Discovery and Design of Novel Small Molecule GSK-3 Inhibitors Targeting the Substrate Binding Site

 , , and
, , and 
Abstract
:
1. Introduction
2. Results
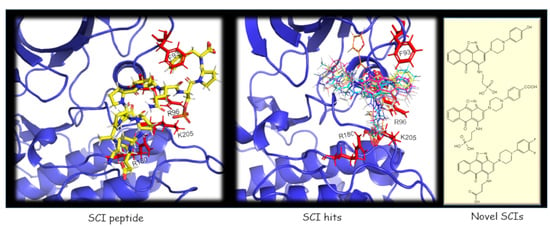
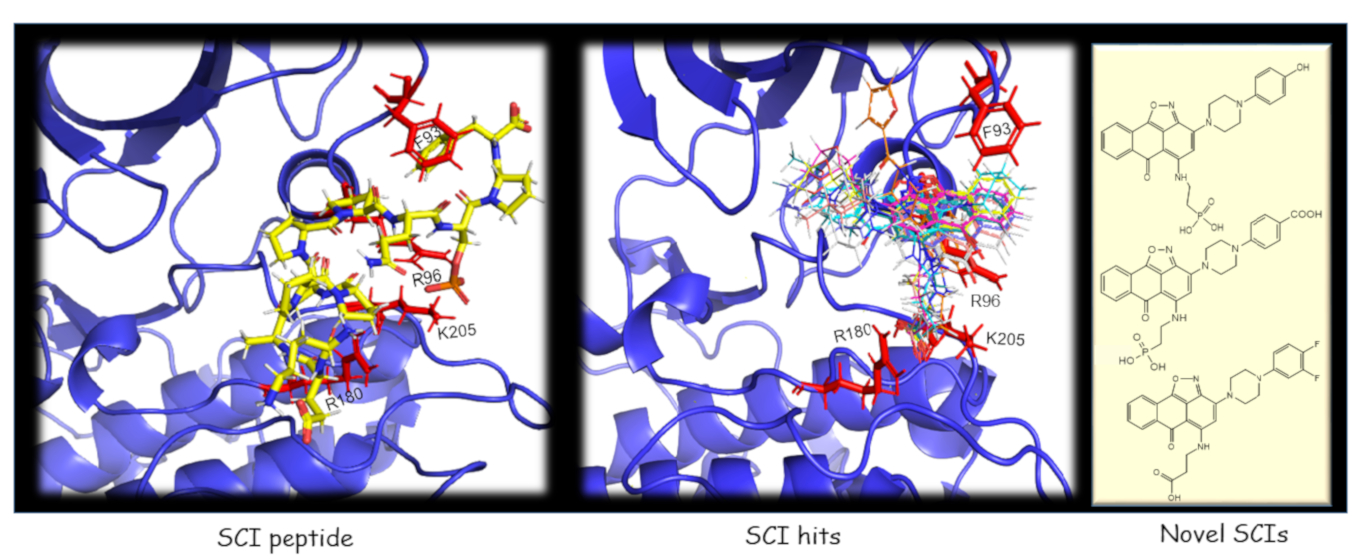
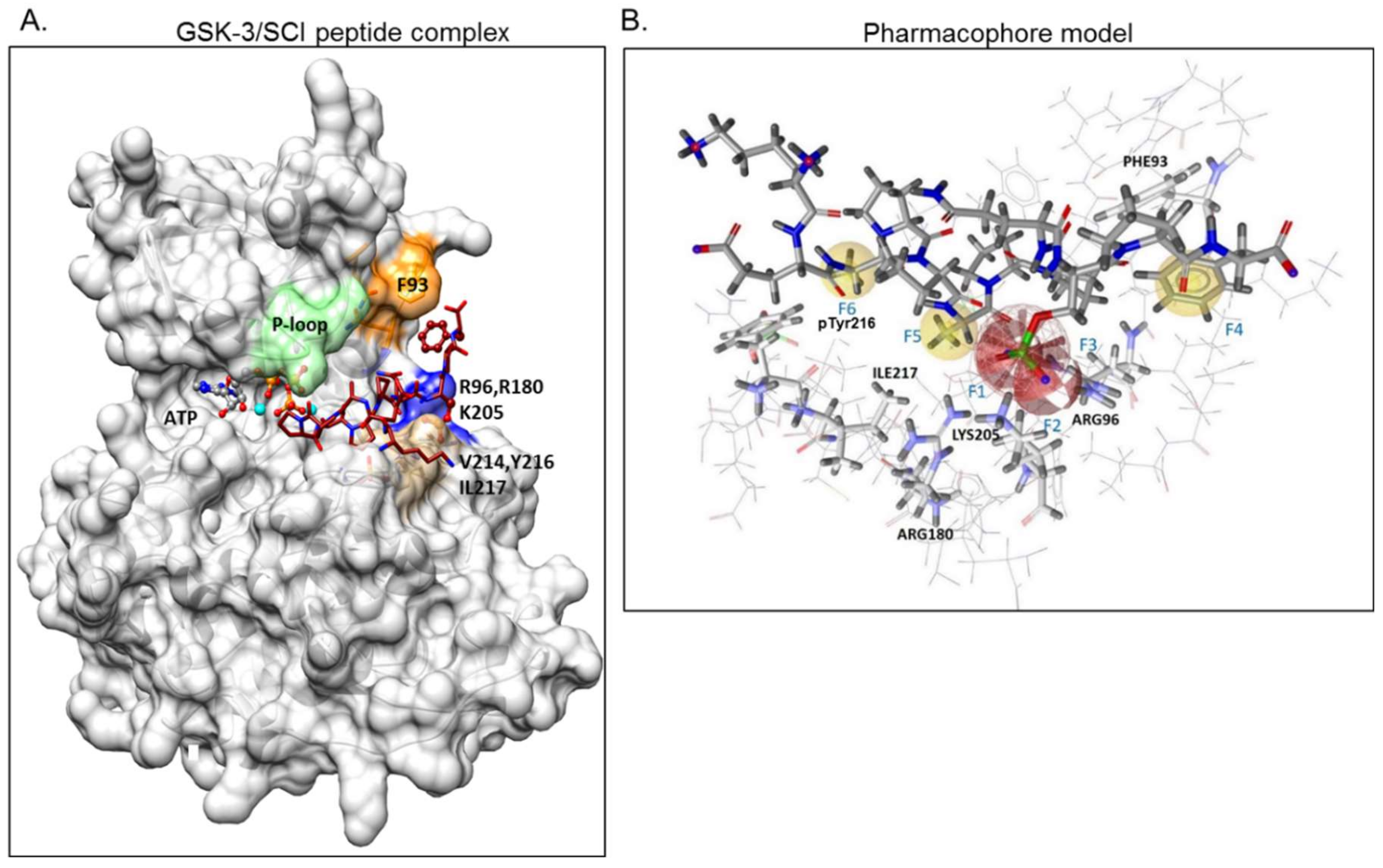
2.1. The Use of GSK-3/SCI Peptide Binding Model for Pharmacophore-Based Virtual Screening
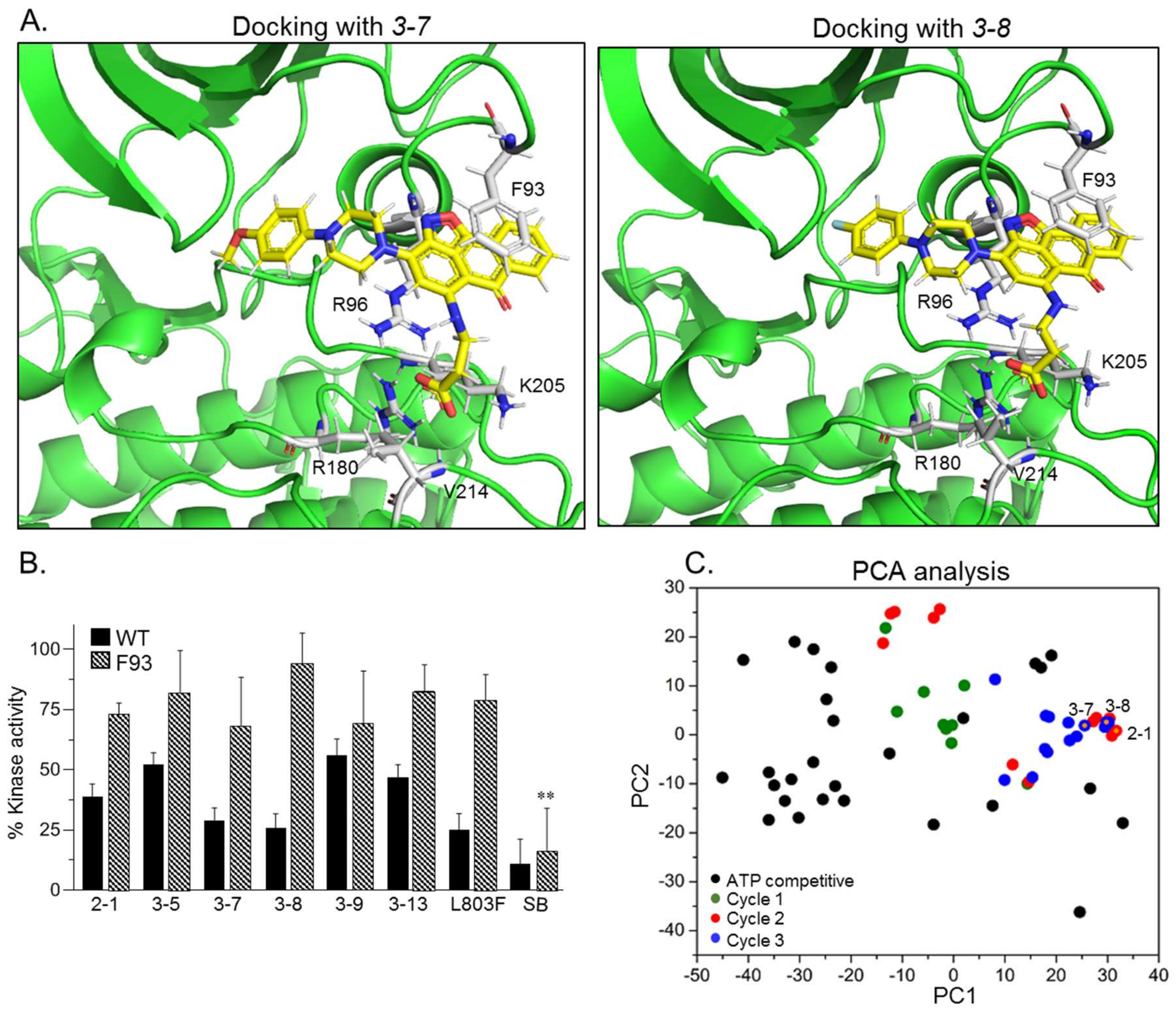
2.2. SCI Hits Interact with the GSK-3β Substrate Binding Site and Are Chemically Unique
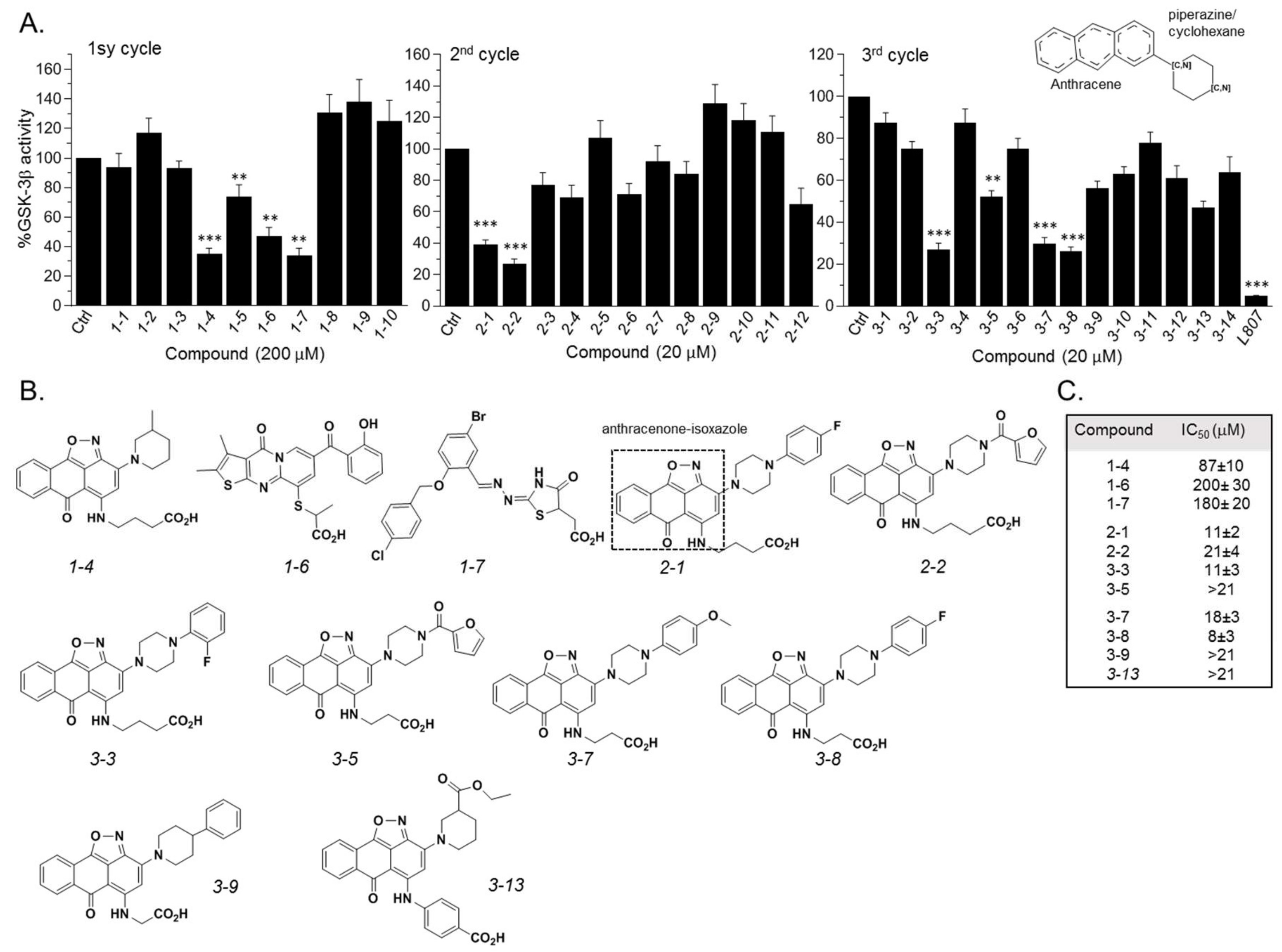
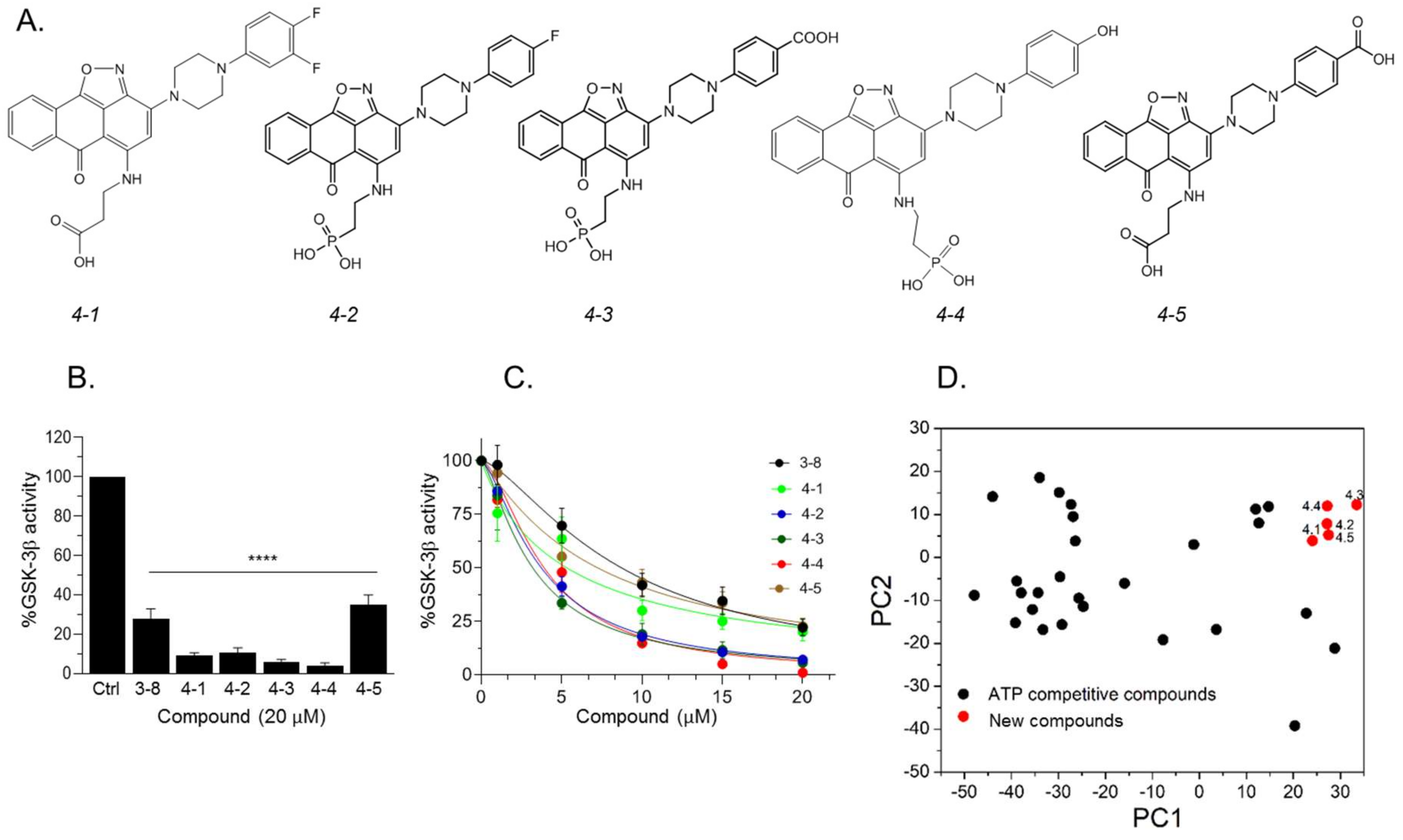
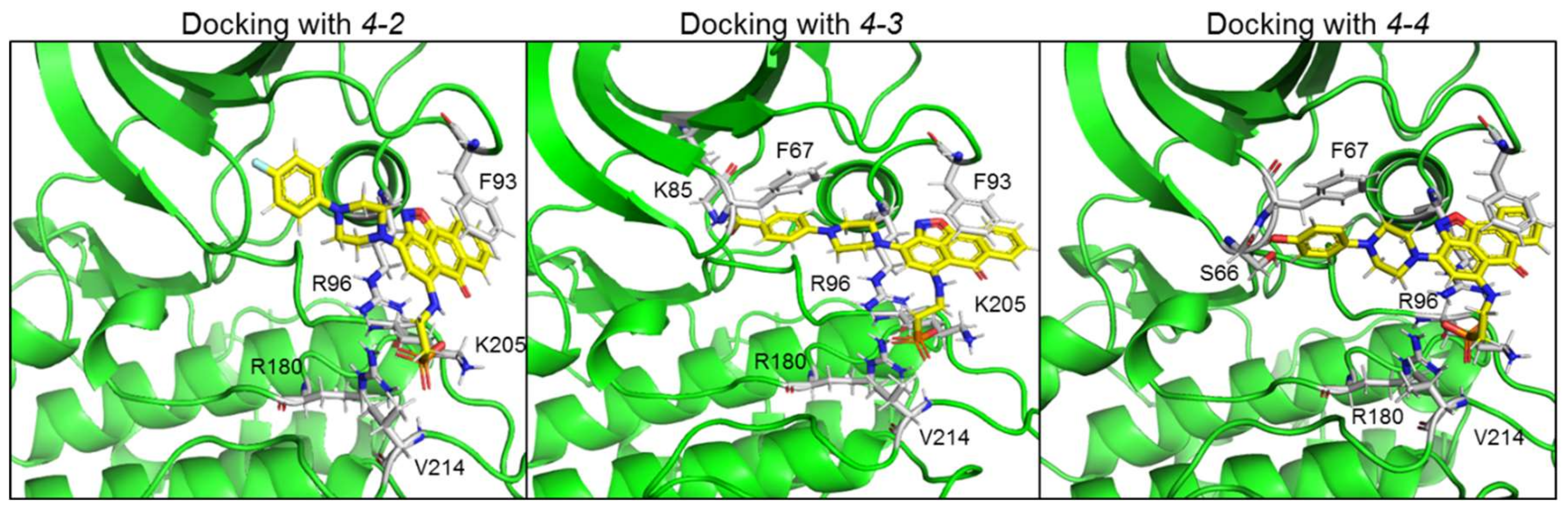
2.3. Design and Synthesis of Novel GSK-3 SCIs
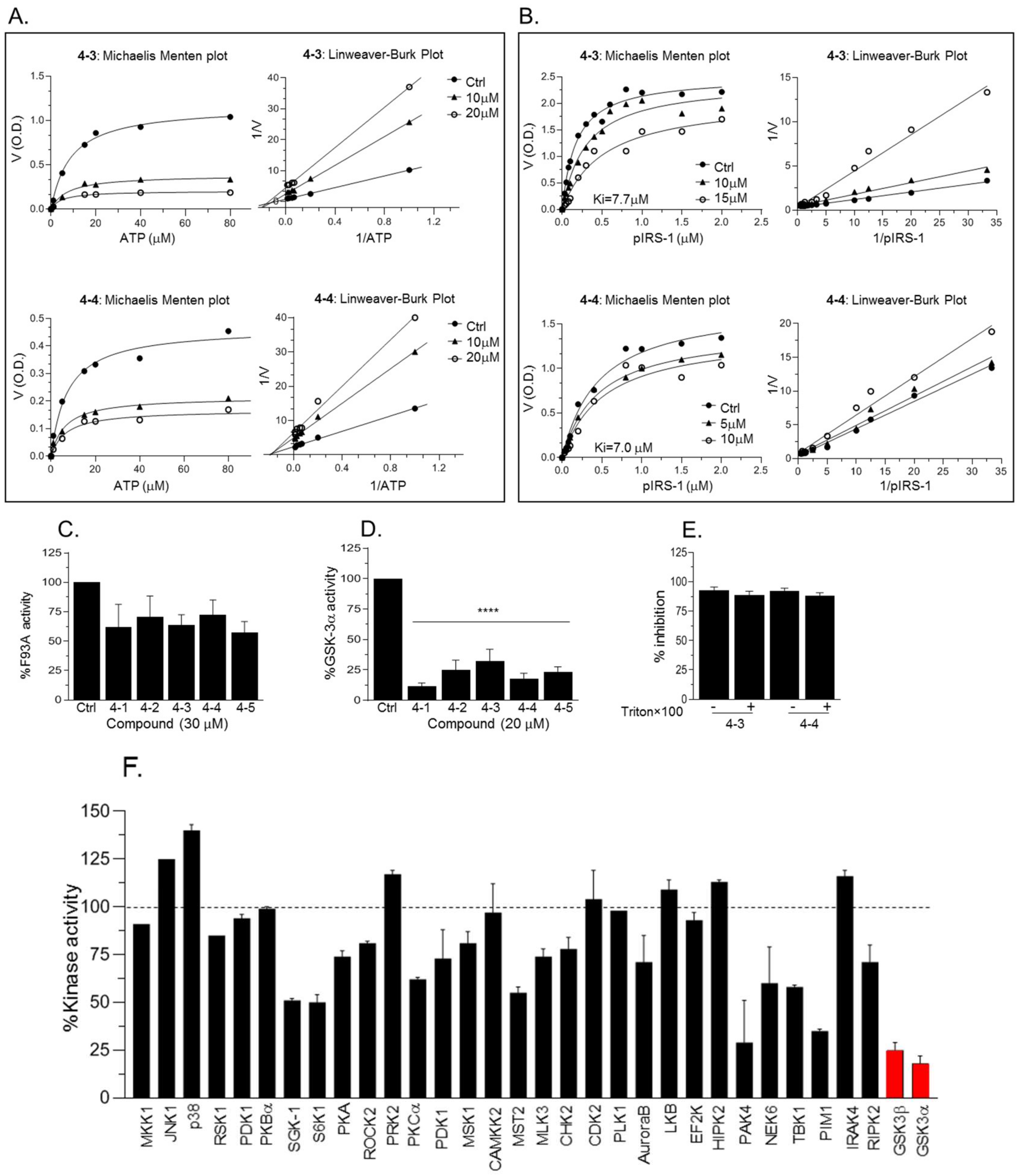
2.4. New Compounds Function as GSK-3 SCIs and Show Selectivity
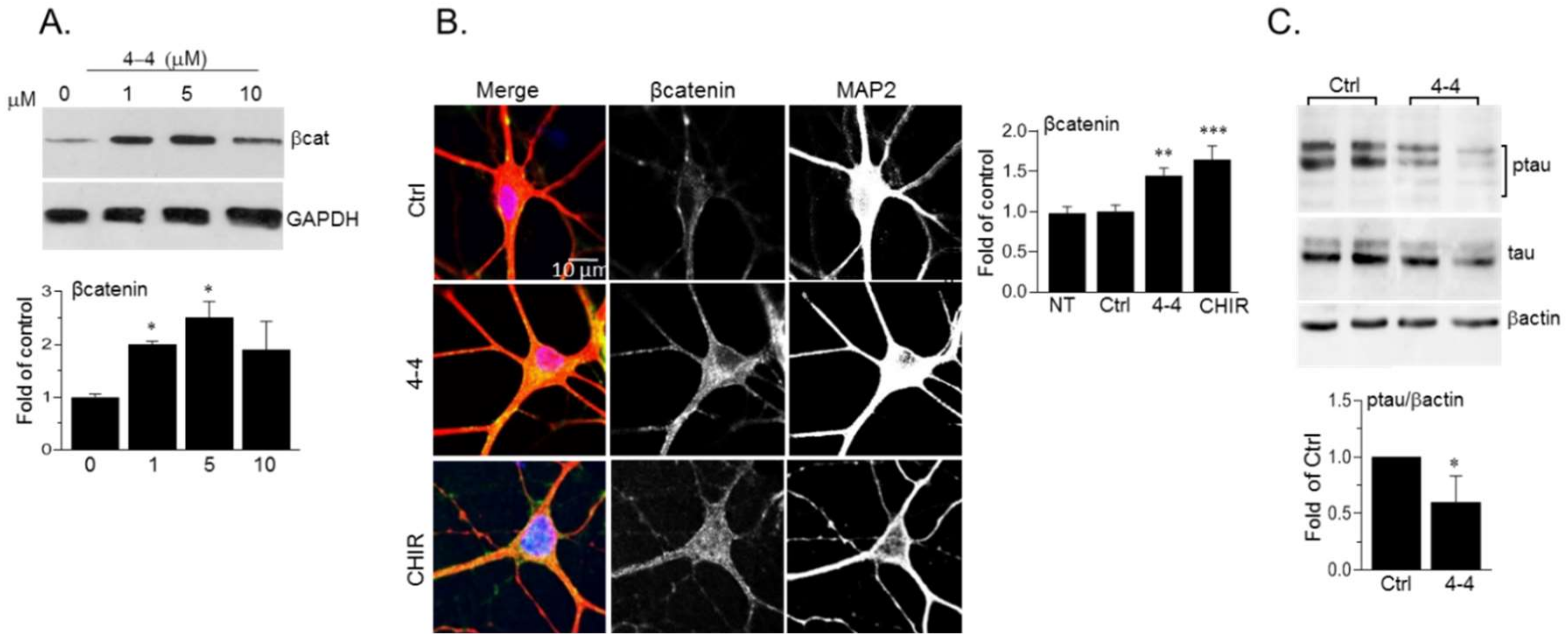
2.5. Biological Activity of the GSK-3 SCI Compounds
3. Discussion
4. Materials and Methods
4.1. Pharmacophore Design and Virtual Screening
4.2. Chemicals
4.3. PCA Analysis
4.4. In Vitro Kinase Assays
4.5. Cell Culture and Cell Extraction
4.6. Primary Neurons
4.7. Western Blot Analysis
4.8. Immunostaining
4.9. Image Processing
4.10. Statistical Analyses
5. Patents
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Manning, G.; Whyte, D.B.; Martinez, R.; Hunter, T.; Sudarsanam, S. The protein kinase complement of the human genome. Science 2002, 298, 1912–1934. [Google Scholar] [CrossRef] [Green Version]
- Hanks, S.K.; Quinn, A.M.; Hunter, T. The protein kinase family: Conserved features and deduced phylogeny of the catalytic domains. Science 1988, 241, 42–52. [Google Scholar] [CrossRef]
- Taylor, S.S.; Radzio-Andzelm, E.; Hunter, T. How do protein kinases discriminate between serine/threonine and tyrosine? Structural insights from the insulin receptor protein-tyrosine kinase. FASEB J. 1995, 9, 1255–1266. [Google Scholar] [CrossRef] [Green Version]
- Bain, J.; Plater, L.; Elliott, M.; Shpiro, N.; Hastie, C.J.; McLauchlan, H.; Klevernic, I.; Arthur, J.S.; Alessi, D.R.; Cohen, P. The selectivity of protein kinase inhibitors: A further update. Biochem. J. 2007, 408, 297–315. [Google Scholar] [CrossRef] [Green Version]
- Davis, M.I.; Hunt, J.P.; Herrgard, S.; Ciceri, P.; Wodicka, L.M.; Pallares, G.; Hocker, M.; Treiber, D.K.; Zarrinkar, P.P. Comprehensive analysis of kinase inhibitor selectivity. Nat. Biotechnol. 2011, 29, 1046–1051. [Google Scholar] [CrossRef]
