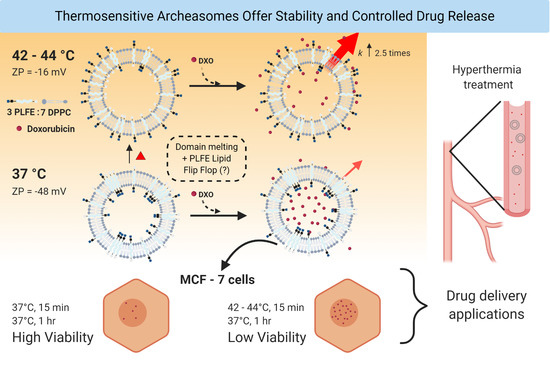
Polar Lipid Fraction E from Sulfolobus acidocaldarius and Dipalmitoylphosphatidylcholine Can Form Stable yet Thermo-Sensitive Tetraether/Diester Hybrid Archaeosomes with Controlled Release Capability

Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results and Discussions
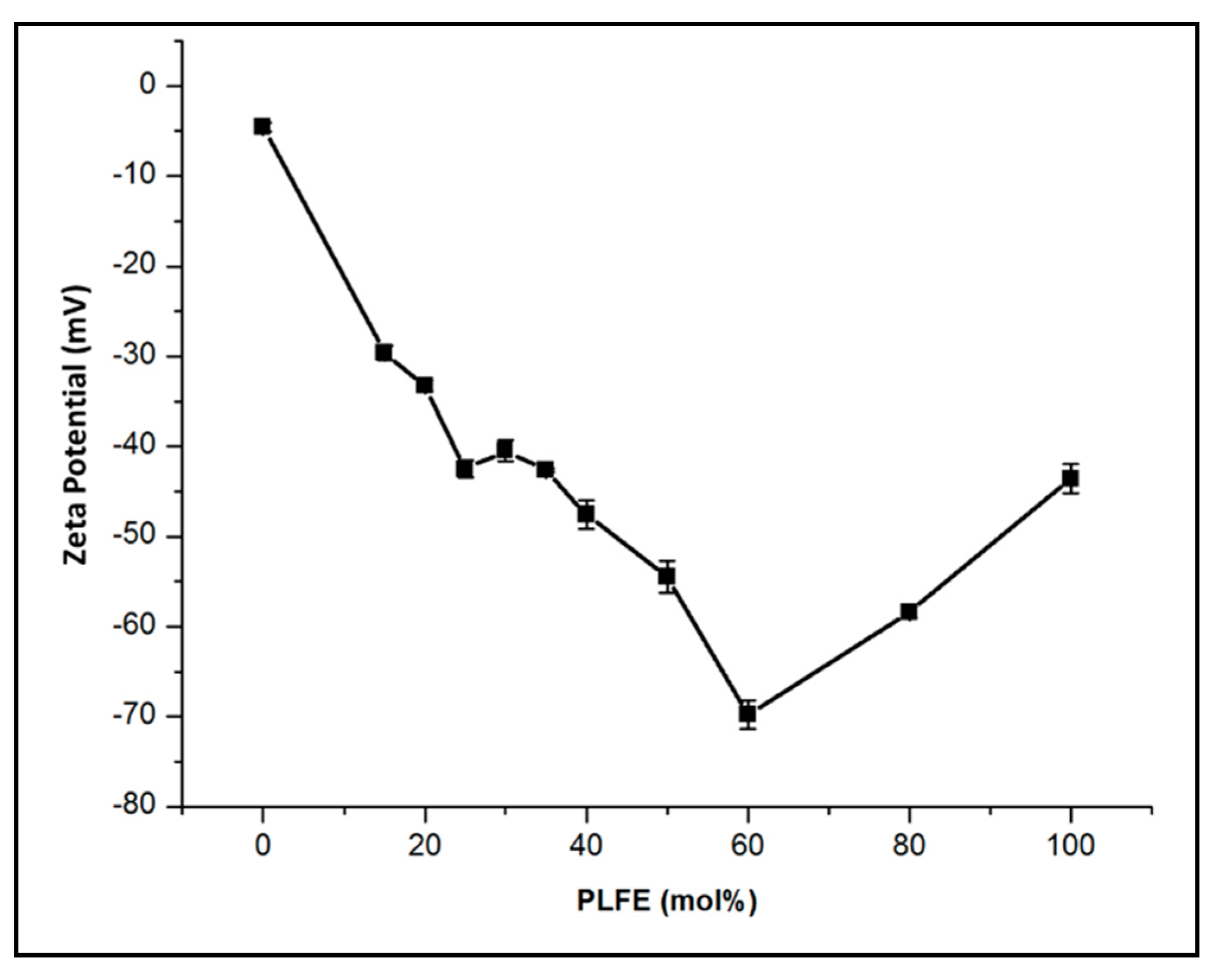
2.1. Effect of PLFE Molar Content on Zeta Potential of DPPC/PLFE Hybrid Archaeosomes
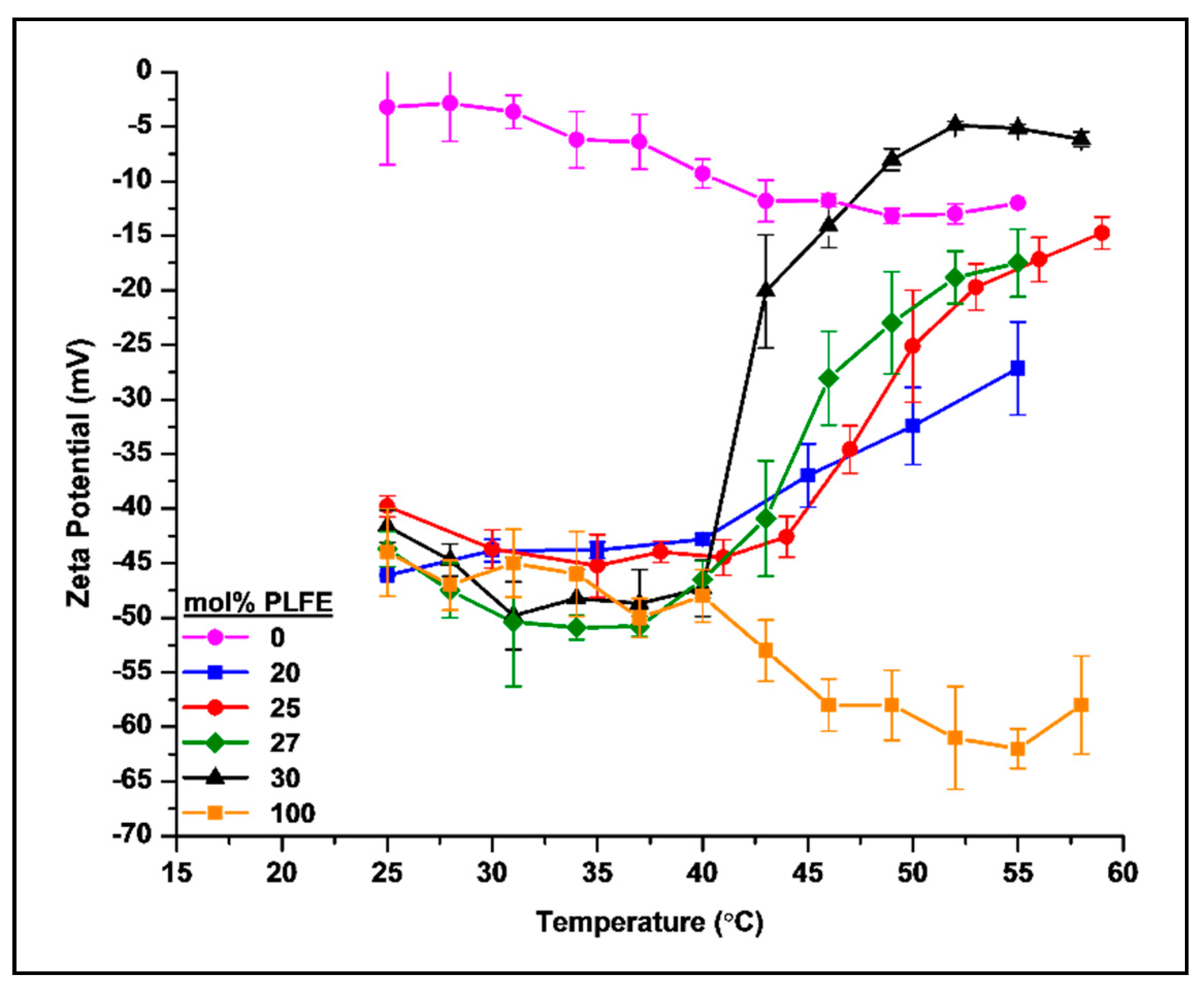
2.2. Thermo-Induced ZP Transition
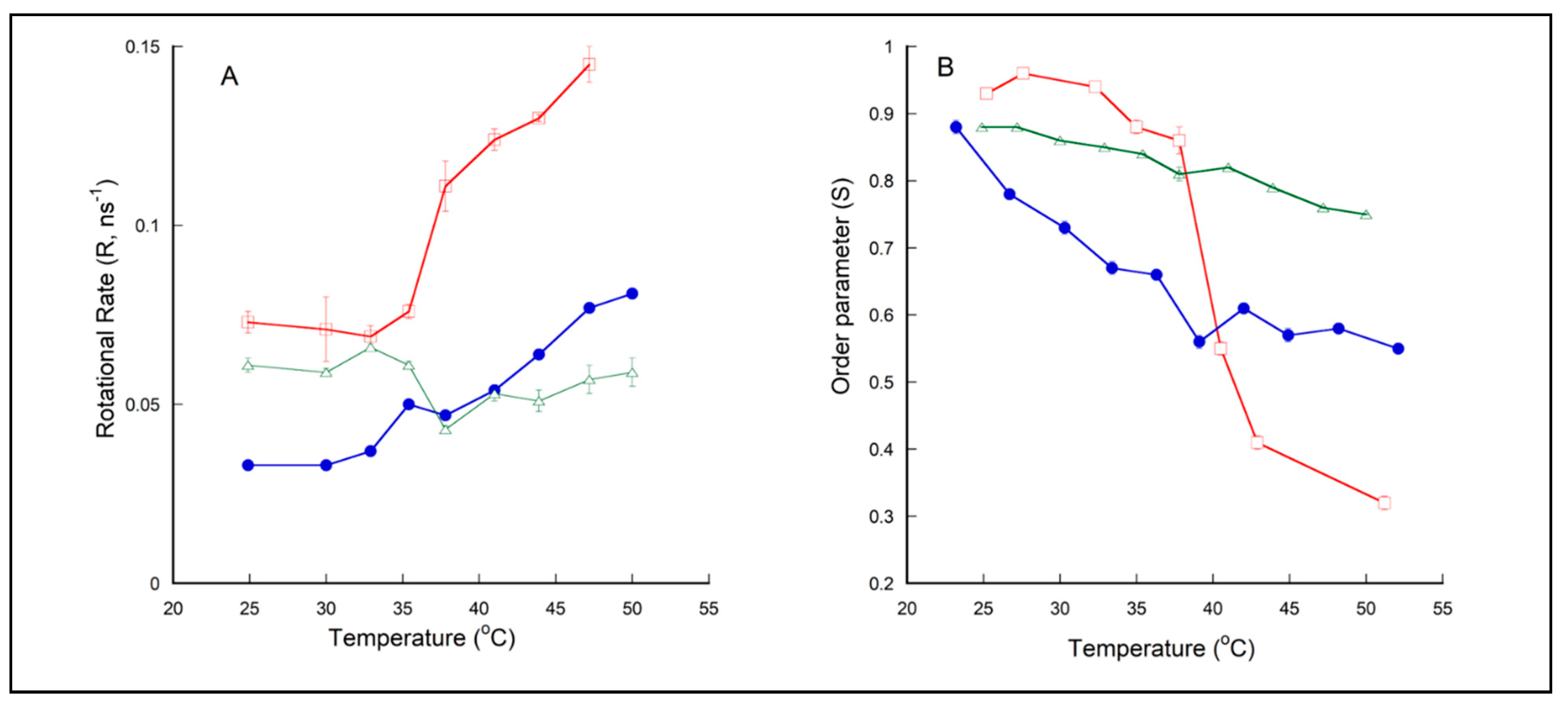
2.3. Membrane Dynamics of PLFE/DPPC(3:7) Archaeosomes as Explored by DPH Nanosecond Fluorometry
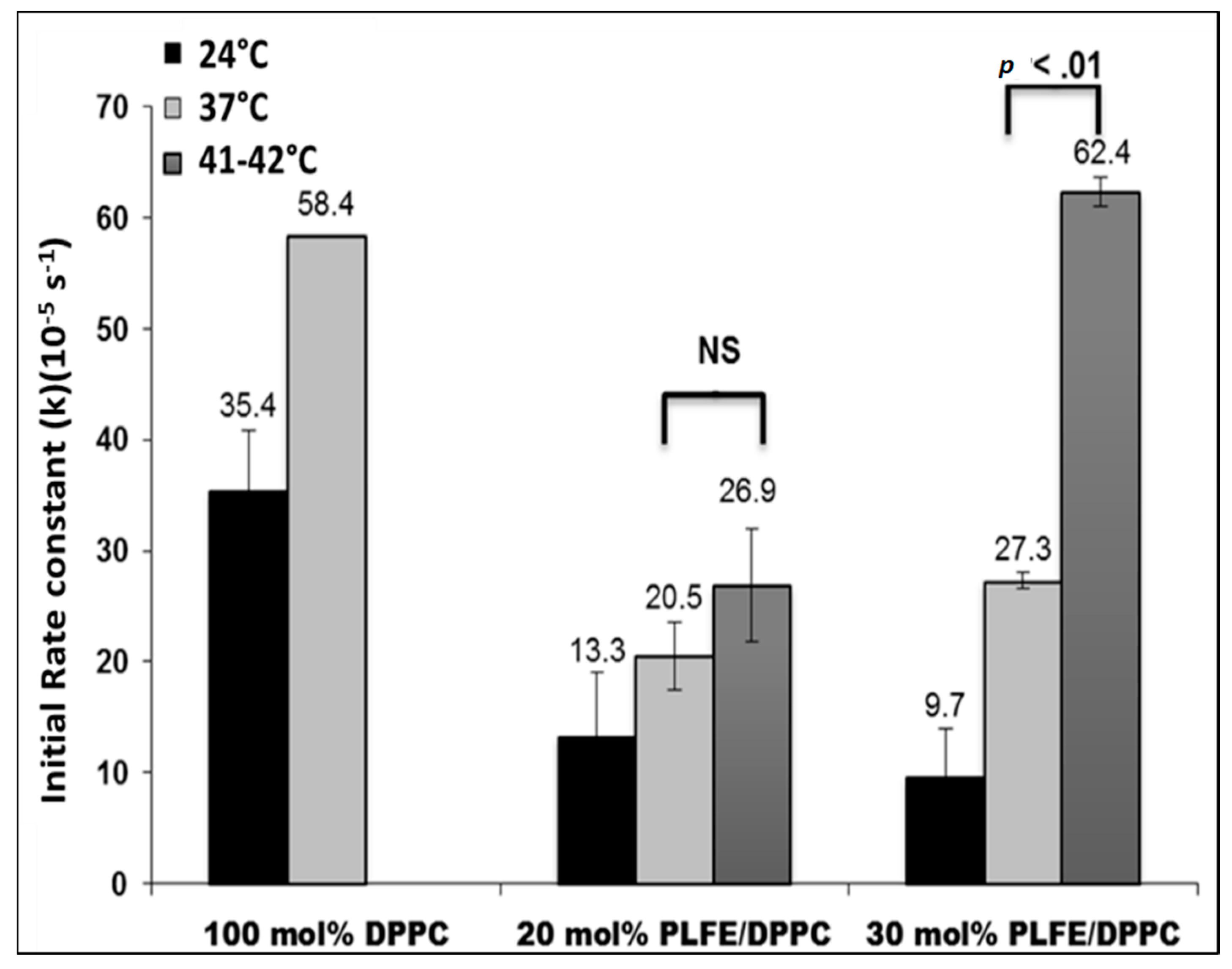
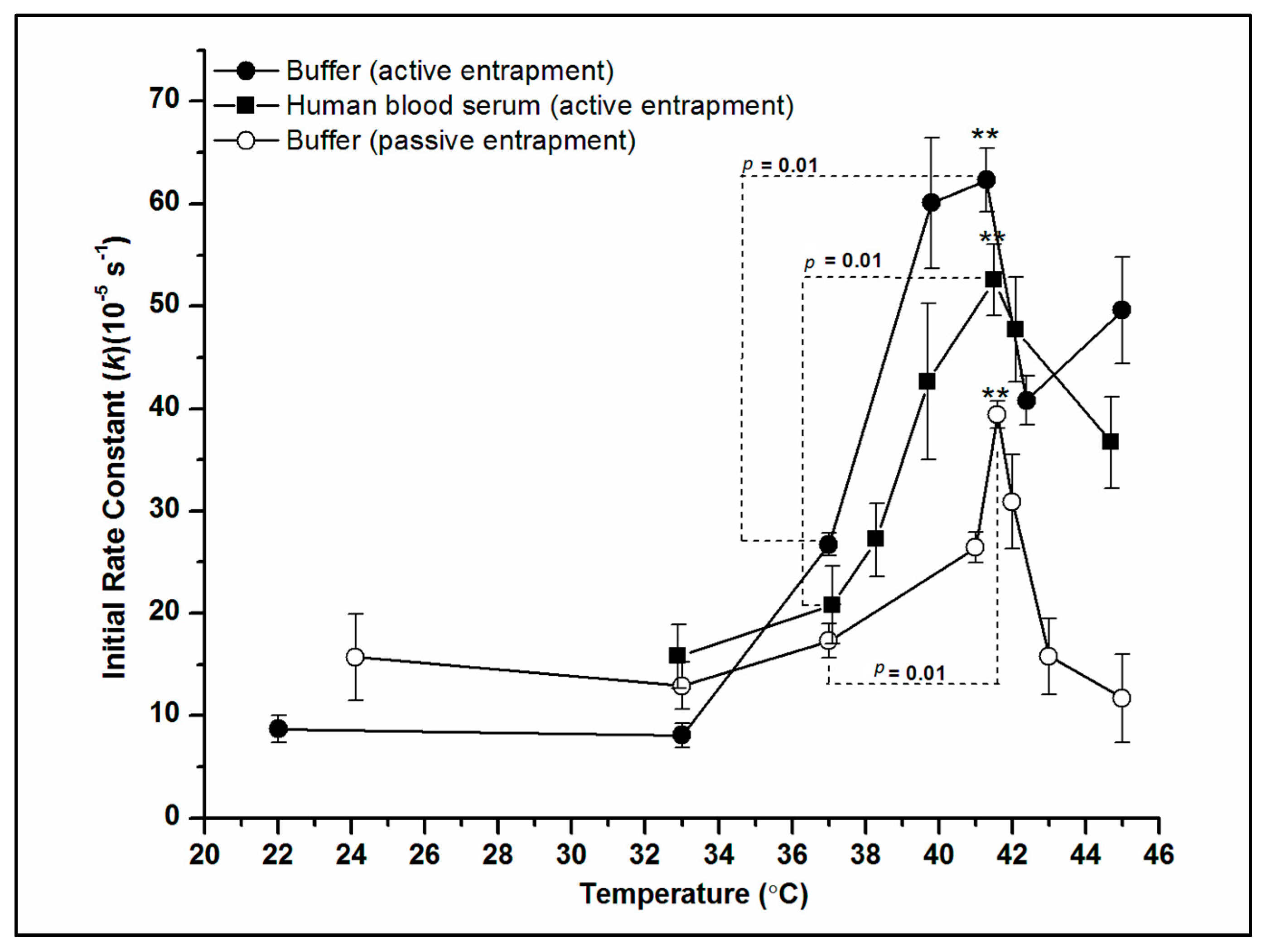
2.4. Effect of Temperature on DXO Release from PLFE/DPPC Hybrid Archaeosomes
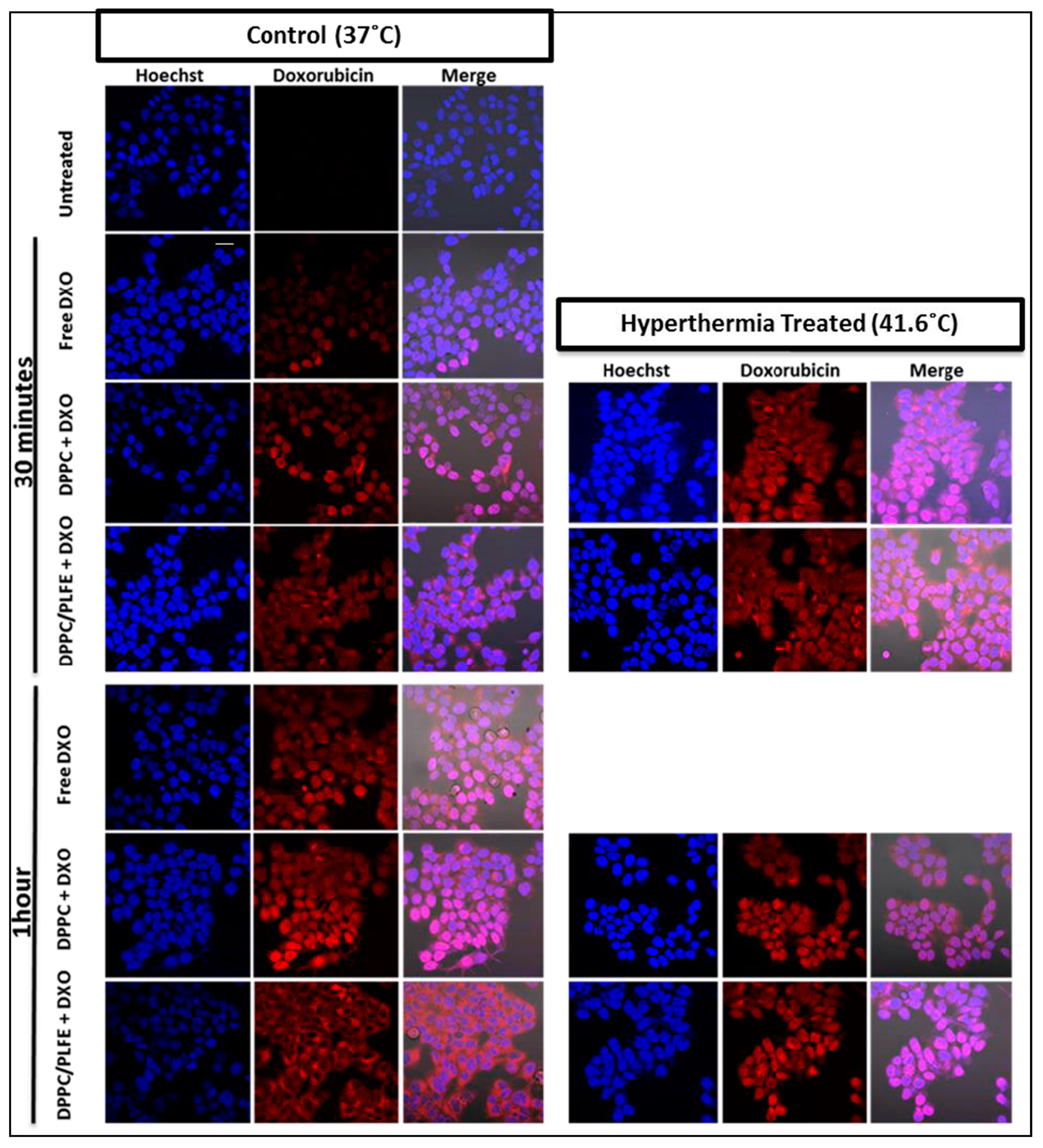
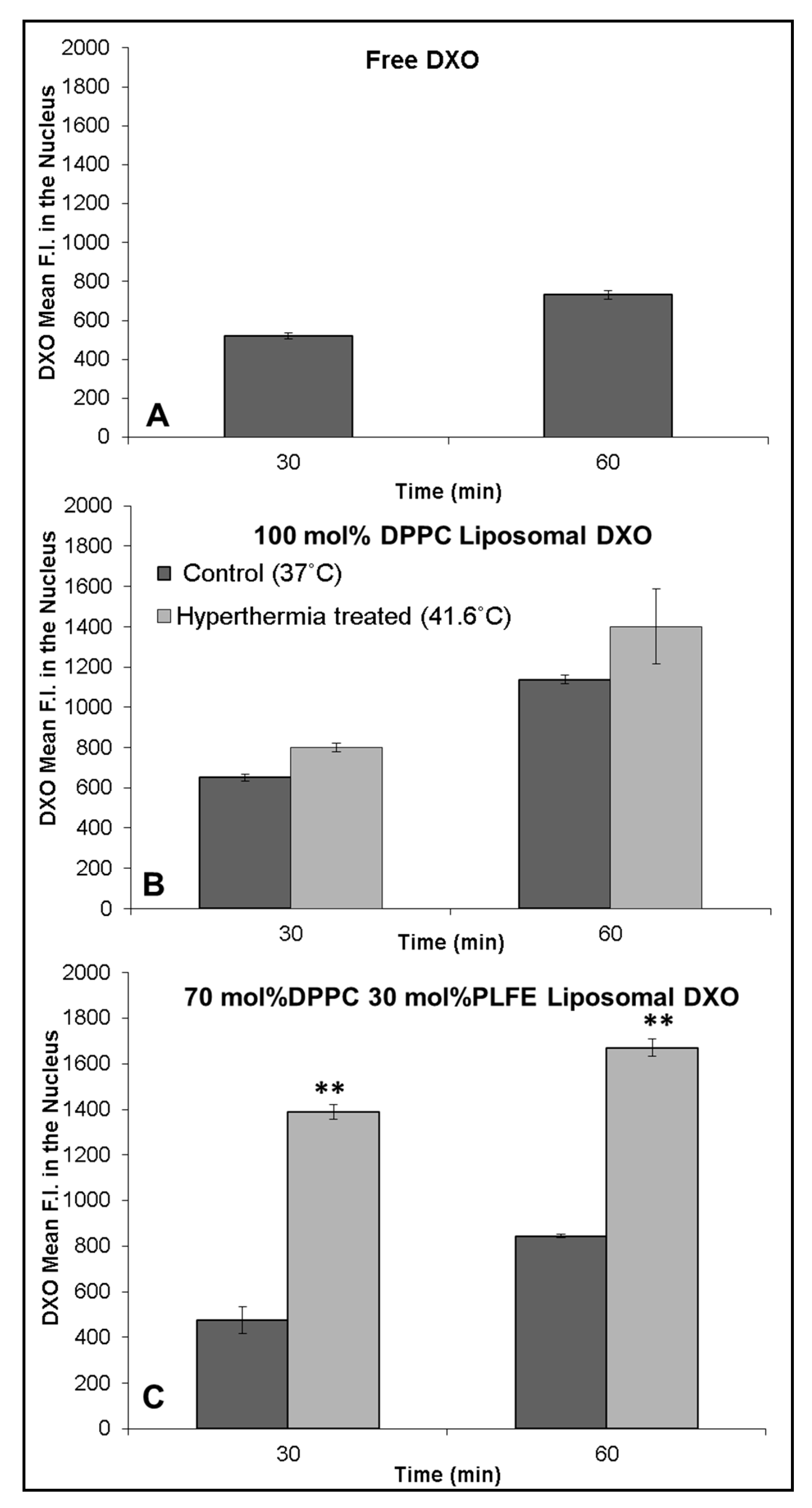
2.5. Interactions of PLFE/DPPC(3:7) Archaeosomal DXO with Live Cells
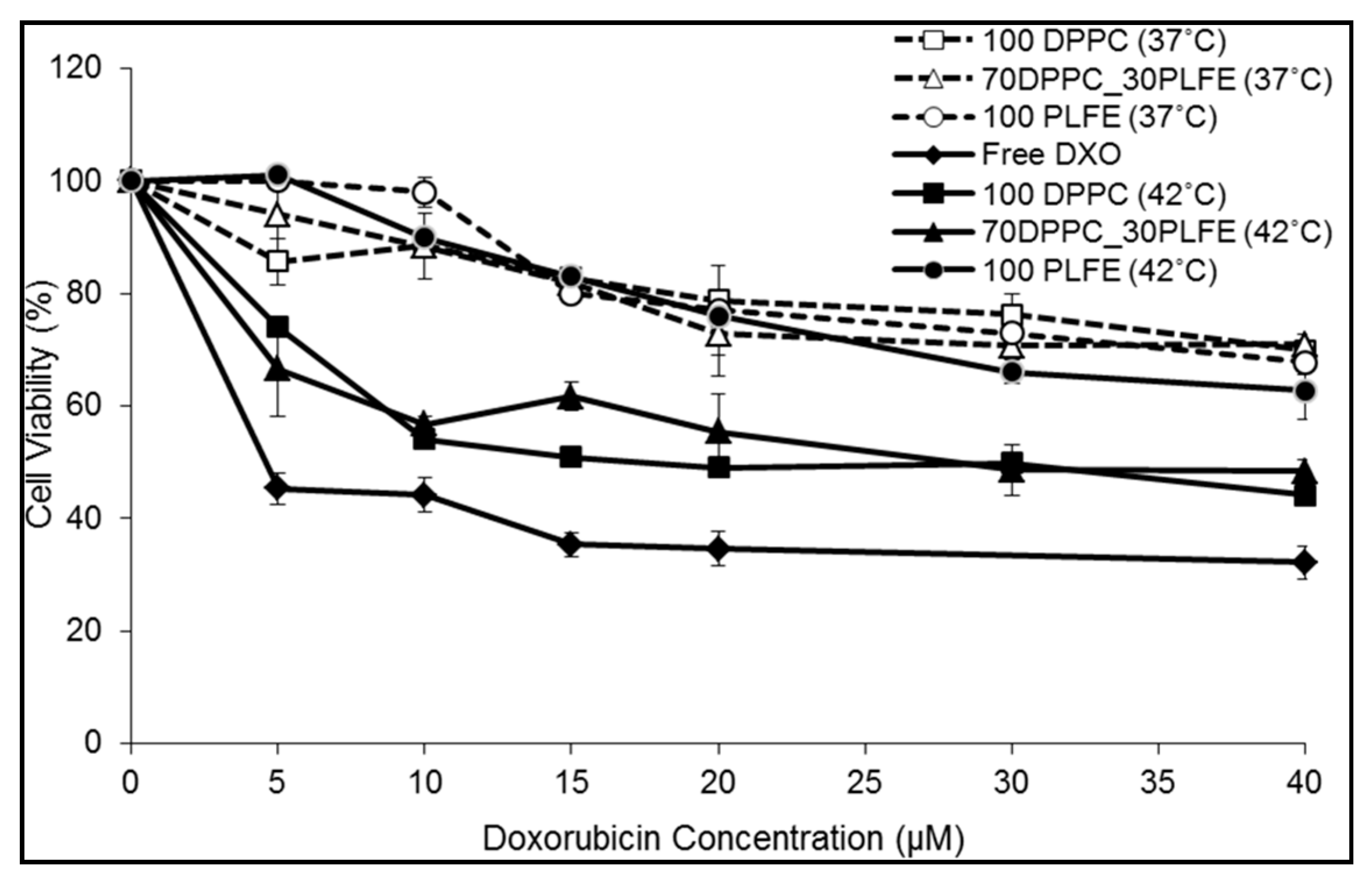
2.6. Cytotoxicity of PLFE/DPPC(3:7) Archaeosomes
3. Materials and Methods
3.1. Archaeal Cells and PLFE Lipids
3.2. Liposome Preparation
3.3. Particle Size and Zeta Potential Measurements
3.4. Doxorubicin Entrapment
3.5. Membrane Phase Transition as Assessed by Generalized Polarization of Laurdan Fluorescence
3.6. Membrane Packing Tightness as Revealed by DPH Anisotropy Decay
3.7. Drug Release
3.8. Mammalian Cell Growth
3.9. Confocal Microscopy
3.10. Cell Proliferation Assay
3.11. Statistical Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| DPH | 1,6-diphenyl-1,3,5-hexatriene |
| DPPC | 1,2-dipalmitoyl-sn-glycero-3-phosphocholine |
| DSPE-PEG-2000 | 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[methoxy(poly(ethylene glycol))-2000] |
| DXO | doxorubicin; |
| GDNT | glycerol dialkylcalditol tetraether |
| GDGT | glycerol dialkyglycerol tetraether |
| Laurdan | 6-lauroyl-1,2-dimethylamino-naphthalene |
| MLV | multilamellar vesicles |
| MSPC | monostearoylphosphatidylcholine |
| PLFE | polar lipid fraction E |
| TSL | thermo-sensitive liposomes |
References
- Tacar, O.; Sriamornsak, P.; Dass, C.R. Doxorubicin: An Update on Anticancer Molecular Action, Toxicity and Novel Drug Delivery Systems. J. Pharm. Pharmacol. 2012, 65, 157–170. [Google Scholar] [CrossRef] [PubMed]
- Carvalho, C.; Santos, R.X.; Cardoso, S.; Correia, S.; Oliveira, P.J.; Santos, M.S.; Moreira, P.I. Doxorubicin: The Good, the Bad and the Ugly Effect. Curr. Med. Chem. 2009, 16, 3267–3285. [Google Scholar] [CrossRef] [PubMed]
- Bulbake, U.; Doppalapudi, S.; Kommineni, N.; Khan, W. Liposomal Formulations in Clinical Use: An Updated Review. Pharmaceutics 2017, 9, 12. [Google Scholar] [CrossRef] [PubMed]
- Zylberberg, C.; Matosevic, S. Pharmaceutical Liposomal Drug Delivery: A Review of New Delivery Systems and a Look at the Regulatory Landscape. Drug Deliv. 2016, 23, 3319–3329. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tagami, T.; May, J.P.; Ernsting, M.J.; Li, S.-D. A Thermosensitive Liposome Prepared with a Cu2+ Gradient Demonstrates Improved Pharmacokinetics, Drug Delivery and Antitumor Efficacy. J. Control. Release 2012, 161, 142–149. [Google Scholar] [CrossRef]
