Combination of Inhibitors of USP7 and PLK1 has a Strong Synergism against Paclitaxel Resistance


Abstract
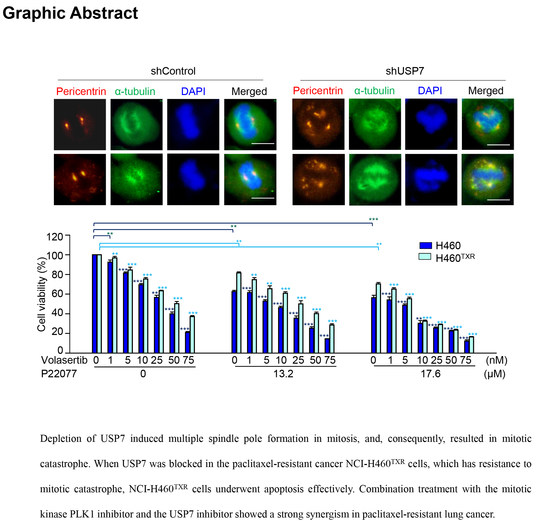
:
1. Introduction
2. Results
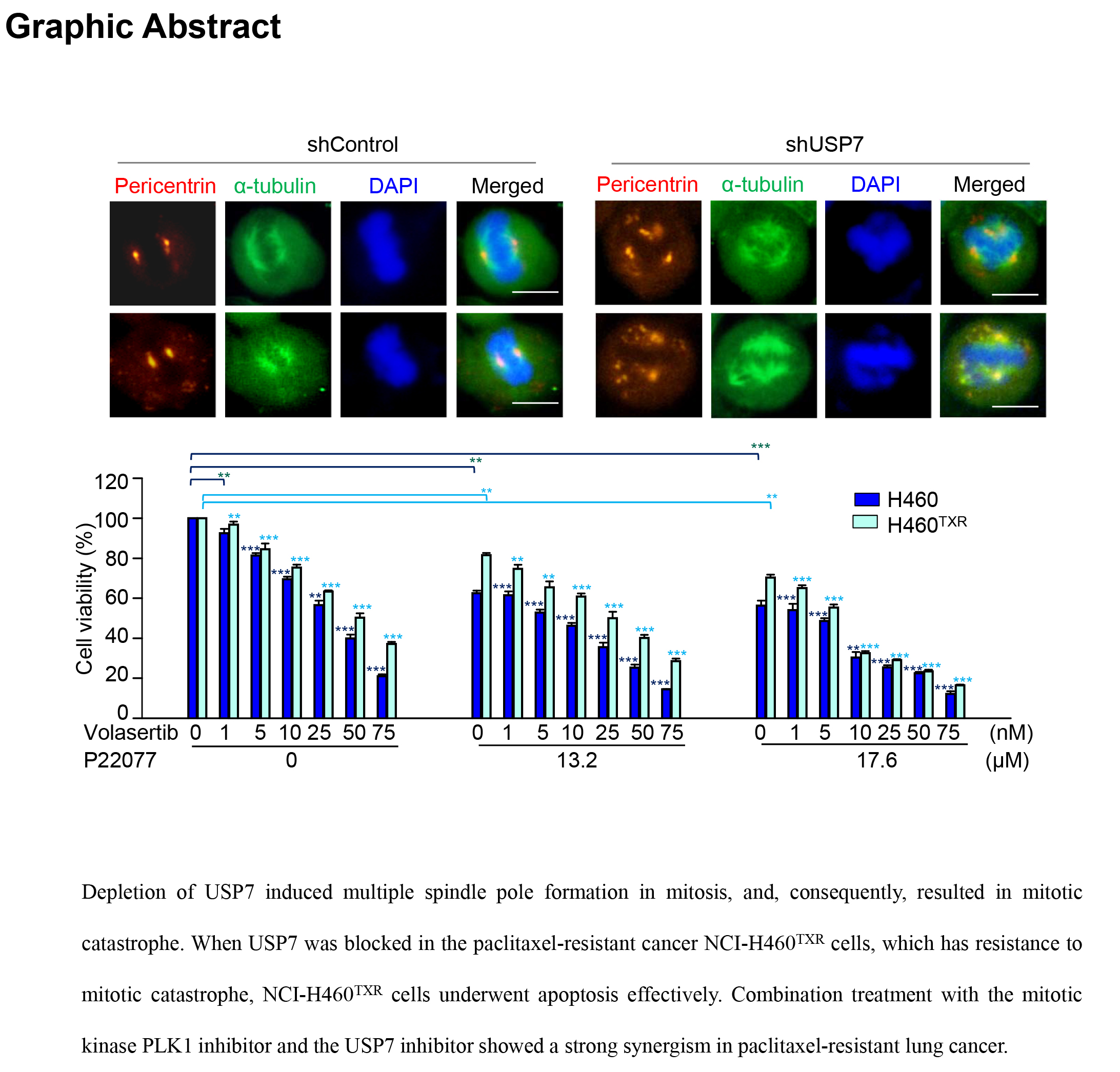
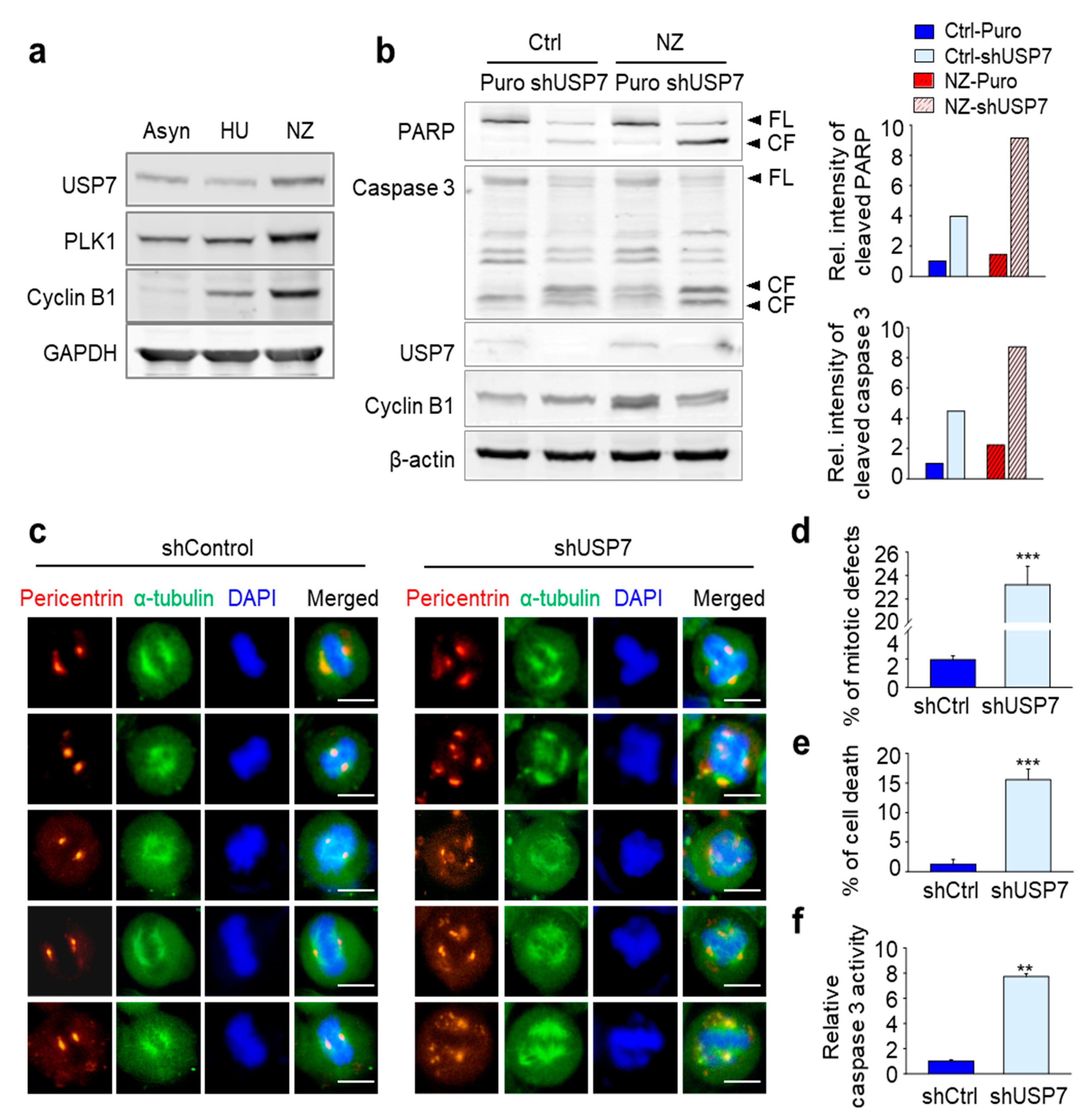
2.1. Depletion of USP7 Induces Apoptosis in Several Carcinoma
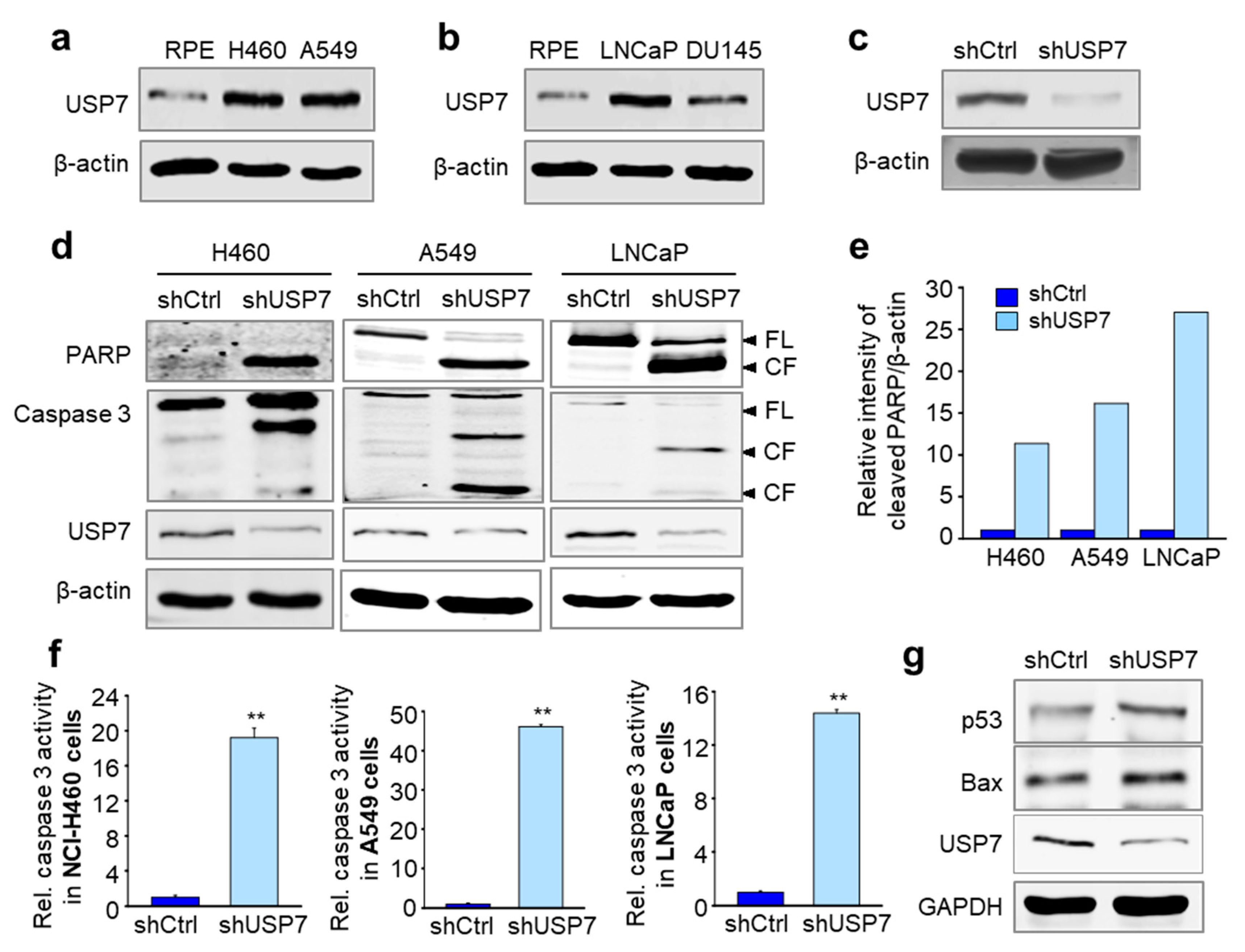
2.2. USP7 Was Highly Expressed Concomitantly with the Mitotic Factors in Tumor Tissues from Prostate and Non-Small Cell Lung Cancer
2.3. Loss of USP7 Results in the Formation of Multiple Spindle Poles, Consequently Inducing Mitotic Catastrophe
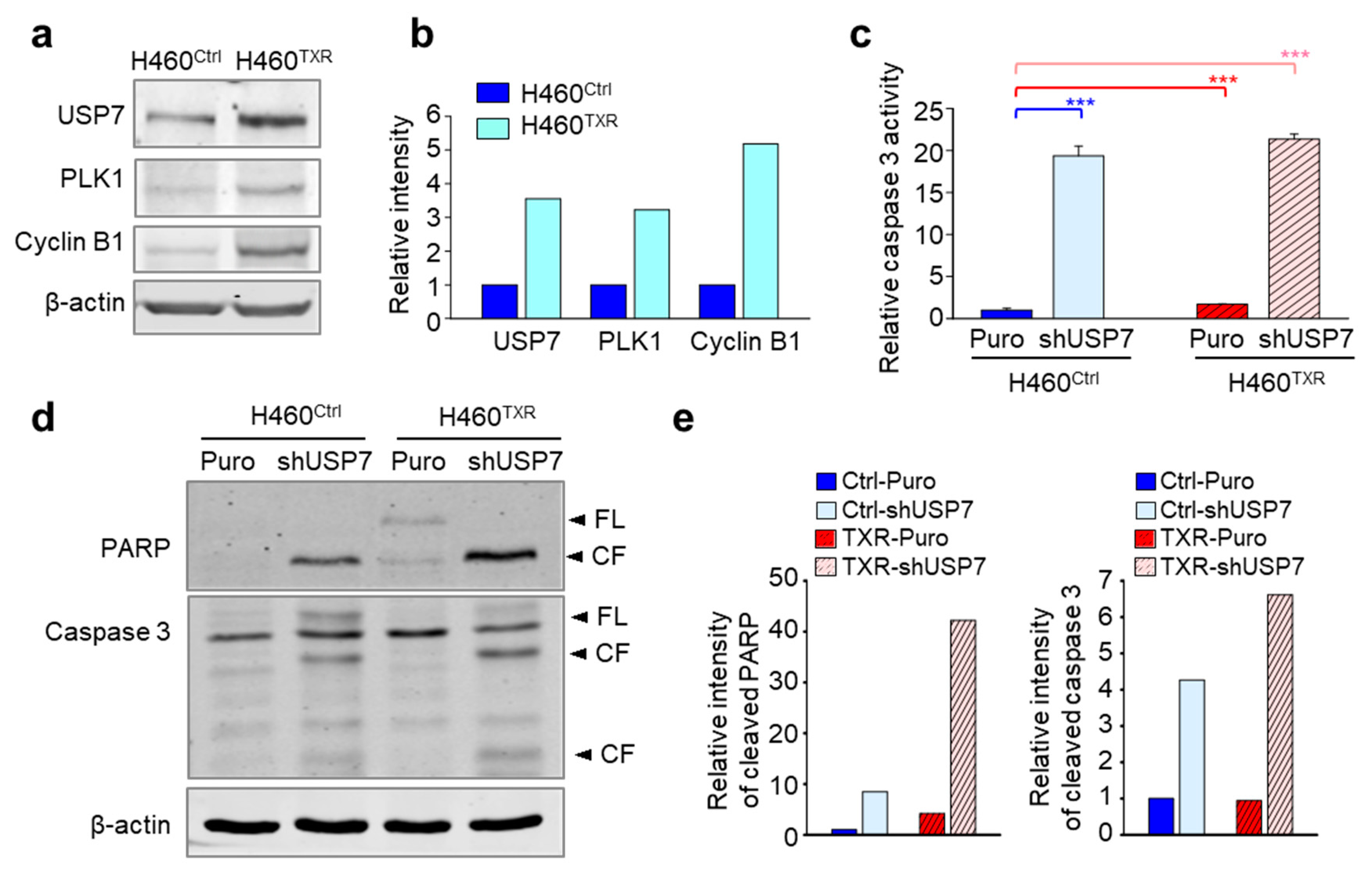
2.4. Targeting USP7 Increases the Sensitivity of Paclitaxel-Resistant Lung Cancer
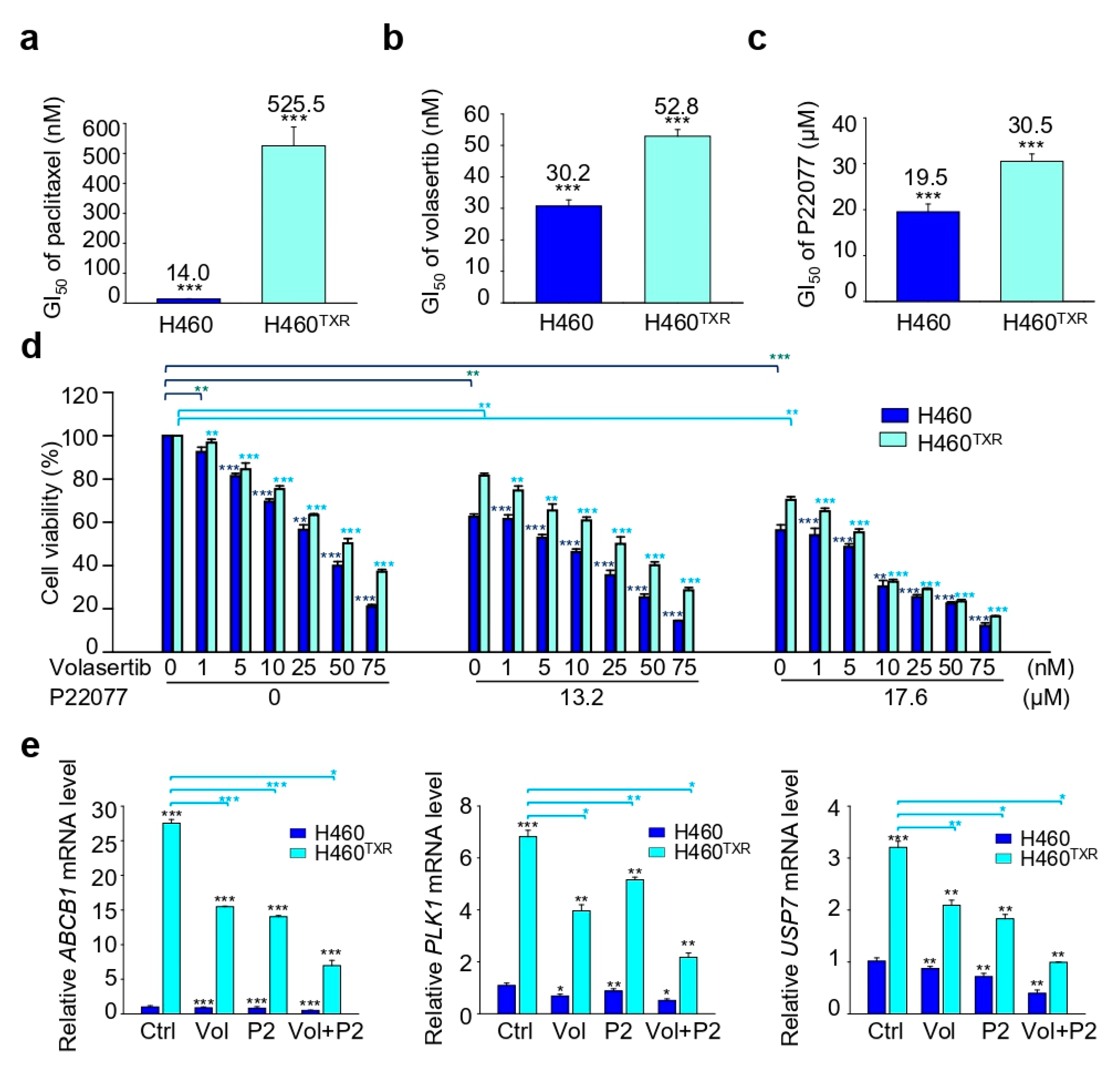
2.5. Combination Treatment with the USP7 Inhibitor P22077 and PLK1 Inhibitor Volasertib Shows Synergic Anticancer Effects in Paclitaxel-Resistant Lung Cancer
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Cell Culture and Establishment of Paclitaxel-Resistant Cancer Cells
4.3. Bioinformatics Analysis
4.4. Lentivirus-Based shRNA Preparation and Selection
4.5. Cell Viability Assay
4.6. Fluorometric Caspase 3 Activity Assay
4.7. Western Blotting
4.8. Immunofluorescence
4.9. Quantitative Reverse Transcription Polymerase Chain Reaction (qRT-PCR)
4.10. Statistical Analysis
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| PLK1 | Polo-like kinase 1 |
| USP7 | Ubiquitin-specific-processing protease 7 |
| TXR | Taxol Resistance |
| GI | The growth inhibitory concentration |
| CI | Combination index |
| RI | Resistance index |
References
