Locus Coeruleus Modulates Neuroinflammation in Parkinsonism and Dementia

 ,
,
Abstract
:1. Introduction
2. Neuroinflammation
3. The Locus Coeruleus
4. Locus Coeruleus Degeneration in Alzheimer’s and Parkinson’s Disease
5. The Role of Neuroinflammation in Neurodegenerative Disorders
5.1. In Vivo and Post-Mortem Evidence for the Occurrence of Neuroinflammation in PD and AD in Humans
5.2. Potential Neuroinflammatory Mechanisms in NDD
5.2.1. Potential Mechanisms in AD
5.2.2. Potential Mechanisms in PD
6. The Potential Role of LC Degeneration in Neuroinflammation Promotion during NDD Pathogenesis
6.1. General Effects of Locus Coeruleus on Neuroinflammation
6.2. Experimental Data on the Role of Locus Coeruleus in Neuroinflammation Occurring in Alzheimer’s Disease
6.3. Experimental Data on the Role of Locus Coeruleus in Neuroinflammation Occurring in Parkinson’s Disease
7. Discussion and Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| Aβ | Amyloid β |
| AD | Alzheimer’s disease |
| AF | Amyloid fibrils |
| α-syn | α-synuclein |
| AO | Amyloid oligomer |
| AP | Amyloid plaque |
| APC | Antigen presenting cell |
| APP23 | Amyloid precursor protein 23 |
| BBB | Blood-brain barrier |
| CSF | Cerebrospinal fluid |
| CNS | Central nervous system |
| DA | Dopamine |
| DSP-4 | N-(2-chloroethyl)-N-ethyl-2-bromobenzylamine |
| HSP-70 | Heat shock protein 70 |
| LB | Lewy body |
| LC | Locus Coeruleus |
| LPS | Lipopolysaccharide |
| MCI | Mild cognitive impairment |
| MTPT | 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine |
| NA | Noradrenaline |
| NDD | Neurodegenerative disorders |
| NFT | Neurofibrillary tangle |
| NM | Neuromelanin |
| PAMP | Pathogen-associated molecular pattern |
| PD | Parkinson’s disease |
| PET | Positron emission tomography |
| PPARγ | Peroxisome proliferator-activated receptor-γ |
| PRR | Pattern recognition receptor |
| p-Tau | Hyperphosphorylated tau |
| ROS | Reactive oxygen species |
| SNpc | Substantia nigra pars compacta |
| TJ | Tight junction |
| TLR | Toll-like receptor |
| TSPO | Translocation protein |
| VNS | Vagus nerve stimulation. |
References
- Yang, Q.Q.; Zhou, J.W. Neuroinflammation in the central nervous system: Symphony of glial cells. Glia 2019, 67, 1017–1035. [Google Scholar] [CrossRef] [PubMed]
- Colonna, M.; Butovsky, O. Microglia function in the central nervous system during health and neurodegeneration. Annu. Rev. Immunol. 2017, 35, 441–468. [Google Scholar] [CrossRef]
- Pawate, S.; Bhat, N.R. Role of Glia in CNS Inflammation. In Handbook of Neurochemistry and Molecular Neurobiology; Springer: Boston, MA, USA, 2008. [Google Scholar]
- Block, M.L.; Zecca, L.; Hong, J.S. Microglia-mediated neurotoxicity: Uncovering the molecular mechanisms. Nat. Rev. Neurosci. 2007, 8, 57–69. [Google Scholar] [CrossRef] [PubMed]
- Mietelska-Porowska, A.; Wojda, U. T Lymphocytes and Inflammatory Mediators in the Interplay between Brain and Blood in Alzheimer’s Disease: Potential Pools of New Biomarkers. J. Immunol. Res. 2017, 2017, 4626540. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Argaw, A.T.; Asp, L.; Zhang, J.; Navrazhina, K.; Pham, T.; Mariani, J.N.; Mahase, S.; Dutta, D.J.; Seto, J.; Kramer, E.G.; et al. Astrocyte-derived VEGF-A drives blood-brain barrier disruption in CNS inflammatory disease. J. Clin. Investig. 2012, 122, 2454–2468. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abbott, N.J.; Rönnbäck, L.; Hansson, E. Astrocyte-endothelial interactions at the blood-brain barrier. Nat. Rev. Neurosci. 2006, 7, 41–53. [Google Scholar] [CrossRef] [PubMed]
- Nelson, A.R.; Sweeney, M.D.; Sagare, A.P.; Zlokovic, B. V Neurovascular dysfunction and neurodegeneration in dementia and Alzheimer’s disease. Biochim. Biophys. Acta 2016, 1862, 887–900. [Google Scholar] [CrossRef]
- Dahlström, A.; Fuxe, K. Localization of monoamines in the lower brain stem. Experientia 1964, 20, 398–399. [Google Scholar] [CrossRef]
- Counts, S.E.; Mufson, E.J. Locus coeruleus. Hum. Nerv. Syst. 2012, 3, 425–438. [Google Scholar]
- Foote, S.L.; Morrison, J.H. Development of the noradrenergic, serotonergic, and dopaminergic innervation of neocortex. Curr. Top. Dev. Biol. 1987, 21, 391–423. [Google Scholar] [CrossRef]
- Beaudet, A.; Descarries, L. The monoamine innervation of rat cerebral cortex: Synaptic and nonsynaptic axon terminals. Neuroscience 1978, 3, 851–860. [Google Scholar] [CrossRef]
- Séguéla, P.; Watkins, K.C.; Geffard, M.; Descarries, L. Noradrenaline axon terminals in adult rat neocortex: An immunocytochemical analysis in serial thin sections. Neuroscience 1990, 35, 249–264. [Google Scholar] [CrossRef]
- Fuxe, K.; Agnati, L.F.; Marcoli, M.; Borroto-Escuela, D.O. Volume Transmission in Central Dopamine and Noradrenaline Neurons and Its Astroglial Targets. Neurochem. Res. 2015, 40, 2600–2614. [Google Scholar] [CrossRef]
- Schwarz, L.A.; Luo, L. Organization of the locus coeruleus-norepinephrine system. Curr. Biol. 2015, 25, R1051–R1056. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weinshenker, D. Long road to ruin: Noradrenergic dysfunction in neurodegenerative disease. Trends Neurosci. 2018, 41, 211–223. [Google Scholar] [CrossRef] [PubMed]