- Head, M.; Jameson, M.B. The Development of the Tumor Vascular-Disrupting Agent ASA404 (vadimezan, DMXAA): Current Status and Future Opportunities. Expert Opin. Investig. Drugs 2010, 19, 295–304. [Google Scholar] [CrossRef] [PubMed]
- Siemann, D.W.; Horsman, M.R. Targeting the Tumor Vasculature: A Strategy to Improve Radiation Therapy. Expert Rev. Anticancer. Ther. 2004, 4, 321–327. [Google Scholar] [CrossRef]
- Uster, P.S.; Working, P.K.; Vaage, J. Pegylated Liposomal Doxorubicin (DOXIL, CAELYX) Distribution in Tumor Models Observed with Confocal Laser Scanning Microscopy. Int. J. Pharm. 1998, 162, 77–86. [Google Scholar] [CrossRef]
- Lyass, O.; Hubert, A.; Gabizon, A. Phase I Study of Doxil-Cisplatin Combination Chemotherapy in Patients with Advanced Malignancies. Clin. Cancer Res. 2001, 7, 3040–3046. [Google Scholar]
- Batist, G.; Ramakrishnan, G.; Rao, C.S.; Chandrasekharan, A.; Gutheil, J.; Guthrie, T.; Shah, P.; Khojasteh, A.; Nair, M.K.; Hoelzer, K.; et al. Reduced Cardiotoxicity and Preserved Antitumor Efficacy of Liposome-Encapsulated Doxorubicin and Cyclophosphamide Compared with Conventional Doxorubicin and Cyclophosphamide in a Randomized, Multicenter Trial of Metastatic Breast Cancer. J. Clin. Oncol. 2001, 19, 1444–1454. [Google Scholar] [CrossRef]
- Al-Jamal, W.T.; Al-Ahmady, Z.S.; Kostarelos, K. Pharmacokinetics & Tissue Distribution of Temperature-sensitive Liposomal Doxorubicin in Tumor-bearing Mice Triggered with Mild Hyperthermia. Biomaterials 2012, 33, 4608–4617. [Google Scholar] [CrossRef]
- Immordino, M.L.; Dosio, F.; Cattel, L. Stealth Liposomes: Review of the Basic Science, Rationale, and Clinical Applications, Existing and Potential. Int. J. Nanomed. 2006, 1, 297–315. [Google Scholar]
- Manzoor, A.; Lindner, L.; Landon, C.; Park, J.-Y.; Simnick, A.; Dreher, M.; Das, S.; Hanna, G.; Park, W.; Chilkoti, A.; et al. Overcoming Limitations in Nanoparticle Drug Delivery: Triggered, Intravascular Release to Improve Drug Penetration into Tumors. Cancer Res. 2012, 72, 5566–5575. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kong, G.; Braun, R.D.; Dewhirst, M.W. Characterization of the Effect of Hyperthermia on Nanoparticle Extravasation from Tumor Vasculature. Cancer Res. 2001, 61, 3027–3032. [Google Scholar]
- Gaber, M.H.; Hong, K.; Huang, S.K.; Papahadjopoulos, D. Thermosensitive Sterically Stabilized Liposomes: Formulation and in Vitro Studies on Mechanism of Doxorubicin Release by Bovine Serum and Human Plasma. Pharm. Res. 1995, 12, 1407–1416. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Hagen, T.L.M.T.; Hossann, M.; Süss, R.; Van Rhoon, G.C.; Eggermont, A.M.; Haemmerich, D.; Koning, G.A. Mild Hyperthermia Triggered Doxorubicin Release from Optimized Stealth Thermosensitive Liposomes Improves Intra-Tumoral Drug Delivery and Efficacy. J. Control. Release 2013, 168, 142–150. [Google Scholar] [CrossRef]