- Fabian, M.A.; Biggs, W.H., III; Treiber, D.K.; Atteridge, C.E.; Azimioara, M.D.; Benedetti, M.G.; Carter, T.A.; Ciceri, P.; Edeen, P.T.; Floyd, M.; et al. A small molecule-kinase interaction map for clinical kinase inhibitors. Nat. Biotechnol. 2005, 23, 329–336. [Google Scholar] [CrossRef]
- Dar, A.C.; Shokat, K.M. The evolution of protein kinase inhibitors from antagonists to agonists of cellular signaling. Annu. Rev. Biochem. 2011, 80, 769–795. [Google Scholar] [CrossRef] [Green Version]
- Rosti, G.; Castagnetti, F.; Gugliotta, G.; Baccarani, M. Tyrosine kinase inhibitors in chronic myeloid leukaemia: Which, when, for whom? Nat. Rev. Clin. Oncol. 2017, 14, 141–154. [Google Scholar] [CrossRef]
- Breen, M.E.; Matthew, B.S. Small molecule substrate phosphorylation site inhibitors of protein kinases: Approaches and challenges. ACS Chem. Biol. 2015, 10, 175–189. [Google Scholar] [CrossRef]
- Eldar-Finkelman, H.; Eisenstein, M. Peptide inhibitors targeting protein kinases. Curr. Pharm. Des. 2009, 15, 2463–2470. [Google Scholar] [CrossRef] [Green Version]
- De Oliveira, P.S.; Ferraz, F.A.; Pena, D.A.; Pramio, D.T.; Morais, F.A.; Schechtman, D. Revisiting protein kinase-substrate interactions: Toward therapeutic development. Sci. Signal. 2016, 9, re3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kolb, P.; Ferreira, R.S.; Irwin, J.J.; Shoichet, B.K. Docking and chemoinformatic screens for new ligands and targets. Curr. Opin. Biotechnol. 2009, 20, 429–436. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lavecchia, A.; Di Giovanni, C. Virtual screening strategies in drug discovery: A critical review. Curr. Med. Chem. 2013, 20, 2839–2860. [Google Scholar] [CrossRef] [PubMed]
- Irwin, J.J.; Shoichet, B.K. Docking Screens for Novel Ligands Conferring New Biology. J. Med. Chem. 2016, 59, 4103–4120. [Google Scholar] [CrossRef] [Green Version]
- Li, Q.; Kang, C. Mechanisms of Action for Small Molecules Revealed by Structural Biology in Drug Discovery. Int. J. Mol. Sci. 2020, 21, 5262. [Google Scholar] [CrossRef]
- Eldar-Finkelman, H.; Martinez, A. GSK-3 Inhibitors: Preclinical and Clinical Focus on CNS. Front. Mol. Neurosci. 2011, 4, 32. [Google Scholar] [CrossRef] [Green Version]
