1. Introduction
The term extracellular vesicle (EV) refers to particles released from cells which are delimited by a lipid bilayer, but do not contain a nucleus [
1]. EVs are heterogeneous in biogenesis [
2], size, and content. They range in size from 30 nm exosomes [
3] to oncosomes up to 10 µM [
4], and contain cargo of all biomolecule categories [
5]. Attempts to categorise EVs primarily by surface marker expression have been confounded by the recognition that many of the markers previously considered to be subset- or derivation-specific, are actually present on multiple or even all classes of EVs [
6]. Delineation and characterisation of specific EV subsets is an essential goal to achieve a better understanding of EV biology [
7], yet there are no techniques that accurately quantify EVs across the full EV size range, or combine quantification with the ability to screen for EV marker expression or EV content.
Small EVs are characteristically isolated by high-speed centrifugation at 100,000×
g [
8], and include both EVs derived intracellularly from late endosomes and released by exocytosis (exosomes), and other small EVs not derived from endosomes (3). Multiple commercial solutions exist for the isolation of exosomes from a variety of biological fluids including tissue culture supernatant, plasma, and urine. In contrast, there are no commercially available solutions for the isolation of large EVs. As a result, isolation methods vary, and knowledge of large EV content and function in biological samples is relatively lacking. Large EVs, defined as >200 nm by recent guidelines set out by the International Society of Extracellular Vesicles [
1], include cancer cell-derived oncosomes, dead cell-derived apoptotic bodies and platelets, and are visible by light microscopy [
9]. In published literature, EVs larger than 1 µm have historically been assumed to be apoptotic bodies [
10]. However, we and others [
4] have demonstrated that viable cell cultures produce large EVs which do not have the ultrastructural features reminiscent of fragments of apoptotic cells.
EVs are released by all cells providing an efficient mechanism of cell to cell communication [
11]. Increasing evidence points to key roles for EVs in cancer diagnosis, prognostication, and surveillance of cancer [
12]. Large EVs from prostate cancer cells were shown to contain tumour-specific biomarkers [
4,
13] and mediate intercellular transfer of bioactive molecules including miRNA [
14]. We previously reported a population of large EVs released by leukaemic cells which were actin-rich and contained intact organelles [
15]. These large EVs could be internalised by normal stromal cells and induced a switch in the preferred metabolic pathway of the recipient cells [
9]. Additionally, we found that leukaemia-derived EVs expressed a surface marker indicative of their parent cell (CD19) and could be detected in the peripheral blood of murine models and patient bone marrow plasma [
9]. Taken together, our previous work and existing large EV literature suggest that large EVs, often discarded in techniques to isolate smaller EVs and exosomes, could be considered as extensive reservoirs of biomolecules useful to study EV biogenesis and function, and to identify clinically relevant biomarkers for disease detection and treatment monitoring [
16,
17].
The principle advantage of characterising large EVs as single events by imaging flow cytometry is the potential for simultaneously identifying parent cell, EV, and tumour markers. We report for the first time a characterisation of size distribution and EV marker expression in this heterogeneous EV population, undertaken in accordance with the most recent international consensus guidelines for EV research from the International Society of Extracellular Vesicles [
1].
In this proof of concept study, we set out to: (1) highlight the abundance of large EVs produced by cells derived from the malignant brain tumour medulloblastoma in vitro; (2) describe variations in the expression of established EV markers in the large EV population; (3) describe how the Imagestream (ISX) can address sample heterogeneity by facilitating high throughput, single event EV analyses. We describe a protocol to isolate intact large EVs without cell contamination, from cells growing in serum-free medium, using gravity flow filtration combined with low-speed centrifugation. Our data show the breadth of heterogeneity in the size and marker expression of large EVs isolated from a single cell line and serves to highlight the importance of sample purity, isolation techniques and experimental controls as we seek to identify tumour-specific EV markers for use in the clinic.
2. Results
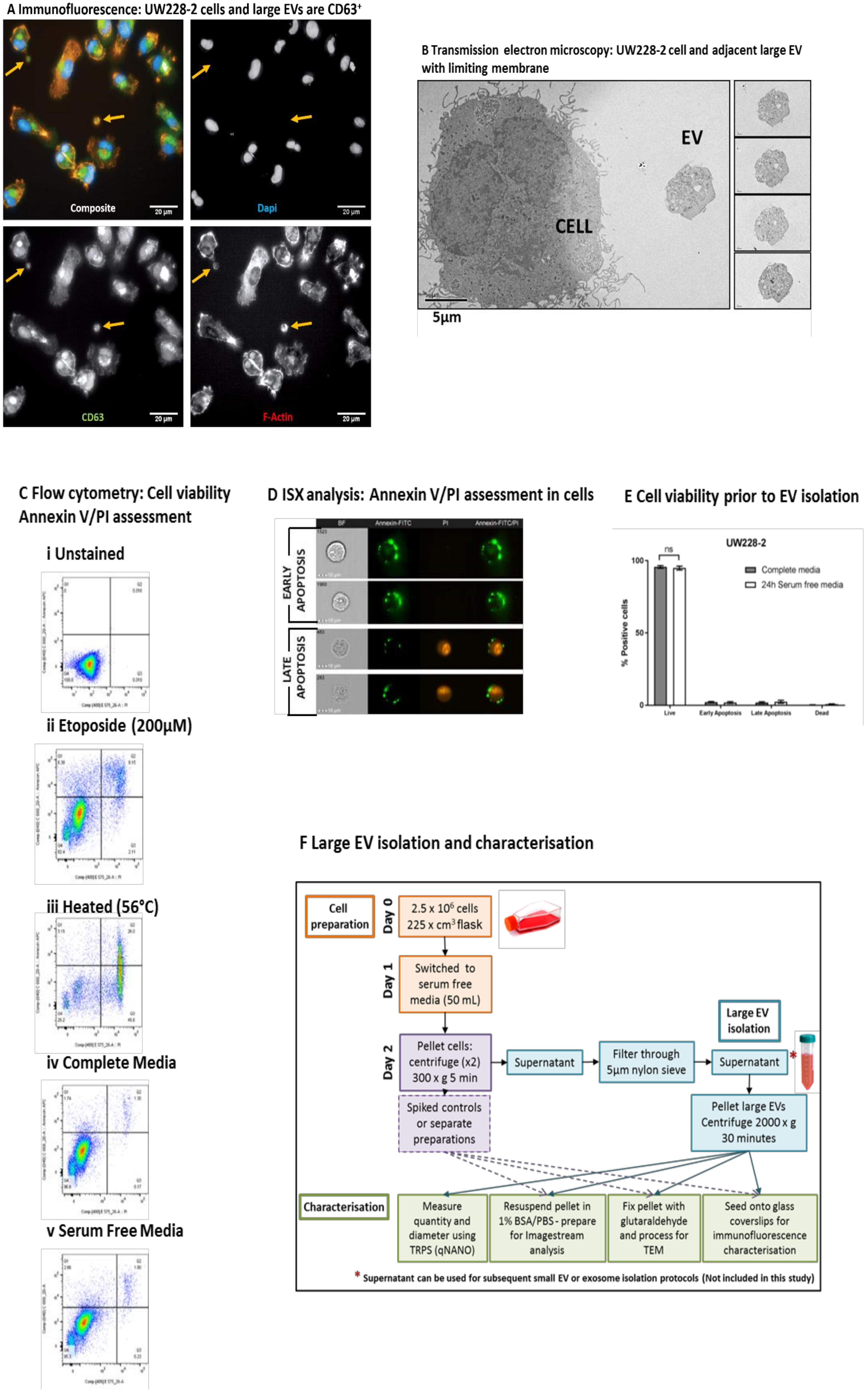
Large EVs from the medulloblastoma cell line UW228-2 are visible with light microscopy, can be isolated from viable, serum-free cell culture supernatant with intact membranes, and contain a polymerised actin cytoskeleton. A proportion of these large EVs express reported EV markers.