- Dreher, M.R.; Liu, W.; Michelich, C.R.; Dewhirst, M.W.; Yuan, F.; Chilkoti, A. Tumor Vascular Permeability, Accumulation, and Penetration of Macromolecular Drug Carriers. J. Natl. Cancer Inst. 2006, 98, 335–344. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; He, C.-Q.; Lin, A.-H.; Gu, W.; Chen, Z.-P.; Li, W.; Cai, B.-C. Thermosensitive Liposomes with Higher Phase Transition Temperature for Targeted Drug Delivery to Tumor. Int. J. Pharm. 2014, 475, 408–415. [Google Scholar] [CrossRef]
- Chiu, G.N.; Abraham, S.A.; Ickenstein, L.M.; Ng, R.; Karlsson, G.; Edwards, K.; Wasan, E.K.; Bally, M.B. Encapsulation of Doxorubicin into Thermosensitive Liposomes via Complexation with the Transition Metal Manganese. J. Control. Release 2005, 104, 271–288. [Google Scholar] [CrossRef]
- Hossann, M.; Kneidl, B.; Peller, M.; Lindner, L.H.; Winter, G. Thermosensitive Liposomal Drug Delivery Systems: State of the Art Review. Int. J. Nanomed. 2014, 9, 4387. [Google Scholar] [CrossRef] [Green Version]
- Ta, T.; Porter, T.M. Thermosensitive Liposomes for Localized Delivery and Triggered Release of Chemotherapy. J. Control. Release 2013, 169, 112–125. [Google Scholar] [CrossRef] [Green Version]
- Lencioni, R.; Cioni, D. RFA Plus Lyso-thermosensitive Liposomal Doxorubicin: In Search of the Optimal Approach to Cure Intermediate-size Hepatocellular Carcinoma. Hepatic Oncol. 2016, 3, 193–200. [Google Scholar] [CrossRef]
- Lyon, P.C.; Griffiths, L.F.; Lee, J.; Chung, D.; Carlisle, R.; Wu, F.; Middleton, M.R.; Gleeson, F.V.; Coussios, C.C. Clinical Trial Protocol for TARDOX: A Phase I Study to Investigate the Feasibility of Targeted Release of Lyso-thermosensitive Liposomal Doxorubicin (ThermoDox®) Using Focused Ultrasound in Patients with Liver Tumours. J. Ther. Ultrasound 2017, 5, 1–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tak, W.Y.; Lin, S.-M.; Wang, Y.; Zheng, J.; Vecchione, A.; Park, S.Y.; Chen, M.H.; Wong, S.; Xu, R.; Peng, C.-Y.; et al. Phase III HEAT Study Adding Lyso-Thermosensitive Liposomal Doxorubicin to Radiofrequency Ablation in Patients with Unresectable Hepatocellular Carcinoma Lesions. Clin. Cancer Res. 2017, 24, 73–83. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gray, M.D.; Lyon, P.C.; Mannaris, C.; Folkes, L.K.; Stratford, M.; Campo, L.; Chung, D.Y.F.; Scott, S.; Anderson, M.; Goldin, R.D.; et al. Focused Ultrasound Hyperthermia for Targeted Drug Release from Thermosensitive Liposomes: Results from a Phase I Trial. Radiology 2019, 291, 232–238. [Google Scholar] [CrossRef]
- Park, S.M.; Kim, M.S.; Park, S.-J.; Park, E.S.; Choi, K.-S.; Kim, Y.-S.; Kim, H.R. Novel Temperature-triggered Liposome with High Stability: Formulation, in vitro Evaluation, and in vivo Study Combined with High-intensity Focused Ultrasound (HIFU). J. Control. Release 2013, 170, 373–379. [Google Scholar] [CrossRef]
- Zagar, T.M.; Vujaskovic, Z.; Formenti, S.; Rugo, H.; Muggia, F.; O’Connor, B.; Myerson, R.; Stauffer, P.; Hsu, I.-C.; Diederich, C.; et al. Two Phase I Dose-escalation/pharmacokinetics Studies of Low Temperature Liposomal Doxorubicin (LTLD) and Mild Local Hyperthermia in Heavily Pretreated Patients with Local Regionally RecurrentBreast Cancer. Int. J. Hyperth. 2014, 30, 285–294. [Google Scholar] [CrossRef]
