Emerging Strategies to Combat β-Lactamase Producing ESKAPE Pathogens

 ,
,  , ,
, ,
Abstract
:1. Introduction
2. Classification of β-Lactam Antibiotics (BLA)
2.1. Penicillins
2.2. Cephalosporins
2.3. Monobactams
2.4. Carbapenems
3. β-Lactamases in Gram-Negative Bacteria
3.1. Origins
3.2. Classification
4. β-Lactamase Inhibitors (BLIs)
4.1. Well Documented BLIs
4.2. Newer BLIs
4.2.1. Ceftazidime-Avibactam (CAZ-AVI)
4.2.2. Ceftolozane/Tazobactam (CEF-TAZ)
4.2.3. Imipenem/Relebactam (IMI-REL) and Meropenem/Vaborbactam (MER-VAB)
4.2.4. Cefepime/Zidebactam (WCK 5222)
4.2.5. MBL Inhibitors (MBLi)
5. Alternative Approaches to Combat ESKAPE Pathogens
5.1. Antimicrobial Peptides (AMPs)
Resistance to AMPs
5.2. Metal Nanoparticles
5.3. Bacteriophages
5.4. CRISPR Cas—An Emergent Strategy in Controlling ESKAPE Pathogens
5.5. Vaccination
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Bush, K.; Bradford, P.A. β-Lactams and β-Lactamase Inhibitors: An Overview. Cold Spring Harb. Perspect. Med. 2016, 6, a025247. [Google Scholar] [CrossRef] [PubMed]
- Tipper, D.J.; Strominger, J.L. Mechanism of action of penicillins: A proposal based on their structural similarity to acyl-D-alanyl-D-alanine. Proc. Natl. Acad. Sci. USA 1965, 54, 1133–1141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frère, J.M.; Joris, B. Penicillin-sensitive enzymes in peptidoglycan biosynthesis. Crit. Rev. Microbiol. 1985, 11, 299–396. [Google Scholar] [CrossRef] [PubMed]
- Otero, L.H.; Rojas-Altuve, A.; Llarrull, L.I.; Carrasco-López, C.; Kumarasiri, M.; Lastochkin, E.; Fishovitz, J.; Dawley, M.; Hesek, D.; Lee, M.; et al. How allosteric control of Staphylococcus aureus penicillin binding protein 2a enables methicillin resistance and physiological function. Proc. Natl. Acad. Sci. USA 2013, 110, 16808–16813. [Google Scholar] [CrossRef] [Green Version]
- Gonzales, P.R.; Pesesky, M.W.; Bouley, R.; Ballard, A.; Biddy, B.A.; Suckow, M.A.; Wolter, W.R.; Schroeder, V.A.; Burnham, C.A.D.; Mobashery, S.; et al. Synergistic, collaterally sensitive β-lactam combinations suppress resistance in MRSA. Nat. Chem. Biol. 2015, 11, 855–861. [Google Scholar] [CrossRef]
- Sykes, J.E.; Papich, M.G. Antibacterial Drugs. Canine Feline Infect. Dis. 2013, 66–86. [Google Scholar] [CrossRef]
- Silhavy, T.J.; Kahne, D.; Walker, S. The bacterial cell envelope. Cold Spring Harb. Perspect. Biol. 2010, 2, a000414. [Google Scholar] [CrossRef]
- May, K.L.; Grabowicz, M. The bacterial outer membrane is an evolving antibiotic barrier. Proc. Natl. Acad. Sci. USA 2018, 115, 8852–8854. [Google Scholar] [CrossRef]
- Miller, S.I. Antibiotic Resistance and Regulation of the Gram-Negative Bacterial Outer Membrane Barrier by Host Innate Immune Molecules. MBio 2016, 7, e01541-16. [Google Scholar] [CrossRef] [Green Version]
- Vergalli, J.; Bodrenko, I.V.; Masi, M.; Moynié, L.; Acosta-Gutiérrez, S.; Naismith, J.H.; Davin-Regli, A.; Ceccarelli, M.; van den Berg, B.; Winterhalter, M.; et al. Porins and small-molecule translocation across the outer membrane of Gram-negative bacteria. Nat. Rev. Microbiol. 2020, 18, 164–176. [Google Scholar] [CrossRef]
- Mulani, M.S.; Kamble, E.E.; Kumkar, S.N.; Tawre, M.S.; Pardesi, K.R. Emerging Strategies to Combat ESKAPE Pathogens in the Era of Antimicrobial Resistance: A Review. Front. Microbiol. 2019, 10, 539. [Google Scholar] [CrossRef] [PubMed]
- Drawz, S.M.; Bonomo, R.A. Three decades of beta-lactamase inhibitors. Clin. Microbiol. Rev. 2010, 23, 160–201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ur Rahman, S.; Ali, T.; Ali, I.; Khan, N.A.; Han, B.; Gao, J. The Growing Genetic and Functional Diversity of Extended Spectrum Beta-Lactamases. Biomed Res. Int. 2018, 2018, 9519718. [Google Scholar] [CrossRef] [PubMed]
- Medeiros, A.A. beta-Lactamases: Quality and resistance. Clin. Microbiol. Infect. Off. Publ. Eur. Soc. Clin. Microbiol. Infect. Dis. 1997, 3, S2–S9. [Google Scholar]
- Bush, K. Past and Present Perspectives on β-Lactamases. Antimicrob. Agents Chemother. 2018, 62, e01076-18. [Google Scholar] [CrossRef] [Green Version]
- Thapa Shrestha, U.; Shrestha, S.; Adhikari, N.; Rijal, K.R.; Shrestha, B.; Adhikari, B.; Banjara, M.R.; Ghimire, P. Plasmid Profiling and Occurrence of β-Lactamase Enzymes in Multidrug-Resistant Uropathogenic Escherichia coli in Kathmandu, Nepal. Infect. Drug Resist. 2020, 13, 1905–1917. [Google Scholar] [CrossRef]
- Massova, I.; Mobashery, S. Kinship and diversification of bacterial penicillin-binding proteins and beta-lactamases. Antimicrob. Agents Chemother. 1998, 42, 1–17. [Google Scholar] [CrossRef] [Green Version]
- Georgopapadakou, N.H.; Liu, F.Y. Binding of beta-lactam antibiotics to penicillin-binding proteins of Staphylococcus aureus and Streptococcus faecalis: Relation to antibacterial activity. Antimicrob. Agents Chemother. 1980, 18, 834–836. [Google Scholar] [CrossRef] [Green Version]
- Miller, E.L. The penicillins: A review and update. J. Midwifery Women’s Health 2002, 47, 426–434. [Google Scholar] [CrossRef]
- Holten, K.B.; Onusko, E.M. Appropriate prescribing of oral beta-lactam antibiotics. Am. Fam. Physician 2000, 62, 611–620. [Google Scholar]
- Schito, G.C. The importance of the development of antibiotic resistance in Staphylococcus aureus. Clin. Microbiol. Infect. Off. Publ. Eur. Soc. Clin. Microbiol. Infect. Dis. 2006, 12, 3–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fishovitz, J.; Hermoso, J.A.; Chang, M.; Mobashery, S. Penicillin-binding protein 2a of methicillin-resistant Staphylococcus aureus. IUBMB Life 2014, 66, 572–577. [Google Scholar] [CrossRef] [Green Version]
- Balsalobre, L.; Blanco, A.; Alarcón, T. Beta-Lactams. In Antibiotic Drug Resistance; John Wiley & Sons, Ltd.: Hoboken, NJ, USA, 2019; pp. 57–72. ISBN 9781119282549. [Google Scholar]
- Gin, A.; Dilay, L.; Karlowsky, J.A.; Walkty, A.; Rubinstein, E.; Zhanel, G.G. Piperacillin-tazobactam: A beta-lactam/beta-lactamase inhibitor combination. Expert Rev. Anti. Infect. Ther. 2007, 5, 365–383. [Google Scholar] [CrossRef] [PubMed]
- Livermore, D.M.; Hope, R.; Mushtaq, S.; Warner, M. Orthodox and unorthodox clavulanate combinations against extended-spectrum beta-lactamase producers. Clin. Microbiol. Infect. 2008, 14, 189–193. [Google Scholar] [CrossRef] [Green Version]
- MacDougall, C. Penicillins, Cephalosporins, and other β-lactam antibiotics. In The Pharmacological Basis of Therapeutics; Brunton, L.L., Hilal-Dandan, R., Knollmannn, B.C., Eds.; McGraw-Hill Education: New York, NY, USA, 2017. [Google Scholar]
- Chaudhry, S.B.; Veve, M.P.; Wagner, J.L. Cephalosporins: A Focus on Side Chains and β-Lactam Cross-Reactivity. Pharmacy 2019, 7, 103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shahid, M.; Sobia, F.; Singh, A.; Malik, A.; Khan, H.M.; Jonas, D.; Hawkey, P.M. Beta-lactams and beta-lactamase-inhibitors in current- or potential-clinical practice: A comprehensive update. Crit. Rev. Microbiol. 2009, 35, 81–108. [Google Scholar] [CrossRef] [PubMed]
- Bratzler, D.W.; Dellinger, E.P.; Olsen, K.M.; Perl, T.M.; Auwaerter, P.G.; Bolon, M.K.; Fish, D.N.; Napolitano, L.M.; Sawyer, R.G.; Slain, D.; et al. Clinical practice guidelines for antimicrobial prophylaxis in surgery. Am. J. Health Pharm. AJHP Off. J. Am. Soc. Health Pharm. 2013, 70, 195–283. [Google Scholar] [CrossRef] [Green Version]
- Livermore, D.M. beta-Lactamases in laboratory and clinical resistance. Clin. Microbiol. Rev. 1995, 8, 557–584. [Google Scholar] [CrossRef]
- Wilson, W.; Taubert, K.A.; Gewitz, M.; Lockhart, P.B.; Baddour, L.M.; Levison, M.; Bolger, A.; Cabell, C.H.; Takahashi, M.; Baltimore, R.S.; et al. Prevention of infective endocarditis: Guidelines from the American Heart Association: A guideline from the American Heart Association Rheumatic Fever, Endocarditis, and Kawasaki Disease Committee, Council on Cardiovascular Disease in the Young, and the C. Circulation 2007, 116, 1736–1754. [Google Scholar] [CrossRef] [Green Version]
- Halperin, J.J.; Shapiro, E.D.; Logigian, E.; Belman, A.L.; Dotevall, L.; Wormser, G.P.; Krupp, L.; Gronseth, G.; Bever, C.T.J. Practice parameter: Treatment of nervous system Lyme disease (an evidence-based review): Report of the Quality Standards Subcommittee of the American Academy of Neurology. Neurology 2007, 69, 91–102. [Google Scholar] [CrossRef] [Green Version]
- Nicolle, L.E.; Gupta, K.; Bradley, S.F.; Colgan, R.; DeMuri, G.P.; Drekonja, D.; Eckert, L.O.; Geerlings, S.E.; Köves, B.; Hooton, T.M.; et al. Clinical Practice Guideline for the Management of Asymptomatic Bacteriuria: 2019 Update by the Infectious Diseases Society of America. Clin. Infect. Dis. 2019, 68, e83–e110. [Google Scholar] [CrossRef] [PubMed]
- Solomkin, J.S.; Mazuski, J.E.; Bradley, J.S.; Rodvold, K.A.; Goldstein, E.J.C.; Baron, E.J.; O’Neill, P.J.; Chow, A.W.; Dellinger, E.P.; Eachempati, S.R.; et al. Diagnosis and management of complicated intra-abdominal infection in adults and children: Guidelines by the Surgical Infection Society and the Infectious Diseases Society of America. Clin. Infect. Dis. 2010, 50, 133–164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Workowski, K.A.; Bolan, G.A. Sexually transmitted diseases treatment guidelines, 2015. MMWR. Recomm. Rep. Morb. Mortal. Wkly. Rep. Recomm. Rep. 2015, 64, 1–137. [Google Scholar]
- Nikaido, H.; Liu, W.; Rosenberg, E.Y. Outer membrane permeability and beta-lactamase stability of dipolar ionic cephalosporins containing methoxyimino substituents. Antimicrob. Agents Chemother. 1990, 34, 337–342. [Google Scholar] [CrossRef] [Green Version]
- Bellido, F.; Pechère, J.C.; Hancock, R.E. Novel method for measurement of outer membrane permeability to new beta-lactams in intact Enterobacter cloacae cells. Antimicrob. Agents Chemother. 1991, 35, 68–72. [Google Scholar] [CrossRef] [Green Version]
- Stevens, D.L.; Bisno, A.L.; Chambers, H.F.; Dellinger, E.P.; Goldstein, E.J.C.; Gorbach, S.L.; Hirschmann, J.V.; Kaplan, S.L.; Montoya, J.G.; Wade, J.C. Practice guidelines for the diagnosis and management of skin and soft tissue infections: 2014 update by the Infectious Diseases Society of America. Clin. Infect. Dis. 2014, 59, e10–e52. [Google Scholar] [CrossRef] [Green Version]
- Karlowsky, J.A.; Adam, H.J.; Decorby, M.R.; Lagacé-Wiens, P.R.S.; Hoban, D.J.; Zhanel, G.G. In vitro activity of ceftaroline against gram-positive and gram-negative pathogens isolated from patients in Canadian hospitals in 2009. Antimicrob. Agents Chemother. 2011, 55, 2837–2846. [Google Scholar] [CrossRef] [Green Version]
- Hebeisen, P.; Heinze-Krauss, I.; Angehrn, P.; Hohl, P.; Page, M.G.; Then, R.L. In vitro and in vivo properties of Ro 63-9141, a novel broad-spectrum cephalosporin with activity against methicillin-resistant staphylococci. Antimicrob. Agents Chemother. 2001, 45, 825–836. [Google Scholar] [CrossRef] [Green Version]
- Davies, T.A.; Shang, W.; Bush, K. Activities of ceftobiprole and other beta-lactams against Streptococcus pneumoniae clinical isolates from the United States with defined substitutions in penicillin-binding proteins PBP 1a, PBP 2b, and PBP 2x. Antimicrob. Agents Chemother. 2006, 50, 2530–2532. [Google Scholar] [CrossRef] [Green Version]
- Lovering, A.L.; Gretes, M.C.; Safadi, S.S.; Danel, F.; de Castro, L.; Page, M.G.P.; Strynadka, N.C.J. Structural insights into the anti-methicillin-resistant Staphylococcus aureus (MRSA) activity of ceftobiprole. J. Biol. Chem. 2012, 287, 32096–32102. [Google Scholar] [CrossRef] [Green Version]
- Queenan, A.M.; Shang, W.; Kania, M.; Page, M.G.P.; Bush, K. Interactions of ceftobiprole with beta-lactamases from molecular classes A to D. Antimicrob. Agents Chemother. 2007, 51, 3089–3095. [Google Scholar] [CrossRef] [Green Version]
- Zhanel, G.G.; Chung, P.; Adam, H.; Zelenitsky, S.; Denisuik, A.; Schweizer, F.; Lagacé-Wiens, P.R.S.; Rubinstein, E.; Gin, A.S.; Walkty, A.; et al. Ceftolozane/tazobactam: A novel cephalosporin/β-lactamase inhibitor combination with activity against multidrug-resistant gram-negative bacilli. Drugs 2014, 74, 31–51. [Google Scholar] [CrossRef] [PubMed]
- Kohira, N.; West, J.; Ito, A.; Ito-Horiyama, T.; Nakamura, R.; Sato, T.; Rittenhouse, S.; Tsuji, M.; Yamano, Y. In Vitro Antimicrobial Activity of a Siderophore Cephalosporin, S-649266, against Enterobacteriaceae Clinical Isolates, Including Carbapenem-Resistant Strains. Antimicrob. Agents Chemother. 2016, 60, 729–734. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhanel, G.G.; Golden, A.R.; Zelenitsky, S.; Wiebe, K.; Lawrence, C.K.; Adam, H.J.; Idowu, T.; Domalaon, R.; Schweizer, F.; Zhanel, M.A.; et al. Cefiderocol: A Siderophore Cephalosporin with Activity Against Carbapenem-Resistant and Multidrug-Resistant Gram-Negative Bacilli. Drugs 2019, 79, 271–289. [Google Scholar] [CrossRef] [PubMed]
- Jacobs, M.R.; Abdelhamed, A.M.; Good, C.E.; Rhoads, D.D.; Hujer, K.M.; Hujer, A.M.; Domitrovic, T.N.; Rudin, S.D.; Richter, S.S.; van Duin, D.; et al. ARGONAUT-I: Activity of Cefiderocol (S-649266), a Siderophore Cephalosporin, against Gram-Negative Bacteria, Including Carbapenem-Resistant Nonfermenters and Enterobacteriaceae with Defined Extended-Spectrum β-Lactamases and Carbapenemases. Antimicrob. Agents Chemother. 2019, 63, e01801-18. [Google Scholar] [CrossRef] [Green Version]
- Hsueh, S.C.; Lee, Y.J.; Huang, Y.T.; Liao, C.H.; Tsuji, M.; Hsueh, P.R. In vitro activities of cefiderocol, ceftolozane/tazobactam, ceftazidime/avibactam and other comparative drugs against imipenem-resistant Pseudomonas aeruginosa and Acinetobacter baumannii, and Stenotrophomonas maltophilia, all associated with bloodstream. J. Antimicrob. Chemother. 2019, 74, 380–386. [Google Scholar] [CrossRef] [PubMed]