- Song, M.S.; Salmena, L.; Carracedo, A.; Egia, A.; Lo-Coco, F.; Teruya-Feldstein, J.; Pandolfi, P.P. The deubiquitinylation and localization of PTEN are regulated by a HAUSP-PML network. Nature 2008, 455, 813–817. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yim, H.; Shin, S.B.; Woo, S.U.; Lee, P.C.; Erikson, R.L. Plk1-mediated stabilization of 53BP1 through USP7 regulates centrosome positioning to maintain bipolarity. Oncogene 2017, 36, 966–978. [Google Scholar] [CrossRef] [PubMed]
- Peng, Y.; Liu, Y.; Gao, Y.; Yuan, B.; Qi, X.; Fu, Y.; Zhu, Q.; Cao, T.; Zhang, S.; Yin, L.; et al. USP7 is a novel Deubiquitinase sustaining PLK1 protein stability and regulating chromosome alignment in mitosis. J. Exp. Clin. Cancer Res. 2019, 38, 468. [Google Scholar] [CrossRef] [PubMed]
- Giovinazzi, S.; Sirleto, P.; Aksenova, V.; Morozov, V.M.; Zori, R.; Reinhold, W.C.; Ishov, A.M. Usp7 protects genomic stability by regulating Bub3. Oncotarget 2014, 5, 3728–3742. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giovinazzi, S.; Morozov, V.M.; Summers, M.K.; Reinhold, W.C.; Ishov, A.M. USP7 and Daxx regulate mitosis progression and taxane sensitivity by affecting stability of Aurora-A kinase. Cell Death Differ. 2013, 20, 721–731. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Glickman, M.H.; Ciechanover, A. The ubiquitin-proteasome proteolytic pathway: Destruction for the sake of construction. Physiol. Rev. 2002, 82, 373–428. [Google Scholar] [CrossRef]
- Yarychkivska, O.; Tavana, O.; Gu, W.; Bestor, T.H. Independent functions of DNMT1 and USP7 at replication foci. Epigenet. Chromatin 2018, 11, 9. [Google Scholar] [CrossRef] [Green Version]
- Epping, M.T.; Meijer, L.A.; Krijgsman, O.; Bos, J.L.; Pandolfi, P.P.; Bernards, R. TSPYL5 suppresses p53 levels and function by physical interaction with USP7. Nat. Cell Biol. 2011, 13, 102–108. [Google Scholar] [CrossRef]