In this study the large EV isolation SOP was applied to the SHH-driven medulloblastoma cell line UW228-2. In accordance with the recently published international consensus MISEV2018 guidelines [
1], we demonstrated the existence and membrane integrity of large EVs using both light and electron microscopy (
Figure 1A,B). The pan-EV marker CD63 was expressed by parent medulloblastoma cells and a proportion of large EVs in viable cultures (
Figure 1A). Large EVs also exhibited an active cytoskeleton indicated by the polymerised actin marker Phalloidin, but did not stain for DAPI, indicating that they did not contain nuclear double-stranded DNA. EV membrane integrity, size, and intra-vesicle content was examined by transmission electron microscopy (TEM) (
Figure 1B). Isolated EVs were spiked with medulloblastoma parent cells for comparison and adhered to ACLAR film coated with CellTak prior to TEM. Serial sections demonstrated that the EVs had a limiting membrane, internal organelles but no nucleus, and were independent from cells. By contrast, cells had internal organelles including a nucleus and cytoplasmic protrusions indicative of filopodia.
Cells were grown in serum-free medium for 24 h prior to EV isolation, staining, and analysis. Experimental conditions were optimised to eliminate false positive membrane labelling or bovine EV contamination [
18]. To assess whether growth in serum-free medium resulted in increased cell apoptosis, phosphatidyl serine exposure on the surface of cells was examined.
Figure 1C indicates the quadrant gating strategy applied using positive controls and the comparison of cells grown in standard culture versus serum-free conditions. Imaging flow cytometry was also performed to visualise Annexin V/PI staining on parent cells (
Figure 1D). In triplicate experiments, there was no significant difference between the viability of cells cultured in complete or serum-free media (
Figure 1E).
Figure 1F provides a workflow for the isolation and characterisation of large EVs used in this study. At the time of each EV isolation, total parent cell count and viability was assessed using trypan blue exclusion and found to be 7.9 × 10
6 cells (98% viable), 8.3 × 10
6 cells (99% viable), and 6.8 × 10
6 cells (98% viable).
2.1. EVs are Highly Heterogeneous and Differentially Express EV Markers
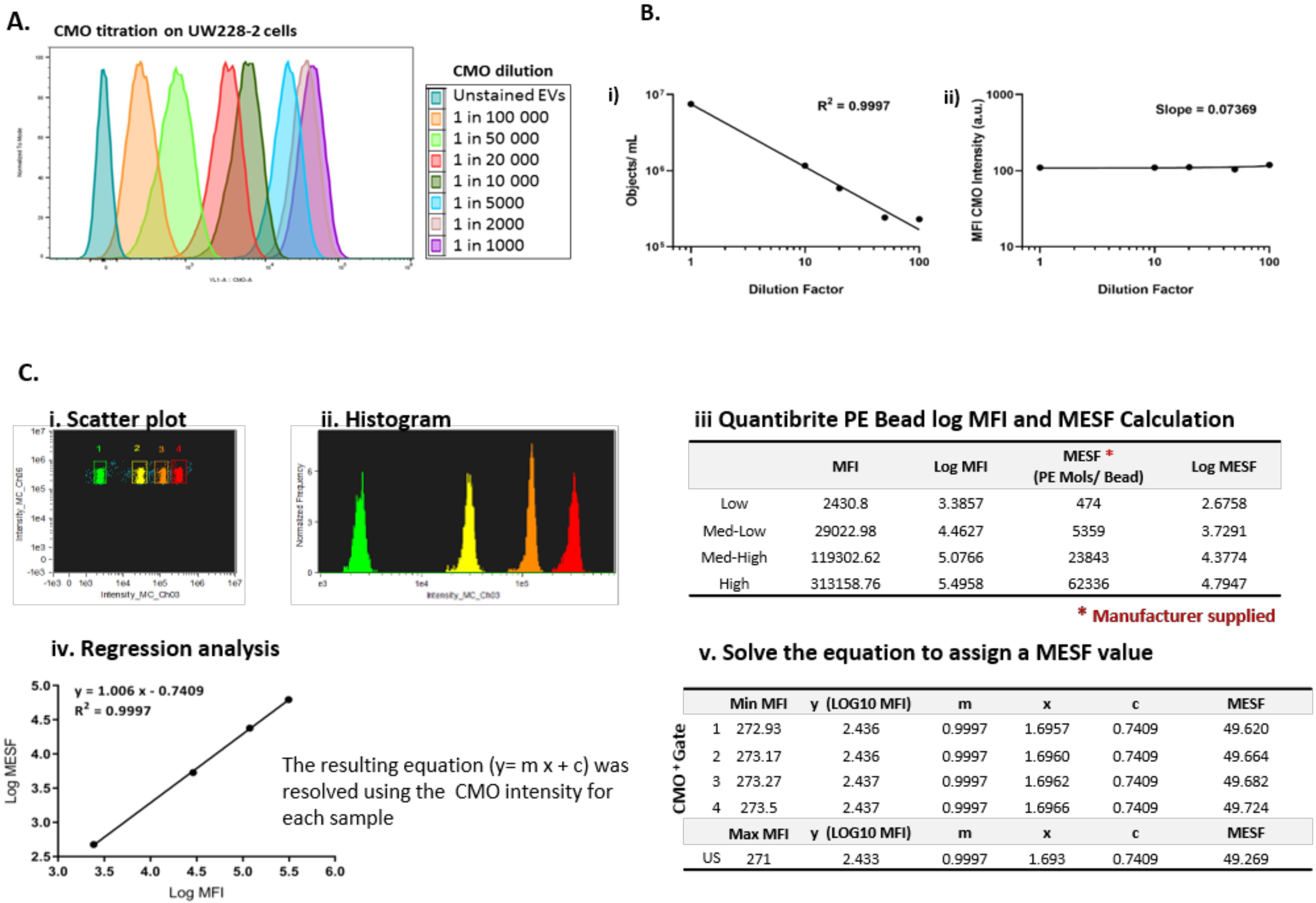
A reliable fluorescence marker was essential to demarcate EVs from background scatter events. Cell mask orange (CMO) is a fluorescent plasma membrane label composed of amphipathic molecules comprising a lipophilic moiety for membrane loading and a negatively charged hydrophilic dye for anchoring of the probe in the plasma membrane. We performed a titration using the parent cells with serial dilutions of the dye in serum-free media ranging from 1 in 1000 (5 µg/mL) to 1 in 100,000 (50 ng/mL) (
Figure 2A) and found that 2.5 µg/mL (1 in 2000) was an optimal concentration providing high median fluorescence intensity without saturation. Using the Imagestream Mark II (ISX) we confirmed that the final concentration of CMO labelled EVs in a typical preparation did not result in coincidence events or swarm which could lead to false positive results when looking at multi-colour labelling. Serial dilutions of CMO labelled EVs showed a linear decrease in objects per mL (
Figure 2B(i)) with an increasing dilution factor, whilst the fluorescence intensity remained stable across the dilutions (
Figure 2B(ii)). To facilitate reproducible reporting across experiments and to offer a means to standardise EV measurements, we determined the minimum MESF value for CMO+ events which would distinguish between unstained and CMO labelled EVs using our staining protocol, as described elsewhere [
19].