- Tomaras, A.P.; Crandon, J.L.; McPherson, C.J.; Banevicius, M.A.; Finegan, S.M.; Irvine, R.L.; Brown, M.F.; O’Donnell, J.P.; Nicolau, D.P. Adaptation-based resistance to siderophore-conjugated antibacterial agents by Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 2013, 57, 4197–4207. [Google Scholar] [CrossRef] [Green Version]
- Sykes, R.B.; Bonner, D.P.; Bush, K.; Georgopapadakou, N.H. Azthreonam (SQ 26,776), a synthetic monobactam specifically active against aerobic gram-negative bacteria. Antimicrob. Agents Chemother. 1982, 21, 85–92. [Google Scholar] [CrossRef] [Green Version]
- Asbel, L.E.; Levison, M.E. Cephalosporins, carbapenems, and monobactams. Infect. Dis. Clin. N. Am. 2000, 14, 435–447. [Google Scholar] [CrossRef]
- Rodríguez-Baño, J.; Gutiérrez-Gutiérrez, B.; Machuca, I.; Pascual, A. Treatment of Infections Caused by Extended-Spectrum-Beta-Lactamase-, AmpC-, and Carbapenemase-Producing Enterobacteriaceae. Clin. Microbiol. Rev. 2018, 31. [Google Scholar] [CrossRef] [Green Version]
- Papp-Wallace, K.M.; Bajaksouzian, S.; Abdelhamed, A.M.; Foster, A.N.; Winkler, M.L.; Gatta, J.A.; Nichols, W.W.; Testa, R.; Bonomo, R.A.; Jacobs, M.R. Activities of ceftazidime, ceftaroline, and aztreonam alone and combined with avibactam against isogenic Escherichia coli strains expressing selected single β-lactamases. Diagn. Microbiol. Infect. Dis. 2015, 82, 65–69. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- El-Shorbagi, A.N.; Chaudhary, S. Monobactams: A Unique Natural Scaffold of Four-Membered Ring Skeleton, Recent Development to Clinically Overcome Infections by Multidrug- Resistant Microbes. Lett. Drug Des. Discov. 2019, 16. [Google Scholar] [CrossRef]
- Page, M.G.P.; Dantier, C.; Desarbre, E. In vitro properties of BAL30072, a novel siderophore sulfactam with activity against multiresistant gram-negative bacilli. Antimicrob. Agents Chemother. 2010, 54, 2291–2302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flanagan, M.E.; Brickner, S.J.; Lall, M.; Casavant, J.; Deschenes, L.; Finegan, S.M.; George, D.M.; Granskog, K.; Hardink, J.R.; Huband, M.D.; et al. Preparation, gram-negative antibacterial activity, and hydrolytic stability of novel siderophore-conjugated monocarbam diols. ACS Med. Chem. Lett. 2011, 2, 385–390. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Papp-Wallace, K.M.; Endimiani, A.; Taracila, M.A.; Bonomo, R.A. Carbapenems: Past, present, and future. Antimicrob. Agents Chemother. 2011, 55, 4943–4960. [Google Scholar] [CrossRef] [Green Version]
- Chahine, E.B.; Ferrill, M.J.; Poulakos, M.N. Doripenem: A new carbapenem antibiotic. Am. J. Heal. Pharm. AJHP Off. J. Am. Soc. Health Pharm. 2010, 67, 2015–2024. [Google Scholar] [CrossRef] [Green Version]
- Mandell, L. Doripenem: A new carbapenem in the treatment of nosocomial infection. Clin. Infect. Dis. 2009, 49, S1–S3. [Google Scholar] [CrossRef]
- Codjoe, F.S.; Donkor, E.S. Carbapenem Resistance: A Review. Med. Sci. 2017, 6, 1. [Google Scholar] [CrossRef] [Green Version]
- Bratu, S.; Landman, D.; Alam, M.; Tolentino, E.; Quale, J. Detection of KPC carbapenem-hydrolyzing enzymes in Enterobacter spp. from Brooklyn, New York. Antimicrob. Agents Chemother. 2005, 49, 776–778. [Google Scholar] [CrossRef] [Green Version]
- Muggeo, A.; Guillard, T.; Klein, F.; Reffuveille, F.; François, C.; Babosan, A.; Bajolet, O.; Bertrand, X.; de Champs, C. Spread of Klebsiella pneumoniae ST395 non-susceptible to carbapenems and resistant to fluoroquinolones in North-Eastern France. J. Glob. Antimicrob. Resist. 2018, 13, 98–103. [Google Scholar] [CrossRef]
- Yigit, H.; Queenan, A.M.; Anderson, G.J.; Domenech-Sanchez, A.; Biddle, J.W.; Steward, C.D.; Alberti, S.; Bush, K.; Tenover, F.C. Novel carbapenem-hydrolyzing beta-lactamase, KPC-1, from a carbapenem-resistant strain of Klebsiella pneumoniae. Antimicrob. Agents Chemother. 2001, 45, 1151–1161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bradford, P.A.; Bratu, S.; Urban, C.; Visalli, M.; Mariano, N.; Landman, D.; Rahal, J.J.; Brooks, S.; Cebular, S.; Quale, J. Emergence of carbapenem-resistant Klebsiella species possessing the class A carbapenem-hydrolyzing KPC-2 and inhibitor-resistant TEM-30 beta-lactamases in New York City. Clin. Infect. Dis. 2004, 39, 55–60. [Google Scholar] [CrossRef] [PubMed]
- Bratu, S.; Landman, D.; Haag, R.; Recco, R.; Eramo, A.; Alam, M.; Quale, J. Rapid spread of carbapenem-resistant Klebsiella pneumoniae in New York City: A new threat to our antibiotic armamentarium. Arch. Intern. Med. 2005, 165, 1430–1435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Z.; Wang, Y.; Tian, L.; Zhu, X.; Li, L.; Zhang, B.; Yan, S.; Sun, Z. First report in China of Enterobacteriaceae clinical isolates coharboring blaNDM-1 and blaIMP-4 drug resistance genes. Microb. Drug Resist. 2015, 21, 167–170. [Google Scholar] [CrossRef]
- Du, H.; Chen, L.; Chavda, K.D.; Pandey, R.; Zhang, H.; Xie, X.; Tang, Y.W.; Kreiswirth, B.N. Genomic Characterization of Enterobacter cloacae Isolates from China That Coproduce KPC-3 and NDM-1 Carbapenemases. Antimicrob. Agents Chemother. 2016, 60, 2519–2523. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Yuan, M.; Chen, H.; Chen, X.; Jia, Y.; Zhu, X.; Bai, L.; Bai, X.; Fanning, S.; Lu, J.; et al. First Report of Klebsiella oxytoca Strain Simultaneously Producing NDM-1, IMP-4, and KPC-2 Carbapenemases. Antimicrob. Agents Chemother. 2017, 61. [Google Scholar] [CrossRef] [Green Version]
- Yang, B.; Feng, Y.; McNally, A.; Zong, Z. Occurrence of Enterobacter hormaechei carrying bla(NDM-1) and bla(KPC-2) in China. Diagn. Microbiol. Infect. Dis. 2018, 90, 139–142. [Google Scholar] [CrossRef]
- Fujimoto, K.; Takemoto, K.; Hatano, K.; Nakai, T.; Terashita, S.; Matsumoto, M.; Eriguchi, Y.; Eguchi, K.; Shimizudani, T.; Sato, K.; et al. Novel carbapenem antibiotics for parenteral and oral applications: In vitro and in vivo activities of 2-aryl carbapenems and their pharmacokinetics in laboratory animals. Antimicrob. Agents Chemother. 2013, 57, 697–707. [Google Scholar] [CrossRef] [Green Version]
- Sawa, T.; Kooguchi, K.; Moriyama, K. Molecular diversity of extended-spectrum β-lactamases and carbapenemases, and antimicrobial resistance. J. Intensive Care 2020, 8, 13. [Google Scholar] [CrossRef] [Green Version]
- Abraham, E.P.; Chain, E. An Enzyme from Bacteria able to Destroy Penicillin. Nature 1940, 146, 837. [Google Scholar] [CrossRef]
- Matthew, M.; Harris, A.M. Identification of beta-lactamases by analytical isoelectric focusing: Correlation with bacterial taxonomy. J. Gen. Microbiol. 1976, 94, 55–67. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murray, B.E.; Mederski-Samaroj, B. Transferable beta-lactamase. A new mechanism for in vitro penicillin resistance in Streptococcus faecalis. J. Clin. Investig. 1983, 72, 1168–1171. [Google Scholar] [CrossRef] [PubMed]
- Hamilton-Miller, J.M.T. β-Lactamases and their clinical significance. J. Antimicrob. Chemother. 1982, 9, 11–19. [Google Scholar] [CrossRef] [PubMed]
- Hall, B.G.; Barlow, M. Evolution of the serine beta-lactamases: Past, present and future. Drug Resist. Updates Rev. Comment. Antimicrob. Anticancer Chemother. 2004, 7, 111–123. [Google Scholar] [CrossRef]
- D’Costa, V.M.; King, C.E.; Kalan, L.; Morar, M.; Sung, W.W.L.; Schwarz, C.; Froese, D.; Zazula, G.; Calmels, F.; Debruyne, R.; et al. Antibiotic resistance is ancient. Nature 2011, 477, 457–461. [Google Scholar] [CrossRef] [PubMed]
- Song, J.S.; Jeon, J.H.; Lee, J.H.; Jeong, S.H.; Jeong, B.C.; Kim, S.J.; Lee, J.H.; Lee, S.H. Molecular characterization of TEM-type beta-lactamases identified in cold-seep sediments of Edison Seamount (south of Lihir Island, Papua New Guinea). J. Microbiol. 2005, 43, 172–178. [Google Scholar]
- Rascovan, N.; Telke, A.; Raoult, D.; Rolain, J.M.; Desnues, C. Exploring divergent antibiotic resistance genes in ancient metagenomes and discovery of a novel beta-lactamase family. Environ. Microbiol. Rep. 2016, 8, 886–895. [Google Scholar] [CrossRef]
- Segawa, T.; Takeuchi, N.; Rivera, A.; Yamada, A.; Yoshimura, Y.; Barcaza, G.; Shinbori, K.; Motoyama, H.; Kohshima, S.; Ushida, K. Distribution of antibiotic resistance genes in glacier environments. Environ. Microbiol. Rep. 2013, 5, 127–134. [Google Scholar] [CrossRef]
- Bartoloni, A.; Pallecchi, L.; Rodríguez, H.; Fernandez, C.; Mantella, A.; Bartalesi, F.; Strohmeyer, M.; Kristiansson, C.; Gotuzzo, E.; Paradisi, F.; et al. Antibiotic resistance in a very remote Amazonas community. Int. J. Antimicrob. Agents 2009, 33, 125–129. [Google Scholar] [CrossRef]
- Knott-Hunziker, V.; Waley, S.G.; Orlek, B.S.; Sammes, P.G. Penicillinase active sites: Labelling of serine-44 in beta-lactamase I by 6beta-bromopenicillanic acid. FEBS Lett. 1979, 99, 59–61. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Hao, Q. Crystal structure of NDM-1 reveals a common β-lactam hydrolysis mechanism. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2011, 25, 2574–2582. [Google Scholar] [CrossRef] [PubMed]
- Sawai, T.; Mitsuhashi, S.; Yamagishi, S. Drug resistance of enteric bacteria. XIV. Comparison of beta-lactamases in gram-negative rod bacteria resistant to alpha-aminobenzylpenicillin. Jpn. J. Microbiol. 1968, 12, 423–434. [Google Scholar] [CrossRef] [PubMed]
- Ambler, R.P. The structure of beta-lactamases. Philos. Trans. R. Soc. Lond. Ser. B Biol. Sci. 1980, 289, 321–331. [Google Scholar] [CrossRef]
- Matthew, M. Plasmid-mediated beta-lactamases of Gram-negative bacteria: Properties and distribution. J. Antimicrob. Chemother. 1979, 5, 349–358. [Google Scholar] [CrossRef] [PubMed]
- Simpson, I.N.; Harper, P.B.; O’Callaghan, C.H. Principal beta-lactamases responsible for resistance to beta-lactam antibiotics in urinary tract infections. Antimicrob. Agents Chemother. 1980, 17, 929–936. [Google Scholar] [CrossRef] [Green Version]
- Roy, C.; Foz, A.; Segura, C.; Tirado, M.; Fuster, C.; Reig, R. Plasmid-determined beta-lactamases identified in a group of 204 ampicillin-resistant Enterobacteriaceae. J. Antimicrob. Chemother. 1983, 12, 507–510. [Google Scholar] [CrossRef]
- Medeiros, A.A. Beta-lactamases. Br. Med. Bull. 1984, 40, 18–27. [Google Scholar] [CrossRef]
- Roy, C.; Segura, C.; Tirado, M.; Reig, R.; Hermida, M.; Teruel, D.; Foz, A. Frequency of plasmid-determined beta-lactamases in 680 consecutively isolated strains of Enterobacteriaceae. Eur. J. Clin. Microbiol. 1985, 4, 146–147. [Google Scholar] [CrossRef]
- Peirano, G.; van der Bij, A.K.; Gregson, D.B.; Pitout, J.D.D. Molecular epidemiology over an 11-year period (2000 to 2010) of extended-spectrum β-lactamase-producing Escherichia coli causing bacteremia in a centralized Canadian region. J. Clin. Microbiol. 2012, 50, 294–299. [Google Scholar] [CrossRef] [Green Version]
- Barrios, H.; Garza-Ramos, U.; Mejia-Miranda, I.; Reyna-Flores, F.; Sánchez-Pérez, A.; Mosqueda-García, D.; Silva-Sanchez, J. ESBL-producing Escherichia coli and Klebsiella pneumoniae: The most prevalent clinical isolates obtained between 2005 and 2012 in Mexico. J. Glob. Antimicrob. Resist. 2017, 10, 243–246. [Google Scholar] [CrossRef]
- Bush, K.; Tanaka, S.K.; Bonner, D.P.; Sykes, R.B. Resistance caused by decreased penetration of beta-lactam antibiotics into Enterobacter cloacae. Antimicrob. Agents Chemother. 1985, 27, 555–560. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, V.T.; Arlet, G.; Ericsson, B.M.; Tammelin, A.; Courvalin, P.; Lambert, T. Emergence of imipenem resistance in Klebsiella pneumoniae owing to combination of plasmid-mediated CMY-4 and permeability alteration. J. Antimicrob. Chemother. 2000, 46, 895–900. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cullmann, W.; Dick, W. Heterogeneity of beta-lactamase production in Pseudomonas maltophilia, a nosocomial pathogen. Chemotherapy 1990, 36, 117–126. [Google Scholar] [CrossRef] [PubMed]
- Nordmann, P.; Mariotte, S.; Naas, T.; Labia, R.; Nicolas, M.H. Biochemical properties of a carbapenem-hydrolyzing beta-lactamase from Enterobacter cloacae and cloning of the gene into Escherichia coli. Antimicrob. Agents Chemother. 1993, 37, 939–946. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Watanabe, M.; Iyobe, S.; Inoue, M.; Mitsuhashi, S. Transferable imipenem resistance in Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 1991, 35, 147–151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lauretti, L.; Riccio, M.L.; Mazzariol, A.; Cornaglia, G.; Amicosante, G.; Fontana, R.; Rossolini, G.M. Cloning and characterization of blaVIM, a new integron-borne metallo-beta-lactamase gene from a Pseudomonas aeruginosa clinical isolate. Antimicrob. Agents Chemother. 1999, 43, 1584–1590. [Google Scholar] [CrossRef] [Green Version]
- Herbert, S.; Halvorsen, D.S.; Leong, T.; Franklin, C.; Harrington, G.; Spelman, D. Large outbreak of infection and colonization with gram-negative pathogens carrying the metallo- beta -lactamase gene blaIMP-4 at a 320-bed tertiary hospital in Australia. Infect. Control Hosp. Epidemiol. 2007, 28, 98–101. [Google Scholar] [CrossRef]
- Turton, J.F.; Wright, L.; Underwood, A.; Witney, A.A.; Chan, Y.T.; Al-Shahib, A.; Arnold, C.; Doumith, M.; Patel, B.; Planche, T.D.; et al. High-Resolution Analysis by Whole-Genome Sequencing of an International Lineage (Sequence Type 111) of Pseudomonas aeruginosa Associated with Metallo-Carbapenemases in the United Kingdom. J. Clin. Microbiol. 2015, 53, 2622–2631. [Google Scholar] [CrossRef] [Green Version]
- Kitchel, B.; Rasheed, J.K.; Patel, J.B.; Srinivasan, A.; Navon-Venezia, S.; Carmeli, Y.; Brolund, A.; Giske, C.G. Molecular epidemiology of KPC-producing Klebsiella pneumoniae isolates in the United States: Clonal expansion of multilocus sequence type 258. Antimicrob. Agents Chemother. 2009, 53, 3365–3370. [Google Scholar] [CrossRef] [Green Version]
