Special Delivery: Potential Mechanisms of Botulinum Neurotoxin Uptake and Trafficking within Motor Nerve Terminals



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
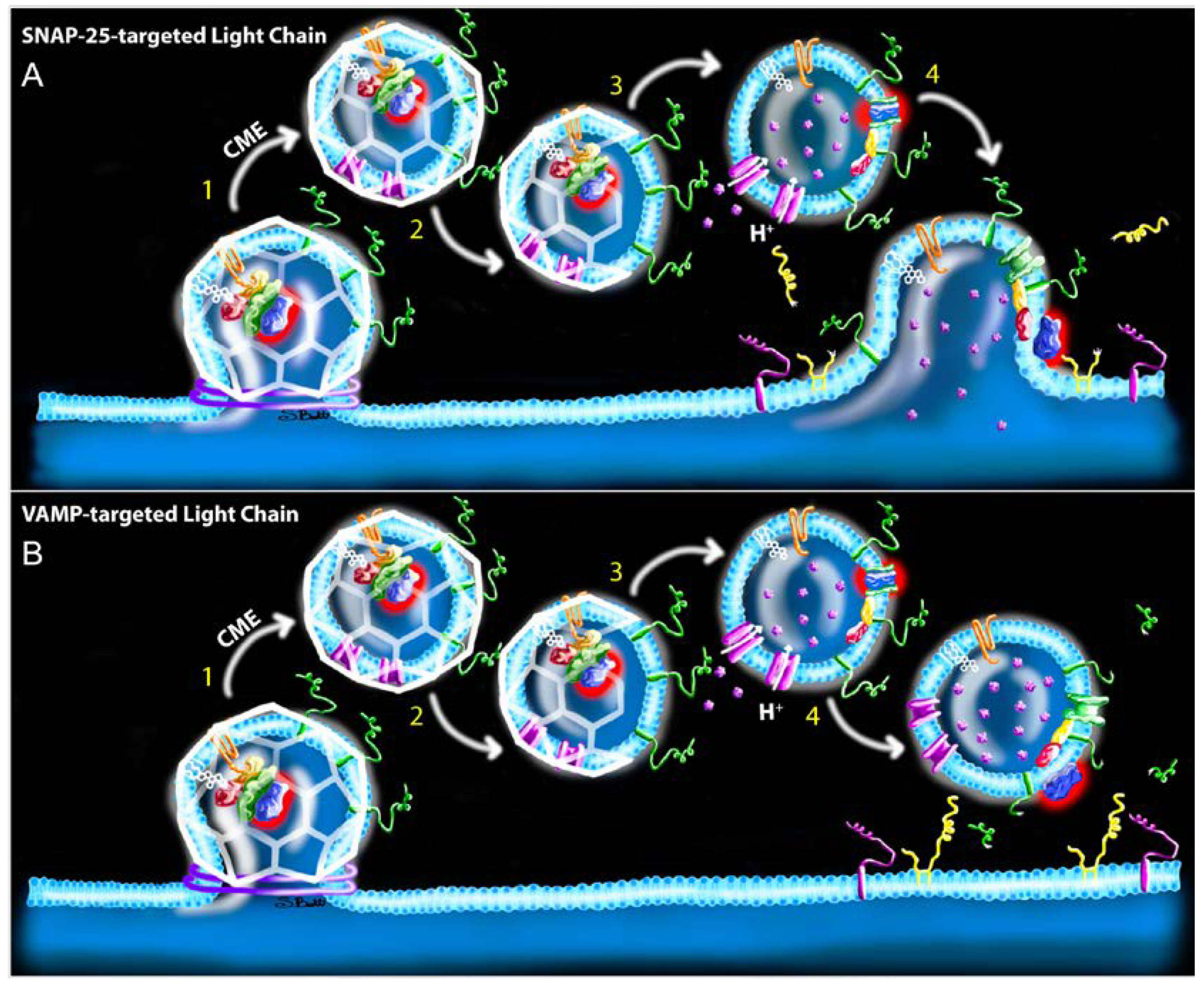
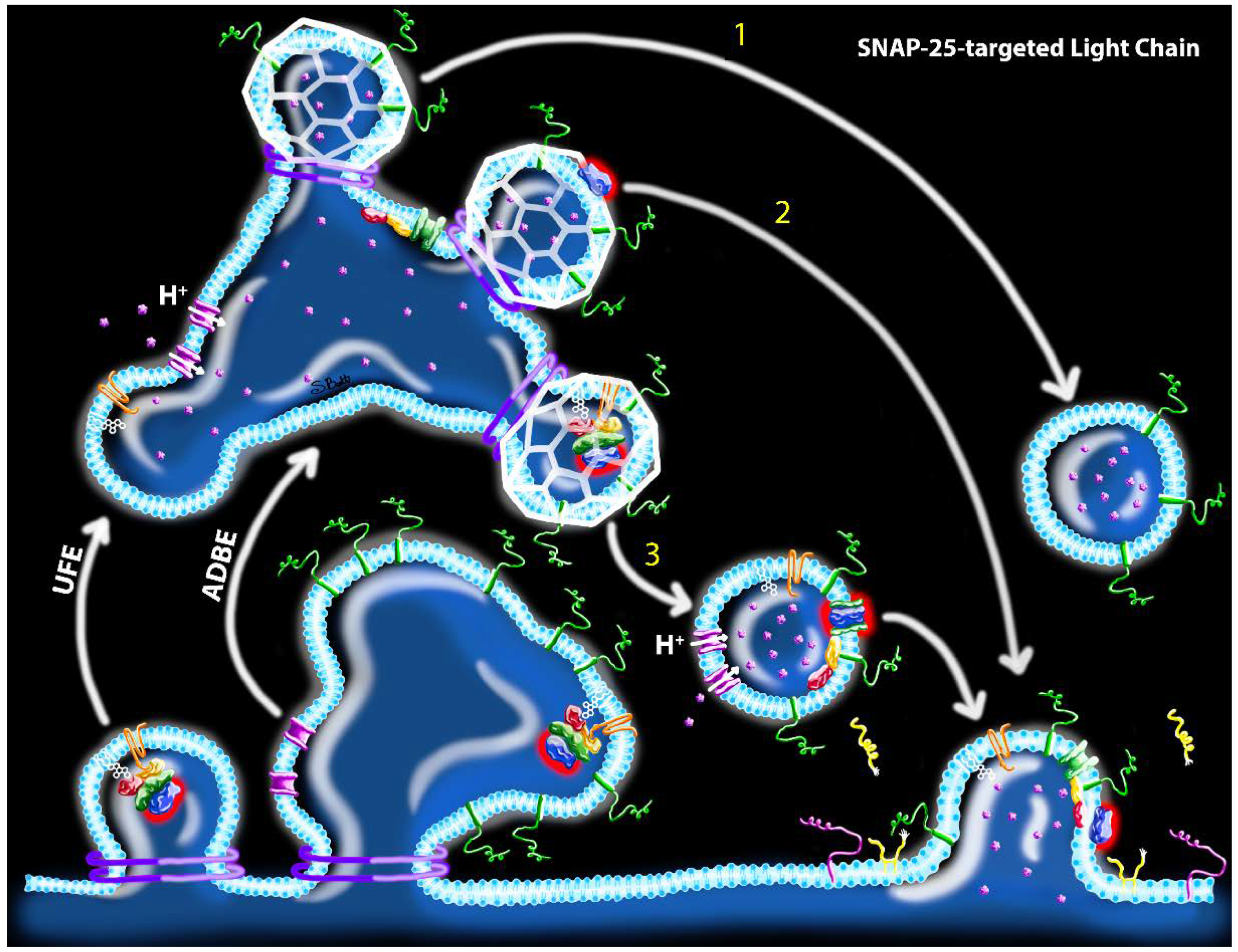
Abstract
:1. Introduction
2. Does the Specific Mechanism of Toxin Internalization into Neurons Affect Toxin Uptake?
2.1. Proposed Routes of Toxin Uptake by Synaptic Endocytosis
2.2. Functional Implications of Endocytosis Mode on Intracellular Toxin Activity
2.3. Mode of Endocytosis May Influence the Rate of Neuronal Intoxication
2.4. Does Mode of Endocytosis Affect the Amount of Toxin Uptake?
3. Conclusions
Funding
Conflicts of Interest
Abbreviations
| BoNT | botulinum neurotoxin |
| SNARE | soluble N-ethylmaleimide-sensitive fusion protein attachment protein receptors |
| HC | heavy chain |
| LC | light chain |
| SV | synaptic vesicle |
| CME | clathrin-mediated endocytosis |
| UFE | ultrafast endocytosis |
| ADBE | activity-dependent bulk endocytosis |
| vLC | VAMP1-3-targeted light chain |
| sLC | SNAP-25-targeted light chain |
| RRP | readily releasable pool |
References
- Pirazzini, M.; Rossetto, O. Challenges in searching for therapeutics against Botulinum Neurotoxins. Expert Opin. Drug Discov. 2017, 12, 497–510. [Google Scholar] [CrossRef] [PubMed]
- Montal, M. Botulinum neurotoxin: A marvel of protein design. Annu Rev. Biochem 2010, 79, 591–617. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sudhof, T.C.; Rizo, J. Synaptic vesicle exocytosis. Cold Spring Harb. Perspect. Biol. 2011, 3, a005637. [Google Scholar] [CrossRef]
- Lacy, D.B.; Tepp, W.; Cohen, A.C.; DasGupta, B.R.; Stevens, R.C. Crystal structure of botulinum neurotoxin type A and implications for toxicity. Nat. Struct. Biol. 1998, 5, 898–902. [Google Scholar] [CrossRef] [PubMed]
- Pirazzini, M.; Rossetto, O.; Eleopra, R.; Montecucco, C. Botulinum Neurotoxins: Biology, Pharmacology, and Toxicology. Pharmacol. Rev. 2017, 69, 200–235. [Google Scholar] [CrossRef] [PubMed]
- Lindstrom, M.; Korkeala, H. Laboratory diagnostics of botulism. Clin. Microbiol. Rev. 2006, 19, 298–314. [Google Scholar] [CrossRef] [Green Version]
- Larsen, J.C. US Army botulinum neurotoxin (BoNT) medical therapeutics research program: Past accomplishments and future directions. Drug Dev. Res. 2009, 70, 266–278. [Google Scholar] [CrossRef]
- Smith, T.J.; Roxas-Duncan, V.I.; Smith, L.A. Botulinum Neurotoxins as Biothreat Agents. J. Bioterrorism Biodefense 2012, S2, 003. [Google Scholar] [CrossRef]