- Viglianti, B.L.; Dewhirst, M.W.; Boruta, R.; Park, J.-Y.; Landon, C.; Fontanella, A.N.; Guo, J.; Manzoor, A.; Hofmann, C.L.; Palmer, G.M. Systemic Anti-tumour Effects of Local Thermally Sensitive Liposome Therapy. Int. J. Hyperth. 2014, 30, 385–392. [Google Scholar] [CrossRef]
- Sandström, M.; Ickenstein, L.; Mayer, L.; Edwards, K. Effects of Lipid Segregation and Lysolipid Dissociation on Drug Release from Thermosensitive Liposomes. J. Control. Release 2005, 107, 131–142. [Google Scholar] [CrossRef]
- Yeh, M.-K.; Chang, H.-I.; Cheng, M.-Y. Clinical Development of Liposome Based Drugs: Formulation, Characterization, and Therapeutic Efficacy. Int. J. Nanomed. 2011, 7, 49–60. [Google Scholar] [CrossRef] [Green Version]
- Anyarambhatla, G.R.; Needham, D. Enhancement of the Phase Transition Permeability of DPPC Liposomes by Incorporation of MPPC: A New Temperature-Sensitive Liposome for Use with Mild Hyperthermia. J. Liposome Res. 1999, 9, 491–506. [Google Scholar] [CrossRef]
- Banno, B.; Ickenstein, L.M.; Chiu, G.N.; Bally, M.B.; Thewalt, J.; Brief, E.; Wasan, E.K. The Functional Roles of Poly(ethylene glycol)-lipid and Lysolipid in the Drug Retention and Release from Lysolipid-Containing Thermosensitive Liposomes in vitro and in vivo. J. Pharm. Sci. 2010, 99, 2295–2308. [Google Scholar] [CrossRef]
- Beck, J.G.; Mathieu, D.; Loudet, C.; Buchoux, S.; Dufourc, E.J. Plant Sterols in “Rafts”: A Better Way to Regulate Membrane Thermal Shocks. FASEB J. 2007, 21, 1714–1723. [Google Scholar] [CrossRef] [Green Version]
- May, J.P.; Li, S.-D. Thermosensitive Liposomes in Cancer Therapy. Recent Patents Biomed. Eng. 2012, 5, 148–158. [Google Scholar] [CrossRef]
- Chang, E.L.; Lo, S.L. Extraction and Purification of Tetraether Lipids from Sulfolobus acidocaldarius. In Protocols for Archaebacterial Research; Fleischmann, E.M., Place, A.R., Robb, R.T., Schreier, H.J., Eds.; Maryland Biotechnology Institute: Baltimore, MD, USA, 1991; pp. 2.3.1–2.3.14. [Google Scholar]
- Lo, S.-L.; Chang, E. Purification and Characterization of a Liposomal-forming Tetraether Lipid Fraction. Biochem. Biophys. Res. Commun. 1990, 167, 238–243. [Google Scholar] [CrossRef]
- Komatsu, H.; Chong, P.L.-G. Low Permeability of Liposomal Membranes Composed of Bipolar Tetraether Lipids from Thermoacidophilic Archaebacterium Sulfolobus acidocaldarius. Biochemistry 1998, 37, 107–115. [Google Scholar] [CrossRef] [PubMed]
- Chang, E. Unusual Thermal Stability of Liposomes Made from Bipolar Tetraether Lipids. Biochem. Biophys. Res. Commun. 1994, 202, 673–679. [Google Scholar] [CrossRef] [PubMed]
- Mathai, J.C.; Sprott, G.D.; Zeidel, M.L. Molecular Mechanisms of Water and Solute Transport across Archaebacterial Lipid Membranes. J. Biol. Chem. 2001, 276, 27266–27271. [Google Scholar] [CrossRef] [Green Version]
- Chong, P.L.-G.; Sulc, M.; Winter, R. Compressibilities and Volume Fluctuations of Archaeal Tetraether Liposomes. Biophys. J. 2010, 99, 3319–3326. [Google Scholar] [CrossRef]
- Zhai, Y.; Chong, P.L.-G.; Taylor, L.J.-A.; Erlkamp, M.; Grobelny, S.; Czeslik, C.; Watkins, E.; Winter, R. Physical Properties of Archaeal Tetraether Lipid Membranes as Revealed by Differential Scanning and Pressure Perturbation Calorimetry, Molecular Acoustics, and Neutron Reflectometry: Effects of Pressure and Cell Growth Temperature. Langmuir 2012, 28, 5211–5217. [Google Scholar] [CrossRef]