- Zucca, F.A.; Bellei, C.; Giannelli, S.; Terreni, M.R.; Gallorini, M.; Rizzio, E.; Pezzoli, G.; Albertini, A.; Zecca, L. Neuromelanin and iron in human locus coeruleus and substantia nigra during aging: Consequences for neuronal vulnerability. J. Neural Transm. 2006, 113, 757–767. [Google Scholar] [CrossRef]
- Liu, K.Y.; Marijatta, F.; Hämmerer, D.; Acosta-Cabronero, J.; Düzel, E.; Howard, R.J. Magnetic resonance imaging of the human locus coeruleus: A systematic review. Neurosci. Biobehav. Rev. 2017, 83, 325–355. [Google Scholar] [CrossRef] [Green Version]
- Pieribone, V.A.; Aston-Jones, G. Adrenergic innervation of the rat nucleus locus coeruleus arises predominantly from the C1 adrenergic cell group in the rostral medulla. Neuroscience 1991, 41, 525–542. [Google Scholar] [CrossRef]
- Rajkowski, J.; Kubiak, P.; Aston-Jones, G. Locus coeruleus activity in monkey: Phasic and tonic changes are associated with altered vigilance. Brain Res. Bull. 1994, 35, 607–616. [Google Scholar] [CrossRef]
- Giorgi, F.S.; Ferrucci, M.; Lazzeri, G.; Pizzanelli, C.; Lenzi, P.; AlessandrÏ, M.G.; Murri, L.; Fornai, F. A damage to locus coeruleus neurons converts sporadic seizures into self-sustaining limbic status epilepticus. Eur. J. Neurosci. 2003, 17, 2593–2601. [Google Scholar] [CrossRef]
- Sara, S.J. The locus coeruleus and noradrenergic modulation of cognition. Nat. Rev. Neurosci. 2009, 10, 211–223. [Google Scholar] [CrossRef] [PubMed]
- Benarroch, E.E. Locus coeruleus. Cell Tissue Res. 2018, 373, 221–232. [Google Scholar] [CrossRef] [PubMed]
- Giorgi, F.S.; Mauceli, G.; Blandini, F.; Ruggieri, S.; Paparelli, A.; Murri, L.; Fornai, F. Locus coeruleus and neuronal plasticity in a model of focal limbic epilepsy. Epilepsia 2006, 47, 21–25. [Google Scholar] [CrossRef]
- Giorgi, F.S.; Blandini, F.; Cantafora, E.; Biagioni, F.; Armentero, M.-T.; Pasquali, L.; Orzi, F.; Murri, L.; Paparelli, A.; Fornai, F. Activation of brain metabolism and fos during limbic seizures: The role of locus coeruleus. Neurobiol. Dis. 2008, 30, 388–399. [Google Scholar] [CrossRef] [PubMed]
- Giorgi, F.S.; Galgani, A.; Puglisi-Allegra, S.; Limanaqi, F.; Busceti, C.L.; Fornai, F. Locus Coeruleus and neurovascular unit: From its role in physiology to its potential role in Alzheimer’s disease pathogenesis. J. Neurosci. Res. 2020, 98, 2406–2434. [Google Scholar] [CrossRef]
- Hornykiewicz, O.; Kish, S.J. Biochemical pathophysiology of Parkinson’s disease. Adv. Neurol. 1987, 45, 19–34. [Google Scholar]
- German, D.C.; Manaye, K.F.; White, C.L.; Woodward, D.J.; McIntire, D.D.; Smith, W.K.; Kalaria, R.N.; Mann, D.M.A. Disease-specific patterns of locus coeruleus cell loss. Ann. Neurol. 1992, 32, 667–676. [Google Scholar] [CrossRef]
- Zarow, C.; Lyness, S.A.; Mortimer, J.A.; Chui, H.C. Neuronal loss is greater in the locus coeruleus than nucleus basalis and substantia nigra in Alzheimer and Parkinson diseases. Arch. Neurol. 2003, 60, 337–341. [Google Scholar] [CrossRef]
- Bertrand, E.; Lechowcz, W.; Szpak, G.M.; Dymecki, J. Qualitative and quantitative analysis of locus coeruleus neurons in Parkinson’s disease. Folia Neuropathol. 1997, 35, 80–86. [Google Scholar]
- Braak, H.; Del Tredici, K.; Rüb, U.; De Vos, R.A.I.; Jansen Steur, E.N.H.; Braak, E. Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol. Aging 2003, 24, 197–211. [Google Scholar] [CrossRef]
- Marien, M.; Briley, M.; Colpaert, F. Noradrenaline depletion exacerbates MPTP-induced striatal dopamine loss in mice. Eur. J. Pharmacol. 1993, 236, 487–489. [Google Scholar] [CrossRef]
- Mavridis, M.; Degryse, A.D.; Lategan, A.J.; Marien, M.R.; Colpaert, F.C. Effects of locus coeruleus lesions on parkinsonian signs, striatal dopamine and substantia nigra cell loss after 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine in monkeys: A possible role for the locus coeruleus in the progression of Parkinson’s disease. Neuroscience 1991, 41, 507–523. [Google Scholar] [CrossRef]
- Fornai, F.; Alessandrì, M.G.; Torracca, M.T.; Bassi, L.; Corsini, G.U. Effects of noradrenergic lesions on MPTP/MPP+ kinetics and MPTP-induced nigrostriatal dopamine depletions. J. Pharmacol. Exp. Ther. 1997, 283, 100–107. [Google Scholar] [PubMed]
- Fornai, F.; Bassi, L.; Torracca, M.T.; Scalori, V.; Corsini, G.U. Norepinephrine loss exacerbates methamphetamine-induced striatal dopamine depletion in mice. Eur. J. Pharmacol. 1995, 283, 99–102. [Google Scholar] [CrossRef]
- Fornai, F.; Giorgi, F.S.; Alessandrí, M.G.; Giusiani, M.; Corsini, G.U. Effects of pretreatment with N-(2-chloroethyl)-N-ethyl-2- bromobenzylamine (DSP-4) on methamphetamine pharmacokinetics and striatal dopamine losses. J. Neurochem. 1999, 72, 777–784. [Google Scholar] [CrossRef] [Green Version]
- Fornai, F.; Bassi, L.; Torracca, M.T.; Alessandrı̀, M.G.; Scalori, V.; Corsini, G.U. Region-and neurotransmitter-dependent species and strain differences in DSP–4–induced monoamine depletion in rodents. Neurodegeneration 1996, 5, 241–249. [Google Scholar] [CrossRef]