- Llorens-Martin, M.; Jurado, J.; Hernandez, F.; Avila, J. GSK-3beta, a pivotal kinase in Alzheimer disease. Front. Mol. Neurosci. 2014, 7, 46. [Google Scholar] [CrossRef] [Green Version]
- Kim, W.Y.; Snider, W.D. Functions of GSK-3 Signaling in Development of the Nervous System. Front. Mol. Neurosci. 2011, 4, 44. [Google Scholar] [CrossRef] [Green Version]
- Lal, H.; Ahmad, F.; Woodgett, J.; Force, T. The GSK-3 family as therapeutic target for myocardial diseases. Circ. Res. 2015, 116, 138–149. [Google Scholar] [CrossRef]
- Lovestone, S.; Killick, R.; Di Forti, M.; Murray, R. Schizophrenia as a GSK-3 dysregulation disorder. Trends Neurosci. 2007, 30, 142–149. [Google Scholar] [CrossRef]
- Lin, R.; Jones, N.C.; Kwan, P. Unravelling the Role of Glycogen Synthase Kinase-3 in Alzheimer’s Disease-Related Epileptic Seizures. Int. J. Mol. Sci. 2020, 21, 3676. [Google Scholar] [CrossRef] [PubMed]
- Woodgett, J.R. Molecular cloning and expression of glycogen synthase kinase-3/factorA. EMBO J. 1990, 9, 2431–2438. [Google Scholar] [CrossRef] [PubMed]
- Hanger, D.P.; Hughes, K.; Woodgett, J.R.; Brion, J.P.; Anderton, B.H. Glycogen synthase kinase-3 induces Alzheimer’s disease-like phosphorylation of tau: Generation of paired helical filament epitopes and neuronal localisation of the kinase. Neurosci. Lett. 1992, 147, 58–62. [Google Scholar] [CrossRef]
- Li, T.; Hawkes, C.; Qureshi, H.Y.; Kar, S.; Paudel, H.K. Cyclin-dependent protein kinase 5 primes microtubule-associated protein tau site-specifically for glycogen synthase kinase 3beta. Biochemistry 2006, 45, 3134–3145. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, S.; Kishida, S.; Yamamoto, H.; Murai, H.; Koyama, S.; Kikuchi, A. Axin, a negative regulator of the Wnt signaling pathway, forms a complex with GSK-3beta and beta-catenin and promotes GSK-3beta-dependent phosphorylation of beta-catenin. EMBO J. 1998, 17, 1371–1384. [Google Scholar] [CrossRef]
- Yost, C.; Torres, M.; Miller, J.; Huang, E.; Kimelman, D.; Moon, R. The axis-inducing activity, stability, and subcellular distribution of beta-catenin is regulated in Xenopus embryos by glycogen synthase kinase 3. Genes Dev. 1996, 10, 1443–1454. [Google Scholar] [CrossRef] [Green Version]
- Hoeflich, K.P.; Luo, J.; Rubie, E.A.; Tsao, M.S.; Jin, O.; Woodgett, J.R. Requirement for glycogen synthase kinase-3beta in cell survival and NF-kappaB activation. Nature 2000, 406, 86–90. [Google Scholar] [CrossRef]