- Pitout, J.D.D.; Nordmann, P.; Poirel, L. Carbapenemase-Producing Klebsiella pneumoniae, a Key Pathogen Set for Global Nosocomial Dominance. Antimicrob. Agents Chemother. 2015, 59, 5873–5884. [Google Scholar] [CrossRef] [Green Version]
- Giacobbe, D.R.; Del Bono, V.; Trecarichi, E.M.; De Rosa, F.G.; Giannella, M.; Bassetti, M.; Bartoloni, A.; Losito, A.R.; Corcione, S.; Bartoletti, M.; et al. Risk factors for bloodstream infections due to colistin-resistant KPC-producing Klebsiella pneumoniae: Results from a multicenter case-control-control study. Clin. Microbiol. Infect. 2015, 21, e1–e8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, M.F.; Lan, C.Y. Antimicrobial resistance in Acinetobacter baumannii: From bench to bedside. World J. Clin. Cases 2014, 2, 787–814. [Google Scholar] [CrossRef] [PubMed]
- Nowak, P.; Paluchowska, P. Acinetobacter baumannii: Biology and drug resistance—Role of carbapenemases. Folia Histochem. Cytobiol. 2016, 54, 61–74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lyon, J.A. Imipenem/cilastatin: The first carbapenem antibiotic. Drug Intell. Clin. Pharm. 1985, 19, 895–899. [Google Scholar]
- Périchon, B.; Goussard, S.; Walewski, V.; Krizova, L.; Cerqueira, G.; Murphy, C.; Feldgarden, M.; Wortman, J.; Clermont, D.; Nemec, A.; et al. Identification of 50 class D β-lactamases and 65 Acinetobacter-derived cephalosporinases in Acinetobacter spp. Antimicrob. Agents Chemother. 2014, 58, 936–949. [Google Scholar] [CrossRef] [Green Version]
- Davandeh, I.; Eraç, B.; Aydemir, S.Ş. Investigation of class-d beta-lactamases causing carbapenem resistance in clinical Acinetobacter baumannii isolates. Turk. J. Med. Sci. 2017, 47, 1661–1666. [Google Scholar] [CrossRef]
- Evans, B.A.; Amyes, S.G.B. OXA β-lactamases. Clin. Microbiol. Rev. 2014, 27, 241–263. [Google Scholar] [CrossRef] [Green Version]
- Donald, H.M.; Scaife, W.; Amyes, S.G.; Young, H.K. Sequence analysis of ARI-1, a novel OXA beta-lactamase, responsible for imipenem resistance in Acinetobacter baumannii 6B92. Antimicrob. Agents Chemother. 2000, 44, 196–199. [Google Scholar] [CrossRef] [Green Version]
- Rouhi, S.; Ramazanzadeh, R. Prevalence of bla (Oxacillinase-23)and bla (Oxacillinase-24/40-)type Carbapenemases in Pseudomonas aeruginosa Species Isolated From Patients with Nosocomial and Non-nosocomial Infections in the West of Iran. Iran. J. Pathol. 2018, 13, 348–356. [Google Scholar]
- Ayoub Moubareck, C.; Hammoudi Halat, D.; Akkawi, C.; Nabi, A.; AlSharhan, M.A.; AlDeesi, Z.O.; Peters, C.C.; Celiloglu, H.; Karam Sarkis, D. Role of outer membrane permeability, efflux mechanism, and carbapenemases in carbapenem-nonsusceptible Pseudomonas aeruginosa from Dubai hospitals: Results of the first cross-sectional survey. Int. J. Infect. Dis. IJID Off. Publ. Int. Soc. Infect. Dis. 2019, 84, 143–150. [Google Scholar] [CrossRef] [Green Version]
- McCracken, M.G.; Adam, H.J.; Blondeau, J.M.; Walkty, A.J.; Karlowsky, J.A.; Hoban, D.J.; Zhanel, G.G.; Mulvey, M.R. Characterization of carbapenem-resistant and XDR Pseudomonas aeruginosa in Canada: Results of the CANWARD 2007-16 study. J. Antimicrob. Chemother. 2019, 74, iv32–iv38. [Google Scholar] [CrossRef] [PubMed]
- Pereira, P.S.; Borghi, M.; Albano, R.M.; Lopes, J.C.O.; Silveira, M.C.; Marques, E.A.; Oliveira, J.C.R.; Asensi, M.D.; Carvalho-Assef, A.P.D. Coproduction of NDM-1 and KPC-2 in Enterobacter hormaechei from Brazil. Microb. Drug Resist. 2015, 21, 234–236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhuang, Z.; Lv, L.; Lu, J.; Lin, J.; Liu, J.H. Emergence of Klebsiella pneumoniae and Enterobacter cloacae producing OXA-48 carbapenemases from retail meats in China, 2018. J. Antimicrob. Chemother. 2019, 74, 3632–3634. [Google Scholar] [CrossRef] [PubMed]
- Peirano, G.; Matsumura, Y.; Adams, M.D.; Bradford, P.; Motyl, M.; Chen, L.; Kreiswirth, B.N.; Pitout, J.D.D. Genomic Epidemiology of Global Carbapenemase-Producing Enterobacter spp., 2008–2014. Emerg. Infect. Dis. 2018, 24, 1010–1019. [Google Scholar] [CrossRef] [Green Version]
- Kollenda, H.; Frickmann, H.; Ben Helal, R.; Wiemer, D.F.; Naija, H.; El Asli, M.S.; Egold, M.; Bugert, J.J.; Handrick, S.; Wölfel, R.; et al. Screening for Carbapenemases in Ertapenem-Resistant Enterobacteriaceae Collected at a Tunisian Hospital Between 2014 and 2018. Eur. J. Microbiol. Immunol. 2019, 9, 9–13. [Google Scholar] [CrossRef]
- Izdebski, R.; Baraniak, A.; Zabicka, D.; Sekowska, A.; Gospodarek-Komkowska, E.; Hryniewicz, W.; Gniadkowski, M. VIM/IMP carbapenemase-producing Enterobacteriaceae in Poland: Epidemic Enterobacter hormaechei and Klebsiella oxytoca lineages. J. Antimicrob. Chemother. 2018, 73, 2675–2681. [Google Scholar] [CrossRef]
- Bush, K.; Jacoby, G.A.; Medeiros, A.A. A functional classification scheme for beta-lactamases and its correlation with molecular structure. Antimicrob. Agents Chemother. 1995, 39, 1211–1233. [Google Scholar] [CrossRef] [Green Version]
- Bush, K.; Jacoby, G.A. Updated functional classification of beta-lactamases. Antimicrob. Agents Chemother. 2010, 54, 969–976. [Google Scholar] [CrossRef] [Green Version]
- Thomson, G.; Turner, D.; Brasso, W.; Kircher, S.; Guillet, T.; Thomson, K. High-Stringency Evaluation of the Automated BD Phoenix CPO Detect and Rapidec Carba NP Tests for Detection and Classification of Carbapenemases. J. Clin. Microbiol. 2017, 55, 3437–3443. [Google Scholar] [CrossRef] [Green Version]
- Lee, D.; Das, S.; Dawson, N.L.; Dobrijevic, D.; Ward, J.; Orengo, C. Novel Computational Protocols for Functionally Classifying and Characterising Serine Beta-Lactamases. PLoS Comput. Biol. 2016, 12, e1004926. [Google Scholar] [CrossRef] [Green Version]
- Bush, K. Proliferation and significance of clinically relevant β-lactamases. Ann. N. Y. Acad. Sci. 2013, 1277, 84–90. [Google Scholar] [CrossRef] [PubMed]
- Moran, R.A.; Anantham, S.; Holt, K.E.; Hall, R.M. Prediction of antibiotic resistance from antibiotic resistance genes detected in antibiotic-resistant commensal Escherichia coli using PCR or WGS. J. Antimicrob. Chemother. 2017, 72, 700–704. [Google Scholar] [CrossRef] [PubMed]
- Reading, C.; Cole, M. Clavulanic acid: A beta-lactamase-inhiting beta-lactam from Streptomyces clavuligerus. Antimicrob. Agents Chemother. 1977, 11, 852–857. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- White, A.R.; Kaye, C.; Poupard, J.; Pypstra, R.; Woodnutt, G.; Wynne, B. Augmentin (amoxicillin/clavulanate) in the treatment of community-acquired respiratory tract infection: A review of the continuing development of an innovative antimicrobial agent. J. Antimicrob. Chemother. 2004, 53, i3–i20. [Google Scholar] [CrossRef] [Green Version]
- Zhanel, G.G.; Lawson, C.D.; Adam, H.; Schweizer, F.; Zelenitsky, S.; Lagacé-Wiens, P.R.S.; Denisuik, A.; Rubinstein, E.; Gin, A.S.; Hoban, D.J.; et al. Ceftazidime-avibactam: A novel cephalosporin/β-lactamase inhibitor combination. Drugs 2013, 73, 159–177. [Google Scholar] [CrossRef] [Green Version]
- Ehmann, D.E.; Jahić, H.; Ross, P.L.; Gu, R.F.; Hu, J.; Kern, G.; Walkup, G.K.; Fisher, S.L. Avibactam is a covalent, reversible, non-β-lactam β-lactamase inhibitor. Proc. Natl. Acad. Sci. USA 2012, 109, 11663–11668. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tuon, F.F.; Rocha, J.L.; Formigoni-Pinto, M.R. Pharmacological aspects and spectrum of action of ceftazidime-avibactam: A systematic review. Infection 2018, 46, 165–181. [Google Scholar] [CrossRef] [PubMed]
- Papp-Wallace, K.M.; Nguyen, N.Q.; Jacobs, M.R.; Bethel, C.R.; Barnes, M.D.; Kumar, V.; Bajaksouzian, S.; Rudin, S.D.; Rather, P.N.; Bhavsar, S.; et al. Strategic Approaches to Overcome Resistance against Gram-Negative Pathogens Using β-Lactamase Inhibitors and β-Lactam Enhancers: Activity of Three Novel Diazabicyclooctanes WCK 5153, Zidebactam (WCK 5107), and WCK 4234. J. Med. Chem. 2018, 61, 4067–4086. [Google Scholar] [CrossRef] [PubMed]
- Moya, B.; Barcelo, I.M.; Bhagwat, S.; Patel, M.; Bou, G.; Papp-Wallace, K.M.; Bonomo, R.A.; Oliver, A. WCK 5107 (Zidebactam) and WCK 5153 Are Novel Inhibitors of PBP2 Showing Potent “β-Lactam Enhancer” Activity against Pseudomonas aeruginosa, Including Multidrug-Resistant Metallo-β-Lactamase-Producing High-Risk Clones. Antimicrob. Agents Chemother. 2017, 61. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Durand-Réville, T.F.; Guler, S.; Comita-Prevoir, J.; Chen, B.; Bifulco, N.; Huynh, H.; Lahiri, S.; Shapiro, A.B.; McLeod, S.M.; Carter, N.M.; et al. ETX2514 is a broad-spectrum β-lactamase inhibitor for the treatment of drug-resistant Gram-negative bacteria including Acinetobacter baumannii. Nat. Microbiol. 2017, 2, 17104. [Google Scholar] [CrossRef]
- Wright, A.J. The penicillins. Mayo Clin. Proc. 1999, 74, 290–307. [Google Scholar] [CrossRef] [PubMed]
- Laws, M.; Shaaban, A.; Rahman, K.M. Antibiotic resistance breakers: Current approaches and future directions. FEMS Microbiol. Rev. 2019, 43, 490–516. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Payne, D.J.; Cramp, R.; Winstanley, D.J.; Knowles, D.J. Comparative activities of clavulanic acid, sulbactam, and tazobactam against clinically important beta-lactamases. Antimicrob. Agents Chemother. 1994, 38, 767–772. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Penwell, W.F.; Shapiro, A.B.; Giacobbe, R.A.; Gu, R.F.; Gao, N.; Thresher, J.; McLaughlin, R.E.; Huband, M.D.; DeJonge, B.L.M.; Ehmann, D.E.; et al. Molecular mechanisms of sulbactam antibacterial activity and resistance determinants in Acinetobacter baumannii. Antimicrob. Agents Chemother. 2015, 59, 1680–1689. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rafailidis, P.I.; Kouranos, V.D.; Christodoulou, C.; Falagas, M.E. Linezolid for patients with neutropenia: Are bacteriostatic agents appropriate? Expert Rev. Anti Infect. Ther. 2009, 7, 415–422. [Google Scholar] [CrossRef]
- Mohanty, S.; Singhal, R.; Sood, S.; Dhawan, B.; Das, B.K.; Kapil, A. Comparative in vitro activity of beta-lactam/beta-lactamase inhibitor combinations against gram negative bacteria. Indian J. Med. Res. 2005, 122, 425–428. [Google Scholar]
- Cho, J.C.; Fiorenza, M.A.; Estrada, S.J. Ceftolozane/Tazobactam: A Novel Cephalosporin/β-Lactamase Inhibitor Combination. Pharmacotherapy 2015, 35, 701–715. [Google Scholar] [CrossRef]
- Melchior, N.H.; Keiding, J. In-vitro evaluation of ampicillin/brobactam and comparison with other beta-lactam antibiotics. J. Antimicrob. Chemother. 1991, 27, 29–40. [Google Scholar] [CrossRef]
- Thomson, K.S.; AbdelGhani, S.; Snyder, J.W.; Thomson, G.K. Activity of Cefepime-Zidebactam against Multidrug-Resistant (MDR) Gram-Negative Pathogens. Antibiotics 2019, 8, 32. [Google Scholar] [CrossRef] [Green Version]
- Moya, B.; Barcelo, I.M.; Bhagwat, S.; Patel, M.; Bou, G.; Papp-Wallace, K.M.; Bonomo, R.A.; Oliver, A. Potent β-Lactam Enhancer Activity of Zidebactam and WCK 5153 against Acinetobacter baumannii, Including Carbapenemase-Producing Clinical Isolates. Antimicrob. Agents Chemother. 2017, 61. [Google Scholar] [CrossRef] [Green Version]
- Isler, B.; Doi, Y.; Bonomo, R.A.; Paterson, D.L. New Treatment Options against Carbapenem-Resistant Acinetobacter baumannii Infections. Antimicrob. Agents Chemother. 2019, 63. [Google Scholar] [CrossRef] [Green Version]
- Castanheira, M.; Rhomberg, P.R.; Flamm, R.K.; Jones, R.N. Effect of the β-Lactamase Inhibitor Vaborbactam Combined with Meropenem against Serine Carbapenemase-Producing Enterobacteriaceae. Antimicrob. Agents Chemother. 2016, 60, 5454–5458. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, L.P.; Pinto, N.A.; Vu, T.N.; Lee, H.; Cho, Y.L.; Byun, J.H.; D’Souza, R.; Yong, D. In Vitro Activity of a Novel Siderophore-Cephalosporin, GT-1 and Serine-Type β-Lactamase Inhibitor, GT-055, against Escherichia coli, Klebsiella pneumoniae and Acinetobacter spp. Panel Strains. Antibiotics 2020, 9, 267. [Google Scholar] [CrossRef] [PubMed]
- Shirley, M. Ceftazidime-Avibactam: A Review in the Treatment of Serious Gram-Negative Bacterial Infections. Drugs 2018, 78, 675–692. [Google Scholar] [CrossRef]
- Lahiri, S.D.; Johnstone, M.R.; Ross, P.L.; McLaughlin, R.E.; Olivier, N.B.; Alm, R.A. Avibactam and class C β-lactamases: Mechanism of inhibition, conservation of the binding pocket, and implications for resistance. Antimicrob. Agents Chemother. 2014, 58, 5704–5713. [Google Scholar] [CrossRef] [Green Version]
- Drawz, S.M.; Papp-Wallace, K.M.; Bonomo, R.A. New β-lactamase inhibitors: A therapeutic renaissance in an MDR world. Antimicrob. Agents Chemother. 2014, 58, 1835–1846. [Google Scholar] [CrossRef] [Green Version]
- Karlowsky, J.A.; Biedenbach, D.J.; Kazmierczak, K.M.; Stone, G.G.; Sahm, D.F. Activity of Ceftazidime-Avibactam against Extended-Spectrum- and AmpC β-Lactamase-Producing Enterobacteriaceae Collected in the INFORM Global Surveillance Study from 2012 to 2014. Antimicrob. Agents Chemother. 2016, 60, 2849–2857. [Google Scholar] [CrossRef] [Green Version]
- Testa, R.; Cantón, R.; Giani, T.; Morosini, M.I.; Nichols, W.W.; Seifert, H.; Stefanik, D.; Rossolini, G.M.; Nordmann, P. In vitro activity of ceftazidime, ceftaroline and aztreonam alone and in combination with avibactam against European Gram-negative and Gram-positive clinical isolates. Int. J. Antimicrob. Agents 2015, 45, 641–646. [Google Scholar] [CrossRef] [Green Version]
- Wagenlehner, F.M.; Sobel, J.D.; Newell, P.; Armstrong, J.; Huang, X.; Stone, G.G.; Yates, K.; Gasink, L.B. Ceftazidime-avibactam Versus Doripenem for the Treatment of Complicated Urinary Tract Infections, Including Acute Pyelonephritis: RECAPTURE, a Phase 3 Randomized Trial Program. Clin. Infect. Dis. 2016, 63, 754–762. [Google Scholar] [CrossRef] [Green Version]