- Arnon, S.S.; Schechter, R.; Maslanka, S.E.; Jewell, N.P.; Hatheway, C.L. Human botulism immune globulin for the treatment of infant botulism. N. Engl. J. Med. 2006, 354, 462–471. [Google Scholar] [CrossRef]
- O’Horo, J.C.; Harper, E.P.; El Rafei, A.; Ali, R.; DeSimone, D.C.; Sakusic, A.; Abu Saleh, O.M.; Marcelin, J.R.; Tan, E.M.; Rao, A.K.; et al. Efficacy of Antitoxin Therapy in Treating Patients With Foodborne Botulism: A Systematic Review and Meta-analysis of Cases, 1923–2016. Clin. Infect. Dis 2017, 66 (Suppl. 1), S43–S56. [Google Scholar]
- Ranney, M.L.; Griffeth, V.; Jha, A.K. Critical Supply Shortages-The Need for Ventilators and Personal Protective Equipment during the Covid-19 Pandemic. N. Engl. J. Med. 2020, 382, e41. [Google Scholar] [CrossRef] [PubMed]
- Fleck-Derderian, S.; Shankar, M.; Rao, A.K.; Chatham-Stephens, K.; Adjei, S.; Sobel, J.; Meltzer, M.I.; Meaney-Delman, D.; Pillai, S.K. The Epidemiology of Foodborne Botulism Outbreaks: A Systematic Review. Clin. Infect. Dis 2017, 66 (Suppl. 1), S73–S81. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McCarty, C.L.; Angelo, K.; Beer, K.D.; Cibulskas-White, K.; Quinn, K.; de Fijter, S.; Bokanyi, R.; St Germain, E.; Baransi, K.; Barlow, K.; et al. Large Outbreak of Botulism Associated with a Church Potluck Meal--Ohio, 2015. Mmwr. Morb. Mortal. Wkly. Rep. 2015, 64, 802–803. [Google Scholar] [CrossRef]
- Chai, Q.; Arndt, J.W.; Dong, M.; Tepp, W.H.; Johnson, E.A.; Chapman, E.R.; Stevens, R.C. Structural basis of cell surface receptor recognition by botulinum neurotoxin B. Nature 2006, 444, 1096–1100. [Google Scholar] [CrossRef]
- Koriazova, L.K.; Montal, M. Translocation of botulinum neurotoxin light chain protease through the heavy chain channel. Nat. Struct. Biol. 2003, 10, 13–18. [Google Scholar] [CrossRef]
- Simpson, L.L.; Coffield, J.A.; Bakry, N. Inhibition of vacuolar adenosine triphosphatase antagonizes the effects of clostridial neurotoxins but not phospholipase A2 neurotoxins. J. Pharm. Exp. 1994, 269, 256–262. [Google Scholar]
- Blasi, J.; Chapman, E.R.; Yamasaki, S.; Binz, T.; Niemann, H.; Jahn, R. Botulinum neurotoxin C1 blocks neurotransmitter release by means of cleaving HPC-1/syntaxin. EMBO J. 1993, 12, 4821–4828. [Google Scholar] [CrossRef]
- Schiavo, G.; Santucci, A.; Dasgupta, B.R.; Mehta, P.P.; Jontes, J.; Benfenati, F.; Wilson, M.C.; Montecucco, C. Botulinum neurotoxins serotypes A and E cleave SNAP-25 at distinct COOH-terminal peptide bonds. FEBS Lett. 1993, 335, 99–103. [Google Scholar] [CrossRef] [Green Version]
