020, 97, 77–79. [Google Scholar] [CrossRef]
- Mastroianni, J.; Stickel, N.; Andrlova, H.; Hanke, K.; Melchinger, W.; Duquesne, S.; Schmidt, D.; Falk, M.; Andrieux, G.; Pfeifer, D.; et al. MiR-146a Controls Immune Response in the Melanoma Microenvironment. Cancer Res. 2019, 79, 183–195. [Google Scholar] [CrossRef] [Green Version]
- Gerloff, D.; Lützkendorf, J.; Moritz, R.K.C.; Wersig, T.; Mäder, K.; Müller, L.P.; Sunderkötter, C. Melanoma-derived exosomal mir-125b-5p educates tumor associated macrophages (TAMs) by targeting lysosomal acid lipase A (LIPA). Cancers 2020, 12, 464. [Google Scholar] [CrossRef] [Green Version]
- Chaudhuri, A.A.; So, A.Y.-L.; Sinha, N.; Gibson, W.S.J.; Taganov, K.D.; O’Connell, R.M.; Baltimore, D. MicroRNA-125b Potentiates Macrophage Activation. J. Immunol. 2011, 187, 5062–5068. [Google Scholar] [CrossRef]
- Audrito, V.; Serra, S.; Stingi, A.; Orso, F.; Gaudino, F.; Bologna, C.; Neri, F.; Garaffo, G.; Nassini, R.; Baroni, G.; et al. PD-L1 up-regulation in melanoma increases disease aggressiveness and is mediated through miR-17-5p. Oncotarget 2017, 8, 15894–15911. [Google Scholar] [CrossRef] [Green Version]
- Motti, M.L.; Minopoli, M.; Carluccio, G.D.; Ascierto, P.A.; Carriero, M.V. MicroRNAs as Key Players in Melanoma Cell Resistance to MAPK and Immune Checkpoint Inhibitors. Int. J. Mol. Sci. 2020, 21, 4544. [Google Scholar] [CrossRef]
- Li, Q.; Johnston, N.; Zheng, X.; Wang, H.; Zhang, X.; Gao, D.; Min, W. miR-28 modulates exhaustive differentiation of T cells through silencing programmed cell death-1 and regulating cytokine secretion. Oncotarget 2016, 7, 53735–53750. [Google Scholar] [CrossRef] [Green Version]
- Romano, G.; Kwong, L.N. MiRNAs, melanoma and microenvironment: An intricate network. Int. J. Mol. Sci. 2017, 18, 2354. [Google Scholar] [CrossRef] [Green Version]
- Mathsyaraja, H.; Thies, K.; Taffany, D.A.; Deighan, C.; Liu, T.; Yu, L.; Fernandez, S.A.; Shapiro, C.; Otero, J.; Timmers, C.; et al. CSF1-ETS2-induced microRNA in myeloid cells promote metastatic tumor growth. Oncogene 2015, 34, 3651–3661. [Google Scholar] [CrossRef] [Green Version]
- Arts, N.; Cané, S.; Hennequart, M.; Lamy, J.; Bommer, G.; Van den Eynde, B.; De Plaen, E. MicroRNA-155, Induced by Interleukin-1β, represses the expression of microphthalmia-associated transcription factor (MITF-M) in melanoma cells. PLoS ONE 2015, 10, 1–18. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Lai, L.; Chen, Q.; Song, Y.; Xu, S.; Ma, F.; Wang, X.; Wang, J.; Yu, H.; Cao, X.; et al. MicroRNA-494 Is Required for the Accumulation and Functions of Tumor-Expanded Myeloid-Derived Suppressor Cells via Targeting of PTEN. J. Immunol. 2012, 188, 5500–5510. [Google Scholar] [CrossRef]
- Heinemann, A.; Zhao, F.; Pechlivanis, S.; Eberle, J.; Steinle, A.; Diederichs, S.; Schadendorf, D.; Paschen, A. Tumor suppressive microRNAs miR-34a/c control cancer cell expression of ULBP2, a stress-induced ligand of the natural killer cell receptor NKG2D. Cancer Res. 2012, 72, 460–471. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cai, X.; Yin, Y.; Li, N.; Zhu, D.; Zhang, J.; Zhang, C.-Y.; Zen, K. Jiangsu Letter to the Editor Re-polarization of tumor-associated macrophages to pro-inflammatory M 1 macrophages. J. Mol. Cell Biol. 2012, 4, 341–343. [Google Scholar] [CrossRef]