- Steinbrecher, K.A.; Wilson, W., III; Cogswell, P.C.; Baldwin, A.S. Glycogen synthase kinase 3beta functions to specify gene-specific, NF-kappaB-dependent transcription. Mol. Cell. Biol. 2005, 25, 8444–8455. [Google Scholar] [CrossRef] [Green Version]
- Jope, R.S.; Cheng, Y.; Lowell, J.A.; Worthen, R.J.; Sitbon, Y.H.; Beurel, E. Stressed and Inflamed, Can GSK3 Be Blamed? Trends Biochem. Sci. 2017, 42, 180–192. [Google Scholar] [CrossRef] [Green Version]
- Azoulay-Alfaguter, I.; Elya, R.; Avrahami, L.; Katz, A.; Eldar-Finkelman, H. Combined regulation of mTORC1 and lysosomal acidification by GSK-3 suppresses autophagy and contributes to cancer cells growth. Oncogene 2015, 34, 4613–4623. [Google Scholar] [CrossRef] [Green Version]
- Avrahami, L.; Farfara, D.; Shaham-Kol, M.; Vassar, R.; Frenkel, D.; Eldar-Finkelman, H. Inhibition of GSK-3 Ameliorates beta-Amyloid (A-beta) Pathology and Restores Lysosomal Acidification and mTOR Activity in the Alzheimer’s Disease Mouse Model. In vivo and In vitro Studies. J. Biol. Chem. 2013, 288, 1295–1306. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Licht-Murava, A.; Paz, R.; Vaks, L.; Avrahami, L.; Plotkin, B.; Eisenstein, M.; Eldar-Finkelman, H. A unique type of GSK-3 inhibitor brings new opportunities to the clinic. Sci. Signal. 2016, 9, ra110. [Google Scholar] [CrossRef] [PubMed]
- Licht-Murava, A.; Plotkin, B.; Eisenstein, M.; Eldar-Finkelman, H. Elucidating substrate and inhibitor binding sites on the surface of GSK-3beta and the refinement of a competitive inhibitor. J. Mol. Biol. 2011, 408, 366–378. [Google Scholar] [CrossRef] [PubMed]
- Chu, B.; Soncin, F.; Price, B.D.; Stevenson, M.A.; Calderwood, S.K. Sequential phosphorylation by mitogen-activated protein kinase and glycogen synthase kinase 3 represses transcriptional activation by heat shock factor-1. J. Biol. Chem. 1996, 271, 30847–30857. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ilouz, R.; Kowelsman, N.; Eisenstein, M.; Eldar-Finkelman, H. Identification of novel glycogen synthase kinase-3beta substrate-interacting residues suggests a common mechanism for substrate recognition. J. Biol. Chem. 2006, 281, 30621–30630. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dajani, R.; Fraser, E.; Roe, S.M.; Young, N.; Good, V.; Dale, T.C.; Pearl, L.H. Crystal structure of glycogen synthase kinase 3 beta: Structural basis for phosphate-primed substrate specificity and autoinhibition. Cell 2001, 105, 721–732. [Google Scholar] [CrossRef]