- Zhang, C.; Lu, J.; Zhang, Q.W.; Zhao, W.; Guo, J.H.; Liu, S.L.; Wu, Y.L.; Jiang, B.; Gao, F.H. USP7 promotes cell proliferation through the stabilization of Ki-67 protein in non-small cell lung cancer cells. Int. J. Biochem. Cell Biol. 2016, 79, 209–221. [Google Scholar] [CrossRef]
- Zhao, G.Y.; Lin, Z.W.; Lu, C.L.; Gu, J.; Yuan, Y.F.; Xu, F.K.; Liu, R.H.; Ge, D.; Ding, J.Y. USP7 overexpression predicts a poor prognosis in lung squamous cell carcinoma and large cell carcinoma. Tumour Biol. 2015, 36, 1721–1729. [Google Scholar] [CrossRef] [Green Version]
- Sun, Y.; Cao, L.; Sheng, X.; Chen, J.; Zhou, Y.; Yang, C.; Deng, T.; Ma, H.; Feng, P.; Liu, J.; et al. WDR79 promotes the proliferation of non-small cell lung cancer cells via USP7-mediated regulation of the Mdm2-p53 pathway. Cell Death Dis. 2017, 8, e2743. [Google Scholar] [CrossRef] [Green Version]
- Wang, Q.; Ma, S.; Song, N.; Li, X.; Liu, L.; Yang, S.; Ding, X.; Shan, L.; Zhou, X.; Su, D.; et al. Stabilization of histone demethylase PHF8 by USP7 promotes breast carcinogenesis. J. Clin. Investig. 2016, 126, 2205–2220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, L.; Wang, H.; Tian, L.; Li, H. Expression of USP7 and MARCH7 Is Correlated with Poor Prognosis in Epithelial Ovarian Cancer. Tohoku J. Exp. Med. 2016, 239, 165–175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, C.; Niu, C.; Yang, Y.; Wang, Y.; Lu, M. Expression of HAUSP in gliomas correlates with disease progression and survival of patients. Oncol. Rep. 2013, 29, 1730–1736. [Google Scholar] [CrossRef] [Green Version]
- Yang, Z.; Huo, S.; Shan, Y.; Liu, H.; Xu, Y.; Yao, K.; Li, X.; Zhang, X. STAT3 repressed USP7 expression is crucial for colon cancer development. FEBS Lett. 2012, 586, 3013–3017. [Google Scholar]