- Wieckowski, E.U.; Visus, C.; Szajnik, M.; Szczepanski, M.J.; Storkus, W.J.; Whiteside, T.L. Tumor-Derived Microvesicles Promote Regulatory T Cell Expansion and Induce Apoptosis in Tumor-Reactive Activated CD8 + T Lymphocytes. J. Immunol. 2009, 183, 3720–3730. [Google Scholar] [CrossRef] [Green Version]
- Zhou, J.; Yang, Y.; Wang, W.W.; Zhang, Y.; Chen, Z.R.; Hao, C.L.; Zhang, J.P. Melanoma-released exosomes directly activate the mitochondrial apoptotic pathway of CD4+ T cells through their microRNA cargo. Exp. Cell Res. 2018, 371, 364–371. [Google Scholar] [CrossRef] [PubMed]
- Gowda, R.; Robertson, B.M.; Iyer, S.; Barry, J.; Dinavahi, S.S.; Robertson, G.P. The role of exosomes in metastasis and progression of melanoma. Cancer Treat. Rev. 2020, 85, 101975. [Google Scholar] [CrossRef]
- Escudier, B.; Dorval, T.; Chaput, N.; André, F.; Caby, M.P.; Novault, S.; Flament, C.; Leboulaire, C.; Borg, C.; Amigorena, S.; et al. Vaccination of metastatic melanoma patients with autologous dendritic cell (DC) derived-exosomes: Results of the first phase 1 clinical trial. J. Transl. Med. 2005, 3, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, L.; Kalimuthu, S.; Gangadaran, P.; Oh, J.M.; Lee, H.W.; Baek, S.H.; Jeong, S.Y.; Lee, S.W.; Lee, J.; Ahn, B.C. Exosomes derived from natural killer cells exert therapeutic effect in melanoma. Theranostics 2017, 7, 2732–2745. [Google Scholar] [CrossRef]
- Böhme, I.; Bosserhoff, A. Extracellular acidosis triggers a senescence-like phenotype in human melanoma cells. Pigment Cell Melanoma Res. 2019, 33, 41–51. [Google Scholar] [CrossRef] [Green Version]
- Pilon-Thomas, S.; Kodumudi, K.N.; El-Kenawi, A.E.; Russell, S.; Weber, A.M.; Luddy, K.; Damaghi, M.; Wojtkowiak, J.W.; Mulé, J.J.; Ibrahim-Hashim, A.; et al. Neutralization of tumor acidity improves antitumor responses to immunotherapy. Cancer Res. 2016, 76, 1381–1390. [Google Scholar] [CrossRef] [Green Version]
- Orimo, A.; Weinberg, R.A. Stromal fibroblasts in cancer: A novel tumor-promoting cell type. Cell Cycle 2006, 5, 1597–1601. [Google Scholar] [CrossRef]
- Ziani, L.; Safta-Saadoun, T.B.; Gourbeix, J.; Cavalcanti, A.; Robert, C.; Favre, G.; Chouaib, S.; Thiery, J. Melanoma-associated fibroblasts decrease tumor cell susceptibility to NK cell-mediated killing through matrix-metalloproteinases secretion. Oncotarget 2017, 8, 19780–19794. [Google Scholar] [CrossRef] [Green Version]
- Fantini, M.C.; Becker, C.; Monteleone, G.; Pallone, F.; Galle, P.R.; Neurath, M.F. Cutting Edge: TGF-β Induces a Regulatory Phenotype in CD4 + CD25 − T Cells through Foxp3 Induction and Down-Regulation of Smad7. J. Immunol. 2004, 172, 5149–5153. [Google Scholar] [CrossRef] [Green Version]