- Brown, D.A.; Venegas, B.; Cooke, P.H.; English, V.; Chong, P.L.-G. Bipolar Tetraether Archaeosomes Exhibit Unusual Stability against Autoclaving as Studied by Dynamic Light Scattering and Electron Microscopy. Chem. Phys. Lipids 2009, 159, 95–103. [Google Scholar] [CrossRef]
- Kanichay, R.; Boni, L.T.; Cooke, P.H.; Khan, T.K.; Chong, P.L.-G. Calcium-induced Aggregation of Archaeal Bipolar Tetraether Liposomes Derived from the Thermoacidophilic Archaeon Sulfolobus acidocaldarius. Archaea 2003, 1, 175–183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Z.; Chen, J.; Sun, W.; Xu, Y. Investigation of Archaeosomes as Carriers for Oral Delivery of Peptides. Biochem. Biophys. Res. Commun. 2010, 394, 412–417. [Google Scholar] [CrossRef]
- Chong, P.L.-G.; Zein, M.; Khan, T.K.; Winter, R. Structure and Conformation of Bipolar Tetraether Lipid Membranes Derived from Thermoacidophilic Archaeon Sulfolobus acidocaldarius as Revealed by Small-Angle X-Ray Scattering and High Pressure FT-IR Spectroscopy. J. Phys. Chem. 2003, 107, 8694–8700. [Google Scholar] [CrossRef]
- Chong, P.L.-G.; Ravindra, R.; Khurana, M.; English, V.; Winter, R. Pressure Perturbation and Differential Scanning Calorimetric Studies of Bipolar Tetraether Liposomes Derived from the Thermoacidophilic Archaeon Sulfolobus acidocaldarius. Biophys. J. 2005, 89, 1841–1849. [Google Scholar] [CrossRef] [Green Version]
- Bagatolli, L.; Gratton, E.; Khan, T.K.; Chong, P.L.-G. Two-Photon Fluorescence Microscopy Studies of Bipolar Tetraether Giant Liposomes from Thermoacidophilic Archaebacteria Sulfolobus acidocaldarius. Biophys. J. 2000, 79, 416–425. [Google Scholar] [CrossRef] [Green Version]
- Sprott, G.; Patel, G.B.; Krishnan, L. Archaeobacterial Ether Lipid Liposomes as Vaccine Adjuvants. Methods Enzymol. 2003, 373, 155–172. [Google Scholar] [CrossRef] [PubMed]
- Gabriel, J.L.; Chong, P.L.G. Molecular Modeling of Archaebacterial Bipolar Tetraether Lipid Membranes. Chem. Phys. Lipids 2000, 105, 193–200. [Google Scholar] [CrossRef]
- Elferink, M.G.; De Wit, J.G.; Driessen, A.J.M.; Konings, W.N. Stability and Proton-Permeability of Liposomes Composed of Archaeal Tetraether Lipids. Biochim. Biophys. Acta (BBA)-Biomembr. 1994, 1193, 247–254. [Google Scholar] [CrossRef] [Green Version]
- Chong, P.L.-G.; Ayesa, U.; Daswani, V.P.; Hur, E.C. On Physical Properties of Tetraether Lipid Membranes: Effects of Cyclopentane Rings. Archaea 2012, 2012, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Kaur, G.; Garg, T.; Rath, G.; Goyal, A.K. Archaeosomes: An Excellent Carrier for Drug and Cell Delivery. Drug Deliv. 2015, 23, 2497–2512. [Google Scholar] [CrossRef]
- Benvegnu, T.; Réthoré, G.; Brard, M.; Richter, W.; Plusquellec, D. Archaeosomes Based on Novel Synthetic Tetraether-type Lipids for the Development of Oral Delivery Systems. Chem. Commun. 2005, 44, 5536. [Google Scholar] [CrossRef]
- Patel, G.B.; Agnew, B.J.; Deschatelets, L.; Fleming, L.; Sprott, G. In vitro Assessment of Archaeosome Stability for Developing Oral Delivery Systems. Int. J. Pharm. 2000, 194, 39–49. [Google Scholar] [CrossRef]