- Colland, F.; Formstecher, E.; Jacq, X.; Reverdy, C.; Planquette, C.; Conrath, S.; Trouplin, V.; Bianchi, J.; Aushev, V.N.; Camonis, J.; et al. Small-molecule inhibitor of USP7/HAUSP ubiquitin protease stabilizes and activates p53 in cells. Mol. Cancer Ther. 2009, 8, 2286–2295. [Google Scholar] [CrossRef] [Green Version]
- Fan, Y.H.; Cheng, J.; Vasudevan, S.A.; Dou, J.; Zhang, H.; Patel, R.H.; Ma, I.T.; Rojas, Y.; Zhao, Y.; Yu, Y.; et al. USP7 inhibitor P22077 inhibits neuroblastoma growth via inducing p53-mediated apoptosis. Cell Death Dis. 2013, 4, e867. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, M.; Zhang, Y.; Wang, T.; Zhang, J.; Zhou, Z.; Sun, Y.; Wang, S.; Shi, Y.; Luan, X.; Zhang, Y.; et al. The USP7 Inhibitor P5091 Induces Cell Death in Ovarian Cancers with Different P53 Status. Cell Physiol. Biochem. 2017, 43, 1755–1766. [Google Scholar] [CrossRef]
- An, T.; Gong, Y.; Li, X.; Kong, L.; Ma, P.; Gong, L.; Zhu, H.; Yu, C.; Liu, J.; Zhou, H.; et al. USP7 inhibitor P5091 inhibits Wnt signaling and colorectal tumor growth. Biochem. Pharmacol. 2017, 131, 29–39. [Google Scholar] [CrossRef]
- Reverdy, C.; Conrath, S.; Lopez, R.; Planquette, C.; Atmanene, C.; Collura, V.; Harpon, J.; Battaglia, V.; Vivat, V.; Sippl, W.; et al. Discovery of specific inhibitors of human USP7/HAUSP deubiquitinating enzyme. Chem. Biol. 2012, 19, 467–477. [Google Scholar] [CrossRef] [Green Version]
- Jackson, J.R.; Patrick, D.R.; Dar, M.M.; Huang, P.S. Targeted anti-mitotic therapies: Can we improve on tubulin agents? Nat. Rev. Cancer 2007, 7, 107–117. [Google Scholar] [CrossRef] [PubMed]
- Jordan, M.A.; Wilson, L. Microtubules as a target for anticancer drugs. Nat. Rev. Cancer 2004, 4, 253–265. [Google Scholar] [CrossRef] [PubMed]