- Ter Haar, E.; Coll, J.T.; Austen, D.A.; Hsiao, H.M.; Swenson, L.; Jain, J. Structure of GSK3beta reveals a primed phosphorylation mechanism. Nat. Struct. Biol. 2001, 8, 593–596. [Google Scholar] [CrossRef]
- Chen, G.; Bower, K.A.; Ma, C.; Fang, S.; Thiele, C.J.; Luo, J. Glycogen synthase kinase 3beta (GSK3beta) mediates 6-hydroxydopamine-induced neuronal death. FASEB J. 2004, 18, 1162–1164. [Google Scholar] [CrossRef]
- Zhu, Q.; Yang, J.; Han, S.; Liu, J.; Holzbeierlein, J.; Thrasher, J.B.; Li, B. Suppression of glycogen synthase kinase 3 activity reduces tumor growth of prostate cancer in vivo. Prostate 2011, 71, 835–845. [Google Scholar] [CrossRef]
- Kaidanovich-Beilin, O.; Milman, A.; Weizman, A.; Pick, C.; Eldar-Finkelman, H. Rapid anti-depressive like activity of specific GSK-3 inhibitor, and its effect on beta-catenin in the mouse hippocampus. Biol. Psychiatry 2004, 55, 781–784. [Google Scholar] [CrossRef]
- Pardo, M.; Cheng, Y.; Velmeshev, D.; Magistri, M.; Eldar-Finkelman, H.; Martinez, A.; Faghihi, M.A.; Jope, R.S.; Beurel, E. Intranasal siRNA administration reveals IGF2 deficiency contributes to impaired cognition in Fragile X syndrome mice. JCI Insight 2017, 2, e91782. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beurel, E.; Kaidanovich-Beilin, O.; Yeh, W.I.; Song, L.; Palomo, V.; Michalek, S.M.; Woodgett, J.R.; Harrington, L.E.; Eldar-Finkelman, H.; Martinez, A.; et al. Regulation of Th1 cells and experimental autoimmune encephalomyelitis by glycogen synthase kinase-3. J. Immunol. 2013, 190, 5000–5011. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Plotkin, B.; Kaidanovich, O.; Talior, I.; Eldar-Finkelman, H. Insulin mimetic action of synthetic phosphorylated peptide inhibitors of glycogen synthase kinase-3. J. Pharmacol. Exp. Ther. 2003, 974–980. [Google Scholar] [CrossRef] [PubMed]