- Laouini, A.; Jaafar-Maalej, C.; Limayem-Blouza, I.; Sfar, S.; Charcosset, C.; Fessi, H. Preparation, Characterization and Applications of Liposomes: State of the Art. J. Colloid Sci. Biotechnol. 2012, 1, 147–168. [Google Scholar] [CrossRef]
- Hunter, R.J.; Midmore, B.R.; Zhang, H. Zeta Potential of Highly Charged Thin Double-Layer Systems. J. Colloid Interface Sci. 2001, 237, 147–149. [Google Scholar] [CrossRef]
- Sugár, I.P. A Generalization of the Shell Theorem. Electric Potential of Charged Spheres and Charged Vesicles Surrounded by Electrolyte. AIMS Biophys. 2020, 7, 76–89. [Google Scholar] [CrossRef]
- Makino, K.; Yamada, T.; Kimura, M.; Oka, T.; Ohshima, H.; Kondo, T. Temperature- and Ionic Strength-induced Conformational Changes in the Lipid Head Group Region of Liposomes as Suggested by Zeta Potential Data. Biophys. Chem. 1991, 41, 175–183. [Google Scholar] [CrossRef]
- Lin, W.-C.; Blanchette, C.D.; Ratto, T.V.; Longo, M.L. Lipid Asymmetry in DLPC/DSPC-Supported Lipid Bilayers: A Combined AFM and Fluorescence Microscopy Study. Biophys. J. 2006, 90, 228–237. [Google Scholar] [CrossRef] [Green Version]
- Hernández-Caselles, T.; Villalaín, J.; Gómez-Fernández, J.C. Influence of Liposome Charge and Composition on their Interaction with Human Blood Serum Proteins. Mol. Cell. Biochem. 1993, 120, 119–126. [Google Scholar] [CrossRef]
- Bonté, F.; Juliano, R. Interactions of Liposomes with Serum Proteins. Chem. Phys. Lipids 1986, 40, 359–372. [Google Scholar] [CrossRef]
- Zhaorigetu, S.; Rodriguez-Aguayo, C.; Sood, A.K.; Lopez-Berestein, G.; Walton, B.L. Delivery of Negatively Charged Liposomes into the Atherosclerotic Plague of Apolipoprotein E-Deficient Mouse Aortic Tissue. J. Liposome Res. 2014, 24, 182–190. [Google Scholar] [CrossRef] [Green Version]
- Gabizon, A.; Papahadjopoulos, D. Liposome Formulations with Prolonged Circulation Time in Blood and Enhanced Uptake by Tumors. Proc. Natl. Acad. Sci. USA 1988, 85, 6949–6953. [Google Scholar] [CrossRef] [Green Version]
- Maruyama, K.; Unezaki, S.; Takahashi, N.; Iwatsuru, M. Enhanced Delivery of Doxorubicin to Tumor by Long-circulating Thermosensitive Liposomes and Local Hyperthermia. Biochim. Biophys. Acta (BBA)-Biomembr. 1993, 1149, 209–216. [Google Scholar] [CrossRef]
- Patel, G.B.; Sprott, G.D. Archaeobacterial Ether Lipid Liposomes (Archaeosomes) as Novel Vaccine and Drug Delivery Systems. Crit. Rev. Biotechnol. 1999, 19, 317–357. [Google Scholar] [CrossRef]
- Weber, G. Limited Rotational Motion: Recognition by Differential Phase Fluorometry. Acta. Phys. Pol. 1978, A54, 859–865. [Google Scholar]
- Lakowicz, J.R.; Prendergast, F.G.; Hogen, D. Differential Polarized Phase Fluorometric Investigations of Diphenylhexatriene in Lipid Bilayers. Quantitation of Hindered Depolarizing Rotations. Biochemistry 1979, 18, 508–519. [Google Scholar] [CrossRef]
- Chen, L.A.; Dale, R.E.; Roth, S.; Brand, L. Nanosecond Time-dependent Fluorescence Depolarization of Diphenylhexatriene in Dimyristoyllecithin Vesicles and the Determination of “Microviscosity”. J. Biol. Chem. 1977, 252, 2163–2169. [Google Scholar]