- Murphy, W.K.; Fossella, F.V.; Winn, R.J.; Shin, D.M.; Hynes, H.E.; Gross, H.M.; Davilla, E.; Leimert, J.; Dhingra, H.; Raber, M.N.; et al. Phase II study of taxol in patients with untreated advanced non-small-cell lung cancer. J. Natl. Cancer Inst. 1993, 85, 384–388. [Google Scholar] [CrossRef] [PubMed]
- Smith, D.C.; Tangen, C.M.; Van Veldhuizen, P.J., Jr.; Harrer, G.W.; Golshayan, A.; Mills, G.M.; Vogelzang, N.J.; Thompson, I.M.; Hussain, M.H. Phase II evaluation of early oral estramustine, oral etoposide, and intravenous paclitaxel combined with hormonal therapy in patients with high-risk metastatic prostate adenocarcinoma: Southwest Oncology Group S0032. Urology 2011, 77, 1172–1176. [Google Scholar] [CrossRef] [Green Version]
- Yoshitomi, S.; Taira, N.; Doihara, H.; Mizoo, T.; Nogami, T.; Iwamoto, T.; Motoki, T.; Shien, T.; Ogasawara, Y.; Matsuoka, J.; et al. A phase 1, dose-finding and pharmacokinetic study of gemcitabine with nab-paclitaxel in patients with metastatic breast cancer. Cancer Chemother. Pharmacol. 2016, 78, 289–294. [Google Scholar] [CrossRef]
- Chan, J.K.; Brady, M.F.; Monk, B.J. Weekly vs. Every-3-Week Paclitaxel for Ovarian Cancer. N. Engl. J. Med. 2016, 374, 2603–2604. [Google Scholar] [CrossRef] [Green Version]
- Chan, J.K.; Brady, M.F.; Penson, R.T.; Huang, H.; Birrer, M.J.; Walker, J.L.; DiSilvestro, P.A.; Rubin, S.C.; Martin, L.P.; Davidson, S.A.; et al. Weekly vs. Every-3-Week Paclitaxel and Carboplatin for Ovarian Cancer. N. Engl. J. Med. 2016, 374, 738–748. [Google Scholar] [CrossRef] [Green Version]
- Orr, G.A.; Verdier-Pinard, P.; McDaid, H.; Horwitz, S.B. Mechanisms of Taxol resistance related to microtubules. Oncogene 2003, 22, 7280–7295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miyashita, T.; Reed, J.C. Tumor suppressor p53 is a direct transcriptional activator of the human bax gene. Cell 1995, 80, 293–299. [Google Scholar] [PubMed] [Green Version]