- Shields, R.K.; Nguyen, M.H.; Chen, L.; Press, E.G.; Potoski, B.A.; Marini, R.V.; Doi, Y.; Kreiswirth, B.N.; Clancy, C.J. Ceftazidime-Avibactam Is Superior to Other Treatment Regimens against Carbapenem-Resistant Klebsiella pneumoniae Bacteremia. Antimicrob. Agents Chemother. 2017, 61. [Google Scholar] [CrossRef] [Green Version]
- Van Duin, D.; Lok, J.J.; Earley, M.; Cober, E.; Richter, S.S.; Perez, F.; Salata, R.A.; Kalayjian, R.C.; Watkins, R.R.; Doi, Y.; et al. Colistin Versus Ceftazidime-Avibactam in the Treatment of Infections Due to Carbapenem-Resistant Enterobacteriaceae. Clin. Infect. Dis. 2018, 66, 163–171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shields, R.K.; Chen, L.; Cheng, S.; Chavda, K.D.; Press, E.G.; Snyder, A.; Pandey, R.; Doi, Y.; Kreiswirth, B.N.; Nguyen, M.H.; et al. Emergence of Ceftazidime-Avibactam Resistance Due to Plasmid-Borne bla(KPC-3) Mutations during Treatment of Carbapenem-Resistant Klebsiella pneumoniae Infections. Antimicrob. Agents Chemother. 2017, 61. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lutgring, J.D.; Limbago, B.M. The Problem of Carbapenemase-Producing-Carbapenem-Resistant-Enterobacteriaceae Detection. J. Clin. Microbiol. 2016, 54, 529–534. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pagès, J.M.; Peslier, S.; Keating, T.A.; Lavigne, J.P.; Nichols, W.W. Role of the Outer Membrane and Porins in Susceptibility of β-Lactamase-Producing Enterobacteriaceae to Ceftazidime-Avibactam. Antimicrob. Agents Chemother. 2015, 60, 1349–1359. [Google Scholar] [CrossRef] [Green Version]
- Escolà-Vergé, L.; Pigrau, C.; Almirante, B. Ceftolozane/tazobactam for the treatment of complicated intra-abdominal and urinary tract infections: Current perspectives and place in therapy. Infect. Drug Resist. 2019, 12, 1853–1867. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moyá, B.; Zamorano, L.; Juan, C.; Ge, Y.; Oliver, A. Affinity of the new cephalosporin CXA-101 to penicillin-binding proteins of Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 2010, 54, 3933–3937. [Google Scholar] [CrossRef] [Green Version]
- Cluck, D.; Lewis, P.; Stayer, B.; Spivey, J.; Moorman, J. Ceftolozane-tazobactam: A new-generation cephalosporin. Am. J. Health Pharm. AJHP Off. J. Am. Soc. Health Pharm. 2015, 72, 2135–2146. [Google Scholar] [CrossRef]
- Pfaller, M.A.; Bassetti, M.; Duncan, L.R.; Castanheira, M. Ceftolozane/tazobactam activity against drug-resistant Enterobacteriaceae and Pseudomonas aeruginosa causing urinary tract and intraabdominal infections in Europe: Report from an antimicrobial surveillance programme (2012-15). J. Antimicrob. Chemother. 2017, 72, 1386–1395. [Google Scholar] [CrossRef]
- Snydman, D.R.; McDermott, L.A.; Jacobus, N.V. Activity of ceftolozane-tazobactam against a broad spectrum of recent clinical anaerobic isolates. Antimicrob. Agents Chemother. 2014, 58, 1218–1223. [Google Scholar] [CrossRef] [Green Version]
- Popejoy, M.W.; Paterson, D.L.; Cloutier, D.; Huntington, J.A.; Miller, B.; Bliss, C.A.; Steenbergen, J.N.; Hershberger, E.; Umeh, O.; Kaye, K.S. Efficacy of ceftolozane/tazobactam against urinary tract and intra-abdominal infections caused by ESBL-producing Escherichia coli and Klebsiella pneumoniae: A pooled analysis of Phase 3 clinical trials. J. Antimicrob. Chemother. 2017, 72, 268–272. [Google Scholar] [CrossRef] [Green Version]
- Wong, D.; van Duin, D. Novel Beta-Lactamase Inhibitors: Unlocking Their Potential in Therapy. Drugs 2017, 77, 615–628. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saravolatz, L.D.; Pawlak, J.; Johnson, L.; Bonilla, H.; Saravolatz, L.D., 2nd; Fakih, M.G.; Fugelli, A.; Olsen, W.M. In vitro activities of LTX-109, a synthetic antimicrobial peptide, against methicillin-resistant, vancomycin-intermediate, vancomycin-resistant, daptomycin-nonsusceptible, and linezolid-nonsusceptible Staphylococcus aureus. Antimicrob. Agents Chemother. 2012, 56, 4478–4482. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lapuebla, A.; Abdallah, M.; Olafisoye, O.; Cortes, C.; Urban, C.; Landman, D.; Quale, J. Activity of Imipenem with Relebactam against Gram-Negative Pathogens from New York City. Antimicrob. Agents Chemother. 2015, 59, 5029–5031. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lob, S.H.; Hackel, M.A.; Kazmierczak, K.M.; Young, K.; Motyl, M.R.; Karlowsky, J.A.; Sahm, D.F. In Vitro Activity of Imipenem-Relebactam against Gram-Negative ESKAPE Pathogens Isolated by Clinical Laboratories in the United States in 2015 (Results from the SMART Global Surveillance Program). Antimicrob. Agents Chemother. 2017, 61. [Google Scholar] [CrossRef] [Green Version]
- Canver, M.C.; Satlin, M.J.; Westblade, L.F.; Kreiswirth, B.N.; Chen, L.; Robertson, A.; Fauntleroy, K.; La Spina, M.; Callan, K.; Jenkins, S.G. Activity of Imipenem-Relebactam and Comparator Agents against Genetically Characterized Isolates of Carbapenem-Resistant Enterobacteriaceae. Antimicrob. Agents Chemother. 2019, 63. [Google Scholar] [CrossRef] [Green Version]
- Haidar, G.; Clancy, C.J.; Chen, L.; Samanta, P.; Shields, R.K.; Kreiswirth, B.N.; Nguyen, M.H. Identifying Spectra of Activity and Therapeutic Niches for Ceftazidime-Avibactam and Imipenem-Relebactam against Carbapenem-Resistant Enterobacteriaceae. Antimicrob. Agents Chemother. 2017, 61. [Google Scholar] [CrossRef] [Green Version]
- Livermore, D.M.; Warner, M.; Mushtaq, S. Activity of MK-7655 combined with imipenem against Enterobacteriaceae and Pseudomonas aeruginosa. J. Antimicrob. Chemother. 2013, 68, 2286–2290. [Google Scholar] [CrossRef] [Green Version]
- Hirsch, E.B.; Ledesma, K.R.; Chang, K.T.; Schwartz, M.S.; Motyl, M.R.; Tam, V.H. In vitro activity of MK-7655, a novel β-lactamase inhibitor, in combination with imipenem against carbapenem-resistant Gram-negative bacteria. Antimicrob. Agents Chemother. 2012, 56, 3753–3757. [Google Scholar] [CrossRef] [Green Version]
- Karlowsky, J.A.; Lob, S.H.; Kazmierczak, K.M.; Hawser, S.P.; Magnet, S.; Young, K.; Motyl, M.R.; Sahm, D.F. In vitro activity of imipenem/relebactam against Gram-negative ESKAPE pathogens isolated in 17 European countries: 2015 SMART surveillance programme. J. Antimicrob. Chemother. 2018, 73, 1872–1879. [Google Scholar] [CrossRef]
- Goldstein, E.J.C.; Citron, D.M.; Tyrrell, K.L.; Leoncio, E.; Merriam, C.V. Comparative In Vitro Activities of Relebactam, Imipenem, the Combination of the Two, and Six Comparator Antimicrobial Agents against 432 Strains of Anaerobic Organisms, Including Imipenem-Resistant Strains. Antimicrob. Agents Chemother. 2018, 62. [Google Scholar] [CrossRef] [Green Version]
- Zhanel, G.G.; Lawrence, C.K.; Adam, H.; Schweizer, F.; Zelenitsky, S.; Zhanel, M.; Lagacé-Wiens, P.R.S.; Walkty, A.; Denisuik, A.; Golden, A.; et al. Imipenem-Relebactam and Meropenem-Vaborbactam: Two Novel Carbapenem-β-Lactamase Inhibitor Combinations. Drugs 2018, 78, 65–98. [Google Scholar] [CrossRef] [PubMed]
- Patel, T.S.; Pogue, J.M.; Mills, J.P.; Kaye, K.S. Meropenem-vaborbactam: A new weapon in the war against infections due to resistant Gram-negative bacteria. Future Microbiol. 2018, 13, 971–983. [Google Scholar] [CrossRef] [PubMed]
- Viehman, J.A.; Nguyen, M.H.; Doi, Y. Treatment options for carbapenem-resistant and extensively drug-resistant Acinetobacter baumannii infections. Drugs 2014, 74, 1315–1333. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pai, H.; Kim, J.; Kim, J.; Lee, J.H.; Choe, K.W.; Gotoh, N. Carbapenem resistance mechanisms in Pseudomonas aeruginosa clinical isolates. Antimicrob. Agents Chemother. 2001, 45, 480–484. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fish, D.N.; Singletary, T.J. Meropenem, a new carbapenem antibiotic. Pharmacotherapy 1997, 17, 644–669. [Google Scholar] [PubMed]
- Sader, H.S.; Castanheira, M.; Huband, M.; Jones, R.N.; Flamm, R.K. WCK 5222 (Cefepime-Zidebactam) Antimicrobial Activity against Clinical Isolates of Gram-Negative Bacteria Collected Worldwide in 2015. Antimicrob. Agents Chemother. 2017, 61. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Almarzoky Abuhussain, S.S.; Avery, L.M.; Abdelraouf, K.; Nicolau, D.P. In Vivo Efficacy of Humanized WCK 5222 (Cefepime-Zidebactam) Exposures against Carbapenem-Resistant Acinetobacter baumannii in the Neutropenic Thigh Model. Antimicrob. Agents Chemother. 2019, 63. [Google Scholar] [CrossRef] [Green Version]
- Rodvold, K.A.; Gotfried, M.H.; Chugh, R.; Gupta, M.; Patel, A.; Chavan, R.; Yeole, R.; Friedland, H.D.; Bhatia, A. Plasma and Intrapulmonary Concentrations of Cefepime and Zidebactam following Intravenous Administration of WCK 5222 to Healthy Adult Subjects. Antimicrob. Agents Chemother. 2018, 62. [Google Scholar] [CrossRef] [Green Version]
- Bhagwat, S.S.; Periasamy, H.; Takalkar, S.S.; Palwe, S.R.; Khande, H.N.; Patel, M.V. The Novel β-Lactam Enhancer Zidebactam Augments the In Vivo Pharmacodynamic Activity of Cefepime in a Neutropenic Mouse Lung Acinetobacter baumannii Infection Model. Antimicrob. Agents Chemother. 2019, 63. [Google Scholar] [CrossRef] [Green Version]
- Lepak, A.J.; Zhao, M.; Andes, D.R. WCK 5222 (Cefepime/Zidebactam) Pharmacodynamic Target Analysis against Metallo-β-lactamase producing Enterobacteriaceae in the Neutropenic Mouse Pneumonia Model. Antimicrob. Agents Chemother. 2019, 63. [Google Scholar] [CrossRef]
- Avery, L.M.; Abdelraouf, K.; Nicolau, D.P. Assessment of the In Vivo Efficacy of WCK 5222 (Cefepime-Zidebactam) against Carbapenem-Resistant Acinetobacter baumannii in the Neutropenic Murine Lung Infection Model. Antimicrob. Agents Chemother. 2018, 62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moya, B.; Bhagwat, S.; Cabot, G.; Bou, G.; Patel, M.; Oliver, A. Effective inhibition of PBPs by cefepime and zidebactam in the presence of VIM-1 drives potent bactericidal activity against MBL-expressing Pseudomonas aeruginosa. J. Antimicrob. Chemother. 2020, 75, 1474–1478. [Google Scholar] [CrossRef] [PubMed]
- Kidd, J.M.; Abdelraouf, K.; Nicolau, D.P. Efficacy of human-simulated bronchopulmonary exposures of cefepime, zidebactam and the combination (WCK 5222) against MDR Pseudomonas aeruginosa in a neutropenic murine pneumonia model. J. Antimicrob. Chemother. 2020, 75, 149–155. [Google Scholar] [CrossRef] [PubMed]
- Khan, Z.; Iregui, A.; Landman, D.; Quale, J. Activity of cefepime/zidebactam (WCK 5222) against Enterobacteriaceae, Pseudomonas aeruginosa and Acinetobacter baumannii endemic to New York City medical centres. J. Antimicrob. Chemother. 2019, 74, 2938–2942. [Google Scholar] [CrossRef] [PubMed]
- Docquier, J.D.; Mangani, S. An update on β-lactamase inhibitor discovery and development. Drug Resist. Updates Rev. Comment. Antimicrob. Anticancer Chemother. 2018, 36, 13–29. [Google Scholar] [CrossRef] [PubMed]
- Ju, L.C.; Cheng, Z.; Fast, W.; Bonomo, R.A.; Crowder, M.W. The Continuing Challenge of Metallo-β-Lactamase Inhibition: Mechanism Matters. Trends Pharmacol. Sci. 2018, 39, 635–647. [Google Scholar] [CrossRef]
- Hinchliffe, P.; González, M.M.; Mojica, M.F.; González, J.M.; Castillo, V.; Saiz, C.; Kosmopoulou, M.; Tooke, C.L.; Llarrull, L.I.; Mahler, G.; et al. Cross-class metallo-β-lactamase inhibition by bisthiazolidines reveals multiple binding modes. Proc. Natl. Acad. Sci. USA 2016, 113, E3745–E3754. [Google Scholar] [CrossRef] [Green Version]
- Hinchliffe, P.; Tanner, C.A.; Krismanich, A.P.; Labbé, G.; Goodfellow, V.J.; Marrone, L.; Desoky, A.Y.; Calvopiña, K.; Whittle, E.E.; Zeng, F.; et al. Structural and Kinetic Studies of the Potent Inhibition of Metallo-β-lactamases by 6-Phosphonomethylpyridine-2-carboxylates. Biochemistry 2018, 57, 1880–1892. [Google Scholar] [CrossRef]
- Fu, H.; Fang, H.; Sun, J.; Wang, H.; Liu, A.; Sun, J.; Wu, Z. Boronic acid-based enzyme inhibitors: A review of recent progress. Curr. Med. Chem. 2014, 21, 3271–3280. [Google Scholar] [CrossRef]
- Rojas, L.J.; Taracila, M.A.; Papp-Wallace, K.M.; Bethel, C.R.; Caselli, E.; Romagnoli, C.; Winkler, M.L.; Spellberg, B.; Prati, F.; Bonomo, R.A. Boronic Acid Transition State Inhibitors Active against KPC and Other Class A β-Lactamases: Structure-Activity Relationships as a Guide to Inhibitor Design. Antimicrob. Agents Chemother. 2016, 60, 1751–1759. [Google Scholar] [CrossRef] [Green Version]
- Hamrick, J.C.; Docquier, J.D.; Uehara, T.; Myers, C.L.; Six, D.A.; Chatwin, C.L.; John, K.J.; Vernacchio, S.F.; Cusick, S.M.; Trout, R.E.L.; et al. VNRX-5133 (Taniborbactam), a Broad-Spectrum Inhibitor of Serine- and Metallo-β-Lactamases, Restores Activity of Cefepime in Enterobacterales and Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 2020, 64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abdelraouf, K.; Almarzoky Abuhussain, S.; Nicolau, D.P. In vivo pharmacodynamics of new-generation β-lactamase inhibitor taniborbactam (formerly VNRX-5133) in combination with cefepime against serine-β-lactamase-producing Gram-negative bacteria. J. Antimicrob. Chemother. 2020. [Google Scholar] [CrossRef] [PubMed]
- Lang, P.A.; Parkova, A.; Leissing, T.M.; Calvopiña, K.; Cain, R.; Krajnc, A.; Panduwawala, T.D.; Philippe, J.; Fishwick, C.W.G.; Trapencieris, P.; et al. Bicyclic Boronates as Potent Inhibitors of AmpC, the Class C β-Lactamase from Escherichia coli. Biomolecules 2020, 10, 899. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Zhao, C.; Wang, Q.; Wang, Z.; Liang, X.; Zhang, F.; Zhang, Y.; Meng, H.; Chen, H.; Li, S.; et al. In vitro activity of the novel β-lactamase inhibitor taniborbactam (VNRX-5133), in combination with cefepime or meropenem, against MDR Gram-negative bacterial isolates from China. J. Antimicrob. Chemother. 2020, 75, 1850–1858. [Google Scholar] [CrossRef]