- Bhat, R.; Xue, Y.; Berg, S.; Hellberg, S.; Ormo, M.; Nilsson, Y.; Radesater, A.C.; Jerning, E.; Markgren, P.O.; Borgegard, T.; et al. Structural insights and biological effects of glycogen synthase kinase 3-specific inhibitor AR-A014418. J. Biol. Chem. 2003, 278, 45937–45945. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, C.A.; Rayment, I. Active site comparisons highlight structural similarities between myosin and other P-loop proteins. Biophys. J. 1996, 70, 1590–1602. [Google Scholar] [CrossRef]
- Coghlan, M.P.; Culbert, A.A.; Cross, D.A.; Corcoran, S.L.; Yates, J.W.; Pearce, N.J.; Rausch, O.L.; Murphy, G.J.; Carter, P.S.; Roxbee Cox, L.; et al. Selective small molecule inhibitors of glycogen synthase kinase-3 modulate glycogen metabolism and gene transcription. Chem. Biol. 2000, 7, 793–803. [Google Scholar] [CrossRef] [Green Version]
- Owen, S.C.; Doak, A.K.; Wassam, P.; Shoichet, M.S.; Shoichet, B.K. Colloidal aggregation affects the efficacy of anticancer drugs in cell culture. ACS Chem. Biol. 2012, 7, 1429–1435. [Google Scholar] [CrossRef]
- Lucas, J.J.; Hernandez, F.; Gomez-Ramos, P.; Moran, M.A.; Hen, R.; Avila, J. Decreased nuclear beta-catenin, tau hyperphosphorylation and neurodegeneration in GSK-3beta conditional transgenic mice. EMBO J. 2001, 20, 27–39. [Google Scholar] [CrossRef]
- Sato, A.K.; Viswanathan, M.; Kent, R.B.; Wood, C.R. Therapeutic peptides: Technological advances driving peptides into development. Curr. Opin. Biotechnol. 2006, 17, 638–642. [Google Scholar] [CrossRef]
- Fiol, C.J.; Mahrenholz, A.M.; Wang, Y.; Roeske, R.W.; Roach, P.J. Formation of protein kinase recognition sites by covalent modification of the substrate. Molecular mechanism for the synergistic action of casein kinase II and glycogen synthase kinase 3. J. Biol. Chem. 1987, 262, 14042–14048. [Google Scholar]
- Wolber, G.; Langer, T. LigandScout: 3-D pharmacophores derived from protein-bound ligands and their use as virtual screening filters. J. Chem. Inf. Model. 2005, 45, 160–169. [Google Scholar] [CrossRef] [PubMed]
- Irwin, J.J.; Shoichet, B.K. ZINC—A free database of commercially available compounds for virtual screening. J. Chem. Inf. Model. 2005, 45, 177–182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hawkins, P.C.; Skillman, A.G.; Warren, G.L.; Ellingson, B.A.; Stahl, M.T. Conformer generation with OMEGA: Algorithm and validation using high quality structures from the Protein Databank and Cambridge Structural Database. J. Chem. Inf. Model. 2010, 50, 572–584. [Google Scholar] [CrossRef] [PubMed]