- Shin, S.B.; Woo, S.U.; Yim, H. Cotargeting Plk1 and androgen receptor enhances the therapeutic sensitivity of paclitaxel-resistant prostate cancer. Ther. Adv. Med. Oncol. 2019, 11, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Yim, H.; Erikson, R.L. Plk1-targeted therapies in TP53- or RAS-mutated cancer. Mutat. Res. Rev. Mutat. Res. 2014, 761, 31–39. [Google Scholar] [CrossRef] [PubMed]
- Gottesman, M.M.; Fojo, T.; Bates, S.E. Multidrug resistance in cancer: Role of ATP-dependent transporters. Nat. Rev. Cancer 2002, 2, 48–58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Komander, D. The emerging complexity of protein ubiquitination. Biochem. Soc. Trans. 2009, 37 Pt 5, 937–953. [Google Scholar] [CrossRef] [Green Version]
- Li, M.; Brooks, C.L.; Kon, N.; Gu, W. A dynamic role of HAUSP in the p53-Mdm2 pathway. Mol. Cell 2004, 13, 879–886. [Google Scholar] [CrossRef]
- Hu, M.; Gu, L.; Li, M.; Jeffrey, P.D.; Gu, W.; Shi, Y. Structural basis of competitive recognition of p53 and MDM2 by HAUSP/USP7: Implications for the regulation of the p53-MDM2 pathway. PLoS Biol. 2006, 4, e27. [Google Scholar] [CrossRef]
- Cummins, J.M.; Rago, C.; Kohli, M.; Kinzler, K.W.; Lengauer, C.; Vogelstein, B. Tumour suppression: Disruption of HAUSP gene stabilizes p53. Nature 2004, 428, 1–2. [Google Scholar] [CrossRef]
- Cummins, J.M.; Vogelstein, B. HAUSP is required for p53 destabilization. Cell Cycle 2004, 3, 689–692. [Google Scholar] [CrossRef] [Green Version]
- Kon, N.; Kobayashi, Y.; Li, M.; Brooks, C.L.; Ludwig, T.; Gu, W. Inactivation of HAUSP in vivo modulates p53 function. Oncogene 2010, 29, 1270–1279. [Google Scholar] [CrossRef] [Green Version]