- Thomas, D.A.; Massagué, J. TGF-β directly targets cytotoxic T cell functions during tumor evasion of immune surveillance. Cancer Cell 2005, 8, 369–380. [Google Scholar] [CrossRef] [Green Version]
- Seeger, P.; Musso, T.; Sozzani, S. The TGF-β superfamily in dendritic cell biology. Cytokine Growth Factor Rev. 2015, 26, 647–657. [Google Scholar] [CrossRef]
- Tirosh, I.; Izar, B.; Prakadan, S.M.; Wadsworth, M.H.; Treacy, D.; Trombetta, J.J.; Rotem, A.; Rodman, C.; Lian, C.; Murphy, G.; et al. Dissecting the multicellular ecosystem of metastatic melanoma by single-cell RNA-seq. Science 2016, 352, 189–196. [Google Scholar] [CrossRef] [Green Version]
- Ohshio, Y.; Teramoto, K.; Hanaoka, J.; Tezuka, N.; Itoh, Y.; Asai, T.; Daigo, Y.; Ogasawara, K. Cancer-associated fibroblast-targeted strategy enhances antitumor immune responses in dendritic cell-based vaccine. Cancer Sci. 2015, 106, 134–142. [Google Scholar] [CrossRef]
- Ohshio, Y.; Hanaoka, J.; Kontani, K.; Teramoto, K. Tranilast Inhibits the Function of Cancer-Associated Fibroblasts Responsible for the Induction of Immune Suppressor Cell Types. Scand. J. Immunol. 2014, 80, 408–416. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khalili, J.S.; Liu, S.; Rodríguez-Cruz, T.G.; Whittington, M.; Wardell, S.; Liu, C.; Zhang, M.; Cooper, Z.A.; Frederick, D.T.; Li, Y.; et al. Oncogenic BRAF(V600E) promotes stromal cell-mediated immunosuppression via induction of interleukin-1 in melanoma. Clin. Cancer Res. 2012, 18, 5329–5340. [Google Scholar] [CrossRef] [Green Version]
- Wong, P.F.; Wei, W.; Gupta, S.; Smithy, J.W.; Zelterman, D.; Kluger, H.M.; Rimm, D.L. Multiplex quantitative analysis of cancer-associated fibroblasts and immunotherapy outcome in metastatic melanoma. J. Immunother. Cancer 2019, 7, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Zhao, F.; Evans, K.; Xiao, C.; DeVito, N.; Theivanthiran, B.; Holtzhausen, A.; Siska, P.J.; Blobe, G.C.; Hanks, B.A. Stromal fibroblasts mediate anti–PD-1 resistance via MMP-9 and dictate TGFb inhibitor sequencing in melanoma. Cancer Immunol. Res. 2018, 6, 1459–1471. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Shklovskaya, E.; Lim, S.Y.; Carlino, M.S.; Menzies, A.M.; Stewart, A.; Pedersen, B.; Irvine, M.; Alavi, S.; Yang, J.Y.H.; et al. Transcriptional downregulation of MHC class I and melanoma de-differentiation in resistance to PD-1 inhibition. Nat. Commun. 2020, 11, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Érsek, B.; Silló, P.; Cakir, U.; Molnár, V.; Bencsik, A.; Mayer, B.; Mezey, E.; Kárpáti, S.; Pós, Z.; Németh, K. Melanoma-associated fibroblasts impair CD8+ T cell function and modify expression of immune checkpoint regulators via increased arginase activity. Cell. Mol. Life Sci. 2020, 1–3. [Google Scholar] [CrossRef] [Green Version]
- Zhou, L.; Yang, K.; Andl, T.; Wickett, R.R.; Zhang, Y. Perspective of Targeting Cancer-Associated Fibroblasts in Melanoma. J. Cancer 2015, 6, 717. [Google Scholar] [CrossRef] [Green Version]