- Kurosaki, H.; Yamaguchi, Y.; Higashi, T.; Soga, K.; Matsueda, S.; Yumoto, H.; Misumi, S.; Yamagata, Y.; Arakawa, Y.; Goto, M. Irreversible inhibition of metallo-beta-lactamase (IMP-1) by 3-(3-mercaptopropionylsulfanyl)propionic acid pentafluorophenyl ester. Angew. Chem. Int. Ed. Engl. 2005, 44, 3861–3864. [Google Scholar] [CrossRef]
- Chiou, J.; Wan, S.; Chan, K.F.; So, P.K.; He, D.; Chan, E.W.; Chan, T.; Wong, K.; Tao, J.; Chen, S. Ebselen as a potent covalent inhibitor of New Delhi metallo-β-lactamase (NDM-1). Chem. Commun. 2015, 51, 9543–9546. [Google Scholar] [CrossRef]
- Chen, C.; Xiang, Y.; Yang, K.W.; Zhang, Y.; Wang, W.M.; Su, J.P.; Ge, Y.; Liu, Y. A protein structure-guided covalent scaffold selectively targets the B1 and B2 subclass metallo-β-lactamases. Chem. Commun. 2018, 54, 4802–4805. [Google Scholar] [CrossRef]
- Vrancianu, C.O.; Gheorghe, I.; Czobor, I.B.; Chifiriuc, M.C. Antibiotic Resistance Profiles, Molecular Mechanisms and Innovative Treatment Strategies of Acinetobacter baumannii. Microorganisms 2020, 8, 935. [Google Scholar] [CrossRef]
- Govender, T.; Dawood, A.; Esterhuyse, A.J.; Katerere, D.R. Antimicrobial properties of the skin secretions of frogs. S. Afr. J. Sci. 2012, 108, 25–30. [Google Scholar] [CrossRef] [Green Version]
- Pfalzgraff, A.; Brandenburg, K.; Weindl, G. Antimicrobial Peptides and Their Therapeutic Potential for Bacterial Skin Infections and Wounds. Front. Pharmacol. 2018, 9, 281. [Google Scholar] [CrossRef]
- Björn, C.; Mahlapuu, M.; Mattsby-Baltzer, I.; Håkansson, J. Anti-infective efficacy of the lactoferrin-derived antimicrobial peptide HLR1r. Peptides 2016, 81, 21–28. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Gómez, S.; Ferrer-Espada, R.; Stewart, P.S.; Pitts, B.; Lohner, K.; Martínez de Tejada, G. Antimicrobial activity of synthetic cationic peptides and lipopeptides derived from human lactoferricin against Pseudomonas aeruginosa planktonic cultures and biofilms. BMC Microbiol. 2015, 15, 137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, S.; Long, Q.; Xu, Y.; Wang, J.; Xu, Z.; Wang, L.; Zhou, M.; Wu, Y.; Chen, T.; Shaw, C. Assessment of antimicrobial and wound healing effects of Brevinin-2Ta against the bacterium Klebsiella pneumoniae in dermally-wounded rats. Oncotarget 2017, 8, 111369–111385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Håkansson, J.; Ringstad, L.; Umerska, A.; Johansson, J.; Andersson, T.; Boge, L.; Rozenbaum, R.T.; Sharma, P.K.; Tollbäck, P.; Björn, C.; et al. Characterization of the in vitro, ex vivo, and in vivo Efficacy of the Antimicrobial Peptide DPK-060 Used for Topical Treatment. Front. Cell. Infect. Microbiol. 2019, 9, 174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Du, H.; Puri, S.; McCall, A.; Norris, H.L.; Russo, T.; Edgerton, M. Human Salivary Protein Histatin 5 Has Potent Bactericidal Activity against ESKAPE Pathogens. Front. Cell. Infect. Microbiol. 2017, 7, 41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, J.; Li, Y.; Li, J.; Yan, Z.; Wang, D.; Guo, X.; Zhang, J.; Zhang, B.; Mou, L.; Yang, W.; et al. Potent effects of amino acid scanned antimicrobial peptide Feleucin-K3 analogs against both multidrug-resistant strains and biofilms of Pseudomonas aeruginosa. Amino Acids 2018, 50, 1471–1483. [Google Scholar] [CrossRef]
- Rishi, P.; Vashist, T.; Sharma, A.; Kaur, A.; Kaur, A.; Kaur, N.; Kaur, I.P.; Tewari, R. Efficacy of designer K11 antimicrobial peptide (a hybrid of melittin, cecropin A1 and magainin 2) against Acinetobacter baumannii-infected wounds. Pathog. Dis. 2018, 76. [Google Scholar] [CrossRef] [Green Version]
- Gaglione, R.; Dell’Olmo, E.; Bosso, A.; Chino, M.; Pane, K.; Ascione, F.; Itri, F.; Caserta, S.; Amoresano, A.; Lombardi, A.; et al. Novel human bioactive peptides identified in Apolipoprotein B: Evaluation of their therapeutic potential. Biochem. Pharmacol. 2017, 130, 34–50. [Google Scholar] [CrossRef]
- Wu, C.L.; Hsueh, J.Y.; Yip, B.S.; Chih, Y.H.; Peng, K.L.; Cheng, J.W. Antimicrobial Peptides Display Strong Synergy with Vancomycin Against Vancomycin-Resistant E. faecium, S. aureus, and Wild-Type E. coli. Int. J. Mol. Sci. 2020, 21, 4578. [Google Scholar] [CrossRef]
- Mishra, B.; Wang, G. Titanium surfaces immobilized with the major antimicrobial fragment FK-16 of human cathelicidin LL-37 are potent against multiple antibiotic-resistant bacteria. Biofouling 2017, 33, 544–555. [Google Scholar] [CrossRef]
- Mwangi, J.; Hao, X.; Lai, R.; Zhang, Z.Y. Antimicrobial peptides: New hope in the war against multidrug resistance. Zool. Res. 2019, 40, 488–505. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Hong, J.; Liu, X.; Yang, H.; Liu, R.; Wu, J.; Wang, A.; Lin, D.; Lai, R. Snake cathelicidin from Bungarus fasciatus is a potent peptide antibiotics. PLoS ONE 2008, 3, e3217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Zhang, Z.; Chen, L.; Guang, H.; Li, Z.; Yang, H.; Li, J.; You, D.; Yu, H.; Lai, R. Cathelicidin-BF, a snake cathelicidin-derived antimicrobial peptide, could be an excellent therapeutic agent for acne vulgaris. PLoS ONE 2011, 6, e22120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dhople, V.; Krukemeyer, A.; Ramamoorthy, A. The human beta-defensin-3, an antibacterial peptide with multiple biological functions. Biochim. Biophys. Acta 2006, 1758, 1499–1512. [Google Scholar] [CrossRef] [Green Version]
- Giacometti, A.; Cirioni, O.; Ghiselli, R.; Mocchegiani, F.; D’Amato, G.; Circo, R.; Orlando, F.; Skerlavaj, B.; Silvestri, C.; Saba, V.; et al. Cathelicidin peptide sheep myeloid antimicrobial peptide-29 prevents endotoxin-induced mortality in rat models of septic shock. Am. J. Respir. Crit. Care Med. 2004, 169, 187–194. [Google Scholar] [CrossRef]
- Guo, Y.; Xun, M.; Han, J. A bovine myeloid antimicrobial peptide (BMAP-28) and its analogs kill pan-drug-resistant Acinetobacter baumannii by interacting with outer membrane protein A (OmpA). Medicine 2018, 97, e12832. [Google Scholar] [CrossRef]
- Falla, T.J.; Karunaratne, D.N.; Hancock, R.E. Mode of action of the antimicrobial peptide indolicidin. J. Biol. Chem. 1996, 271, 19298–19303. [Google Scholar] [CrossRef] [Green Version]
- Mensa, B.; Howell, G.L.; Scott, R.; DeGrado, W.F. Comparative mechanistic studies of brilacidin, daptomycin, and the antimicrobial peptide LL16. Antimicrob. Agents Chemother. 2014, 58, 5136–5145. [Google Scholar] [CrossRef] [Green Version]
- Sader, H.S.; Dale, G.E.; Rhomberg, P.R.; Flamm, R.K. Antimicrobial Activity of Murepavadin Tested against Clinical Isolates of Pseudomonas aeruginosa from the United States, Europe, and China. Antimicrob. Agents Chemother. 2018, 62. [Google Scholar] [CrossRef] [Green Version]
- Olivieri, C.; Bugli, F.; Menchinelli, G.; Veglia, G.; Buonocore, F.; Scapigliati, G.; Stocchi, V.; Ceccacci, F.; Papi, M.; Sanguinetti, M.; et al. Design and characterization of chionodracine-derived antimicrobial peptides with enhanced activity against drug-resistant human pathogens. RSC Adv. 2018, 8, 41331–41346. [Google Scholar] [CrossRef] [Green Version]
- Spencer, J.D.; Schwaderer, A.L.; Dirosario, J.D.; McHugh, K.M.; McGillivary, G.; Justice, S.S.; Carpenter, A.R.; Baker, P.B.; Harder, J.; Hains, D.S. Ribonuclease 7 is a potent antimicrobial peptide within the human urinary tract. Kidney Int. 2011, 80, 174–180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iijima, N.; Tanimoto, N.; Emoto, Y.; Morita, Y.; Uematsu, K.; Murakami, T.; Nakai, T. Purification and characterization of three isoforms of chrysophsin, a novel antimicrobial peptide in the gills of the red sea bream, Chrysophrys major. Eur. J. Biochem. 2003, 270, 675–686. [Google Scholar] [CrossRef] [PubMed]
- Panteleev, P.V.; Myshkin, M.Y.; Shenkarev, Z.O.; Ovchinnikova, T.V. Dimerization of the antimicrobial peptide arenicin plays a key role in the cytotoxicity but not in the antibacterial activity. Biochem. Biophys. Res. Commun. 2017, 482, 1320–1326. [Google Scholar] [CrossRef] [PubMed]
- Lee, E.; Shin, A.; Jeong, K.W.; Jin, B.; Jnawali, H.N.; Shin, S.; Shin, S.Y.; Kim, Y. Role of phenylalanine and valine10 residues in the antimicrobial activity and cytotoxicity of piscidin-1. PLoS ONE 2014, 9, e114453. [Google Scholar] [CrossRef] [Green Version]
- Ma, J.; Wang, H.; Wang, Y.; Zhang, S. Endotoxin-neutralizing activity of hen egg phosvitin. Mol. Immunol. 2013, 53, 355–362. [Google Scholar] [CrossRef]
- Zhong, J.; Wang, W.; Yang, X.; Yan, X.; Liu, R. A novel cysteine-rich antimicrobial peptide from the mucus of the snail of Achatina fulica. Peptides 2013, 39, 1–5. [Google Scholar] [CrossRef]
- Al Akeel, R.; Mateen, A.; Syed, R.; Alqahtani, M.S.; Alqahtani, A.S. Alanine rich peptide from Populus trichocarpa inhibit growth of Staphylococcus aureus via targetting its extracellular domain of Sensor Histidine Kinase YycGex protein. Microb. Pathog. 2018, 121, 115–122. [Google Scholar] [CrossRef]
- Wu, G.; Wu, P.; Xue, X.; Yan, X.; Liu, S.; Zhang, C.; Shen, Z.; Xi, T. Application of S-thanatin, an antimicrobial peptide derived from thanatin, in mouse model of Klebsiella pneumoniae infection. Peptides 2013, 45, 73–77. [Google Scholar] [CrossRef]
- Mahlapuu, M.; Håkansson, J.; Ringstad, L.; Björn, C. Antimicrobial Peptides: An Emerging Category of Therapeutic Agents. Front. Cell. Infect. Microbiol. 2016, 6, 194. [Google Scholar] [CrossRef] [Green Version]
- Brunetti, J.; Roscia, G.; Lampronti, I.; Gambari, R.; Quercini, L.; Falciani, C.; Bracci, L.; Pini, A. Immunomodulatory and Anti-inflammatory Activity in Vitro and in Vivo of a Novel Antimicrobial Candidate. J. Biol. Chem. 2016, 291, 25742–25748. [Google Scholar] [CrossRef] [Green Version]
- Chung, P.Y.; Khanum, R. Antimicrobial peptides as potential anti-biofilm agents against multidrug-resistant bacteria. J. Microbiol. Immunol. Infect. 2017, 50, 405–410. [Google Scholar] [CrossRef]
- Di, Y.P.; Lin, Q.; Chen, C.; Montelaro, R.C.; Doi, Y.; Deslouches, B. Enhanced therapeutic index of an antimicrobial peptide in mice by increasing safety and activity against multidrug-resistant bacteria. Sci. Adv. 2020, 6, eaay6817. [Google Scholar] [CrossRef] [PubMed]
- Ballantine, R.D.; McCallion, C.E.; Nassour, E.; Tokajian, S.; Cochrane, S.A. Tridecaptin-inspired antimicrobial peptides with activity against multidrug-resistant Gram-negative bacteria. Medchemcomm 2019, 10, 484–487. [Google Scholar] [CrossRef] [PubMed]
- Mukhopadhyay, S.; Bharath Prasad, A.S.; Mehta, C.H.; Nayak, U.Y. Antimicrobial peptide polymers: No escape to ESKAPE pathogens—A review. World J. Microbiol. Biotechnol. 2020, 36, 131. [Google Scholar] [CrossRef] [PubMed]
- Gheorghe, I.; Saviuc, C.; Ciubuca, B.; Lazar, V.; Chifiriuc, M.C. Chapter 8—Nanodrug delivery. In Nanomaterials for Drug Delivery and Therapy; Grumezescu, A.M., Ed.; William Andrew Publishing: Norwich, NY, USA, 2019; pp. 225–244. ISBN 978-0-12-816505-8. [Google Scholar]
- Sun, H.; Hong, Y.; Xi, Y.; Zou, Y.; Gao, J.; Du, J. Synthesis, Self-Assembly, and Biomedical Applications of Antimicrobial Peptide-Polymer Conjugates. Biomacromolecules 2018, 19, 1701–1720. [Google Scholar] [CrossRef] [PubMed]
- Lee, N.Y.; Ko, W.C.; Hsueh, P.R. Nanoparticles in the Treatment of Infections Caused by Multidrug-Resistant Organisms. Front. Pharmacol. 2019, 10, 1153. [Google Scholar] [CrossRef] [Green Version]
- Hemeg, H.A. Nanomaterials for alternative antibacterial therapy. Int. J. Nanomed. 2017, 12, 8211–8225. [Google Scholar] [CrossRef] [Green Version]
- Chatzimitakos, T.G.; Stalikas, C.D. Qualitative Alterations of Bacterial Metabolome after Exposure to Metal Nanoparticles with Bactericidal Properties: A Comprehensive Workflow Based on (1)H NMR, UHPLC-HRMS, and Metabolic Databases. J. Proteome Res. 2016, 15, 3322–3330. [Google Scholar] [CrossRef]
- Zhao, L.; Ashraf, M.A. Influence of Silver-hydroxyapatite Nanocomposite Coating on Biofilm Formation of Joint Prosthesis and Its Mechanism. West Indian Med. J. 2015, 64, 506–513. [Google Scholar] [CrossRef] [Green Version]
- Xu, Y.; Wei, M.T.; Ou-Yang, H.D.; Walker, S.G.; Wang, H.Z.; Gordon, C.R.; Guterman, S.; Zawacki, E.; Applebaum, E.; Brink, P.R.; et al. Exposure to TiO2 nanoparticles increases Staphylococcus aureus infection of HeLa cells. J. Nanobiotechnol. 2016, 14, 34. [Google Scholar] [CrossRef] [Green Version]
- Garza-Cervantes, J.A.; Mendiola-Garza, G.; de Melo, E.M.; Dugmore, T.I.J.; Matharu, A.S.; Morones-Ramirez, J.R. Antimicrobial activity of a silver-microfibrillated cellulose biocomposite against susceptible and resistant bacteria. Sci. Rep. 2020, 10, 7281. [Google Scholar] [CrossRef] [PubMed]
- Bankalgi, S.C.; Londonkar, R.L.; Madire, U.; Tukappa, N.K.A. Biosynthesis, Characterization and Antibacterial Effect of Phenolics-Coated Silver Nanoparticles Using Cassia javanica L. J. Clust. Sci. 2016, 27, 1485–1497. [Google Scholar] [CrossRef]
- Kedziora, A.; Korzekwa, K.; Strek, W.; Pawlak, A.; Doroszkiewicz, W.; Bugla-Ploskonska, G. Silver Nanoforms as a Therapeutic Agent for Killing Escherichia coli and Certain ESKAPE Pathogens. Curr. Microbiol. 2016, 73, 139–147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baptista, P.V.; McCusker, M.P.; Carvalho, A.; Ferreira, D.A.; Mohan, N.M.; Martins, M.; Fernandes, A.R. Nano-Strategies to Fight Multidrug Resistant Bacteria-“A Battle of the Titans”. Front. Microbiol. 2018, 9, 1441. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Su, H.L.; Lin, S.H.; Wei, J.C.; Pao, I.C.; Chiao, S.H.; Huang, C.C.; Lin, S.Z.; Lin, J.J. Novel nanohybrids of silver particles on clay platelets for inhibiting silver-resistant bacteria. PLoS ONE 2011, 6, e21125. [Google Scholar] [CrossRef]
- Singh, K.; Panghal, M.; Kadyan, S.; Chaudhary, U.; Yadav, J.P. Green silver nanoparticles of Phyllanthus amarus: As an antibacterial agent against multi drug resistant clinical isolates of Pseudomonas aeruginosa. J. Nanobiotechnol. 2014, 12, 40. [Google Scholar] [CrossRef] [Green Version]