- Pirazzini, M.; Azarnia Tehran, D.; Zanetti, G.; Megighian, A.; Scorzeto, M.; Fillo, S.; Shone, C.C.; Binz, T.; Rossetto, O.; Lista, F.; et al. Thioredoxin and its reductase are present on synaptic vesicles, and their inhibition prevents the paralysis induced by botulinum neurotoxins. Cell Rep. 2014, 8, 1870–1878. [Google Scholar] [CrossRef]
- Hanig, J.P.; Lamanna, C. Toxicity of botulinum toxin: A stoichiometric model for the locus of its extraordinary potency and persistence at the neuromuscular junction. J. Biol. 1979, 77, 107–113. [Google Scholar] [CrossRef]
- Van der Kloot, W. The regulation of quantal size. Prog. Neurobiol. 1991, 36, 93–130. [Google Scholar] [CrossRef]
- Black, J.D.; Dolly, J.O. Interaction of 125I-labeled botulinum neurotoxins with nerve terminals. II. Autoradiographic evidence for its uptake into motor nerves by acceptor-mediated endocytosis. J. Cell Biol. 1986, 103, 535–544. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simpson, L.L. Kinetic studies on the interaction between botulinum toxin type A and the cholinergic neuromuscular junction. J. Pharm. Exp. 1980, 212, 16–21. [Google Scholar]
- Mori, Y.; Takamori, S. Molecular Signatures Underlying Synaptic Vesicle Cargo Retrieval. Front. Cell. Neurosci. 2017, 11, 422. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Watanabe, S.; Boucrot, E. Fast and ultrafast endocytosis. Curr. Opin. Cell Biol. 2017, 47, 64–71. [Google Scholar] [CrossRef] [Green Version]
- Kononenko, N.L.; Haucke, V. Molecular mechanisms of presynaptic membrane retrieval and synaptic vesicle reformation. Neuron 2015, 85, 484–496. [Google Scholar] [CrossRef] [Green Version]
- Milosevic, I. Revisiting the Role of Clathrin-Mediated Endoytosis in Synaptic Vesicle Recycling. Front. Cell. Neurosci. 2018, 12, 27. [Google Scholar] [CrossRef] [Green Version]
- Takei, K.; Mundigl, O.; Daniell, L.; De Camilli, P. The synaptic vesicle cycle: A single vesicle budding step involving clathrin and dynamin. J. Cell Biol. 1996, 133, 1237–1250. [Google Scholar] [CrossRef] [Green Version]
- Bednarek, S.Y.; Orci, L.; Schekman, R. Traffic COPs and the formation of vesicle coats. Trends Cell Biol. 1996, 6, 468–473. [Google Scholar] [CrossRef]
- Royle, S.J.; Lagnado, L. Clathrin-mediated endocytosis at the synaptic terminal: Bridging the gap between physiology and molecules. Traffic 2010, 11, 1489–1497. [Google Scholar] [CrossRef] [Green Version]