Figure 2C shows a representative bivariate plot (i) of the low, medium-low, medium-high, and high fluorescence bead populations. The histogram (ii) was used to determine the MFI of each peak and log fluorescence intensities were converted to MESF values using information supplied by the manufacturer (iii). Linear regression of log MFI versus log MESF (iv) was used to calculate the MESF corresponding to the minimum fluorescence intensity of events within the designated CMO+ gate as follows. The maximum MESF values of the unstained EVs in each experiment were: Replicate 1: 271.42 = MESF 48.23; Replicate 2: 271.0 = MESF 49.27; Replicate 3: 272.88 = MESF 42.48. Therefore, to set a standardised lower threshold of detection which could be used across replicate experiments we assigned a lower MESF threshold for CMO
+ events of 50 (Example shown in
Figure 2C(v)). We therefore report here CMO
+ events as number of events >MESF 50.
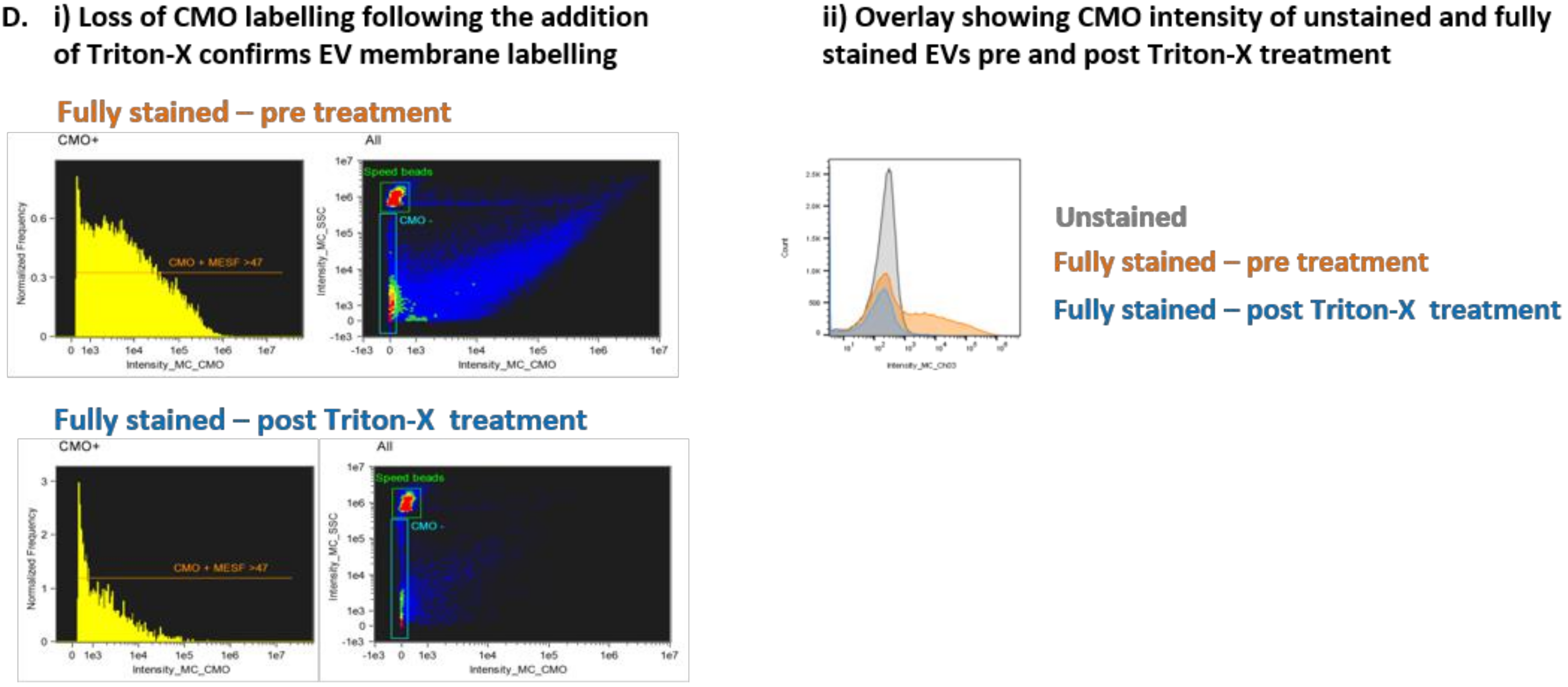
Previous reports have identified that lipid dye aggregates can mimic EVs when using fluorescence lipid markers [
20]. We used Triton-X 100 treatment [
21] of fully labeled EVs to disrupt EV signals demonstrated by a loss of CMO + events (
Figure 2D(i)).
Figure 2D(ii) shows an overlay of unstained EVs, fully stained EVs and fully stained EVs treated with Triton-X 100. The MFI of the post-Triton-X 100 treated sample is reduced to the level of the US-EVs.