- Feroze, N.; Arshad, B.; Younas, M.; Afridi, M.I.; Saqib, S.; Ayaz, A. Fungal mediated synthesis of silver nanoparticles and evaluation of antibacterial activity. Microsc. Res. Tech. 2020, 83, 72–80. [Google Scholar] [CrossRef]
- Gunputh, U.F.; Le, H.; Lawton, K.; Besinis, A.; Tredwin, C.; Handy, R.D. Antibacterial properties of silver nanoparticles grown in situ and anchored to titanium dioxide nanotubes on titanium implant against Staphylococcus aureus. Nanotoxicology 2020, 14, 97–110. [Google Scholar] [CrossRef]
- Algebaly, A.S.; Mohammed, A.E.; Abutaha, N.; Elobeid, M.M. Biogenic synthesis of silver nanoparticles: Antibacterial and cytotoxic potential. Saudi J. Biol. Sci. 2020, 27, 1340–1351. [Google Scholar] [CrossRef]
- Nazer, S.; Andleeb, S.; Ali, S.; Gulzar, N.; Iqbal, T.; Khan, M.A.R.; Raza, A. Synergistic Antibacterial Efficacy of Biogenic Synthesized Silver Nanoparticles using Ajuga bractosa with Standard Antibiotics: A Study Against Bacterial Pathogens. Curr. Pharm. Biotechnol. 2020, 21, 206–218. [Google Scholar] [CrossRef]
- JankauskaitĿ, V.; VitkauskienĿ, A.; Lazauskas, A.; Baltrusaitis, J.; ProsyĿevas, I.; AndruleviĿius, M. Bactericidal effect of graphene oxide/Cu/Ag nanoderivatives against Escherichia coli, Pseudomonas aeruginosa, Klebsiella pneumoniae, Staphylococcus aureus and Methicillin-resistant Staphylococcus aureus. Int. J. Pharm. 2016, 511, 90–97. [Google Scholar] [CrossRef] [PubMed]
- Brown, A.N.; Smith, K.; Samuels, T.A.; Lu, J.; Obare, S.O.; Scott, M.E. Nanoparticles functionalized with ampicillin destroy multiple-antibiotic-resistant isolates of Pseudomonas aeruginosa and Enterobacter aerogenes and methicillin-resistant Staphylococcus aureus. Appl. Environ. Microbiol. 2012, 78, 2768–2774. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, Y.; Chen, Z.; Chen, Y.; Xu, J.; Li, J.; Jiang, X. Synergy of non-antibiotic drugs and pyrimidinethiol on gold nanoparticles against superbugs. J. Am. Chem. Soc. 2013, 135, 12940–12943. [Google Scholar] [CrossRef]
- Ramasamy, M.; Lee, J.H.; Lee, J. Direct one-pot synthesis of cinnamaldehyde immobilized on gold nanoparticles and their antibiofilm properties. Colloids Surf. B Biointerfaces 2017, 160, 639–648. [Google Scholar] [CrossRef]
- Yang, X.; Yang, J.; Wang, L.; Ran, B.; Jia, Y.; Zhang, L.; Yang, G.; Shao, H.; Jiang, X. Pharmaceutical Intermediate-Modified Gold Nanoparticles: Against Multidrug-Resistant Bacteria and Wound-Healing Application via an Electrospun Scaffold. ACS Nano 2017, 11, 5737–5745. [Google Scholar] [CrossRef] [PubMed]
- Yang, P.; Pageni, P.; Rahman, M.A.; Bam, M.; Zhu, T.; Chen, Y.P.; Nagarkatti, M.; Decho, A.W.; Tang, C. Gold Nanoparticles with Antibiotic-Metallopolymers toward Broad-Spectrum Antibacterial Effects. Adv. Healthc. Mater. 2019, 8, e1800854. [Google Scholar] [CrossRef]
- Elbehiry, A.; Al-Dubaib, M.; Marzouk, E.; Moussa, I. Antibacterial effects and resistance induction of silver and gold nanoparticles against Staphylococcus aureus-induced mastitis and the potential toxicity in rats. Microbiologyopen 2019, 8, e00698. [Google Scholar] [CrossRef] [Green Version]
- Aswathanarayan, J.B.; Vittal, R.R. Antimicrobial, Biofilm Inhibitory and Anti-infective Activity of Metallic Nanoparticles Against Pathogens MRSA and Pseudomonas aeruginosa PA01. Pharm. Nanotechnol. 2017, 5, 148–153. [Google Scholar] [CrossRef]
- García-Lara, B.; Saucedo-Mora, M.Á.; Roldán-Sánchez, J.A.; Pérez-Eretza, B.; Ramasamy, M.; Lee, J.; Coria-Jimenez, R.; Tapia, M.; Varela-Guerrero, V.; García-Contreras, R. Inhibition of quorum-sensing-dependent virulence factors and biofilm formation of clinical and environmental Pseudomonas aeruginosa strains by ZnO nanoparticles. Lett. Appl. Microbiol. 2015, 61, 299–305. [Google Scholar] [CrossRef]
- Friedman, A.; Blecher, K.; Sanchez, D.; Tuckman-Vernon, C.; Gialanella, P.; Friedman, J.M.; Martinez, L.R.; Nosanchuk, J.D. Susceptibility of Gram-positive and -negative bacteria to novel nitric oxide-releasing nanoparticle technology. Virulence 2011, 2, 217–221. [Google Scholar] [CrossRef] [Green Version]
- Hetrick, E.M.; Shin, J.H.; Stasko, N.A.; Johnson, C.B.; Wespe, D.A.; Holmuhamedov, E.; Schoenfisch, M.H. Bactericidal efficacy of nitric oxide-releasing silica nanoparticles. ACS Nano 2008, 2, 235–246. [Google Scholar] [CrossRef] [PubMed]
- Khalil, A.T.; Ovais, M.; Ullah, I.; Ali, M.; Shinwari, Z.K.; Maaza, M. Physical properties, biological applications and biocompatibility studies on biosynthesized single phase cobalt oxide (Co3O4) nanoparticles via Sageretia thea (Osbeck.). Arab. J. Chem. 2020, 13, 606–619. [Google Scholar] [CrossRef]
- Dogra, V.; Kaur, G.; Jindal, S.; Kumar, R.; Kumar, S.; Singhal, N.K. Bactericidal effects of metallosurfactants based cobalt oxide/hydroxide nanoparticles against Staphylococcus aureus. Sci. Total Environ. 2019, 681, 350–364. [Google Scholar] [CrossRef] [PubMed]
- Khataminejad, M.R.; Mirnejad, R.; Sharif, M.; Hashemi, M.; Sajadi, N.; Piranfar, V. Antimicrobial Effect of Imipenem-Functionalized Fe(2)O(3) Nanoparticles on Pseudomonas aeruginosa Producing Metallo β-lactamases. Iran. J. Biotechnol. 2015, 13, 43–47. [Google Scholar] [CrossRef] [Green Version]
- Twort, F.W. An investigation on the nature of ultra-microscopic viruses. Lancet 1915, 186, 1241–1243. [Google Scholar] [CrossRef] [Green Version]
- D’Herelle, F. On an invisible microbe antagonistic toward dysenteric bacilli: Brief note by Mr. F. D’Herelle, presented by Mr. Roux. 1917. Res. Microbiol. 2007, 158, 553–554. [Google Scholar] [CrossRef]
- Latz, S.; Wahida, A.; Arif, A.; Häfner, H.; Hoß, M.; Ritter, K.; Horz, H.P. Preliminary survey of local bacteriophages with lytic activity against multi-drug resistant bacteria. J. Basic Microbiol. 2016, 56, 1117–1123. [Google Scholar] [CrossRef]
- Domingo-Calap, P.; Delgado-Martínez, J. Bacteriophages: Protagonists of a Post-Antibiotic Era. Antibiotics 2018, 7, 66. [Google Scholar] [CrossRef] [Green Version]
- Pirnay, J.P.; Verbeken, G.; Ceyssens, P.J.; Huys, I.; De Vos, D.; Ameloot, C.; Fauconnier, A. The Magistral Phage. Viruses 2018, 10, 64. [Google Scholar] [CrossRef] [Green Version]
- Fadlallah, A.; Chelala, E.; Legeais, J.M. Corneal Infection Therapy with Topical Bacteriophage Administration. Open Ophthalmol. J. 2015, 9, 167–168. [Google Scholar] [CrossRef] [Green Version]
- Schooley, R.T.; Biswas, B.; Gill, J.J.; Hernandez-Morales, A.; Lancaster, J.; Lessor, L.; Barr, J.J.; Reed, S.L.; Rohwer, F.; Benler, S.; et al. Development and Use of Personalized Bacteriophage-Based Therapeutic Cocktails To Treat a Patient with a Disseminated Resistant Acinetobacter baumannii Infection. Antimicrob. Agents Chemother. 2017, 61. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fish, R.; Kutter, E.; Wheat, G.; Blasdel, B.; Kutateladze, M.; Kuhl, S. Bacteriophage treatment of intransigent diabetic toe ulcers: A case series. J. Wound Care 2016, 25, S27–S33. [Google Scholar] [CrossRef]
- Leitner, L.; Sybesma, W.; Chanishvili, N.; Goderdzishvili, M.; Chkhotua, A.; Ujmajuridze, A.; Schneider, M.P.; Sartori, A.; Mehnert, U.; Bachmann, L.M.; et al. Bacteriophages for treating urinary tract infections in patients undergoing transurethral resection of the prostate: A randomized, placebo-controlled, double-blind clinical trial. BMC Urol. 2017, 17, 90. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ujmajuridze, A.; Chanishvili, N.; Goderdzishvili, M.; Leitner, L.; Mehnert, U.; Chkhotua, A.; Kessler, T.M.; Sybesma, W. Adapted Bacteriophages for Treating Urinary Tract Infections. Front. Microbiol. 2018, 9, 1832. [Google Scholar] [CrossRef] [Green Version]
- Markoishvili, K.; Tsitlanadze, G.; Katsarava, R.; Morris, J.G.J.; Sulakvelidze, A. A novel sustained-release matrix based on biodegradable poly(ester amide)s and impregnated with bacteriophages and an antibiotic shows promise in management of infected venous stasis ulcers and other poorly healing wounds. Int. J. Dermatol. 2002, 41, 453–458. [Google Scholar] [CrossRef] [PubMed]
- Chan, B.K.; Abedon, S.T.; Loc-Carrillo, C. Phage cocktails and the future of phage therapy. Future Microbiol. 2013, 8, 769–783. [Google Scholar] [CrossRef] [PubMed]
- Rohde, C.; Wittmann, J.; Kutter, E. Bacteriophages: A Therapy Concept against Multi-Drug-Resistant Bacteria. Surg. Infect. 2018, 19, 737–744. [Google Scholar] [CrossRef] [Green Version]
- Casey, E.; van Sinderen, D.; Mahony, J. In Vitro Characteristics of Phages to Guide “Real Life” Phage Therapy Suitability. Viruses 2018, 10, 163. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Zheng, P.; Ji, W.; Fu, Q.; Wang, H.; Yan, Y.; Sun, J. SLPW: A Virulent Bacteriophage Targeting Methicillin-Resistant Staphylococcus aureus In vitro and In vivo. Front. Microbiol. 2016, 7, 934. [Google Scholar] [CrossRef] [Green Version]
- Zhou, W.; Feng, Y.; Zong, Z. Two New Lytic Bacteriophages of the Myoviridae Family Against Carbapenem-Resistant Acinetobacter baumannii. Front. Microbiol. 2018, 9, 850. [Google Scholar] [CrossRef]
- Cooper, C.J.; Koonjan, S.; Nilsson, A.S. Enhancing Whole Phage Therapy and Their Derived Antimicrobial Enzymes through Complex Formulation. Pharmaceuticals 2018, 11, 34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, M.; Hu, K.; Xie, Y.; Liu, Y.; Mu, D.; Guo, H.; Zhang, Z.; Zhang, Y.; Chang, D.; Shi, Y. A Novel Phage PD-6A3, and Its Endolysin Ply6A3, With Extended Lytic Activity Against Acinetobacter baumannii. Front. Microbiol. 2018, 9, 3302. [Google Scholar] [CrossRef] [PubMed]
- Malik, D.J.; Sokolov, I.J.; Vinner, G.K.; Mancuso, F.; Cinquerrui, S.; Vladisavljevic, G.T.; Clokie, M.R.J.; Garton, N.J.; Stapley, A.G.F.; Kirpichnikova, A. Formulation, stabilisation and encapsulation of bacteriophage for phage therapy. Adv. Colloid Interface Sci. 2017, 249, 100–133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Biswas, B.; Adhya, S.; Washart, P.; Paul, B.; Trostel, A.N.; Powell, B.; Carlton, R.; Merril, C.R. Bacteriophage therapy rescues mice bacteremic from a clinical isolate of vancomycin-resistant Enterococcus faecium. Infect. Immun. 2002, 70, 204–210. [Google Scholar] [CrossRef] [Green Version]
- Manohar, P.; Nachimuthu, R.; Lopes, B.S. The therapeutic potential of bacteriophages targeting gram-negative bacteria using Galleria mellonella infection model. BMC Microbiol. 2018, 18, 97. [Google Scholar] [CrossRef] [Green Version]
- Uchiyama, J.; Matsui, H.; Murakami, H.; Kato, S.I.; Watanabe, N.; Nasukawa, T.; Mizukami, K.; Ogata, M.; Sakaguchi, M.; Matsuzaki, S.; et al. Potential Application of Bacteriophages in Enrichment Culture for Improved Prenatal Streptococcus agalactiae Screening. Viruses 2018, 10, 552. [Google Scholar] [CrossRef] [Green Version]
- Tinoco, J.M.; Buttaro, B.; Zhang, H.; Liss, N.; Sassone, L.; Stevens, R. Effect of a genetically engineered bacteriophage on Enterococcus faecalis biofilms. Arch. Oral Biol. 2016, 71, 80–86. [Google Scholar] [CrossRef] [Green Version]
- Khalifa, L.; Brosh, Y.; Gelman, D.; Coppenhagen-Glazer, S.; Beyth, S.; Poradosu-Cohen, R.; Que, Y.A.; Beyth, N.; Hazan, R. Targeting Enterococcus faecalis biofilms with phage therapy. Appl. Environ. Microbiol. 2015, 81, 2696–2705. [Google Scholar] [CrossRef] [Green Version]
- Barros, J.; Melo, L.D.R.; Poeta, P.; Igrejas, G.; Ferraz, M.P.; Azeredo, J.; Monteiro, F.J. Lytic bacteriophages against multidrug-resistant Staphylococcus aureus, Enterococcus faecalis and Escherichia coli isolates from orthopaedic implant-associated infections. Int. J. Antimicrob. Agents 2019, 54, 329–337. [Google Scholar] [CrossRef] [Green Version]
- Prazak, J.; Iten, M.; Cameron, D.R.; Save, J.; Grandgirard, D.; Resch, G.; Goepfert, C.; Leib, S.L.; Takala, J.; Jakob, S.M.; et al. Bacteriophages Improve Outcomes in Experimental Staphylococcus aureus Ventilator-associated Pneumonia. Am. J. Respir. Crit. Care Med. 2019, 200, 1126–1133. [Google Scholar] [CrossRef]
- Kaur, S.; Harjai, K.; Chhibber, S. In Vivo Assessment of Phage and Linezolid Based Implant Coatings for Treatment of Methicillin Resistant S. aureus (MRSA) Mediated Orthopaedic Device Related Infections. PLoS ONE 2016, 11, e0157626. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petrovic Fabijan, A.; Lin, R.C.Y.; Ho, J.; Maddocks, S.; Ben Zakour, N.L.; Iredell, J.R. Safety of bacteriophage therapy in severe Staphylococcus aureus infection. Nat. Microbiol. 2020, 5, 465–472. [Google Scholar] [CrossRef] [PubMed]
- Thiry, D.; Passet, V.; Danis-Wlodarczyk, K.; Lood, C.; Wagemans, J.; De Sordi, L.; van Noort, V.; Dufour, N.; Debarbieux, L.; Mainil, J.G.; et al. New Bacteriophages against Emerging Lineages ST23 and ST258 of Klebsiella pneumoniae and Efficacy Assessment in Galleria mellonella Larvae. Viruses 2019, 11, 411. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patel, D.R.; Bhartiya, S.K.; Kumar, R.; Shukla, V.K.; Nath, G. Use of Customized Bacteriophages in the Treatment of Chronic Nonhealing Wounds: A Prospective Study. Int. J. Low. Extrem. Wounds 2019. [Google Scholar] [CrossRef]
- Manohar, P.; Tamhankar, A.J.; Lundborg, C.S.; Nachimuthu, R. Therapeutic Characterization and Efficacy of Bacteriophage Cocktails Infecting Escherichia coli, Klebsiella pneumoniae, and Enterobacter Species. Front. Microbiol. 2019, 10, 574. [Google Scholar] [CrossRef] [Green Version]
- Chaturvedi, A.; Nath, G. Oral administration of Klebsiella pneumoniae-specific bacteriophage eradicates the bacteria in albino mice. Indian J. Med. Microbiol. 2018, 36, 293–294. [Google Scholar]
- Cano, E.J.; Caflisch, K.M.; Bollyky, P.L.; Van Belleghem, J.D.; Patel, R.; Fackler, J.; Brownstein, M.J.; Horne, B.; Biswas, B.; Henry, M.; et al. Phage Therapy for Limb-threatening Prosthetic Knee Klebsiella pneumoniae Infection: Case Report and In Vitro Characterization of Anti-biofilm Activity. Clin. Infect. Dis. 2020. [Google Scholar] [CrossRef]