- Friesner, R.A.; Murphy, R.B.; Repasky, M.P.; Frye, L.L.; Greenwood, J.R.; Halgren, T.A.; Sanschagrin, P.C.; Mainz, D.T. Extra precision glide: Docking and scoring incorporating a model of hydrophobic enclosure for protein-ligand complexes. J. Med. Chem. 2006, 49, 6177–6196. [Google Scholar] [CrossRef] [Green Version]
- Yap, C.W. PaDEL-descriptor: An open source software to calculate molecular descriptors and fingerprints. J. Comput. Chem. 2011, 32, 1466–1474. [Google Scholar] [CrossRef]
- Duan, J.; Dixon, S.L.; Lowrie, J.F.; Sherman, W. Analysis and comparison of 2D fingerprints: Insights into database screening performance using eight fingerprint methods. J. Mol. Graph. Model. 2010, 29, 157–170. [Google Scholar] [CrossRef]
- Liberman, Z.; Eldar-Finkelman, H. Serine 332 phosphorylation of insulin receptor substrate-1 by glycogen synthase kinase-3 attenuates insulin signaling. J. Biol. Chem. 2005, 280, 4422–4428. [Google Scholar] [CrossRef] [Green Version]
- Eldar-Finkelman, H.; Agrast, G.M.; Foord, O.; Fischer, E.H.; Krebs, E.G. Expression and Characterization of GSK-3 mutants and their effect on glycogen synthase activity. Proc. Natl. Acad. Sci. USA 1996, 93, 10228–10233. [Google Scholar] [CrossRef] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | GSK-3 Interacting Residues | |||||
|---|---|---|---|---|---|---|
| Arg 96 | Arg 180 | Lys 205 | Phe 93 | Val 214 | Others | |
| 1-4 | * Carbonyl of anthracenone * O of CO2H | ** 2O of CO2H | ** O of CO2H | * Gly 202 with amine at the CO2H chain | ||
| 1-6 | ** thiophene ring Carbonyl of anthracenone, * O of CO2H | *O of CO2H | *O of CO2H | * Glut 97 with phenol hydroxyl and ketone, * Asn 95 with phenol hydroxyl | ||
| 1-7 | **** Carbonyl at thiophene ring 2O of CO2H O of CO2H | * O of CO2H | * Bromophenyl ring | * O of CO2H | ||
| 2-1 | *** 2O of CO2H N of isoxazole | *** 2O of CO2H O of CO2H | * O of CO2H | *** Isoxazole and aromatic rings of anthracenone | * O of CO2H | |
| 2-2 | *** Carbonyl at the anthracenone benzoic acid O of CO2H | ** O of CO2H benzoic acid | ** 2O of CO2H | * Gly 202 with amine attached to benzoic acid * Lys 85 with O of ether group | ||
| 3-7 | *** O of CO2H | ** 2O of CO2H * O of CO2H | * O of CO2H * O of CO2H | ** Isoxazole and aromatic rings of anthracenone | * O of CO2H | |