- Farsi, Z.; Gowrisankaran, S.; Krunic, M.; Rammner, B.; Woehler, A.; Lafer, E.M.; Mim, C.; Jahn, R.; Milosevic, I. Clathrin coat controls synaptic vesicle acidification by blocking vacuolar ATPase activity. Elife 2018, 7, e32569. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clayton, E.L.; Evans, G.J.; Cousin, M.A. Bulk synaptic vesicle endocytosis is rapidly triggered during strong stimulation. J. Neurosci. 2008, 28, 6627–6632. [Google Scholar] [CrossRef] [PubMed]
- Heuser, J.E.; Reese, T.S. Evidence for recycling of synaptic vesicle membrane during transmitter release at the frog neuromuscular junction. J. Cell Biol. 1973, 57, 315–344. [Google Scholar] [CrossRef] [PubMed]
- Delvendahl, I.; Vyleta, N.P.; von Gersdorff, H.; Hallermann, S. Fast, Temperature-Sensitive and Clathrin-Independent Endocytosis at Central Synapses. Neuron 2016, 90, 492–498. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kononenko, N.L.; Puchkov, D.; Classen, G.A.; Walter, A.M.; Pechstein, A.; Sawade, L.; Kaempf, N.; Trimbuch, T.; Lorenz, D.; Rosenmund, C.; et al. Clathrin/AP-2 mediate synaptic vesicle reformation from endosome-like vacuoles but are not essential for membrane retrieval at central synapses. Neuron 2014, 82, 981–988. [Google Scholar] [CrossRef] [Green Version]
- Harper, C.B.; Martin, S.; Nguyen, T.H.; Daniels, S.J.; Lavidis, N.A.; Popoff, M.R.; Hadzic, G.; Mariana, A.; Chau, N.; McCluskey, A.; et al. Dynamin inhibition blocks botulinum neurotoxin type A endocytosis in neurons and delays botulism. J. Biol. Chem. 2011, 286, 35966–35976. [Google Scholar] [CrossRef] [Green Version]
- Meng, J.; Wang, J.; Lawrence, G.W.; Dolly, J.O. Molecular components required for resting and stimulated endocytosis of botulinum neurotoxins by glutamatergic and peptidergic neurons. FASEB J. 2013, 27, 3167–3180. [Google Scholar] [CrossRef] [Green Version]
- Granseth, B.; Odermatt, B.; Royle, S.J.; Lagnado, L. Clathrin-mediated endocytosis is the dominant mechanism of vesicle retrieval at hippocampal synapses. Neuron 2006, 51, 773–786. [Google Scholar] [CrossRef] [Green Version]
- Watanabe, S.; Trimbuch, T.; Camacho-Perez, M.; Rost, B.R.; Brokowski, B.; Sohl-Kielczynski, B.; Felies, A.; Davis, M.W.; Rosenmund, C.; Jorgensen, E.M. Clathrin regenerates synaptic vesicles from endosomes. Nature 2014, 515, 228–233. [Google Scholar] [CrossRef] [Green Version]
- Gan, Q.; Watanabe, S. Synaptic Vesicle Endocytosis in Different Model Systems. Front. Cell. Neurosci. 2018, 12, 171. [Google Scholar] [CrossRef]