- Corbellino, M.; Kieffer, N.; Kutateladze, M.; Balarjishvili, N.; Leshkasheli, L.; Askilashvili, L.; Tsertsvadze, G.; Rimoldi, S.G.; Nizharadze, D.; Hoyle, N.; et al. Eradication of a Multidrug-Resistant, Carbapenemase-Producing Klebsiella pneumoniae Isolate Following Oral and Intra-rectal Therapy With a Custom Made, Lytic Bacteriophage Preparation. Clin. Infect. Dis. 2020, 70, 1998–2001. [Google Scholar] [CrossRef]
- Duplessis, C.; Biswas, B.; Hanisch, B.; Perkins, M.; Henry, M.; Quinones, J.; Wolfe, D.; Estrella, L.; Hamilton, T. Refractory Pseudomonas Bacteremia in a 2-Year-Old Sterilized by Bacteriophage Therapy. J. Pediatr. Infect. Dis. Soc. 2018, 7, 253–256. [Google Scholar] [CrossRef] [Green Version]
- Jault, P.; Leclerc, T.; Jennes, S.; Pirnay, J.P.; Que, Y.A.; Resch, G.; Rousseau, A.F.; Ravat, F.; Carsin, H.; Le Floch, R.; et al. Efficacy and tolerability of a cocktail of bacteriophages to treat burn wounds infected by Pseudomonas aeruginosa (PhagoBurn): A randomised, controlled, double-blind phase 1/2 trial. Lancet Infect. Dis. 2019, 19, 35–45. [Google Scholar] [CrossRef]
- Cha, K.; Oh, H.K.; Jang, J.Y.; Jo, Y.; Kim, W.K.; Ha, G.U.; Ko, K.S.; Myung, H. Characterization of Two Novel Bacteriophages Infecting Multidrug-Resistant (MDR) Acinetobacter baumannii and Evaluation of Their Therapeutic Efficacy in Vivo. Front. Microbiol. 2018, 9, 696. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jeon, J.; Park, J.H.; Yong, D. Efficacy of bacteriophage treatment against carbapenem-resistant Acinetobacter baumannii in Galleria mellonella larvae and a mouse model of acute pneumonia. BMC Microbiol. 2019, 19, 70. [Google Scholar] [CrossRef] [PubMed]
- Jasim, H.N.; Hafidh, R.R.; Abdulamir, A.S. Formation of therapeutic phage cocktail and endolysin to highly multi-drug resistant Acinetobacter baumannii: In vitro and in vivo study. Iran. J. Basic Med. Sci. 2018, 21, 1100–1108. [Google Scholar] [CrossRef] [PubMed]
- Maddocks, S.; Fabijan, A.P.; Ho, J.; Lin, R.C.Y.; Ben Zakour, N.L.; Dugan, C.; Kliman, I.; Branston, S.; Morales, S.; Iredell, J.R. Bacteriophage Therapy of Ventilator-associated Pneumonia and Empyema Caused by Pseudomonas aeruginosa. Am. J. Respir. Crit. Care Med. 2019, 200, 1179–1181. [Google Scholar] [CrossRef]
- Bikard, D.; Euler, C.W.; Jiang, W.; Nussenzweig, P.M.; Goldberg, G.W.; Duportet, X.; Fischetti, V.A.; Marraffini, L.A. Exploiting CRISPR-Cas nucleases to produce sequence-specific antimicrobials. Nat. Biotechnol. 2014, 32, 1146–1150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crawley, A.B.; Henriksen, E.D.; Stout, E.; Brandt, K.; Barrangou, R. Characterizing the activity of abundant, diverse and active CRISPR-Cas systems in lactobacilli. Sci. Rep. 2018, 8, 11544. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walker, F.C.; Hatoum-Aslan, A. Conjugation Assay for Testing CRISPR-Cas Anti-plasmid Immunity in Staphylococci. Bio-Protocol 2017, 7. [Google Scholar] [CrossRef] [Green Version]
- Mangas, E.L.; Rubio, A.; Álvarez-Marín, R.; Labrador-Herrera, G.; Pachón, J.; Pachón-Ibáñez, M.E.; Divina, F.; Pérez-Pulido, A.J. Pangenome of Acinetobacter baumannii uncovers two groups of genomes, one of them with genes involved in CRISPR/Cas defence systems associated with the absence of plasmids and exclusive genes for biofilm formation. Microb. Genom. 2019, 5. [Google Scholar] [CrossRef]
- Wang, Y.; Wang, Z.; Chen, Y.; Hua, X.; Yu, Y.; Ji, Q. A Highly Efficient CRISPR-Cas9-Based Genome Engineering Platform in Acinetobacter baumannii to Understand the H(2)O(2)-Sensing Mechanism of OxyR. Cell Chem. Biol. 2019, 26, 1732–1742. [Google Scholar] [CrossRef] [Green Version]
- Tagliaferri, T.L.; Guimarães, N.R.; de Paula Martins Pereira, M.; Vilela, L.F.F.; Horz, H.P.; Dos Santos, S.G.; Mendes, T.A. Exploring the Potential of CRISPR-Cas9 Under Challenging Conditions: Facing High-Copy Plasmids and Counteracting Beta-Lactam Resistance in Clinical Strains of Enterobacteriaceae. Front. Microbiol. 2020, 11, 578. [Google Scholar] [CrossRef]
- Sun, Q.; Wang, Y.; Dong, N.; Shen, L.; Zhou, H.; Hu, Y.; Gu, D.; Chen, S.; Zhang, R.; Ji, Q. Application of CRISPR/Cas9-Based Genome Editing in Studying the Mechanism of Pandrug Resistance in Klebsiella pneumoniae. Antimicrob. Agents Chemother. 2019, 63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Wang, S.; Chen, W.; Song, L.; Zhang, Y.; Shen, Z.; Yu, F.; Li, M.; Ji, Q. CRISPR-Cas9 and CRISPR-Assisted Cytidine Deaminase Enable Precise and Efficient Genome Editing in Klebsiella pneumoniae. Appl. Environ. Microbiol. 2018, 84. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hao, M.; He, Y.; Zhang, H.; Liao, X.P.; Liu, Y.H.; Sun, J.; Du, H.; Kreiswirth, B.N.; Chen, L. CRISPR-Cas9-Mediated Carbapenemase Gene and Plasmid Curing in Carbapenem-Resistant Enterobacteriaceae. Antimicrob. Agents Chemother. 2020, 64. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Jiang, Y.; Shao, L.; Yang, P.; Sun, B.; Yang, S.; Chen, D. CRISPR/Cas9-based efficient genome editing in Staphylococcus aureus. Acta Biochim. Biophys. Sin. 2017, 49, 764–770. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, W.; Zhang, Y.; Zhang, Y.; Pi, Y.; Gu, T.; Song, L.; Wang, Y.; Ji, Q. CRISPR/Cas9-based Genome Editing in Pseudomonas aeruginosa and Cytidine Deaminase-Mediated Base Editing in Pseudomonas Species. iScience 2018, 6, 222–231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pokharel, K.; Dawadi, B.R.; Bhatt, C.P.; Gupte, S. Prevalence of Pseudomonas Aeruginosa and its Antibiotic Sensitivity Pattern. J. Nepal Health Res. Counc. 2019, 17, 109–113. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Li, M.; Li, Y.; Cao, H.; Miao, L.; Xu, Z.; Higuchi, Y.; Yamasaki, S.; Nishino, K.; Woo, P.C.Y.; et al. Native CRISPR-Cas-Mediated Genome Editing Enables Dissecting and Sensitizing Clinical Multidrug-Resistant P. aeruginosa. Cell Rep. 2019, 29, 1707. [Google Scholar] [CrossRef]
- Rosini, R.; Nicchi, S.; Pizza, M.; Rappuoli, R. Vaccines Against Antimicrobial Resistance. Front. Immunol. 2020, 11, 1048. [Google Scholar] [CrossRef]
- Shu, M.H.; MatRahim, N.; NorAmdan, N.; Pang, S.P.; Hashim, S.H.; Phoon, W.H.; AbuBakar, S. An Inactivated Antibiotic-Exposed Whole-Cell Vaccine Enhances Bactericidal Activities Against Multidrug-Resistant Acinetobacter baumannii. Sci. Rep. 2016, 6, 22332. [Google Scholar] [CrossRef] [Green Version]
- Badmasti, F.; Ajdary, S.; Bouzari, S.; Fooladi, A.A.I.; Shahcheraghi, F.; Siadat, S.D. Immunological evaluation of OMV(PagL)+Bap(1-487aa) and AbOmpA(8-346aa)+Bap(1-487aa) as vaccine candidates against Acinetobacter baumannii sepsis infection. Mol. Immunol. 2015, 67, 552–558. [Google Scholar] [CrossRef]
- Pulido, M.R.; García-Quintanilla, M.; Pachón, J.; McConnell, M.J. Immunization with lipopolysaccharide-free outer membrane complexes protects against Acinetobacter baumannii infection. Vaccine 2018, 36, 4153–4156. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Yang, T.; Cao, J.; Sun, J.; Dai, W.; Zhang, L. Mucosal immunization with purified OmpA elicited protective immunity against infections caused by multidrug-resistant Acinetobacter baumannii. Microb. Pathog. 2016, 96, 20–25. [Google Scholar] [CrossRef] [PubMed]
- Gerritzen, M.J.H.; Salverda, M.L.M.; Martens, D.E.; Wijffels, R.H.; Stork, M. Spontaneously released Neisseria meningitidis outer membrane vesicles as vaccine platform: Production and purification. Vaccine 2019, 37, 6978–6986. [Google Scholar] [CrossRef] [PubMed]
- Klimentová, J.; Stulík, J. Methods of isolation and purification of outer membrane vesicles from gram-negative bacteria. Microbiol. Res. 2015, 170, 1–9. [Google Scholar] [CrossRef]
- Gerritzen, M.J.H.; Maas, R.H.W.; van den Ijssel, J.; van Keulen, L.; Martens, D.E.; Wijffels, R.H.; Stork, M. High dissolved oxygen tension triggers outer membrane vesicle formation by Neisseria meningitidis. Microb. Cell Fact. 2018, 17, 157. [Google Scholar] [CrossRef]
- Chen, G.; Bai, Y.; Li, Z.; Wang, F.; Fan, X.; Zhou, X. Bacterial extracellular vesicle-coated multi-antigenic nanovaccines protect against drug-resistant Staphylococcus aureus infection by modulating antigen processing and presentation pathways. Theranostics 2020, 10, 7131–7149. [Google Scholar] [CrossRef]
- Bahey-El-Din, M.; Mohamed, S.A.; Sheweita, S.A.; Haroun, M.; Zaghloul, T.I. Recombinant N-terminal outer membrane porin (OprF) of Pseudomonas aeruginosa is a promising vaccine candidate against both P. aeruginosa and some strains of Acinetobacter baumannii. Int. J. Med. Microbiol. 2020, 310, 151415. [Google Scholar] [CrossRef]
- Hernandez, D.N.; Tam, K.; Shopsin, B.; Radke, E.E.; Law, K.; Cardozo, T.; Torres, V.J.; Silverman, G.J. Convergent Evolution of Neutralizing Antibodies to Staphylococcus aureus γ-Hemolysin C That Recognize an Immunodominant Primary Sequence-Dependent B-Cell Epitope. MBio 2020, 11. [Google Scholar] [CrossRef]
- Soltan, M.A.; Magdy, D.; Solyman, S.M.; Hanora, A. Design of Staphylococcus aureus New Vaccine Candidates with B and T Cell Epitope Mapping, Reverse Vaccinology, and Immunoinformatics. OMICS 2020, 24, 195–204. [Google Scholar] [CrossRef]
- Rostamian, M.; Farasat, A.; Chegene Lorestani, R.; Nemati Zargaran, F.; Ghadiri, K.; Akya, A. Immunoinformatics and molecular dynamics studies to predict T-cell-specific epitopes of four Klebsiella pneumoniae fimbriae antigens. J. Biomol. Struct. Dyn. 2020. [Google Scholar] [CrossRef]
- Rodrigues, M.X.; Yang, Y.; de Souza Meira, E.B.J.; do Carmo Silva, J.; Bicalho, R.C. Development and evaluation of a new recombinant protein vaccine (YidR) against Klebsiella pneumoniae infection. Vaccine 2020, 38, 4640–4648. [Google Scholar] [CrossRef] [PubMed]
- Malachowa, N.; Kobayashi, S.D.; Porter, A.R.; Freedman, B.; Hanley, P.W.; Lovaglio, J.; Saturday, G.A.; Gardner, D.J.; Scott, D.P.; Griffin, A.; et al. Vaccine Protection against Multidrug-Resistant Klebsiella pneumoniae in a Nonhuman Primate Model of Severe Lower Respiratory Tract Infection. MBio 2019, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Creech, C.B.; Frenck, R.W.J.; Sheldon, E.A.; Seiden, D.J.; Kankam, M.K.; Zito, E.T.; Girgenti, D.; Severs, J.M.; Immermann, F.W.; McNeil, L.K.; et al. Safety, tolerability, and immunogenicity of a single dose 4-antigen or 3-antigen Staphylococcus aureus vaccine in healthy older adults: Results of a randomised trial. Vaccine 2017, 35, 385–394. [Google Scholar] [CrossRef] [PubMed]
- Creech, C.B.; Frenck, R.W.; Fiquet, A.; Feldman, R.; Kankam, M.K.; Pathirana, S.; Baber, J.; Radley, D.; Cooper, D.; Eiden, J.; et al. Persistence of Immune Responses Through 36 Months in Healthy Adults After Vaccination with a Novel Staphylococcus aureus 4-Antigen Vaccine (SA4Ag). Open Forum Infect. Dis. 2019, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seeberger, P.H.; Pereira, C.L.; Khan, N.; Xiao, G.; Diago-Navarro, E.; Reppe, K.; Opitz, B.; Fries, B.C.; Witzenrath, M. A Semi-Synthetic Glycoconjugate Vaccine Candidate for Carbapenem-Resistant Klebsiella pneumoniae. Angew. Chem. Int. Ed. Engl. 2017, 56, 13973–13978. [Google Scholar] [CrossRef] [Green Version]
- Hoggarth, A.; Weaver, A.; Pu, Q.; Huang, T.; Schettler, J.; Chen, F.; Yuan, X.; Wu, M. Mechanistic research holds promise for bacterial vaccines and phage therapies for Pseudomonas aeruginosa. Drug Des. Devel. Ther. 2019, 13, 909–924. [Google Scholar] [CrossRef] [Green Version]
- Ainsworth, S.; Ketter, P.M.; Yu, J.J.; Grimm, R.C.; May, H.C.; Cap, A.P.; Chambers, J.P.; Guentzel, M.N.; Arulanandam, B.P. Vaccination with a live attenuated Acinetobacter baumannii deficient in thioredoxin provides protection against systemic Acinetobacter infection. Vaccine 2017, 35, 3387–3394. [Google Scholar] [CrossRef]
- Bidmos, F.A.; Siris, S.; Gladstone, C.A.; Langford, P.R. Bacterial Vaccine Antigen Discovery in the Reverse Vaccinology 2.0 Era: Progress and Challenges. Front. Immunol. 2018, 9, 2315. [Google Scholar] [CrossRef] [Green Version]





{kind=link}
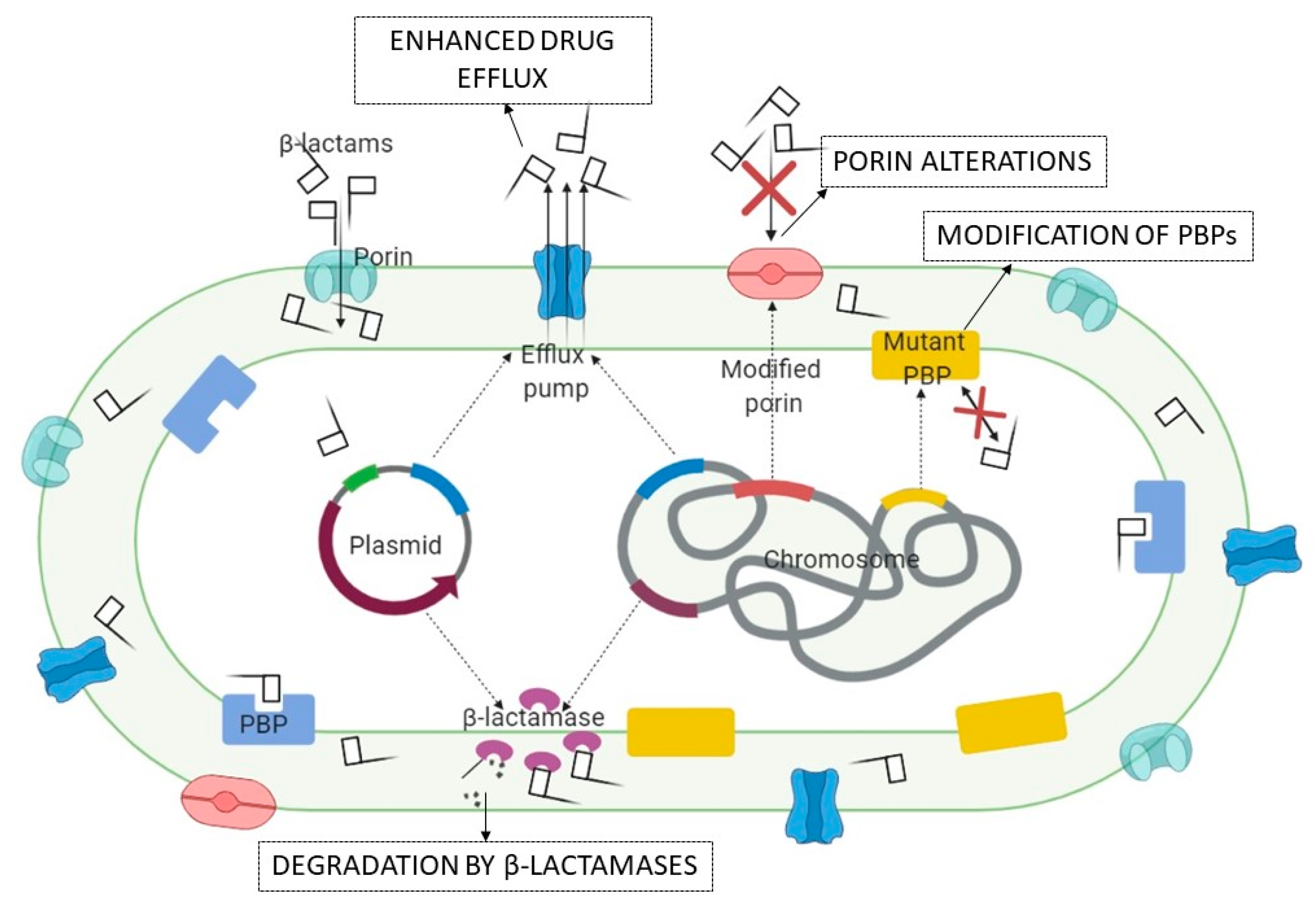{kind=link}
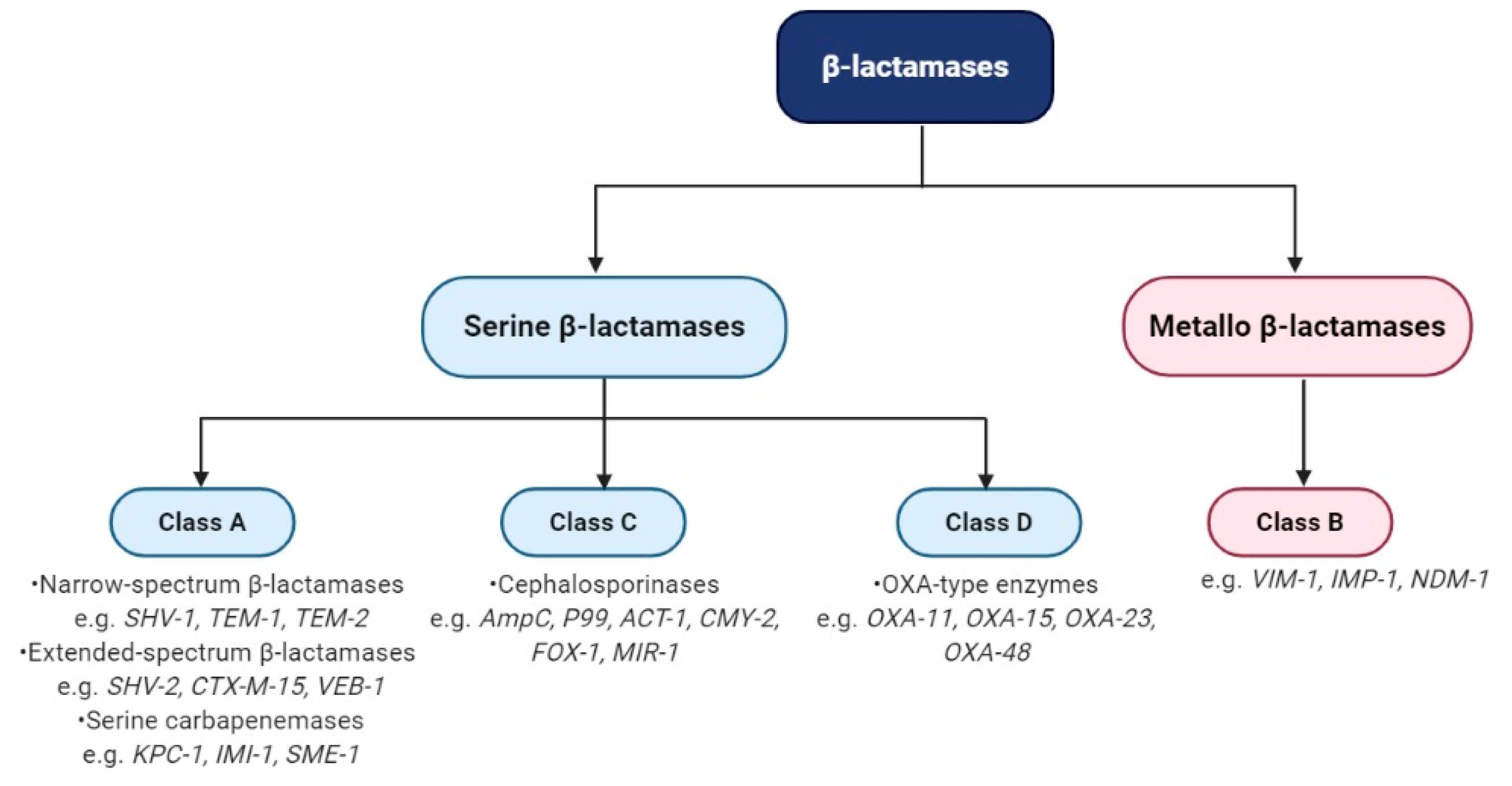{kind=link}
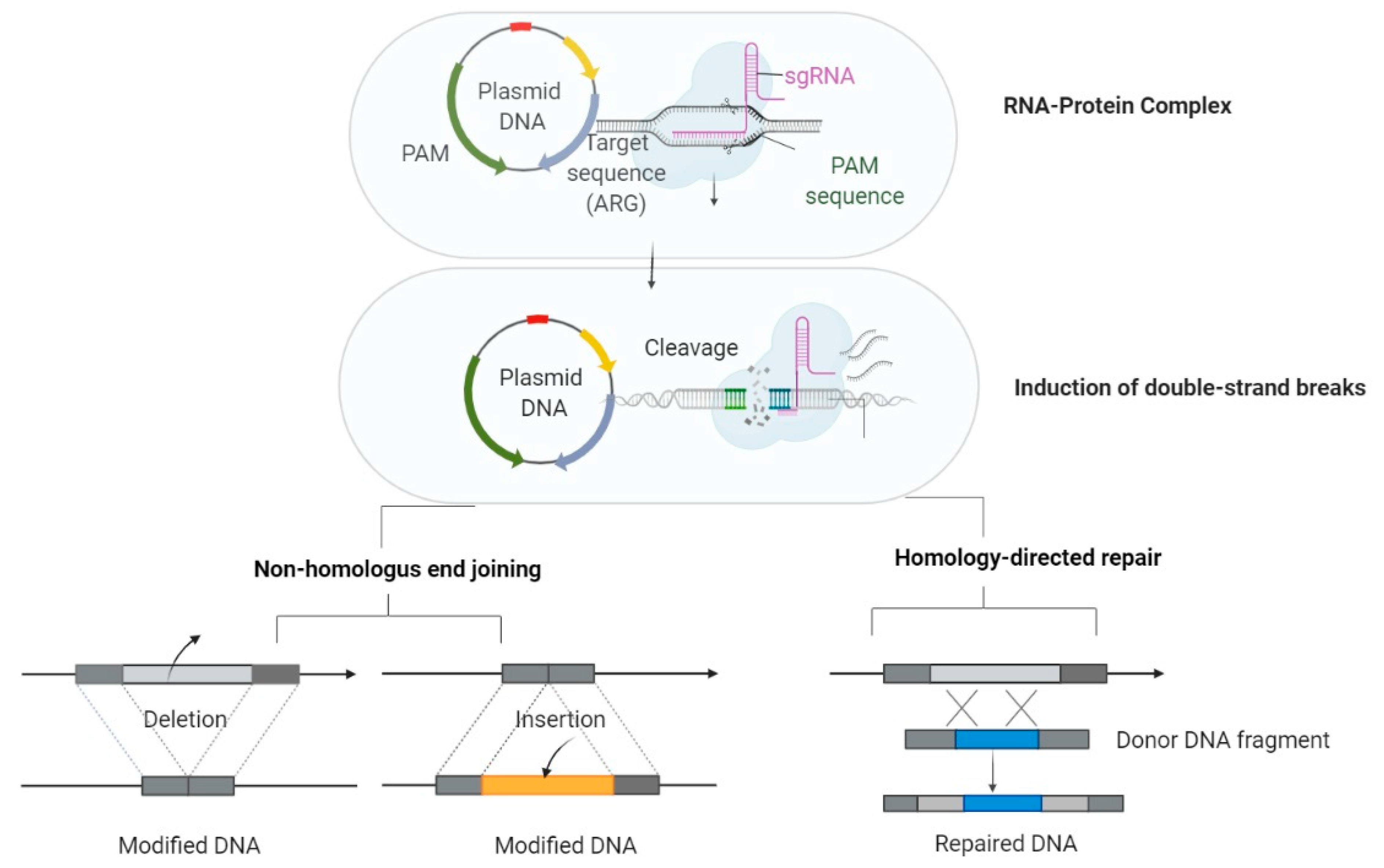{kind=link}
| Agent(s). | Class A | Class B | Class C | Class D |
|---|---|---|---|---|
| CAZ-AVI | ||||
| MER-VAB | ||||
| IMI-REL | ||||
| CEF-TAZ | ||||
| Cefiderocol |
| AMPs | Main Activity | Other Effects | Animal Models | References |
|---|---|---|---|---|
| HLR1–human derived lactoferin peptide | in vitro—microbicidal effect against S. aureus | anti-inflammatory properties non-cytotoxic effect | mice, rats, and pig skin infected with S. aureus | [203] |
| Lactoferrin and Lactoferrin derived AMPs | in vitro—antibacterial activity against E. coli, S. aureus, Acinetobacter spp., P. aeruginosa | anti-biofilm against P. aeruginosa strains | mice | [204] |
| Brevinin-2Ta (B-2Ta) | in vitro—antimicrobial activities against S. aureus, E. coli | low cytotoxicity inflammatory effect in vivo using K. pneumoniae-infected Sprague-Dawley rats | rats | [205] |
| DPK-060 structurally derived from human protein kininogen | in vitro—antimicrobial activity against S. aureus including MRSA | ex vivo pig skin in vivo—mouses | [206] | |
| Histatin 5—human salivary AMP | in vitro—antibacterial activity against S. aureus, A. baumannii, E. cloacae, K. pneumoniae and P. aeruginosa | anti-biofilm activity | [207] | |
| Feleucin-K3 AMP and his analogue FK-1D | in vitro antimicrobial activity against P. aeruginosa | low-toxicity anti-biofilm activity | in vivo against clinical infections caused by P. aeruginosa | [208] |
| K11 hybrid AMP | in vivo—antimicrobial activity against A. baumannii-infected wounds (murine excision) | [209] | ||