- Volk, V.; Cathomas, R.; Mark, M.; von Moos, R.; Klingbiel, D.; Brossart, P.; Mey, U. Weekly carboplatin in combination with weekly paclitaxel in the treatment of metastatic non-small cell lung cancer: A single center 10-year experience. Support. Care Cancer 2016, 24, 2119–2128. [Google Scholar] [CrossRef]
- Socinski, M.A.; Bondarenko, I.; Karaseva, N.A.; Makhson, A.M.; Vynnychenko, I.; Okamoto, I.; Hon, J.K.; Hirsh, V.; Bhar, P.; Zhang, H.; et al. Weekly nab-paclitaxel in combination with carboplatin versus solvent-based paclitaxel plus carboplatin as first-line therapy in patients with advanced non-small-cell lung cancer: Final results of a phase III trial. J. Clin. Oncol. 2012, 30, 2055–2062. [Google Scholar] [CrossRef] [Green Version]
- Sandler, A.; Gray, R.; Perry, M.C.; Brahmer, J.; Schiller, J.H.; Dowlati, A.; Lilenbaum, R.; Johnson, D.H. Paclitaxel-carboplatin alone or with bevacizumab for non-small-cell lung cancer. N. Engl. J. Med. 2006, 355, 2542–2550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosell, R.; Gatzemeier, U.; Betticher, D.C.; Keppler, U.; Macha, H.N.; Pirker, R.; Berthet, P.; Breau, J.L.; Lianes, P.; Nicholson, M.; et al. Phase III randomised trial comparing paclitaxel/carboplatin with paclitaxel/cisplatin in patients with advanced non-small-cell lung cancer: A cooperative multinational trial. Ann. Oncol. 2002, 13, 1539–1549. [Google Scholar] [CrossRef] [PubMed]
- Riordan, J.R.; Deuchars, K.; Kartner, N.; Alon, N.; Trent, J.; Ling, V. Amplification of P-glycoprotein genes in multidrug-resistant mammalian cell lines. Nature 1985, 316, 817–819. [Google Scholar] [CrossRef] [PubMed]
- Stefan, S.M. Multi-target ABC transporter modulators: What next and where to go? Future Med. Chem. 2019, 11, 2353–2358. [Google Scholar] [CrossRef]