- Watanabe, S.; Liu, Q.; Davis, M.W.; Hollopeter, G.; Thomas, N.; Jorgensen, N.B.; Jorgensen, E.M. Ultrafast endocytosis at Caenorhabditis elegans neuromuscular junctions. Elife 2013, 2, e00723. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, S.; Rost, B.R.; Camacho-Perez, M.; Davis, M.W.; Sohl-Kielczynski, B.; Rosenmund, C.; Jorgensen, E.M. Ultrafast endocytosis at mouse hippocampal synapses. Nature 2013, 504, 242–247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soykan, T.; Kaempf, N.; Sakaba, T.; Vollweiter, D.; Goerdeler, F.; Puchkov, D.; Kononenko, N.L.; Haucke, V. Synaptic Vesicle Endocytosis Occurs on Multiple Timescales and Is Mediated by Formin-Dependent Actin Assembly. Neuron 2017, 93, 854–866.e4. [Google Scholar] [PubMed] [Green Version]
- Clayton, E.L.; Cousin, M.A. The molecular physiology of activity-dependent bulk endocytosis of synaptic vesicles. J. Neurochem. 2009, 111, 901–914. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheung, G.; Jupp, O.J.; Cousin, M.A. Activity-dependent bulk endocytosis and clathrin-dependent endocytosis replenish specific synaptic vesicle pools in central nerve terminals. J. Neurosci. 2010, 30, 8151–8161. [Google Scholar] [CrossRef]
- Cousin, M.A. Activity-dependent bulk synaptic vesicle endocytosis--a fast, high capacity membrane retrieval mechanism. Mol. Neurobiol. 2009, 39, 185–189. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Q.; Li, Y.; Tsien, R.W. The dynamic control of kiss-and-run and vesicular reuse probed with single nanoparticles. Science 2009, 323, 1448–1453. [Google Scholar] [CrossRef] [Green Version]
- Ceccarelli, B.; Hurlbut, W.P.; Mauro, A. Turnover of transmitter and synaptic vesicles at the frog neuromuscular junction. J. Cell Biol. 1973, 57, 499–524. [Google Scholar] [CrossRef] [Green Version]
- Aravanis, A.M.; Pyle, J.L.; Harata, N.C.; Tsien, R.W. Imaging single synaptic vesicles undergoing repeated fusion events: Kissing, running, and kissing again. Neuropharmacology 2003, 45, 797–813. [Google Scholar] [CrossRef]
- Gandhi, S.P.; Stevens, C.F. Three modes of synaptic vesicular recycling revealed by single-vesicle imaging. Nature 2003, 423, 607–613. [Google Scholar] [CrossRef]
- Ales, E.; Tabares, L.; Poyato, J.M.; Valero, V.; Lindau, M.; Alvarez de Toledo, G. High calcium concentrations shift the mode of exocytosis to the kiss-and-run mechanism. Nat. Cell Biol 1999, 1, 40–44. [Google Scholar] [CrossRef] [PubMed]
- Jackson, M.B.; Chapman, E.R. Fusion pores and fusion machines in Ca2+-triggered exocytosis. Annu. Rev. Biophys. Biomol. Struct. 2006, 35, 135–160. [Google Scholar] [CrossRef] [PubMed]