| (P)ApoBL and r(P)ApoBS—Apolipoproin B human defence AMPs | in vitro antimicrobial activity aginst MRSA and P. aeruginosa | anti-biofilm activity anti-inflamatory activity | murine | [210] |
| Bip-P113 [Bip: β-(4.4′-biphenyl)alanine] AMP | in vitro antimicrobial activity against S. aureus and E. faecium | [211] | ||
| LL-37, a 37-residue AMP derived from human cathelicidin and his derivate FK-16 titanium coated | in vitro antimicrobial activity against ESKAPE patrogens particularly microbicidal effect on P. aeruginosa, MRSA and A. baumannii | anti-adhesion anti-biofilm activities against S. aureus, P. aeruginosa, and A. baumannii | mice model | [212,213] |
| Cathelicidin-BF | in vitro antimicrobial activity against S. aureus and P. aeruginosa | low hemolytic activity on red blood cells; therapeutic potential against acne vulgaris | [214,215] | |
| hBD-3-human-β defensin 3; AMP-29- a sheep myeloid peptide; rCRAMP- a rat cathelin-derived AMP; BMAP-27- a bovine myeloid AMP- 27 | in vitro microbicidal activity against A. baumannii, P. aeruginosa, and MRSA | anti-biofilm activity anti- immunomodulatory activity | [216,217,218] | |
| Indolicidin | in vitro bactericidal activity against P. aeruginosa and S. aureus | [219] | ||
| PMX-30063 (brilacidin) | in vitro bactericidal activity against S. aureus | [220] | ||
| POL7080 (murepavadin) | in vitro antimicrobial activity against MDR and XDR P. aeruginosa | [221] | ||
| LTX-109 (lytixar) | in vitro bactericidal activity against S. aureus | mouse skin infection model | [164] | |
| chionodracine-derivatives AMPs | in vitro bactericidal activity against K. pneumoniae, A. baumannii, MRSA and P. aeruginosa | [222] | ||
| Ribonuclease 7 AMP | in vitro antimicrobial activity against P. aeruginosa, S. aureus, and VRE | [223] | ||
| Chrysophsin-1 isolated from the gill cells of Chrysophrys major | in vitro antimicrobial activity against MRSA | antiendotoxin properties | [224] | |
| Arenicins-1 isolated from Arenicola marina and one of his variants Ar-1[V8R] | in vitro antimicrobial activity against P. aeruginosa, K. pneumoniae and S. aureus | Ar-1[V8R]—cytotoxicity against mammalian cells | [225] | |
| Pardaxins isolated from mucous glands of Pardachirus marmoratus | in vitro antimicrobial activity against S. aureus, A. calcoaceticus and P. aeruginosa | [226] | ||
| Phosvitin from zebrafish | in vitro antimicrobial activity against S. aureus | immunomodulatory activity; non-cytotoxic and non-hemolytic | mice model | [227] |
| Mytimacin-AF, isolated from marine mollusks | in vitro antimicrobial activity against S. aureus and K. pneumoniae | [228] | ||
| PT-3 Populus trichocarpa crude extract derived AMP | in vitro antimicrobial activity against S. aureus | in vivo antibacterial activity in S. aureus infected G. mellonella model | [229] | |
| Thanatin and its analog, S-thanatin | in vitro antimicrobial activity against K. pneumoniae | low hemolytic activity | mice model | [230] |
| Pexiganan—a synthetic analog of magainin isolated from Xenopus laevis | in vitro bactericidal effect against P. aeruginosa | [231] | ||
| SET-M33 a synthetic AMPs (similar with colistin regarding the mechanism of action) | in vitro microbicidal activity against P. aeruginosa and K. pneumoniae | anti-inflammatory and immunomodulatory activities | mice model | [232] |
| Oritavancin, a synthetic selectively targeted AMPs | bactericidal effects against MRSA and VRSA | anti-biofilm activity | [233] | |
| WLBU2—engineered cationic AMP and his D-enantiomers (D8) | in vitro antimicrobial activity against A. baumannii and P. aeruginosa | anti-inflamatory activities | mice model | [234] |
| Oct-TriA2 (2,8-D-Orn, 7-Orn) and Oct-TriA1 based on the tridecaptins | antimicrobial activity against A. baumannii, K. pneumoniae, and E. cloacae | Oct-TriA1 lower haemolytic activity | [235] |
| MNPs Type and Mechanism of Action (MOA) | Agent Used | Targeted Microorganisms and Advantages | References |
|---|---|---|---|
| Silver (Ag) NPs: MOA—inhibition of peptidoglycan synthesis, structural modification in the membrane permeability, reactive oxygen species (ROS) generation, lipid peroxidation, interaction with DNA affecting DNA’s replication and finally the cell death | AgNPs-microfibrillated cellulose biocomposite | in vitro antimicrobial activity against S. aureus and P. aeruginosa | [244] |
| Phenolics-coated AgNPs | in vitro antimicrobial effects against P. aeruginosa and Enterobacter aerogenes | [245] | |
| Ag nanoform complexed with amorphous TiO2 | in vitro antimicrobial activity against S. aureus and K. pneumoniae | [246] | |
| Ag-containing Hydrofiber® dressing and nanocrystalline Ag-containing dressing | in vitro antimicrobial activity against MRSA and VRE | [247] | |
| AgNPs immobilized on the surface of nanoscale silicate platelets (AgNP/NSPs) | in vitro antimicrobial activity against MRSA | [248] | |
| AgNPs from Phyllanthus amarus extract | in vitro antimicrobial activity against MDR P. aeruginosa | [249] | |
| Fungal biosynthesis of AgNPs | antibacterial activity against S. aureus; nontoxic, safe, inorganic agent. | [250] | |
| TiO2 nanotubes covered with AgNPs | enhanced antimicrobial activity of the bone/dental implants against S. aureus; >80% biocidal activity | [251] | |
| Calligonum comosum and Azadirachta indica leaf extracts as stabilizing AgNPs | antibacterial ability against P. aeruginosa and S. aureus, by causing apoptosis | [252] | |
| AgNPs synthetized using Ajuga bracteosa extract | bactericidal activity against K. pneumoniae, S. aureus, and P. aeruginosa; antioxidant potential effects; pharmacological importance | [253] | |
| Cu/Ag NPs | Graphene oxide/Cu/Ag NPs | in vitro bactericidal activity against P. aeruginosa, K. pneumoniae, and MRSA | [254] |
| (Golden) AuNPs less toxic than Ag | AuNPs functionalized with ampicillin | in vitro bactericidal activity against P. aeruginosa and E. aerogenes | [255] |
| Pyrimidinethiol-modified AuNPs | in vitro antimicrobial activity against MDR E. faecium, P. aeruginosa, MRSA, K. pneumoniae, A. baumannii | [256] | |
| CGNPs (cinnamaldehyde immobilized on AuNPs) | in vitro and in vivo antibiofilm of MRSA and P. aeruginosa | [257] | |
| 6-aminopenicillanic acid-coated AuNPs doped into electrospun fibers of poly(ε-caprolactone) | in vitro and in vivo antimicrobial activity against MDR K. pneumoniae infections | [258] | |
| Metallopolymer-antibiotic bioconjugates on AuNPS | antimicrobial activity against K. pneumoniae and S. aureus | [259] | |
| AuNPs | in vitro and in vivo bactericidal activity against mastitis-causing S. aureus | [260] | |
| Metal oxide NPs | |||
| ZnO NPs—ROS generation; bactericidal effect, by disrupting the cell membrane; glycolysis and transmembrane proton translocation inhibition | ZnO | antimicrobial activity against MRSA and P. aeruginosa; anti-biofilm formation and production of quorum-sensing- in P. aeruginosa; anti-biofilm formation MRSA | [261,262] |
| Nitric oxide (NO)— RNS generation | NO-releasing NP | in vitro antimicrobial activity against MRSA, A. baumannii, K. pneumoniae, and P. aeruginosa | [263] |
| NO-releasing silica NPs | in vivo bactericidal activity against intracellular P. aeruginosa in L929 mouse fibroblasts | [264] | |
| Cobalt oxide NPs—oxidative mechanisms< membrane permeability changes; inhibition of DNA replication | Co3O4 | in vitro antimicrobial activity against S. aureus | [265] |
| Bis hexa decyl trimethyl ammonium cobalt tetrachloride | antimicrobial activity against MDR S. aureus | [266] | |
| Fe2O3 NPs—affect the functionality of porin pumps; occupy the active sites of MBLs | Functionalized Fe2O3 NPs with antibiotics | inhibition growth of P. aeruginosa; reducing overcoming resistance and acute toxicity; low cost; synergistic effects with antibiotics | [267] |
| Phage | Targeted Bacteria | Type of Study | Model Application | In vivo Efficacy; Advantages and Survival of Host | Route of Administration | References |
|---|---|---|---|---|---|---|
| Phage ENB6 and C3 (A2 morphotype group) | Ef | in vivo | Murine bacteremia model | Immunocompatible; 100% survival with multiple doses | Intraperitoneal (IP) | [287] |
| Cocktail of E. coli phage ECP311, K. pneumoniae phage KPP235, and Enterobacter phage ELP140 | K & E | in vivo | Galeria mellonella infection model | 100% reduction after 5 doses; 90% survival | - | [288] |
| Enterococcus phiEF24C, phiEF17H, and phiM1EF22 phages | E | in vitro | - | Inhibition of growth | Co-culture with phages mixture | [289] |
| phage ϕEf11/ϕFL1C(Δ36)PnisA | E | in vitro | - | 10–100-fold decrease in viable cells (CFU/biofilm); biofilm eradication | Inoculation with phage | [290] |
| anti E. faecium EFDG1 phage | Ef | ex vivo | Human root canal model | 5-log growth reduction in stationary cultures; reducing 2-week old biofilm | - | [291] |
| vB_SauM_LM12, vB_EfaS_LM99 and vB_EcoM_JB75 | S | ex vivo | orthopaedic implant infection model | Great antimicrobial activity; growth reduction | Paper strip | [292] |
| 2003, 2002, 3A and K phage cocktail | S | in vivo | Ventilator-associated pneumonia rat model | Reduced lung damage; 100% survival at 12 h after infection; 58% survival until the end of the experiment | Intravenous (IV) | [293] |
| Phage coated implant | S | in vivo | Murine model of joint infection | Normal locomotor activity by 10 day; decreasing bacterial adherence | K-wire implant delivery system | [294] |
| SATA-8505 (ATCC PTA-9476) | S | in vivo | 65-year-old woman with Corneal abscess | stabilization of ocular signs; pathogen eradication | Topical (eye drops and nasal spray) and intravenous (IV) | [273] |
| Staphylococcal phage Sb-1 | S | in vivo | Case series (human subjects with diabetic foot ulcer) | Wound healing within 7 weeks | Topical | [275] |
| Myoviridae bacteriophages (AB-SA01) | S | in vivo | Human single-arm non-comparative trial (13 patients) | 8/13 patients showed clinical improvement; 5 patients died within the first 28 days | IV | [295] |
| vB_KpnP_KL106-ULIP47; vB_KpnP_KL106-ULIP54; vB_KpnP_K1-ULIP33; | K | in vivo | Galleria mellonella larvae infection model | Mortality rate reduced with 20% upon treatment with phage | Phage inoculation | [296] |
| K. pneumoniae isolated phage | K | in vivo | Case series (48 patients with nonhealing chronic wounds) | significant decrease in the mean depth of the wound; improved score of epithelialization; 39/48 patients had a complete cure | Topical | [297] |
| Klebsiella myPSH1235 and Enterobacter myPSH1140 phage | K & E | in vitro | - | Strong bactericidal activity; bacterial density reached to 0 with no viable cells at 24 h after infection | Incubation with phage | [298] |
| K. pneumoniae bacteriophage | K | in vivo | Swiss albino mouse model | gradual reduction of colony-forming unit; complet eradication after 6 days of treatment | Oral | [299] |
| KpJH46ø2 | K | in vivo | Case study (62 year-old diabetic man with prosthetic knee infections) | The restraining of local symptoms, signs of infection, and recovery of function | IV | [300] |
| Lytic bacteriophage | K | in vivo | Case study (57-year patient with Crohn’ disease) | Bacterial eradication | Oral Intrarectal | [301] |
| Phage PEV20 | P | in vivo | Murine infection model | 5-log reduction of bacterial cells | Intranasal; Intratracheal | [285] |
| US Navy library of bacteriophages | P | in vivo | Case study (2-year-old patient with Di George syndrome) | Bacterial eradication after phage therapy | IV | [302] |
| 12 natural lytic anti-P. aeruginosa bacteriophages (PP1131) | P | in vivo | Randomised phase ½ trial (27 patients with wound infections) | Reduced bacterial burden; minor adverse effects | Topical | [303] |
| PB AB08 PB AB25 | A | in vivo | Mice infection model | 35% survival rate | Intranasal | [304] |
| WCHABP1 | A | in vivo | Galleria mellonella infection model | 75% survival rate after phage administration | [283] | |
| PD-6A3 and phage cocktail | A | in vivo | Sepsis mouse model | 60% and 50% survival rate after phage therapy and phage cocktail | IP | [285] |
| Βϕ-R2096 sewage phage | A | in vivo | Galleria mellonella infection model | 80% and 50% survival rate at 96 and 48 h. | Injection | [305] |
| A | in vivo | Mouse model acute pneumonia | 100%, 60% and 30% survival rate at day 12 | Intranasal | ||
| AB3P1, AB3P2, AB3P3, AB3P4, AB3P5 | A | in vivo | Mice infection model | Bactericidal activity; 100% survival rate | IP | [306] |
| AB-PA01 lytic phages | P | in vivo | Case report (77-year old patient with adenocarcinoma) | Improved oxygenation; sedation ceased; bacterial eradication | IV Nebulisation | [307] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vrancianu, C.O.; Gheorghe, I.; Dobre, E.-G.; Barbu, I.C.; Cristian, R.E.; Popa, M.; Lee, S.H.; Limban, C.; Vlad, I.M.; Chifiriuc, M.C. Emerging Strategies to Combat β-Lactamase Producing ESKAPE Pathogens. Int. J. Mol. Sci. 2020, 21, 8527. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21228527
Vrancianu CO, Gheorghe I, Dobre E-G, Barbu IC, Cristian RE, Popa M, Lee SH, Limban C, Vlad IM, Chifiriuc MC. Emerging Strategies to Combat β-Lactamase Producing ESKAPE Pathogens. International Journal of Molecular Sciences. 2020; 21(22):8527. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21228527
Chicago/Turabian StyleVrancianu, Corneliu Ovidiu, Irina Gheorghe, Elena-Georgiana Dobre, Ilda Czobor Barbu, Roxana Elena Cristian, Marcela Popa, Sang Hee Lee, Carmen Limban, Ilinca Margareta Vlad, and Mariana Carmen Chifiriuc. 2020. "Emerging Strategies to Combat β-Lactamase Producing ESKAPE Pathogens" International Journal of Molecular Sciences 21, no. 22: 8527. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21228527