- Repakova, J.; Holopainen, J.M.; Morrow, M.R.; McDonald, M.C.; Čapková, P.; Vattulainen, I. Influence of DPH on the Structure and Dynamics of a DPPC Bilayer. Biophys. J. 2005, 88, 3398–3410. [Google Scholar] [CrossRef] [Green Version]
- Jähnig, F. Structural Order of Lipids and Proteins in Membranes: Evaluation of Fluorescence Anisotropy Data. Proc. Natl. Acad. Sci. USA 1979, 76, 6361–6365. [Google Scholar] [CrossRef] [Green Version]
- Kao, Y.L.; Chong, P.L.; Huang, C.H. Dynamic Motions of 1,6-Diphenyl-1,3,5-hexatriene in Interdigitated C(18):C(10)phosphatidylcholine Bilayers. Biophys. J. 1990, 58, 947–956. [Google Scholar] [CrossRef] [Green Version]
- Lakowicz, J.R.; Prendergast, F.G. Detection of Hindered Rotations of 1,6-Diphenyl-1,3,5-Hexatriene in Lipid Bilayers by Differential Polarized Phase Fluorometry. Biophys. J. 1978, 24, 213–231. [Google Scholar] [CrossRef] [Green Version]
- Chong, P.L.-G. Archaebacterial Bipolar Tetraether Lipids: Physico-chemical and Membrane Properties. Chem. Phys. Lipids 2010, 163, 253–265. [Google Scholar] [CrossRef] [PubMed]
- Dufourc, E.J. Sterols and Membrane Dynamics. J. Chem. Biol. 2008, 1, 63–77. [Google Scholar] [CrossRef] [Green Version]
- Repáková, J.; Čapková, P.; Holopainen, J.M.; Vattulainen, I. Distribution, Orientation, and Dynamics of DPH Probes in DPPC Bilayer. J. Phys. Chem. B 2004, 108, 13438–13448. [Google Scholar] [CrossRef]
- Falck, E.; Patra, M.; Karttunen, M.; Hyvönen, M.T.; Vattulainen, I. Impact of Cholesterol on Voids in Phospholipid Membranes. J. Chem. Phys. 2004, 121, 12676–12689. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lo, S.L.; Montague, C.E.; Chang, E.L. Purification of Glycerol Dialkyl Nonitol Tetraether from Sulfolobus acidocaldarius. J. Lipid Res. 1989, 30, 944–949. [Google Scholar]
- Bartlett, G.R. Phosphorus Assay in Column Chromatography. J. Biol. Chem. 1959, 234, 466–468. [Google Scholar]
- Haran, G.; Cohen, R.; Bar, L.K.; Barenholz, Y. Transmembrane Ammonium Sulfate Gradients in Liposomes Produce Efficient and Stable Entrapment of Amphipathic Weak Bases. Biochim. Biophys. Acta (BBA)-Biomembr. 1993, 1151, 201–215. [Google Scholar] [CrossRef]
- Gabizon, A.; Shmeeda, H.; Barenholz, Y. Pharmacokinetics of Pegylated Liposomal Doxorubicin. Clin. Pharmacokinet. 2003, 42, 419–436. [Google Scholar] [CrossRef]
- Li, X.; Hirsh, D.J.; Cabral-Lilly, D.; Zirkel, A.; Gruner, S.M.; Janoff, A.S.; Perkins, W.R. Doxorubicin Physical State in Solution and Inside Liposomes Loaded via a pH Gradient. Biochim. Biophys. Acta (BBA)-Biomembr. 1998, 1415, 23–40. [Google Scholar] [CrossRef] [Green Version]
- Etrych, T.; Mrkvan, T.; Chytil, P.; Koňák, C.; Říhová, B.; Ulbrich, K. N-(2-hydroxypropyl) methacrylamide-based Polymer Conjugates with pH-controlled Activation of Doxorubicin. I. New Synthesis, Physicochemical Characterization and Preliminary Biological Evaluation. J. Appl. Polym. Sci. 2008, 109, 3050–3060. [Google Scholar] [CrossRef]
- Parasassi, T.; De Stasio, G.; Ravagnan, G.; Rusch, R.; Gratton, E. Quantitation of Lipid Phases in Phospholipid Vesicles by the Generalized Polarization of Laurdan Fluorescence. Biophys. J. 1991, 60, 179–189. [Google Scholar] [CrossRef] [Green Version]