2.2. Large EVs Size Distribution and Quantity was Assessed Using Tunable Resistance Pulse Sensing (TRPS)
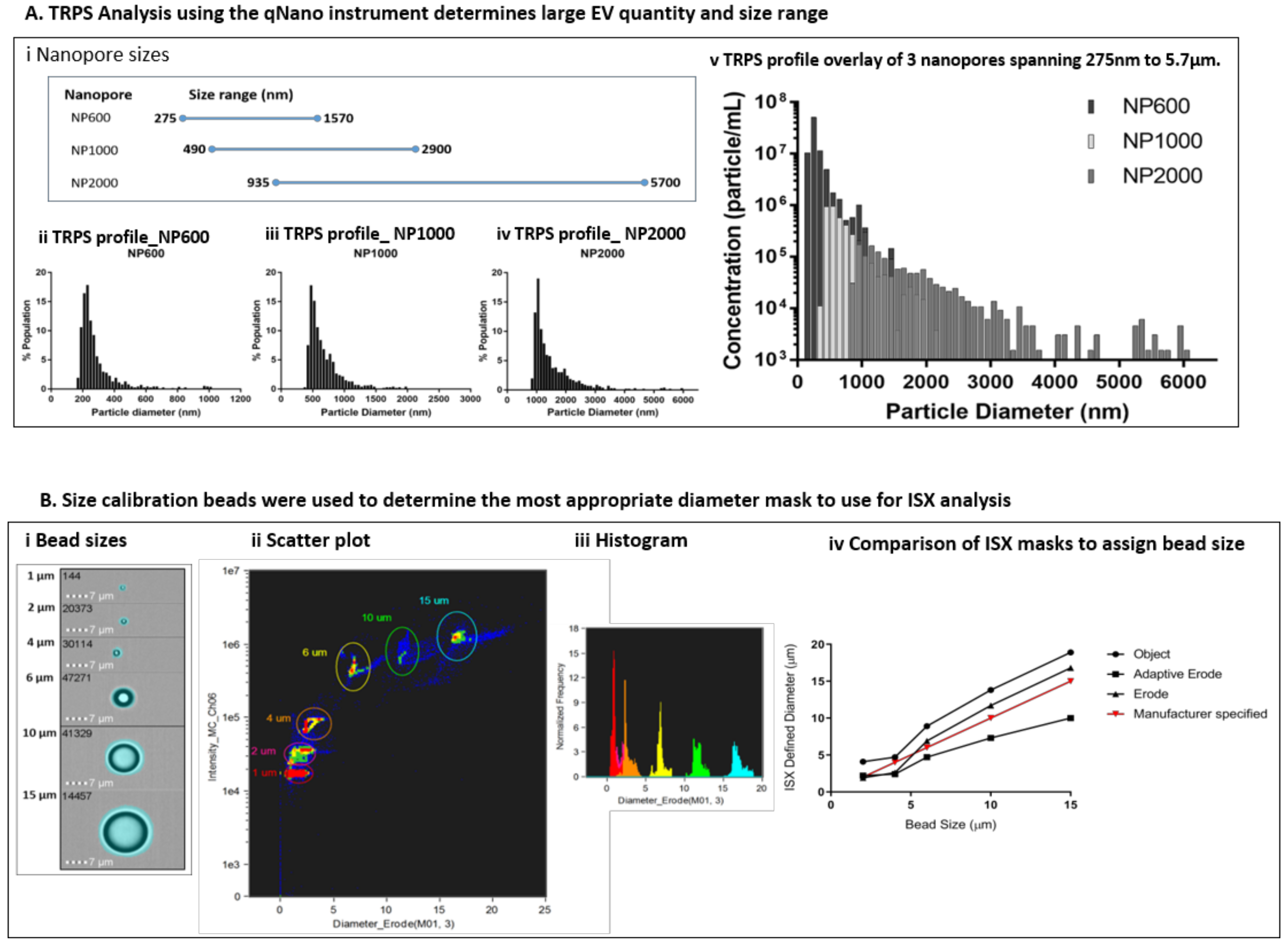
At the time of each preparation, an aliquot of freshly isolated EVs was assessed using the qNano GOLD particle counter to ascertain EV size distribution (diameter) in terms of percentage population (%) and concentration (particles/mL) prior to labelling for ISX analysis. Using a series of 3 overlapping Nanopores (
Figure 3A(i)), each with an optimal size range which spans a total size of approximately 275 nm to 5.7 µm, we assessed the size range of particles in the EV preparations. Representative profiles of diameter against percentage population are shown for a single sample using NP600, NP1000, and NP2000 Nanopores (
Figure 3A(ii–iv)). By selecting overlapping Nanopore sizes, the same calibrator beads (1000 nm) could be used and therefore it was possible to overlay the resulting profiles to visualise the total large EV population within each sample (
Figure 3A(v)). As expected, the EVs present in each biological replicate were heterogeneous in size. However, the size range of EVs across biological replicates was consistent (250 nm to 6 µm); with some variation in the median diameter per sample. Size is presented with a bin of 100 (nm) and in each case the most prevalent large EVs were detected using the NP600. The median size across the replicates using the NP600 for comparison were 250–350 nm 5.15 × 10
7; 450–550 nm 1.7 × 10
7, and 250–350 nm 2.8 × 10
8. The starting volume of EV-containing media used per replicate was 100 mL. The resulting pellet was re-suspended in a total of 700 µL PBS (prior to labelling). As an example, applying this dilution factor (142.86) to the first replicate equates to 7.4 × 10
9 EVs specifically of the 250–350 nm size range that were released by 7.9 × 10
6 UW228-2 cells (NB cell count at harvest) in 24 h into 100 mL serum-free media. It should be noted that due to the number of washes and centrifugation steps during the isolation, this can only be considered as an illustration of the quantity of EVs produced. The total larger EVs as counted using the NP2000 (935 nm − 5.7 µm) were less common but are nevertheless abundantly present in concentrations of 2.3 × 10
8, 6.9 × 10
8 and 2.1 × 10
9 in the 100 mL harvested media from this particular cell line. The total particle count for each Nanopore size, across 3 biological replicates is shown in
Table 1. It should be noted that the counts shown are not cumulative as they represent the same sample analysed across 3 Nanopore sizes.
Size calibration beads were used to determine the most appropriate diameter mask to use for ISX analysis of EV diameter. The bead sizes provided by the manufacturer were 1, 2, 4, 6, 10, and 15 µm. We used the feature tool in IDEAS to apply a range of different diameter masks to the bright field image of the beads (
Figure 3B(i)). We selected bright field for this using non-labelled beads because the Quantibrite beads showed over exaggeration of EV size, possibly due to saturation and flare of fluorescence. The beads were visualised on bivariate plots of diameter versus side scatter (channel 06). Density plots (
Figure 3B(ii)) allowed the bead populations to be gated individually and subsequent histograms (
Figure 3B(iii)) to be viewed. We compared the diameters for each bead population assigned using the Object, Adaptive Erode and Erode Masks against the manufacturer specified size (
Figure 3B(iv)) and found the Erode mask on the bright field image, using 3 pixel reduction to be the most comparable.