- He, L.; Wu, X.S.; Mohan, R.; Wu, L.G. Two modes of fusion pore opening revealed by cell-attached recordings at a synapse. Nature 2006, 444, 102–105. [Google Scholar] [CrossRef] [PubMed]
- He, L.; Wu, L.G. The debate on the kiss-and-run fusion at synapses. Trends Neurosci 2007, 30, 447–455. [Google Scholar] [CrossRef] [PubMed]
- Donovan, J.J.; Middlebrook, J.L. Ion-conducting channels produced by botulinum toxin in planar lipid membranes. Biochemistry 1986, 25, 2872–2876. [Google Scholar] [CrossRef]
- Okamoto, Y.; Lipstein, N.; Hua, Y.; Lin, K.H.; Brose, N.; Sakaba, T.; Midorikawa, M. Distinct modes of endocytotic presynaptic membrane and protein uptake at the calyx of Held terminal of rats and mice. Elife 2016, 5, e14643. [Google Scholar] [CrossRef] [Green Version]
- Cheung, G.; Cousin, M.A. Synaptic vesicle generation from activity-dependent bulk endosomes requires calcium and calcineurin. J. Neurosci. 2013, 33, 3370–3379. [Google Scholar] [CrossRef] [Green Version]
- Nicholson-Fish, J.C.; Smillie, K.J.; Cousin, M.A. Monitoring activity-dependent bulk endocytosis with the genetically-encoded reporter VAMP4-pHluorin. J. Neurosci. Methods 2016, 266, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Reshetniak, S.; Ussling, J.E.; Perego, E.; Rammner, B.; Schikorski, T.; Fornasiero, E.F.; Truckenbrodt, S.; Koster, S.; Rizzoli, S.O. A comparative analysis of the mobility of 45 proteins in the synaptic bouton. EMBO J. 2020, 39, e104596. [Google Scholar] [CrossRef]
- Fernandez-Salas, E.; Steward, L.E.; Ho, H.; Garay, P.E.; Sun, S.W.; Gilmore, M.A.; Ordas, J.V.; Wang, J.; Francis, J.; Aoki, K.R. Plasma membrane localization signals in the light chain of botulinum neurotoxin. Proc. Natl. Acad. Sci. USA 2004, 101, 3208–3213. [Google Scholar] [CrossRef] [Green Version]
- Tao-Cheng, J.H.; Du, J.; McBain, C.J. Snap-25 is polarized to axons and abundant along the axolemma: An immunogold study of intact neurons. J. Neurocytol. 2000, 29, 67–77. [Google Scholar] [CrossRef] [PubMed]
- Wilhelm, B.G.; Mandad, S.; Truckenbrodt, S.; Krohnert, K.; Schafer, C.; Rammner, B.; Koo, S.J.; Classen, G.A.; Krauss, M.; Haucke, V.; et al. Composition of isolated synaptic boutons reveals the amounts of vesicle trafficking proteins. Science 2014, 344, 1023–1028. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Broadie, K.; Prokop, A.; Bellen, H.J.; O’Kane, C.J.; Schulze, K.L.; Sweeney, S.T. Syntaxin and synaptobrevin function downstream of vesicle docking in Drosophila. Neuron 1995, 15, 663–673. [Google Scholar] [CrossRef]