- Terai, M.; Eto, M.; Young, G.D.; Berd, D.; Mastrangelo, M.J.; Tamura, Y.; Harigaya, K.; Sato, T. Interleukin 6 mediates production of interleukin 10 in metastatic melanoma. Cancer Immunol. Immunother. 2012, 61, 145–155. [Google Scholar] [CrossRef]
- Balsamo, M.; Scordamaglia, F.; Pietra, G.; Manzini, C.; Cantoni, C.; Boitano, M.; Queirolo, P.; Vermi, W.; Facchetti, F.; Moretta, A.; et al. Melanoma-associated fibroblasts modulate NK cell phenotype and antitumor cytotoxicity. Proc. Natl. Acad. Sci. USA 2009, 106, 20847–20852. [Google Scholar] [CrossRef] [Green Version]
- Calvani, M.; Pelon, F.; Comito, G.; Taddei, M.L.; Moretti, S.; Innocenti, S.; Nassini, R.; Gerlini, G.; Borgognoni, L.; Bambi, F.; et al. Norepinephrine promotes tumor microenvironment reactivity through β3-adrenoreceptors during melanoma progression. Oncotarget 2015, 6, 4615–4632. [Google Scholar] [CrossRef] [Green Version]
- Calvani, M.; Bruno, G.; Dabraio, A.; Subbiani, A.; Bianchini, F.; Fontani, F.; Casazza, G.; Vignoli, M.; De Logu, F.; Frenos, S.; et al. β3-Adrenoreceptor Blockade Induces Stem Cells Differentiation in Melanoma Microenvironment. Int. J. Mol. Sci. 2020, 21, 1420. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wagner, M.; Bjerkvig, R.; Wiig, H.; Melero-Martin, J.M.; Lin, R.Z.; Klagsbrun, M.; Dudley, A.C. Inflamed tumor-associated adipose tissue is a depot for macrophages that stimulate tumor growth and angiogenesis. Angiogenesis 2012, 15, 481–495. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jung, J.I.; Cho, H.J.; Jung, Y.J.; Kwon, S.H.; Her, S.; Choi, S.S.; Shin, S.H.; Lee, K.W.; Park, J.H.Y. High-fat diet-induced obesity increases lymphangiogenesis and lymph node metastasis in the B16F10 melanoma allograft model: Roles of adipocytes and M2-macrophages. Int. J. Cancer 2015, 136, 258–270. [Google Scholar] [CrossRef] [Green Version]
- Sun, K.; Kusminski, C.M.; Scherer, P.E.; Sun, K.; Kusminski, C.M.; Scherer, P.E. Adipose tissue remodeling and obesity Find the latest version: Review series Adipose tissue remodeling and obesity. J. Clin. Investig. 2011, 121, 2094–2101. [Google Scholar] [CrossRef] [Green Version]
- Sun, Y.; Lodish, H.F. Adiponectin deficiency promotes tumor growth in mice by reducing macrophage infiltration. PLoS ONE 2010, 5, e11987. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Zhang, C.; Du, J.X.; Zhao, J.; Shi, M.T.; Jin, M.W.; Liu, H. Adipocytes promote tumor progression and induce PD-L1 expression via TNF-α/IL-6 signaling. Cancer Cell Int. 2020, 20, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Iwata, T.; Kuwajima, M.; Sukeno, A.; Ishimaru, N.; Hayashi, Y.; Wabitsch, M.; Mizusawa, N.; Itakura, M.; Yoshimoto, K. YKL-40 secreted from adipose tissue inhibits degradation of type I collagen. Biochem. Biophys. Res. Commun. 2009, 388, 511–516. [Google Scholar] [CrossRef]
- Catalán, V.; Gómez-Ambrosi, J.; Rodríguez, A.; Ramírez, B.; Rotellar, F.; Valentí, V.; Silva, C.; Gil, M.J.; Salvador, J.; Frühbeck, G. Increased circulating and visceral adipose tissue expression levels of YKL-40 in obesity-associated type 2 diabetes are related to inflammation: Impact of conventional weight loss and gastric bypass. J. Clin. Endocrinol. Metab. 2011, 96, 200–209. [Google Scholar] [CrossRef]