2.3. Fluorescence Membrane Labelling Can Be Used to Distinguish EVs from Background and Speed Beads
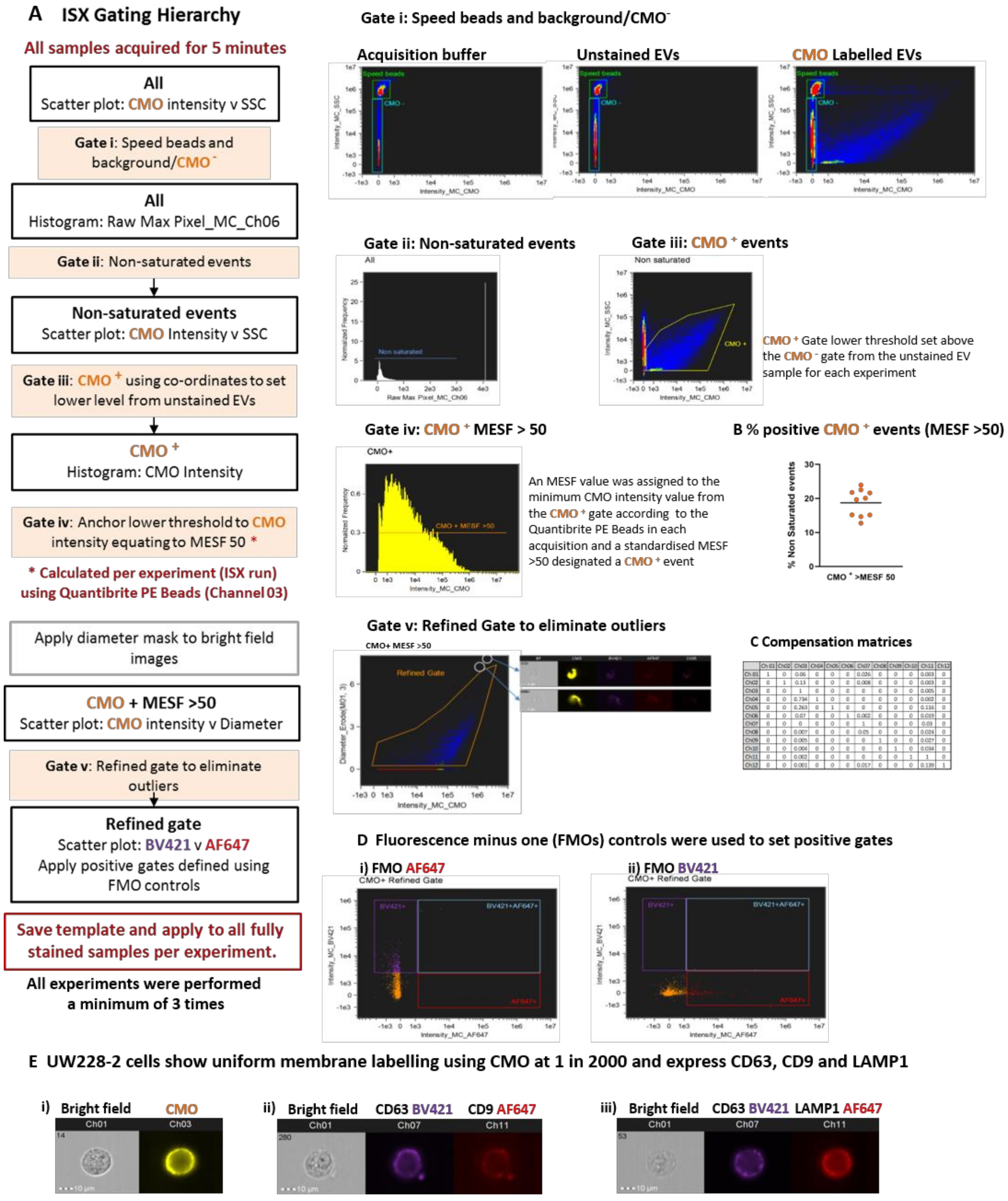
We devised an analysis template within IDEAS which comprised a hierarchical gating strategy aimed at characterising heterogeneous large EVs (
Figure 4A). All buffers were filtered using a 0.2 µm filter and samples were acquired for 5 min to avoid recording different amounts of background per acquisition. Speed beads were included. The speed beads and CMO
− events were defined using density plots of channel 03 (CMO) versus channel 06 (side scatter) intensity (Gate i). Running acquisition buffer only (left side) and unstained EVs (centre) showed the instrument detected a high level of background with low to medium side scatter. Labelling with the cell membrane dye (CMO) therefore helped to distinguish between the low CMO intensity/low side scatter EVs and background detected in both the acquisition buffer and unstained sample (right side). Applying a gate to capture low Raw Max Pixel events (channel 06—SSC: Gate ii) eliminated those events with saturated side scatter, including the speed beads. Outliers in the side scatter versus CMO intensity plots were individually inspected and found to be dual events comprising a speed bead (CMO
−, high SSC) and an EV (CMO+, low SSC) which occupied the flow chamber at the same time. This resulted in aberrant events with both high CMO intensity and high SSC and was therefore excluded from further analysis.
Events classed as CMO+ at this point were included in the initial CMO+ gate (Gate iii). Within the same acquisition, we ran the Quantibrite PE beads which enabled a minimum threshold for CMO+ events to be calculated and converted to MESF units, as described. The lower MESF cut off for this experiment was defined at >50 and a further gate on the CMO intensity histogram (Gate iv) captured all CMO+ events with an MESF >50 for downstream analysis. The proportion of CMO+ events as a percentage of all events acquired over the 5 min period varied across 10 replicates (mean 18.73% ± 1.2 SEM) CMO+ MESF > 50;
Figure 4B). A final gate was included to eliminate outliers which were either high CMO intensity but appeared on the bright field image to be membrane fragments (Gate v) or were not assigned a diameter due to lack of bright field image focus. This stringent gating strategy was designed to eliminate any events which could not be further analysed for EV marker expression or diameter.
We tested a number of approaches to apply compensation to our data in consultation with the manufacturer’s specialists. Initially, we attempted to construct a matrix using single stained EVs; however, the single pixel fluorescence was insufficient for the in-built compensation Wizard to assign a matrix. Next, we tried single stained parent cells, but the compensation matrix formulated by the wizard resulted in over-compensation and negative fluorescence intensity events. Over-compensation was thought to be due to the imbalance between the strong fluorescence signal resulting from cell mask orange, a membrane marker which indiscriminately labels lipids, and relatively weak fluorescence signal from target specific antibodies. These experiments were originally performed using a FITC conjugated CD63 antibody, however we found the level of adjustment required between channel 02 (FITC) and channel 03 (CMO) contributed to the negative populations. We re-optimised using the same CD63 antibody clone (HSC6) conjugated to BV421 (channel 07) which is spectrally more distinct from CMO. Finally, we found that using commercially available compensation beads labelled with our antibodies and acquired in the channels used for our study, in conjunction with assisted manual adjustment, provided the most reproducible compensation matrix.
We combined CMO with BV421 and AF647 and Fluorescence Minus One (FMO) controls for each fluorophore were used to set positive gates (
Figure 4D). The parent UW228-2 cells were screened for expression of the EV markers chosen for this study: CD63, CD9, and LAMP1 as defined in the latest MISEV guidelines [
1]. Multiplex labelling was performed using the following combinations: CMO with CD63 BV421 and CD9 AF647; or CMO with CD63 BV421 and LAMP1 AF647.