- Gesi, M.; Soldani, P.; Giorgi, F.S.; Santinami, A.; Bonaccorsi, I.; Fornai, F. The role of the locus coeruleus in the development of Parkinson’s disease. Neurosci. Biobehav. Rev. 2000, 24, 655–668. [Google Scholar] [CrossRef]
- Tomlinson, B.E.; Irving, D.; Blessed, G. Cell loss in the locus coeruleus in senile dementia of Alzheimer type. J. Neurol. Sci. 1981, 49, 419–428. [Google Scholar] [CrossRef]
- Mann, D.M.; Yates, P.O.; Hawkes, J. The noradrenergic system in Alzheimer and multi-infarct dementias. J. Neurol. Neurosurg. Psychiatry 1982, 45, 113–119. [Google Scholar] [CrossRef] [Green Version]
- Mann, D.M.; Yates, P.O.; Marcyniuk, B. A comparison of changes in the nucleus basalis and locus caeruleus in Alzheimer’s disease. J. Neurol. Neurosurg. Psychiatry 1984, 47, 201–203. [Google Scholar] [CrossRef] [Green Version]
- Bondareff, W.; Mountjoy, C.Q.; Roth, M. Loss of neurons of origin of the adrenergic projection to cerebral cortex (nucleus locus ceruleus) in senile dementia. Neurology 1982, 32, 164–168. [Google Scholar] [CrossRef] [PubMed]
- Kelly, S.C.; He, B.; Perez, S.E.; Ginsberg, S.D.; Mufson, E.J.; Counts, S.E. Locus coeruleus cellular and molecular pathology during the progression of Alzheimer’s disease. Acta Neuropathol. Commun. 2017, 5, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Braak, H.; Thal, D.R.; Ghebremedhin, E.; Del Tredici, K. Stages of the Pathologic Process in Alzheimer Disease: Age Categories From 1 to 100 Years. J. Neuropathol. Exp. Neurol. 2011, 70, 960–969. [Google Scholar] [CrossRef] [PubMed]
- Heneka, M.T.; Ramanathan, M.; Jacobs, A.H.; Dumitrescu-Ozimek, L.; Bilkei-Gorzo, A.; Debeir, T.; Sastre, M.; Galldiks, N.; Zimmer, A.; Hoehn, M.; et al. Locus Ceruleus degeneration promotes Alzheimer pathogenesis in amyloid precursor protein 23 transgenic mice. J. Neurosci. 2006, 26, 1343–1354. [Google Scholar] [CrossRef] [Green Version]
- Heneka, M.T.; Nadrigny, F.; Regen, T.; Martinez-Hernandez, A.; Dumitrescu-Ozimek, L.; Terwel, D.; Jardanhazi-Kurutz, D.; Walter, J.; Kirchhoff, F.; Hanisch, U.K.; et al. Locus ceruleus controls Alzheimer’s disease pathology by modulating microglial functions through norepinephrine. Proc. Natl. Acad. Sci. USA 2010, 107, 6058–6063. [Google Scholar] [CrossRef] [Green Version]
- Poser, C.M.; Huntley, C.J.; Poland, J.D. Para-Encephalitic Parkinsonism. Acta Neurol. Scand. 1969, 45, 199–215. [Google Scholar] [CrossRef]
- Bojinov, S. Encephalitis with acute Parkinsonian syndrome and bilateral inflammatory necrosis of the substantia nigra. J. Neurol. Sci. 1971, 12, 383–415. [Google Scholar] [CrossRef]
- Bauer, J.; Strauss, S.; Schreiter-Gasser, U.; Ganter, U.; Schlegel, P.; Witt, I.; Yolk, B.; Berger, M. Interleukin-6 and α-2-macroglobulin indicate an acute-phase state in Alzheimer’s disease cortices. FEBS Lett. 1991, 285, 111–114. [Google Scholar] [CrossRef] [Green Version]
- Vandenabeele, P.; Fiers, W. Is amyloidogenesis during Alzheimer’s disease due to an IL-1-/IL-6-mediated ‘acute phase response’ in the brain? Immunol. Today 1991, 12, 217–219. [Google Scholar] [CrossRef]
- Potter, H. Chapter 38: The involvement of astrocytes and an acute phase response in the amyloid deposition of Alzheimer’s disease. In Neuronal-Astrocytic Interactions; Yu, A.C.H., Hertz, L., Norenberg, M.D., Waxman, S.G., Syková, E., Eds.; Elsevier: Amsterdam, The Netherlands, 1992; Volume 94, pp. 447–458. ISBN 0079-6123. [Google Scholar]
- Aloe, L.; Fiore, M. TNF-α expressed in the brain of transgenic mice lowers central tyroxine hydroxylase immunoreactivity and alters grooming behavior. Neurosci. Lett. 1997, 238, 65–68. [Google Scholar] [CrossRef]
- Hirsch, E.C.; Hunot, S.; Damier, P.; Faucheux, B. Glial cells and inflammation in Parkinson’s disease: A role in neurodegeneration? Ann. Neurol. 1998, 44, S115–S120. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Le, W.D.; Xie, W.J.; Alexianu, M.E.; Engelhardt, J.I.; Siklós, L.; Appel, S.H. Experimental Destruction of Substantia Nigra Initiated by Parkinson Disease Immunoglobulins. Arch. Neurol. 1998, 55, 1075–1080. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hampel, H.; Caraci, F.; Cuello, A.C.; Caruso, G.; Nisticò, R.; Corbo, M.; Baldacci, F.; Toschi, N.; Garaci, F.; Chiesa, P.A.; et al. A Path Toward Precision Medicine for Neuroinflammatory Mechanisms in Alzheimer’s Disease. Front. Immunol. 2020, 11, 456. [Google Scholar] [CrossRef] [PubMed]
- Schain, M.; Kreisl, W.C. Neuroinflammation in Neurodegenerative Disorders—A Review. Curr. Neurol. Neurosci. Rep. 2017, 17, 25. [Google Scholar] [CrossRef] [PubMed]
- Werry, E.L.; Bright, F.M.; Piguet, O.; Ittner, L.M.; Halliday, G.M.; Hodges, J.R.; Kiernan, M.C.; Loy, C.T.; Kril, J.J.; Kassiou, M. Recent Developments in TSPO PET Imaging as A Biomarker of Neuroinflammation in Neurodegenerative Disorders. Int. J. Mol. Sci. 2019, 20, 3161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kreisl, W.C.; Lyoo, C.H.; Liow, J.-S.; Wei, M.; Snow, J.; Page, E.; Jenko, K.J.; Morse, C.L.; Zoghbi, S.S.; Pike, V.W.; et al. 11C-PBR28 binding to translocator protein increases with progression of Alzheimer’s disease. Neurobiol. Aging 2016, 44, 53–61. [Google Scholar] [CrossRef] [Green Version]