- Toonen, R.F.; Kochubey, O.; de Wit, H.; Gulyas-Kovacs, A.; Konijnenburg, B.; Sorensen, J.B.; Klingauf, J.; Verhage, M. Dissecting docking and tethering of secretory vesicles at the target membrane. EMBO J. 2006, 25, 3725–3737. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neale, E.A.; Bowers, L.M.; Jia, M.; Bateman, K.E.; Williamson, L.C. Botulinum neurotoxin A blocks synaptic vesicle exocytosis but not endocytosis at the nerve terminal. J. Cell Biol. 1999, 147, 1249–1260. [Google Scholar] [CrossRef]
- Ruiz, R.; Cano, R.; Casanas, J.J.; Gaffield, M.A.; Betz, W.J.; Tabares, L. Active zones and the readily releasable pool of synaptic vesicles at the neuromuscular junction of the mouse. J. Neurosci. 2011, 31, 2000–2008. [Google Scholar] [CrossRef] [Green Version]
- Nagwaney, S.; Harlow, M.L.; Jung, J.H.; Szule, J.A.; Ress, D.; Xu, J.; Marshall, R.M.; McMahan, U.J. Macromolecular connections of active zone material to docked synaptic vesicles and presynaptic membrane at neuromuscular junctions of mouse. J. Comp. Neurol. 2009, 513, 457–468. [Google Scholar] [CrossRef] [Green Version]
- Rizzoli, S.O.; Betz, W.J. Synaptic vesicle pools. Nat. Rev. Neurosci. 2005, 6, 57–69. [Google Scholar] [CrossRef]
- Kokotos, A.C.; Peltier, J.; Davenport, E.C.; Trost, M.; Cousin, M.A. Activity-dependent bulk endocytosis proteome reveals a key presynaptic role for the monomeric GTPase Rab11. Proc. Natl. Acad. Sci. USA 2018, 115, E10177–E10186. [Google Scholar] [CrossRef] [Green Version]
- Black, J.D.; Dolly, J.O. Interaction of 125I-labeled botulinum neurotoxins with nerve terminals. I. Ultrastructural autoradiographic localization and quantitation of distinct membrane acceptors for types A and B on motor nerves. J. Cell Biol. 1986, 103, 521–534. [Google Scholar] [CrossRef]
- Williamson, L.C.; Neale, E.A. Syntaxin and 25-kDa synaptosomal-associated protein: Differential effects of botulinum neurotoxins C1 and A on neuronal survival. J. Neurosci. Res. 1998, 52, 569–583. [Google Scholar] [CrossRef]
- Aikawa, Y.; Lynch, K.L.; Boswell, K.L.; Martin, T.F. A second SNARE role for exocytic SNAP25 in endosome fusion. Mol. Biol. Cell 2006, 17, 2113–2124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eleopra, R.; Tugnoli, V.; De Grandis, D. The variability in the clinical effect induced by botulinum toxin type A: The role of muscle activity in humans. Mov. Disord. 1997, 12, 89–94. [Google Scholar] [CrossRef]