2.4. Multiplex Labelling Reveals Heterogeneity in EV Marker Expression
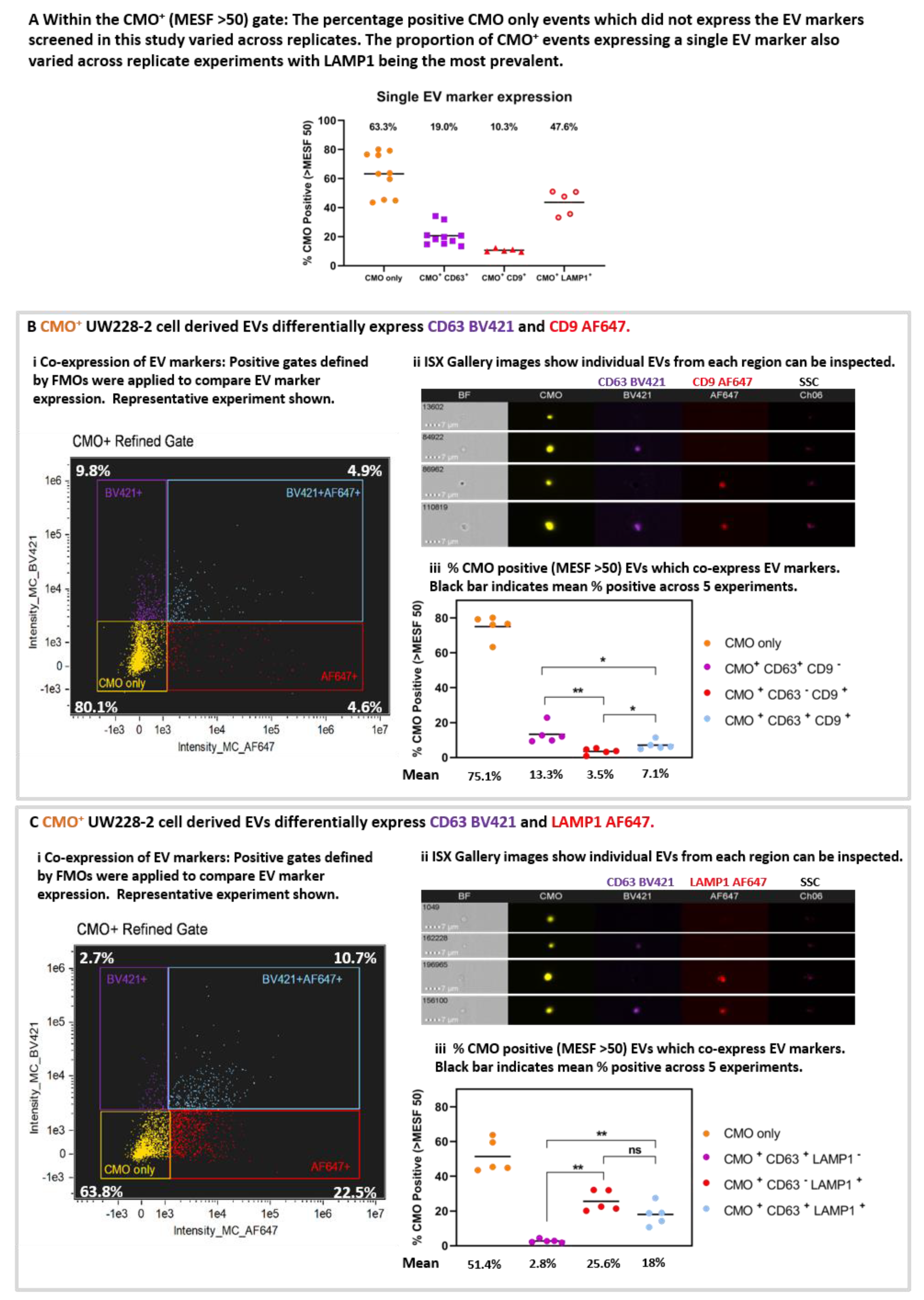
The proportion of CMO+ only events varied across the 10 replicates (
Figure 5A). The mean percentage positive CMO+ (MESF > 50) events (63.3% ± 4.6 SEM) which did not demonstrate expression of any EV markers included in this study were designated CMO only. Considering the single EV markers, the proportion of EVs positive for each EV marker varied CD63 19.0%, (± 2.2 SEM), CD9 mean 10.3% (± 0.5 SEM); LAMP1 47.6% (± 3.8 SEM). These data shown LAMP1 to be the most prevalent EV marker screened in this study.
Differential expression and co-expression of EV markers was examined between five replicate experiments. Representative bivariate plots of AF647 intensity against BV421 intensity (
Figure 5B(i) and
Figure 5C(i) for CD63/CD9 and CD63/LAMP1, respectively), and representative galleries (
Figure 5B(ii) and
Figure 5C(ii)) of EVs displaying each labelling combination taken from the quadrant plots are shown. The proportion of EVs which co-express EV markers within each gate for the five replicates is shown in
Figure 5B(iii) and
Figure 5C(iii). Where the EVs were labelled for CMO, CD63 BV421, and CD9 AF647 (
Figure 5B(iii)): 75.1% (± 3.0 SEM; yellow) were CMO only, 13.3% (± 2.4 SEM; purple) were CMO
+ CD63
+ CD9
-; 3.5% (± 0.8 SEM; red) were CMO
+ CD63
– CD9
+ and 7.1% (± 1.1 SEM; blue) were CMO
+ CD63
+ CD9
+. There were significantly fewer CD63
− CD9
+ EVs compared with CD63
+ (
p = 0.0079) only or CD63
+ CD9
+ EVs (
p = 0.0159). Similarly, there were fewer EV co-expressing CD63 and CD9 compared with CD63
+ alone (
p = 0.0317). (Mann Whitney U test). Considering the EVs labelled with CMO, CD63 BV421 and LAMP1 AF647 (
Figure 5C(iii)). 51.4% (+/− 4.3 SEM; yellow) were CMO+ only, a comparatively low 2.8% (± 0.4 SEM; purple) were CMO
+ CD63
+ LAMP1
–; whilst 25.6% (± 2.7 SEM; red) were CMO
+ CD63
− LAMP1
+ and 18% (± 2.8 SEM; blue) co-expressed both CD63 and LAMP1. There were significantly more EVs which expressed LAMP1 compared with CD63 alone (
p = 0.0079) and more expressing both CD63 and LAMP1 compared with CD63 alone (
p = 0.0079).