- Hamelin, L.; Lagarde, J.; Dorothée, G.; Leroy, C.; Labit, M.; Comley, R.A.; de Souza, L.C.; Corne, H.; Dauphinot, L.; Bertoux, M.; et al. Early and protective microglial activation in Alzheimer’s disease: A prospective study using 18 F-DPA-714 PET imaging. Brain 2016, 139, 1252–1264. [Google Scholar] [CrossRef] [Green Version]
- Fan, Z.; Okello, A.A.; Brooks, D.J.; Edison, P. Longitudinal influence of microglial activation and amyloid on neuronal function in Alzheimer’s disease. Brain 2015, 138, 3685–3698. [Google Scholar] [CrossRef] [Green Version]
- Edison, P.; Ahmed, I.; Fan, Z.; Hinz, R.; Gelosa, G.; Ray Chaudhuri, K.; Walker, Z.; Turkheimer, F.E.; Brooks, D.J. Microglia, Amyloid, and Glucose Metabolism in Parkinson’s Disease with and without Dementia. Neuropsychopharmacology 2013, 38, 938–949. [Google Scholar] [CrossRef]
- Iannaccone, S.; Cerami, C.; Alessio, M.; Garibotto, V.; Panzacchi, A.; Olivieri, S.; Gelsomino, G.; Moresco, R.M.; Perani, D. In vivo microglia activation in very early dementia with Lewy bodies, comparison with Parkinson’s disease. Parkinsonism Relat. Disord. 2013, 19, 47–52. [Google Scholar] [CrossRef] [Green Version]
- Gerhard, A.; Pavese, N.; Hotton, G.; Turkheimer, F.; Es, M.; Hammers, A.; Eggert, K.; Oertel, W.; Banati, R.B.; Brooks, D.J. In vivo imaging of microglial activation with [11C](R)-PK11195 PET in idiopathic Parkinson’s disease. Neurobiol. Dis. 2006, 21, 404–412. [Google Scholar] [CrossRef] [PubMed]
- Brosseron, F.; Krauthausen, M.; Kummer, M.; Heneka, M.T. Body fluid cytokine levels in mild cognitive impairment and Alzheimer’s disease: A comparative overview. Mol. Neurobiol. 2014, 50, 534–544. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taipa, R.; Ferreira, V.; Brochado, P.; Robinson, A.; Reis, I.; Marques, F.; Mann, D.M.; Melo-Pires, M.; Sousa, N. Inflammatory pathology markers (activated microglia and reactive astrocytes) in early and late onset Alzheimer disease: A post mortem study. Neuropathol. Appl. Neurobiol. 2018, 44, 298–313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Osborn, L.M.; Kamphuis, W.; Wadman, W.J.; Hol, E.M. Astrogliosis: An integral player in the pathogenesis of Alzheimer’s disease. Prog. Neurobiol. 2016, 144, 121–141. [Google Scholar] [CrossRef] [PubMed]
- Arranz, A.M.; De Strooper, B. The role of astroglia in Alzheimer’s disease: Pathophysiology and clinical implications. Lancet Neurol. 2019, 18, 406–414. [Google Scholar] [CrossRef]
- Croisier, E.; Moran, L.B.; Dexter, D.T.; Pearce, R.K.B.; Graeber, M.B. Microglial inflammation in the parkinsonian substantia nigra: Relationship to alpha-synuclein deposition. J. Neuroinflamm. 2005, 2, 14. [Google Scholar] [CrossRef] [Green Version]
- Gelders, G.; Baekelandt, V.; Van der Perren, A. Linking neuroinflammation and neurodegeneration in parkinson’s disease. J. Immunol. Res. 2018, 2018, 4784268. [Google Scholar] [CrossRef] [Green Version]
- Heneka, M.T.; Carson, M.J.; El Khoury, J.; Landreth, G.E.; Brosseron, F.; Feinstein, D.L.; Jacobs, A.H.; Wyss-Coray, T.; Vitorica, J.; Ransohoff, R.M.; et al. Neuroinflammation in Alzheimer’s disease. Lancet Neurol. 2015, 14, 388–405. [Google Scholar] [CrossRef] [Green Version]
- Haass, C.; Kaether, C.; Thinakaran, G.; Sisodia, S. Trafficking and proteolytic processing of APP. Cold Spring Harb. Perspect. Med. 2012, 2, a006270. [Google Scholar] [CrossRef]
- Ludewig, S.; Korte, M. Novel Insights into the Physiological Function of the APP (Gene) Family and Its Proteolytic Fragments in Synaptic Plasticity. Front. Mol. Neurosci. 2017, 9, 161. [Google Scholar] [CrossRef] [Green Version]
- Hansen, D.V.; Hanson, J.E.; Sheng, M. Microglia in Alzheimer’s disease. J. Cell Biol. 2017, 217, 459–472. [Google Scholar] [CrossRef] [PubMed]
- Wes, P.D.; Sayed, F.A.; Bard, F.; Gan, L. Targeting microglia for the treatment of Alzheimer’s Disease. Glia 2016, 64, 1710–1732. [Google Scholar] [CrossRef] [PubMed]
- Cai, Z.; Hussain, M.D.; Yan, L.-J. Microglia, neuroinflammation, and beta-amyloid protein in Alzheimer’s disease. Int. J. Neurosci. 2014, 124, 307–321. [Google Scholar] [CrossRef] [PubMed]
- Marr, R.; Hafez, D. Amyloid beta and Alzheimer’s Disease: The role of neprilysin-2 in amyloid-beta clearance. Front. Aging Neurosci. 2014, 6, 187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kelley, N.; Jeltema, D.; Duan, Y.; He, Y. The NLRP3 inflammasome: An overview of mechanisms of activation and regulation. Int. J. Mol. Sci. 2019, 20, 3328. [Google Scholar] [CrossRef] [Green Version]
- Guerreiro, R.; Wojtas, A.; Bras, J.; Carrasquillo, M.; Rogaeva, E.; Majounie, E.; Cruchaga, C.; Sassi, C.; Kauwe, J.S.K.; Lupton, M.K.; et al. TREM2 Variants in Alzheimer’s Disease. N. Engl. J. Med. 2013, 368, 117–127. [Google Scholar] [CrossRef] [Green Version]
- Ulrich, J.D.; Ulland, T.K.; Colonna, M.; Holtzman, D.M. Elucidating the Role of TREM2 in Alzheimer’s Disease. Neuron 2017, 94, 237–248. [Google Scholar] [CrossRef] [Green Version]
- Wolfe, C.M.; Fitz, N.F.; Nam, K.N.; Lefterov, I.; Koldamova, R. The role of APOE and TREM2 in Alzheimer’s disease—Current understanding and perspectives. Int. J. Mol. Sci. 2019, 20, 81. [Google Scholar] [CrossRef] [Green Version]