- Steinhauser, K.; Kloble, P.; Kreis, N.N.; Ritter, A.; Friemel, A.; Roth, S.; Reichel, J.M.; Michaelis, J.; Rieger, M.A.; Louwen, F.; et al. Deficiency of RITA results in multiple mitotic defects by affecting microtubule dynamics. Oncogene 2017, 36, 2146–2159. [Google Scholar] [CrossRef]
- Liu, Y.; Parry, J.A.; Chin, A.; Duensing, S.; Duensing, A. Soluble histone H2AX is induced by DNA replication stress and sensitizes cells to undergo apoptosis. Mol. Cancer 2008, 7, 61. [Google Scholar] [CrossRef] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| H460Ctrl | H460TXR | |||
|---|---|---|---|---|
| GI50 | CI | GI50 | CI | |
| P22077 (µM) | 19.5 | 30.5 | ||
| Volasertib (nM) | 30.2 | 52.8 | ||
| Volasertib (in combination 13.2 µM P22077) | 6.4 | 0.889 | 22.9 | 0.866 |
| Volasertib (in combination 17.6 µM P22077) | 2.4 | 0.982 | 5.8 | 0.687 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shin, S.-B.; Kim, C.-H.; Jang, H.-R.; Yim, H. Combination of Inhibitors of USP7 and PLK1 has a Strong Synergism against Paclitaxel Resistance. Int. J. Mol. Sci. 2020, 21, 8629. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21228629
Shin S-B, Kim C-H, Jang H-R, Yim H. Combination of Inhibitors of USP7 and PLK1 has a Strong Synergism against Paclitaxel Resistance. International Journal of Molecular Sciences. 2020; 21(22):8629. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21228629
Chicago/Turabian StyleShin, Sol-Bi, Chang-Hyeon Kim, Hay-Ran Jang, and Hyungshin Yim. 2020. "Combination of Inhibitors of USP7 and PLK1 has a Strong Synergism against Paclitaxel Resistance" International Journal of Molecular Sciences 21, no. 22: 8629. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21228629