2.5. ISX Allow Correlative Analyses of Diameter with EV Marker Expression for Phenotyping
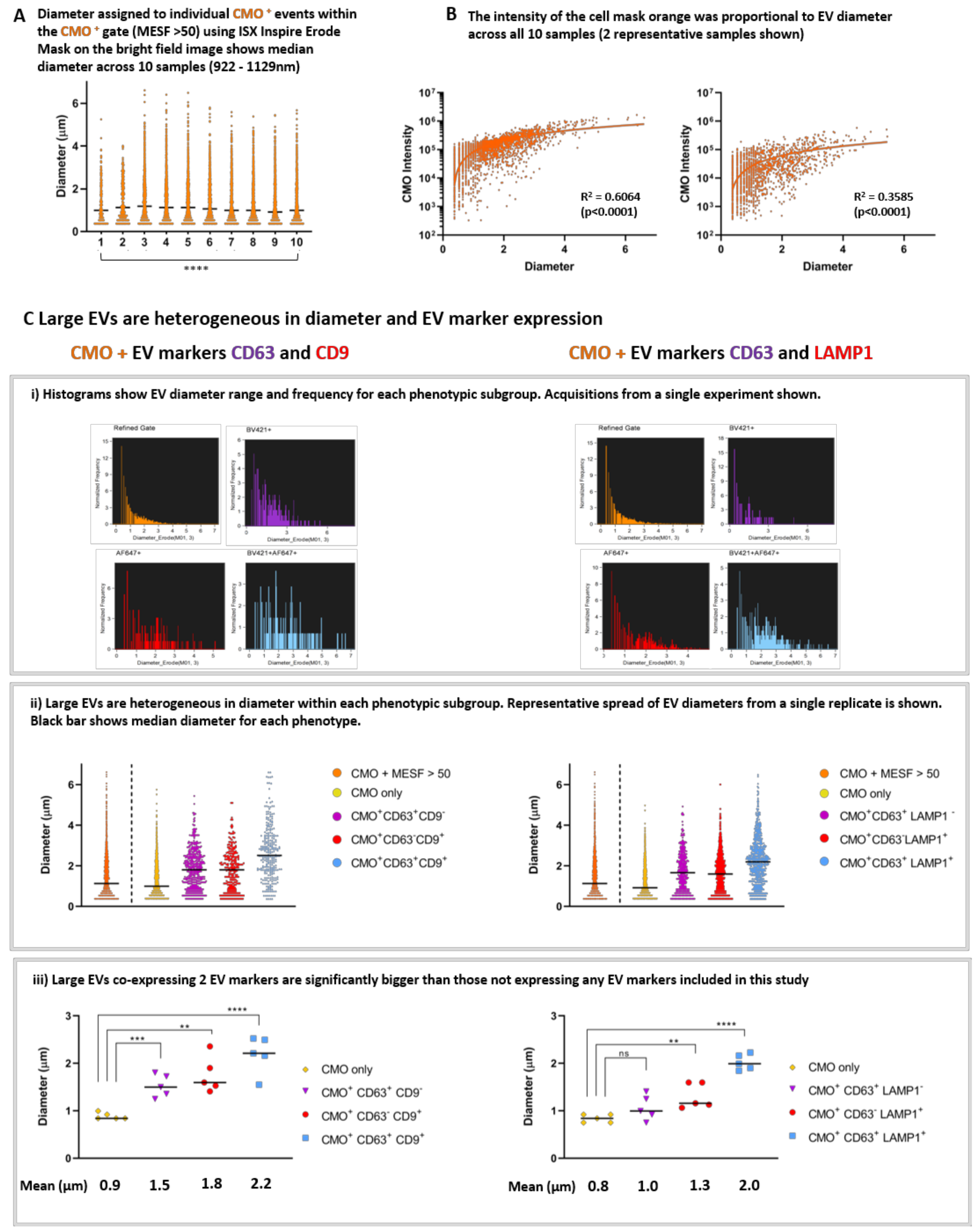
We applied the Diameter Mask; Erode (M01, 3) in IDEAS software to all fully labelled replicates using the Batch analysis function and compared the range of diameters present within the CMO+ MESF> 50 gate.
Figure 6A shows single event diameters within the 10 replicates. This median diameter (indicated with the black line) varied significantly across the replicates (922–1129 nm: Kruskal-Wallis;
p > 0.0001). However, these data represent EVs isolated as 3 biological replicates and when each replicate is analysed using ANOVA there was no significant difference between the median diameters of each EV preparation. There was a clear correlation between EV diameter and CMO intensity as shown in
Figure 6B. Regression analysis was performed on the individual exported feature values from each replicate (2 representative plots are shown from different biological replicates) (
p < 0.0001).
We found that large EVs are heterogeneous in both diameter and EV marker expression. Histogram plots of individual events exported from the quadrant positive gates show the different diameter range and frequency within each phenotypic subgroup (
Figure 6C(i)). Representative acquisitions from a single replicate are shown. ISX analysis facilitates the comparison of individual EV diameters from within each phenotype (
Figure 6C(ii)). A representative plot for all CMO
+ (MESF > 50) in a single replicate of each phenotype is shown and the black bars represent the median diameter for each subgroup. Within this typical replicate, the median diameter and range for each subgroup was as follows: All CMO >MESF 50 1.1 µm (362 nm–6.2 µm), CMO only 1.0 µm (362 nm−5.4 µm), CMO+ CD63
+ CD9
− 1.8 µm (362 nm–5.1 µm), CMO+ CD63
− CD9
+ 1.8 µm (362 nm–4.7 µm), and CMO+ CD63
+ CD9
+ 2.5 µm (362 nm–6.2 µm). When considering the second phenotype: All CMO >MESF 50 1.4 µm (362 nm–6.2 µm), CMO only 1.1 µm (362 nm–4.6 µm), CMO+ CD63
+ CD9
- 1.7 µm (362 nm–4.5 µm), CMO+ CD63
− CD9
+ 1.6 µm (362 nm–5.6 µm), and CMO+ CD63
+ CD9
+ 2.2 µm (362 nm–6.1 µm). In each case the median diameter increases in size with accumulating EV marker expression.
When considering all 5 replicates; we compared the mean diameter for each subgroup (
Figure 6C(iii)). For the first group (left panel): CMO only 0.89 µm (±0.03 SEM), CMO+ CD63
+ CD9
− 1.53 µm (± 0.1 SEM), CMO+ CD63
− CD9
+ 1.76 µm (±0.17 SEM) and CMO+ CD63
+ CD9
+ 2.19 µm (±0.18 SEM). Those EVs expressing either CD63 (
p < 0.0004) or CD9 (
p < 0.0010) were significantly larger than EV which did not (CMO only). For the second subgroup (right panel) the mean diameters were as follows: CMO only 0.84 µm (±0.04 SEM), CMO+ CD63
+ CD9
− 1.06 µm (±0.12 SEM), CMO+ CD63
− CD9
+ 1.31 µm (±0.12 SEM), and CMO+ CD63
+ CD9
+ 2.02 µm (±0.07 SEM). In this group, those EVs expressing CD63 only were not significantly larger than those which did not. However, EVs expressing LAMP1 were larger (
p < 0.005). In both cases, those CMO
+ EVs co-expressing 2 EVs markers: either CD63 and CD9, or CD63 and LAMP1 (blue) were significantly larger than those which do not carry the markers screened in this study. (****
p < 0.0001. Un-paired
t-test).
7. Discussion
Our principal aim was to develop a standardised method for the isolation and characterisation of individual large EVs, which could be further developed for phenotyping large EVs from clinical samples. The value of the Imagestream to the field of EV characterisation has been explored elsewhere [
19,
24,
25]. However, reports focussing on the large EV populations, frequently discarded during small EV isolation protocols, are rarely present in the current literature.