- Becher, B.; Spath, S.; Goverman, J. Cytokine networks in neuroinflammation. Nat. Rev. Immunol. 2017, 17, 49–59. [Google Scholar] [CrossRef]
- Iadecola, C. The Neurovascular Unit Coming of Age: A Journey through Neurovascular Coupling in Health and Disease. Neuron 2017, 96, 17–42. [Google Scholar] [CrossRef] [Green Version]
- Zlokovic, B.V. Neurovascular pathways to neurodegeneration in Alzheimer’s disease and other disorders. Nat. Rev. Neurosci. 2011, 12, 723–738. [Google Scholar] [CrossRef] [PubMed]
- Shen, Y.; Yang, L.; Li, R. What does complement do in Alzheimer’s disease? Old molecules with new insights. Transl. Neurodegener. 2013, 2, 21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fischer, B.; Schmoll, H.; Platt, D.; Popa-Wagner, A.; Riederer, P.; Bauer, J. Complement C1q and C3 mRNA expression in the frontal cortex of Alzheimer’s patients. J. Mol. Med. 1995, 73, 465–471. [Google Scholar] [CrossRef]
- Laurent, C.; Buée, L.; Blum, D. Tau and neuroinflammation: What impact for Alzheimer’s Disease and Tauopathies? Biomed. J. 2018, 41, 21–33. [Google Scholar] [CrossRef]
- Rocha, E.M.; De Miranda, B.; Sanders, L.H. Alpha-synuclein: Pathology, mitochondrial dysfunction and neuroinflammation in Parkinson’s disease. Neurobiol. Dis. 2018, 109, 249–257. [Google Scholar] [CrossRef]
- Vivekanantham, S.; Shah, S.; Dewji, R.; Dewji, A.; Khatri, C.; Ologunde, R. Neuroinflammation in Parkinson’s disease: Role in neurodegeneration and tissue repair. Int. J. Neurosci. 2015, 125, 717–725. [Google Scholar] [CrossRef] [PubMed]
- Qin, L.; Wu, X.; Block, M.L.; Liu, Y.; Breese, G.R.; Hong, J.S.; Knapp, D.J.; Crews, F.T. Systemic LPS causes chronic neuroinflammation and progressive neurodegeneration. Glia 2007, 55, 453–462. [Google Scholar] [CrossRef] [Green Version]
- Hartmann, A.; Hunot, S.; Hirsch, E.C. Inflammation and dopaminergic neuronal loss in Parkinson’s disease: A complex matter. Exp. Neurol. 2003, 184, 561–564. [Google Scholar] [CrossRef]
- Dionísio, P.E.A.; Oliveira, S.R.; Amaral, J.S.J.D.; Rodrigues, C.M.P. Loss of Microglial Parkin Inhibits Necroptosis and Contributes to Neuroinflammation. Mol. Neurobiol. 2019, 56, 2990–3004. [Google Scholar] [CrossRef]
- Russo, I.; Bubacco, L.; Greggio, E. LRRK2 and neuroinflammation: Partners in crime in Parkinson’s disease? J. Neuroinflamm. 2014, 11, 52. [Google Scholar] [CrossRef] [Green Version]
- Giorgi, F.S.; Saccaro, L.F.; Galgani, A.; Busceti, C.L.; Biagioni, F.; Frati, A.; Fornai, F. The role of Locus Coeruleus in neuroinflammation occurring in Alzheimer’s disease. Brain Res. Bull. 2019, 153, 47–58. [Google Scholar] [CrossRef] [PubMed]
- Szabó, C.; Haskó, G.; Zingarelli, B.; Németh, Z.H.; Salzman, A.L.; Kvetan, V.; Pastores, S.M.; Vizi, E.S. Isoproterenol regulates tumour necrosis factor, interleukin-10, interleukin-6 and nitric oxide production and protects against the development of vascular hyporeactivity in endotoxaemia. Immunology 1997, 90, 95–100. [Google Scholar] [CrossRef] [PubMed]
- Agac, D.; Estrada, L.D.; Maples, R.; Hooper, L.V.; Farrar, J.D. The beta2-adrenergic receptor controls inflammation by driving rapid IL-10 secretion. Brain Behav. Immun. 2018, 74, 176–185. [Google Scholar] [CrossRef] [PubMed]
- Klotz, L.; Sastre, M.; Kreutz, A.; Gavrilyuk, V.; Klockgether, T.; Feinstein, D.L.; Heneka, M.T. Noradrenaline induces expression of peroxisome proliferator activated receptor gamma (PPARγ) in murine primary astrocytes and neurons. J. Neurochem. 2003, 86, 907–916. [Google Scholar] [CrossRef]
- Feinstein, D.L.; Heneka, M.T.; Gavrilyuk, V.; Dello Russo, C.; Weinberg, G.; Galea, E. Noradrenergic regulation of inflammatory gene expression in brain. Neurochem. Int. 2002, 41, 357–365. [Google Scholar] [CrossRef]
- Feinstein, D.L.; Kalinin, S.; Braun, D. Causes, consequences, and cures for neuroinflammation mediated via the locus coeruleus: Noradrenergic signaling system. J. Neurochem. 2016, 139, 154–178. [Google Scholar] [CrossRef] [Green Version]
- Frohman, E.M.; Vayuvegula, B.; Gupta, S.; Van Den Noort, S. Norepinephrine inhibits gamma-interferon-induced major histocompatibility class II (Ia) antigen expression on cultured astrocytes via beta-2-adrenergic signal transduction mechanisms. Proc. Natl. Acad. Sci. USA 1988, 85, 1292–1296. [Google Scholar] [CrossRef] [Green Version]
- Fornai, F.; Giorgi, F.S.; Gesi, M.; Chen, K.; Alessr, M.G.; Shih, J.C. Biochemical effects of the monoamine neurotoxins DSP-4 and MDMA in specific brain regions of MAO-B-deficient mice. Synapse 2001, 39, 213–221. [Google Scholar] [CrossRef]
- Heneka, M.T.; Galea, E.; Gavriluyk, V.; Dumitrescu-Ozimek, L.; Daeschner, J.; O’Banion, M.K.; Weinberg, G.; Klockgether, T.; Feinstein, D.L. Noradrenergic depletion potentiates beta -amyloid-induced cortical inflammation: Implications for Alzheimer’s disease. J. Neurosci. 2002, 22, 2434–2442. [Google Scholar] [CrossRef]
- Heneka, M.T.; Gavrilyuk, V.; Landreth, G.E.; O’Banion, M.K.; Weinberg, G.; Feinstein, D.L. Noradrenergic depletion increases inflammatory responses in brain: Effects on IκB and HSP70 expression. J. Neurochem. 2003, 85, 387–398. [Google Scholar] [CrossRef]