| 3-8 | ** 2O of CO2H | ** 2O of CO2H * O of CO2H | ** O of CO2H | ** Isoxazole and aromatic rings of anthracenone | * O of CO2H | |
| Name | GSK-3 Interacting Residues | |||||||
|---|---|---|---|---|---|---|---|---|
| Arg 96 | Arg 180 | Lys 205 | Phe 93 | Val 214 | Phe 67 | Lys 85 | Ser 66 | |
| 4-1 | ** O of CO2H | ** 2O of CO2H | * O of CO2H | ** Isoxazole and aromatic rings of anthracenone | * O of CO2H | |||
| 4-2 (-2) | ** 2O of PO3H2 ** aromatic rings of anthracenone | *** 2O of PO3H2 | ** 2O of PO3H2 | ** Isoxazole and aromatic rings of anthracenone | * O of PO3H2 | |||
| 4-3 (-3) | ** N of isoxazole and O of PO3H2 | *** 2O of PO3H2 | *** 2O of P34H2 | ** Isoxazole and aromatic rings of anthracenone | * O of PO3H2 | * O of CO2H | ** 2O of CO2H | |
| 4-4 (-2) | * O of isoxazole, ** aromatic rings of anthracenone, *** 2O of PO3H | *** O of PO3H2 * aromatic rings of anthracenone | ** O of PO4H2 | ** Isoxazole and aromatic rings of anthracenone | * O of PO3H2 | * Phenol ring | * OH in phen-ol | |
| 4-5 | ** O of CO2H | *** 2O of CO2H | ** O of CO2H | ** Isoxazole and aromatic rings of anthracenone | * O of CO2H | ** O of CO2H at benzoic acid | ** 2O of CO2H | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rippin, I.; Khazanov, N.; Ben Joseph, S.; Kudinov, T.; Berent, E.; Arciniegas Ruiz, S.M.; Marciano, D.; Levy, L.; Gruzman, A.; Senderowitz, H.; et al. Discovery and Design of Novel Small Molecule GSK-3 Inhibitors Targeting the Substrate Binding Site. Int. J. Mol. Sci. 2020, 21, 8709. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21228709
Rippin I, Khazanov N, Ben Joseph S, Kudinov T, Berent E, Arciniegas Ruiz SM, Marciano D, Levy L, Gruzman A, Senderowitz H, et al. Discovery and Design of Novel Small Molecule GSK-3 Inhibitors Targeting the Substrate Binding Site. International Journal of Molecular Sciences. 2020; 21(22):8709. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21228709
Chicago/Turabian StyleRippin, Ido, Netaly Khazanov, Shirley Ben Joseph, Tania Kudinov, Eva Berent, Sara Melisa Arciniegas Ruiz, Daniele Marciano, Laura Levy, Arie Gruzman, Hanoch Senderowitz, and et al. 2020. "Discovery and Design of Novel Small Molecule GSK-3 Inhibitors Targeting the Substrate Binding Site" International Journal of Molecular Sciences 21, no. 22: 8709. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21228709