By using in vitro cultures, we were able to use the ideal conditions to generate EVs and optimise experiments. Specifically, by culturing in serum-free media, we eliminated contamination from bovine EVs present in FBS [
18] and subsequent false positive fluorescence signals from serum lipoproteins [
20]. Nevertheless, harvesting large EVs from any source presents challenges as cells or cell debris, including intracellular vesicles released due to parent cell membrane rupture from early centrifugation steps, can contaminate the subsequent EV pellets. For the EVs to be truly extracellular prior to isolation, the outer membrane of accompanying cells must not be ruptured by mechanical or chemical means during initial harvest. The centrifugation speeds we have used here are low compared to some commonly reported EV isolation protocols [
26] to specifically preserve large EV membrane integrity. Electron microscopy remains the sole technique that can examine individual EVs and EV preparations for sheared cell fragments, but it is neither quantitative nor high throughput. In addition, whilst TEM is widely used for EV investigation, it cannot perhaps distinguish between bone fide EVs and other particles. In this case, cryoEM or CLEM (correlative light and electron microscopy) which facilitates overlaid fluorescence or gold labelled antibody binding and EM would prove more informative. It is necessary to use a combination of techniques to explore the quantity, quality, and biology of EVs. All techniques, many originally designed for analysing cells, have technical challenges when applied to considerably smaller entities. For flow cytometry, background scatter events due to particles in the sheath fluid are an anticipated phenomenon which is rarely reported. In the work we report here, a high level of background appeared within the same gate as unstained EVs and persisted despite 0.2 µm filtration of sheath fluid. Our protocol is therefore reliant on strong and uniform, membrane-bound fluorescence labelling in order to assign an initial gate that separates potential EVs from speed beads or background scatter events. However, we found that the lower fluorescence intensity threshold to define CMO positive events was not clearly distinct from the instrument background. This was likely due to a combination of EV size and relative fluorescence intensity. As recommended elsewhere [
19] we used commercially available fluorescent beads of known intensity (Quantibrite) to provide a means to assign standardised units (MESF) and therefore a mathematical cut-off for our CMO+ gate. We acknowledge that calibration beads differ in the physical properties of EVs, in particular the refractive index, which can affect the intensity of scattered light, a particularly important consideration for the study of smaller EVs. PE was the closest available fluorophore to CMO and used as a standard for channel 03 on the ISX. CMO is a membrane label incorporated into the plasma membrane, and emits a greater fluorescence compared with a target-specific, conjugated antibody. This hampered the use of FITC alongside CMO as the spectrally close fluorophores led to overcompensation between channels 02 (FITC) and channel 03 (CMO). BV421 was a successful alternative but the difficulties encountered raised concerns about trying to further multiplex with additional fluorophores using this platform.
The challenges for choosing the correct technique(s) to analyse EVs have been well described elsewhere [
27]. For detailed advice on considerations for EV separation or enrichment methodologies and recommended steps for EV characterisation, we recommend referring to the MISEV guidelines; a position paper prepared by the international EV community to support EV research [
1]. A major challenge now is to adapt the protocol we describe for the analysis of large EVs from biological fluids. Clinical samples are more complex: EVs from a single cell type as described here offer the ideal model for characterisation, however clinical samples contain heterogeneous EVs from numerous cells [
10]. EV isolation has been reported from peripheral blood with some correlations to clinical outcome in other cancers [
28,
29]. We have previously identified leukaemic EVs in patients’ bone marrow plasma. This was possible using a marker which identified the cell of origin (B cell marker CD19) and understanding that the bone marrow of a leukemia patient is primarily composed of malignant B cells [
9]. However, surface markers for Medulloblastoma cells are less well known, rather molecular signatures define subgroups in this cancer type [
30]. Investigating surface marker expression on medulloblastoma cell-derived EVs would help to develop a more rapid screening tools. The value of a liquid biopsy to diagnose brain tumours in the clinic is clear and some suitable candidates have been identified in Glioblastoma [
31,
32]. A next step could be to investigate EVs released by primary medulloblastoma cells although the requirements for optimal culture conditions to generate sufficient material will add additional complexity. In the case of medulloblastoma, ideally, EVs circulating in either the cerebrospinal fluid or peripheral blood for comparison to matched primary tumour-derived EVs would be of considerable interest.
Our SOP is likely to exclude most small EVs and exosomes, expected to be present in the supernatant discarded at the final step (2000×
g). Experiments to isolate these for comparison are on-going. One classification we examined in this study was the distinction from apoptotic bodies. Demonstration of intact EV membranes, a lack of fragmented nuclei staining (DAPI) and evidence that EVs were derived from viable cells supported our assertion that the EVs analysed were not apoptotic bodies or cell debris [
33]. It might be possible to further distinguish these populations using molecular profiling. Other studies suggest that these EV subgroups display distinct RNA profiles [
34] an approach which is reliant on pure populations and therefore robust and meticulous isolation protocols.
Our data show that large EVs are ubiquitous and whilst absolute quantification is not yet within reach, we demonstrated similar size profiles using two independent techniques: TRPS and ISX. We have previously identified EVs of up to 6 µM using immunofluorescence, ISX and TEM [
15]. However, validating the quantity and size range has only been possible using the qNANO instrument. The qNANO employs TRPS technology to quantify EV count in a given sample and assign a size relative to a calibration bead of known diameter. It is currently the only instrument which can provide this information across the large EV population which spans 250 nm up to 6 µm (from our cells). Other platforms are restricted to small EVs (<1 µM) due to the measurements being reliant on Brownian movement (e.g., Nanosight and Zetaview). We found the most prevalent EV populations to be around 250–450 nm; however, we consistently detect EVs with a much larger diameter range in every cell line we have screened to date. We remain cautious not to define these as oncosomes; as although derived from cancer cells, we have not yet demonstrated their oncogenic potential [
4].
Whilst fluorescence intensity alone cannot be used to quantify protein expression levels due to low level antigen expression on EVs, we did observe patterns of differential expression. In EV literature, 3 principle markers are used to define EVs: CD63, CD9, and CD81. CD63, however, has been identified as a pan-EV marker, present in all defined EV subgroups to date [
6] and therefore CD63 was our preferred initial marker. However, large EVs are as yet poorly characterised and we found that CD63 was not the most abundant EV marker in our study. CD63 is a tetraspanin which has been used as a selection tool for immuno-capture experiments and also for tracking EV release [
35,
36]. Based on our observations, if a full EV repertoire was of interest, then a cocktail of multiple markers should be considered as screening with CD63 alone will likely fail to capture a significant proportion of EVs. We found LAMP1, previously identified on exosomes and EVs from a variety our cell types and biological fluids [
37], was significantly more prevalent.
We also show here that the majority of large EVs did not express any of the three markers screened. We did observe a significant increase in the median diameter of individual EVs which expressed 1 or more markers compared to none (CMO only), in each of the experiments performed. Further, across all replicates, those EVs which co-expressed 2 markers (CD63 + CD9, or CD63 + LAMP1) were significantly larger than those without. Others have suggested that larger EVs are likely to accommodate a greater number of tumour-derived molecules than exosomes [
13] and data presented here would support that hypothesis. Further investigations to define a broader panel of large EV markers followed by more comprehensive techniques, such as proteomic profiling, would help to fully phenotype the large EVs population. From a clinical perspective, it is likely that large EVs will be a rich source of biomarkers of benefit to the study of human disease. Standardised protocols and instruments capable of measuring multiple markers are key to moving the field forward and expanding the interest from exosomes only.
The research we report here demonstrates that high resolution, high throughput imaging flow cytometry is an exceptional tool offering the unique ability to quantify and analyse individual events within heterogeneous EV populations. We set out to develop an isolation protocol consisting of minimal manipulation and processing which may abrogate, mask, or indeed elicit changes in EV structure or biology, which could impact on any functional read outs in downstream experiments. Indeed, fully understanding the biological consequence(s) of EV release or uptake by recipient cells is an essential part of the field and will advance with new technologies and innovation.

 ,
, 







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}