- Wenk, G.L.; Mcgann, K.; Hauss-Wegrzyniak, B.; Rosi, S. The toxicity of tumor necrosis factor-α upon cholinergic neurons within the nucleus basalis and the role of norepinephrine in the regulation of inflammation: Implications for Alzheimer’s disease. Neuroscience 2003, 121, 719–729. [Google Scholar] [CrossRef]
- Heneka, M.T.; O’Banion, M.K.; Terwel, D.; Kummer, M.P. Neuroinflammatory processes in Alzheimer’s disease. J. Neural Transm. 2010, 117, 919–947. [Google Scholar] [CrossRef] [PubMed]
- Kalinin, S.; Gavrilyuk, V.; Polak, P.E.; Vasser, R.; Zhao, J.; Heneka, M.T.; Feinstein, D.L. Noradrenaline deficiency in brain increases β-amyloid plaque burden in an animal model of Alzheimer’s disease. Neurobiol. Aging 2007, 28, 1206–1214. [Google Scholar] [CrossRef] [PubMed]
- Pugh, P.L.; Vidgeon-Hart, M.P.; Ashmeade, T.; Culbert, A.A.; Seymour, Z.; Perren, M.J.; Joyce, F.; Bate, S.T.; Babin, A.; Virley, D.J.; et al. Repeated administration of the noradrenergic neurotoxin N-(2-chloroethyl)-N-ethyl-2-bromobenzylamine (DSP-4) modulates neuroinflammation and amyloid plaque load in mice bearing amyloid precursor protein and presenilin-1 mutant transgenes. J. Neuroinflamm. 2007, 4, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jardanhazi-Kurutz, D.; Kummer, M.P.; Terwel, D.; Vogel, K.; Thiele, A.; Heneka, M.T. Distinct adrenergic system changes and neuroinflammation in response to induced locus ceruleus degeneration in APP/PS1 transgenic mice. Neuroscience 2011, 176, 396–407. [Google Scholar] [CrossRef]
- Jardanhazi-Kurutz, D.; Kummer, M.P.; Terwel, D.; Vogel, K.; Dyrks, T.; Thiele, A.; Heneka, M.T. Induced LC degeneration in APP/PS1 transgenic mice accelerates early cerebral amyloidosis and cognitive deficits. Neurochem. Int. 2010, 57, 375–382. [Google Scholar] [CrossRef]
- Kalinin, S.; Feinstein, D.L.; Xu, H.; Huesa, G.; Pelligrino, D.A.; Galea, E. Degeneration of noradrenergic fibres from the locus coeruleus causes tight-junction disorganisation in the rat brain. Eur. J. Neurosci. 2006, 24, 3393–3400. [Google Scholar] [CrossRef]
- Harik, S.I.; McGunigal, T. The protective influence of the locus ceruleus on the blood-brain barrier. Ann. Neurol. 1984, 15, 568–574. [Google Scholar] [CrossRef]
- Harik, S.I. Blood-brain barrier sodium/potassium pump: Modulation by central noradrenergic innervation. Proc. Natl. Acad. Sci. USA 1986, 83, 4067–4070. [Google Scholar] [CrossRef] [Green Version]
- Scullion, G.A.; Kendall, D.A.; Marsden, C.A.; Sunter, D.; Pardon, M.C. Chronic treatment with the α 2-adrenoceptor antagonist fluparoxan prevents age-related deficits in spatial working memory in APP × PS1 transgenic mice without altering β-amyloid plaque load or astrocytosis. Neuropharmacology 2011, 60, 223–234. [Google Scholar] [CrossRef]
- Gutiérrez, I.L.; González-Prieto, M.; Caso, J.R.; García-Bueno, B.; Leza, J.C.; Madrigal, J.L.M. Reboxetine Treatment Reduces Neuroinflammation and Neurodegeneration in the 5xFAD Mouse Model of Alzheimer’s Disease: Role of CCL2. Mol. Neurobiol. 2019, 56, 8628–8642. [Google Scholar] [CrossRef] [PubMed]
- Kang, S.S.; Liu, X.; Ahn, E.H.; Xiang, J.; Manfredsson, F.P.; Yang, X.; Luo, H.R.; Liles, L.C.; Weinshenker, D.; Ye, K. Norepinephrine metabolite DOPEGAL activates AEP and pathological Tau aggregation in locus coeruleus. J. Clin. Investig. 2020, 130, 422–437. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iba, M.; McBride, J.D.; Guo, J.L.; Zhang, B.; Trojanowski, J.Q.; Lee, V.M.-Y. Tau pathology spread in PS19 tau transgenic mice following locus coeruleus (LC) injections of synthetic tau fibrils is determined by the LC’s afferent and efferent connections. Acta Neuropathol. 2015, 130, 349–362. [Google Scholar] [CrossRef] [PubMed]
- Braak, H.; Del Tredici, K. Alzheimer’s pathogenesis: Is there neuron-to-neuron propagation? Acta Neuropathol. 2011, 121, 589–595. [Google Scholar] [CrossRef] [PubMed]
- Mravec, B.; Lejavova, K.; Vargovic, P.; Ondicova, K.; Horvathova, L.; Novak, P.; Manz, G.; Filipcik, P.; Novak, M.; Kvetnansky, R. Tauopathy in transgenic (SHR72) rats impairs function of central noradrenergic system and promotes neuroinflammation. J. Neuroinflamm. 2016, 13, 15. [Google Scholar] [CrossRef] [Green Version]
- Chalermpalanupap, T.; Schroeder, J.P.; Rorabaugh, J.M.; Liles, L.C.; Lah, J.J.; Levey, A.I.; Weinshenker, D. Locus Coeruleus Ablation Exacerbates Cognitive Deficits, Neuropathology, and Lethality in P301S Tau Transgenic Mice. J. Neurosci. 2018, 38, 74–92. [Google Scholar] [CrossRef] [Green Version]
- Duffy, K.B.; Ray, B.; Lahiri, D.K.; Tilmont, E.M.; Tinkler, G.P.; Herbert, R.L.; Greig, N.H.; Ingram, D.K.; Ottinger, M.A.; Mattison, J.A. Effects of Reducing Norepinephrine Levels via DSP4 Treatment on Amyloid-β Pathology in Female Rhesus Macaques (Macaca Mulatta). J. Alzheimers Dis. 2019, 68, 115–126. [Google Scholar] [CrossRef]
- Fornai, F.; Torracca, M.T.; Bassi, L.; D’Errigo, D.A.; Scalori, V.; Corsini, G.U. Norepinephrine loss selectively enhances chronic nigrostriatal dopamine depletion in mice and rats. Brain Res. 1996, 735, 349–353. [Google Scholar] [CrossRef]