- Bonanno, A.P.; Ii, R.C.B.; Chong, P.L.-G. Sulfolobus acidocaldarius Microvesicles Exhibit Unusually Tight Packing Properties as Revealed by Optical Spectroscopy. Int. J. Mol. Sci. 2019, 20, 5308. [Google Scholar] [CrossRef] [Green Version]
- Venegas, B.; Zhu, W.; Haloupek, N.B.; Lee, J.; Zellhart, E.; Sugar, I.P.; Kiani, M.; Chong, P.L.-G. Cholesterol Supelattice Modulates Combretastatin A4 Disodium Phosphate (CA4P) Release from Liposomes and CA4P Cytotoxicity on Mammary Cancer Cells. Biophys. J. 2012, 102, 2086–2094. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Freisleben, H.-J.; Bormann, J.; Litzinger, D.C.; Lehr, F.; Rudolph, P.; Schatton, W.; Huang, L. Toxicity and Biodistribution of Liposomes of the Main Phospholipid from the Archaebacterium Thermoplasma Acidophilum in Mice. J. Liposome Res. 1995, 5, 215–223. [Google Scholar] [CrossRef]
- Freisleben, H.-J. Tetraether Lipid Liposomes. In Membrane Structure in Disease and Drug Therapy; Zimmer, G., Ed.; Marcel Dekker Inc.: New York, NY, USA, 2000; pp. 127–152. [Google Scholar]
- Whitfield, D.M.; Eichler, E.E.; Sprott, G.D. Synthesis of Archaeal Glycolipid Adjuvants-What is the Optimum Number of Sugars? Carbohydr. Res. 2008, 343, 2349–2360. [Google Scholar] [CrossRef]
- Daswani, V.P.; Ayesa, U.; Venegas, B.; Chong, P.L.-G. Concentration-Induced J-Aggregate Formation Causes a Biphasic Change in the Release of trans-Combretastatin A4 Disodium Phosphate from Archaeosomes and the Subsequent Cytotoxicity on Mammary Cancer Cells. Mol. Pharm. 2015, 12, 3724–3734. [Google Scholar] [CrossRef]
- Lombardo, D.; Calandra, P.; Barreca, D.; Magazù, S.; Kiselev, M.A. Soft Interaction in Liposome Nanocarriers for Therapeutic Drug Delivery. Nanomaterials 2016, 6, 125. [Google Scholar] [CrossRef]
- Jeworrek, C.; Evers, F.; Erlkamp, M.; Grobelny, S.; Tolan, M.; Chong, P.L.-G.; Winter, R. Structure and Phase Behavior of Archaeal Lipid Monolayers. Langmuir 2011, 27, 13113–13121. [Google Scholar] [CrossRef] [PubMed]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ayesa, U.; Chong, P.L.-G. Polar Lipid Fraction E from Sulfolobus acidocaldarius and Dipalmitoylphosphatidylcholine Can Form Stable yet Thermo-Sensitive Tetraether/Diester Hybrid Archaeosomes with Controlled Release Capability. Int. J. Mol. Sci. 2020, 21, 8388. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21218388
Ayesa U, Chong PL-G. Polar Lipid Fraction E from Sulfolobus acidocaldarius and Dipalmitoylphosphatidylcholine Can Form Stable yet Thermo-Sensitive Tetraether/Diester Hybrid Archaeosomes with Controlled Release Capability. International Journal of Molecular Sciences. 2020; 21(21):8388. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21218388
Chicago/Turabian StyleAyesa, Umme, and Parkson Lee-Gau Chong. 2020. "Polar Lipid Fraction E from Sulfolobus acidocaldarius and Dipalmitoylphosphatidylcholine Can Form Stable yet Thermo-Sensitive Tetraether/Diester Hybrid Archaeosomes with Controlled Release Capability" International Journal of Molecular Sciences 21, no. 21: 8388. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21218388