- McNutt, P.; Celver, J.; Hamilton, T.; Mesngon, M. Embryonic stem cell-derived neurons are a novel, highly sensitive tissue culture platform for botulinum research. Biochem. Biophys. Res. Commun. 2011, 405, 85–90. [Google Scholar] [CrossRef] [PubMed]
- Enoka, R.M.; Fuglevand, A.J. Motor unit physiology: Some unresolved issues. Muscle Nerve 2001, 24, 4–17. [Google Scholar] [CrossRef]
- Wagner, J.A.; Carlson, S.S.; Kelly, R.B. Chemical and physical characterization of cholinergic synaptic vesicles. Biochemistry 1978, 17, 1199–1206. [Google Scholar] [CrossRef] [PubMed]
- Takamori, S.; Holt, M.; Stenius, K.; Lemke, E.A.; Gronborg, M.; Riedel, D.; Urlaub, H.; Schenck, S.; Brugger, B.; Ringler, P.; et al. Molecular anatomy of a trafficking organelle. Cell 2006, 127, 831–846. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cousin, M.A. Integration of Synaptic Vesicle Cargo Retrieval with Endocytosis at Central Nerve Terminals. Front. Cell. Neurosci. 2017, 11, 234. [Google Scholar] [CrossRef] [Green Version]
- Mutch, S.A.; Kensel-Hammes, P.; Gadd, J.C.; Fujimoto, B.S.; Allen, R.W.; Schiro, P.G.; Lorenz, R.M.; Kuyper, C.L.; Kuo, J.S.; Bajjalieh, S.M.; et al. Protein quantification at the single vesicle level reveals that a subset of synaptic vesicle proteins are trafficked with high precision. J. Neurosci. 2011, 31, 1461–1470. [Google Scholar] [CrossRef] [Green Version]
- Mutch, S.A.; Gadd, J.C.; Fujimoto, B.S.; Kensel-Hammes, P.; Schiro, P.G.; Bajjalieh, S.M.; Chiu, D.T. Determining the number of specific proteins in cellular compartments by quantitative microscopy. Nat. Protoc. 2011, 6, 1953–1968. [Google Scholar] [CrossRef] [Green Version]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Winner, B.M.; Bodt, S.M.L.; McNutt, P.M. Special Delivery: Potential Mechanisms of Botulinum Neurotoxin Uptake and Trafficking within Motor Nerve Terminals. Int. J. Mol. Sci. 2020, 21, 8715. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21228715
Winner BM, Bodt SML, McNutt PM. Special Delivery: Potential Mechanisms of Botulinum Neurotoxin Uptake and Trafficking within Motor Nerve Terminals. International Journal of Molecular Sciences. 2020; 21(22):8715. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21228715
Chicago/Turabian StyleWinner, Brittany M., Skylar M. L. Bodt, and Patrick M. McNutt. 2020. "Special Delivery: Potential Mechanisms of Botulinum Neurotoxin Uptake and Trafficking within Motor Nerve Terminals" International Journal of Molecular Sciences 21, no. 22: 8715. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21228715