- Krogh, M.; Christensen, I.; Bouwhuis, M.; Johansen, J.S.; Nørgaard, P.; Schmidt, H.; Hansson, J.; Suciu, S.; Eggermont, A.M.M.; Bastholt, L. Prognostic and predictive value of YKL-40 in stage IIB-III melanoma. Melanoma Res. 2016, 26, 367–376. [Google Scholar] [CrossRef]
- Clement, E.; Lazar, I.; Muller, C.; Nieto, L. Obesity and melanoma: Could fat be fueling malignancy? Pigment Cell Melanoma Res. 2017, 30, 294–306. [Google Scholar] [CrossRef] [Green Version]
- Ma, B.; Herzog, E.L.; Moore, M.; Lee, C.M.; Na, S.H.; Lee, C.G.; Elias, J.A. RIG-like Helicase Regulation of Chitinase 3-like 1 Axis and Pulmonary Metastasis. Sci. Rep. 2016, 6, 1–13. [Google Scholar] [CrossRef]
- Fehniger, T.A.; Caligiuri, M.A. Interleukin 15: Biology and relevance to human disease. Blood 2001, 97, 14–32. [Google Scholar] [CrossRef]
- Carson, B.W.E.; Giri, J.G.; Lindemann, M.J.; Linett, M.L.; Ahdieh, M.; Paxton, R.; Anderson, D.; Eisenmann, J.; Grabstein, K.; Caligiuri, M.A. Interleukln (IL) 15 Is a Novel Cytoklne That Activates Human Natural Killer Cells via Components of the IL-2 Receptor. J. Exp. Med. 1994, 180, 1395–1403. [Google Scholar] [CrossRef]
- Xiao, R.; Mansour, A.G.; Huang, W.; Chrislip, L.A.; Wilkins, R.K.; Queen, N.J.; Youssef, Y.; Mao, H.C.; Caligiuri, M.A.; Cao, L. Adipocytes: A Novel Target for IL-15/IL-15Rα Cancer Gene Therapy. Mol. Ther. 2019, 27, 922–932. [Google Scholar] [CrossRef]
- Mohania, D.; Chandel, S.; Kumar, P.; Verma, V.; Digvijay, K.; Tripathi, D.; Choudhury, K.; Mitten, S.K.; Shah, D. Ultraviolet radiations: Skin defense-damage mechanism. In Advances in Experimental Medicine and Biology; Springer: New York, NY, USA, 2017; Volume 996, pp. 71–87. [Google Scholar]
- Bald, T.; Quast, T.; Landsberg, J.; Rogava, M.; Glodde, N.; Lopez-Ramos, D.; Kohlmeyer, J.; Riesenberg, S.; Van Den Boorn-Konijnenberg, D.; Hömig-Hölzel, C.; et al. Ultraviolet-radiation-induced inflammation promotes angiotropism and metastasis in melanoma. Nature 2014, 507, 109–113. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Simiczyjew, A.; Dratkiewicz, E.; Mazurkiewicz, J.; Ziętek, M.; Matkowski, R.; Nowak, D. The Influence of Tumor Microenvironment on Immune Escape of Melanoma. Int. J. Mol. Sci. 2020, 21, 8359. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21218359
Simiczyjew A, Dratkiewicz E, Mazurkiewicz J, Ziętek M, Matkowski R, Nowak D. The Influence of Tumor Microenvironment on Immune Escape of Melanoma. International Journal of Molecular Sciences. 2020; 21(21):8359. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21218359
Chicago/Turabian StyleSimiczyjew, Aleksandra, Ewelina Dratkiewicz, Justyna Mazurkiewicz, Marcin Ziętek, Rafał Matkowski, and Dorota Nowak. 2020. "The Influence of Tumor Microenvironment on Immune Escape of Melanoma" International Journal of Molecular Sciences 21, no. 21: 8359. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21218359