- Butkovich, L.M.; Houser, M.C.; Tansey, M.G. α-synuclein and noradrenergic modulation of immune cells in Parkinson’s disease pathogenesis. Front. Neurosci. 2018, 12, 626. [Google Scholar] [CrossRef]
- Hou, L.; Sun, F.; Sun, W.; Zhang, L.; Wang, Q. Lesion of the Locus Coeruleus Damages Learning and Memory Performance in Paraquat and Maneb-induced Mouse Parkinson’s Disease Model. Neuroscience 2019, 419, 129–140. [Google Scholar] [CrossRef]
- af Bjerkén, S.; Stenmark Persson, R.; Barkander, A.; Karalija, N.; Pelegrina-Hidalgo, N.; Gerhardt, G.A.; Virel, A.; Strömberg, I. Noradrenaline is crucial for the substantia nigra dopaminergic cell maintenance. Neurochem. Int. 2019, 131, 104551. [Google Scholar] [CrossRef] [PubMed]
- Herrera, A.J.; Castaño, A.; Venero, J.L.; Cano, J.; Machado, A. The single intranigral injection of LPS as a new model for studying the selective effects of inflammatory reactions on dopaminergic system. Neurobiol. Dis. 2000, 7, 429–447. [Google Scholar] [CrossRef] [Green Version]
- Bharani, K.L.; Derex, R.; Granholm, A.C.; Ledreux, A. A noradrenergic lesion aggravates the effects of systemic inflammation on the hippocampus of aged rats. PLoS ONE 2017, 12, e0189821. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, S.; Jiang, L.; Oyarzabal, E.A.; Wilson, B.; Li, Z.; Shih, Y.Y.I.; Wang, Q.; Hong, J.S. Loss of Brain Norepinephrine Elicits Neuroinflammation-Mediated Oxidative Injury and Selective Caudo-Rostral Neurodegeneration. Mol. Neurobiol. 2019, 56, 2653–2669. [Google Scholar] [CrossRef] [PubMed]
- Song, S.; Wang, Q.; Jiang, L.; Oyarzabal, E.; Riddick, N.V.; Wilson, B.; Moy, S.S.; Shih, Y.Y.I.; Hong, J.S. Noradrenergic dysfunction accelerates LPS-elicited inflammation-related ascending sequential neurodegeneration and deficits in non-motor/motor functions. Brain Behav. Immun. 2019, 81, 374–387. [Google Scholar] [CrossRef] [PubMed]
- Fornai, F.; Ruffoli, R.; Giorgi, F.S.; Paparelli, A. The role of locus coeruleus in the antiepileptic activity induced by vagus nerve stimulation. Eur. J. Neurosci. 2011, 33, 2169–2178. [Google Scholar] [CrossRef] [PubMed]
- Farrand, A.Q.; Helke, K.L.; Gregory, R.A.; Gooz, M.; Hinson, V.K.; Boger, H.A. Vagus nerve stimulation improves locomotion and neuronal populations in a model of Parkinson’s disease. Brain Stimul. 2017, 10, 1045–1054. [Google Scholar] [CrossRef]
- Farrand, A.Q.; Verner, R.S.; McGuire, R.M.; Helke, K.L.; Hinson, V.K.; Boger, H.A. Differential effects of vagus nerve stimulation paradigms guide clinical development for Parkinson’s disease. Brain Stimul. 2020, 13, 1323–1332. [Google Scholar] [CrossRef]
- Betts, M.J.; Kirilina, E.; Otaduy, M.C.G.; Ivanov, D.; Acosta-Cabronero, J.; Callaghan, M.F.; Lambert, C.; Cardenas-Blanco, A.; Pine, K.; Passamonti, L.; et al. Locus coeruleus imaging as a biomarker for noradrenergic dysfunction in neurodegenerative diseases. Brain 2019, 142, 2558–2571. [Google Scholar] [CrossRef]
- Brumberg, J.; Tran-Gia, J.; Lapa, C.; Isaias, I.U.; Samnick, S. PET imaging of noradrenaline transporters in Parkinson’s disease: Focus on scan time. Ann. Nucl. Med. 2019, 33, 69–77. [Google Scholar] [CrossRef] [Green Version]
- Baldacci, F.; Mazzucchi, S.; Della Vecchia, A.; Giampietri, L.; Giannini, N.; Koronyo-Hamaoui, M.; Ceravolo, R.; Siciliano, G.; Bonuccelli, U.; Elahi, F.M.; et al. The path to biomarker-based diagnostic criteria for the spectrum of neurodegenerative diseases. Expert Rev. Mol. Diagn. 2020, 20, 421–441. [Google Scholar] [CrossRef] [PubMed]
- Hampel, H.; Toschi, N.; Baldacci, F.; Zetterberg, H.; Blennow, K.; Kilimann, I.; Teipel, S.J.; Cavedo, E.; Melo dos Santos, A.; Epelbaum, S.; et al. Alzheimer’s disease biomarker-guided diagnostic workflow using the added value of six combined cerebrospinal fluid candidates: Aβ1–42, total-tau, phosphorylated-tau, NFL, neurogranin, and YKL-40. Alzheimer’s Dement. 2018, 14, 492–501. [Google Scholar] [CrossRef] [PubMed]
- Matsuda, H.; Shigemoto, Y.; Sato, N. Neuroimaging of Alzheimer’s disease: Focus on amyloid and tau PET. Jpn. J. Radiol. 2019, 37, 735–749. [Google Scholar] [CrossRef] [PubMed]
- Farmer, P.; Pugin, J. β-Adrenergic agonists exert their “anti-inflammatory” effects in monocytic cells through the IκB/NF-κB pathway. Am. J. Physiol. Cell. Mol. Physiol. 2000, 279, L675–L682. [Google Scholar] [CrossRef]
- Kong, Y.; Ruan, L.; Qian, L.; Liu, X.; Le, Y. Norepinephrine promotes microglia to uptake and degrade amyloid beta peptide through upregulation of mouse formyl peptide receptor 2 and induction of insulin-degrading enzyme. J. Neurosci. 2010, 30, 11848–11857. [Google Scholar] [CrossRef] [Green Version]
- Mittal, S.; Bjørnevik, K.; Im, D.S.; Flierl, A.; Dong, X.; Locascio, J.J.; Abo, K.M.; Long, E.; Jin, M.; Xu, B.; et al. β2-Adrenoreceptor is a regulator of the α-synuclein gene driving risk of Parkinson’s disease. Science 2017, 357, 891–898. [Google Scholar] [CrossRef] [Green Version]
- Schrag, A.; Sauerbier, A.; Chaudhuri, K.R. New clinical trials for nonmotor manifestations of Parkinson’s disease. Mov. Disord. 2015, 30, 1490–1504. [Google Scholar] [CrossRef]
- Carreno, F.R.; Frazer, A. Vagal Nerve Stimulation for Treatment-Resistant Depression. Neurotherapeutics 2017, 14, 716–727. [Google Scholar] [CrossRef]
- González, H.F.J.; Yengo-Kahn, A.; Englot, D.J. Vagus Nerve Stimulation for the Treatment of Epilepsy. Neurosurg. Clin. N. Am. 2019, 1, 477–482. [Google Scholar] [CrossRef]
- Chang, C.H.; Lane, H.Y.; Lin, C.H. Brain stimulation in Alzheimer’s disease. Front. Psychiatry 2018, 9, 201. [Google Scholar] [CrossRef] [Green Version]
- Morris, R.; Yarnall, A.J.; Hunter, H.; Taylor, J.P.; Baker, M.R.; Rochester, L. Noninvasive vagus nerve stimulation to target gait impairment in Parkinson’s disease. Mov. Disord. 2019, 34, 918–919. [Google Scholar] [CrossRef] [PubMed]
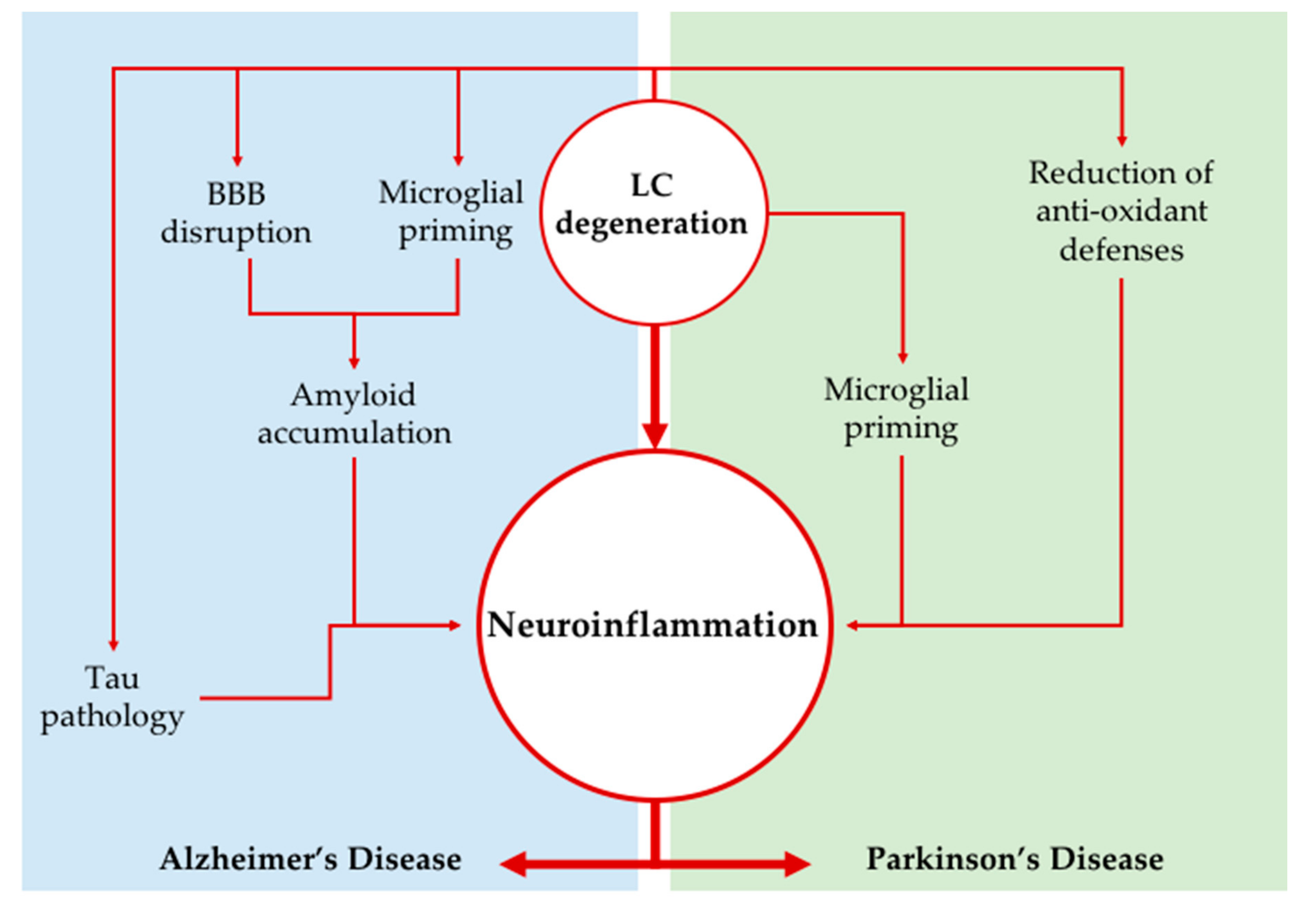

{kind=link}
| Authors and Year [Ref.] | Disease | Animalspecies | LC Manipulation | Neuroinflammatory Changes |
|---|---|---|---|---|
| Heneka et al. 2002 | AD | Rats | DSP-4 lesion | Increased inflammatory response after intracortical amyloid peptides injection |
| Heneka et al. 2003 | AD | Rats | DSP-4 lesion | Reduced basal levels of NF-kβ inhibitory Ikβ protein and expression of HSP-70 |
| Wenk et al. 2003 | AD | Mice | DSP-4 lesion | TNF-α injection within BF caused cholinergic cells death and evoked neuroinflammation, with no differences between LC-lesioned and control mice |
| Heneka et al. 2006 | AD | APP23 Tg Mice | DSP-4 lesion | Increased amyloid deposition and elevation of microglial/astrocytic activation |
| Kalinin et al. 2007 | AD | APP V717F Tg Mice | DSP-4 lesion | Increased amyloid deposition and astroglial activation |
| Pugh et al. 2007 | AD | APP/PS1 Tg Mice | DSP-4 lesion | Increased level of IL-1β and reduced level of NF-kβ inhibitory Ikβ protein |
| Heneka et al. 2010 | AD | APP V717F Tg Mice | DSP-4 lesion | Increased amyloid deposition—independently of APP processing; increased pro-inflammatory chemokines and cytokines concentration; impaired microglial amyloid clearance |
| L-threo-DOPS | Restoration of microglial activity and reduction of cytokines production | |||
| Jardanhazi-Kurutz et al. 2010 | AD | APP/PS1 Tg Mice | DSP-4 lesion | Increased amyloid accumulation and macrophages activation |
| Jardanhazi-Kurutz et al. 2011 | AD | APP/PS1 Tg Mice | DSP-4 lesion | Increased amyloid accumulation and elevation of IL-1β level |
| Scullion et al. 2011 | AD | APP/PS1 Tg Mice | Fluparoxan | The administration of the α-2 blocker was associated with better cognitive performances, while it did not reduce neuroinflammatory or amyloid burden |
| Mravec et al. 2016 | AD | SHR72 Tg Mice | \ | Induced tauopathy causes LC degeneration, which is associated with reduction of NA level and increased concentration of IL6, TNF-α, and iNOS |
| Chalermpalanupap et al. 2018 | AD | P301S Tg Mice | DSP-4 lesion | Increased tau pathology burden in the hippocampus, accompanied by higher microglial activation |
| Gutierrez et al. 2019 | AD | 5xFAD Mice | Reboxetine | Reduced neuroinflammatory and amyloid burden |
| Bharani et al. 2017 | \ | Rats | DSP-4 lesion | Increased astroglial activation and serum interleukin concentration after LPS exposure |
| Farrand et al. 2017 | PD | 6-OHDA treated Rats | DSP-4 lesion/VNS | Both in DSP-4 lesioned and control rats, VNS was associated with better motor performance, reduced neuroinflammation, and increased NA markers in LC, SN, and striatum |
| Bjerken et al. 2019 | PD | Rats | DSP-4 lesion | LC degeneration caused DA cell degeneration; activated microglial observed in impaired substantia nigra |
| Hou et al. 2019 | PD | PQ/MB treated Mice | DSP-4 lesion | Increased degeneration of DA cells; reduction of glutathione contents; increased microglia activation and M1 polarization |
| Song et al. 2019 (a) | PD | gp91phox−/− Mice | DSP-4 lesion | Increased microglial activation and oxidative stress in DA cells |
| Song et al. 2019 (b) | PD | Mice | DSP-4 lesion | Increased neurodegeneration, neuroinflammation, and oxidative stress induced by LPS exposure |
| Farrand et al. 2020 | PD | 6-OHDA treated Rats | DSP-4 lesion/VNS | High frequency VNS was associated with better motor performances, reduced glial activation, and NA impairment |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Giorgi, F.S.; Biagioni, F.; Galgani, A.; Pavese, N.; Lazzeri, G.; Fornai, F. Locus Coeruleus Modulates Neuroinflammation in Parkinsonism and Dementia. Int. J. Mol. Sci. 2020, 21, 8630. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21228630
Giorgi FS, Biagioni F, Galgani A, Pavese N, Lazzeri G, Fornai F. Locus Coeruleus Modulates Neuroinflammation in Parkinsonism and Dementia. International Journal of Molecular Sciences. 2020; 21(22):8630. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21228630
Chicago/Turabian StyleGiorgi, Filippo Sean, Francesca Biagioni, Alessandro Galgani, Nicola Pavese, Gloria Lazzeri, and Francesco Fornai. 2020. "Locus Coeruleus Modulates Neuroinflammation in Parkinsonism and Dementia" International Journal of Molecular Sciences 21, no. 22: 8630. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21228630