Mutual Interplay of Host Immune System and Gut Microbiota in the Immunopathology of Atherosclerosis

 , and
, and
Abstract
:1. Introduction
2. Roles of the Oral and Gut Microbiota in Cardiovascular Diseases (CVD)
3. Role of Inflammation in Atherosclerosis
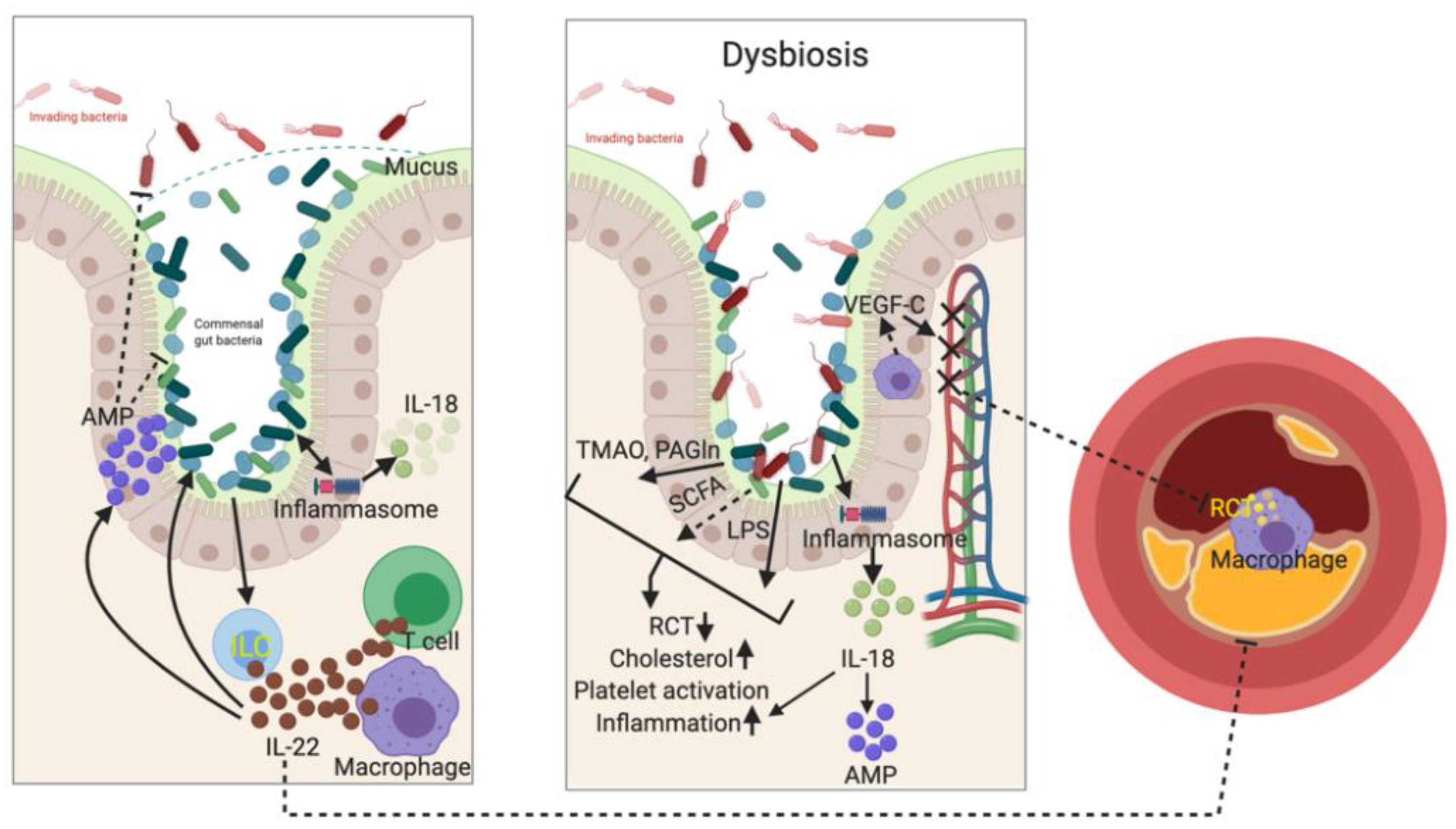
4. Gut Barrier Regulates both Local and Systemic Immune Responses
5. Immune System Shapes Gut Microbiome in Regulating Atherogenesis
5.1. IL-22 and IL-23 Signaling
5.2. Inflammasomes
6. Gut Microbiota Regulates Immune Response in Atherogenesis
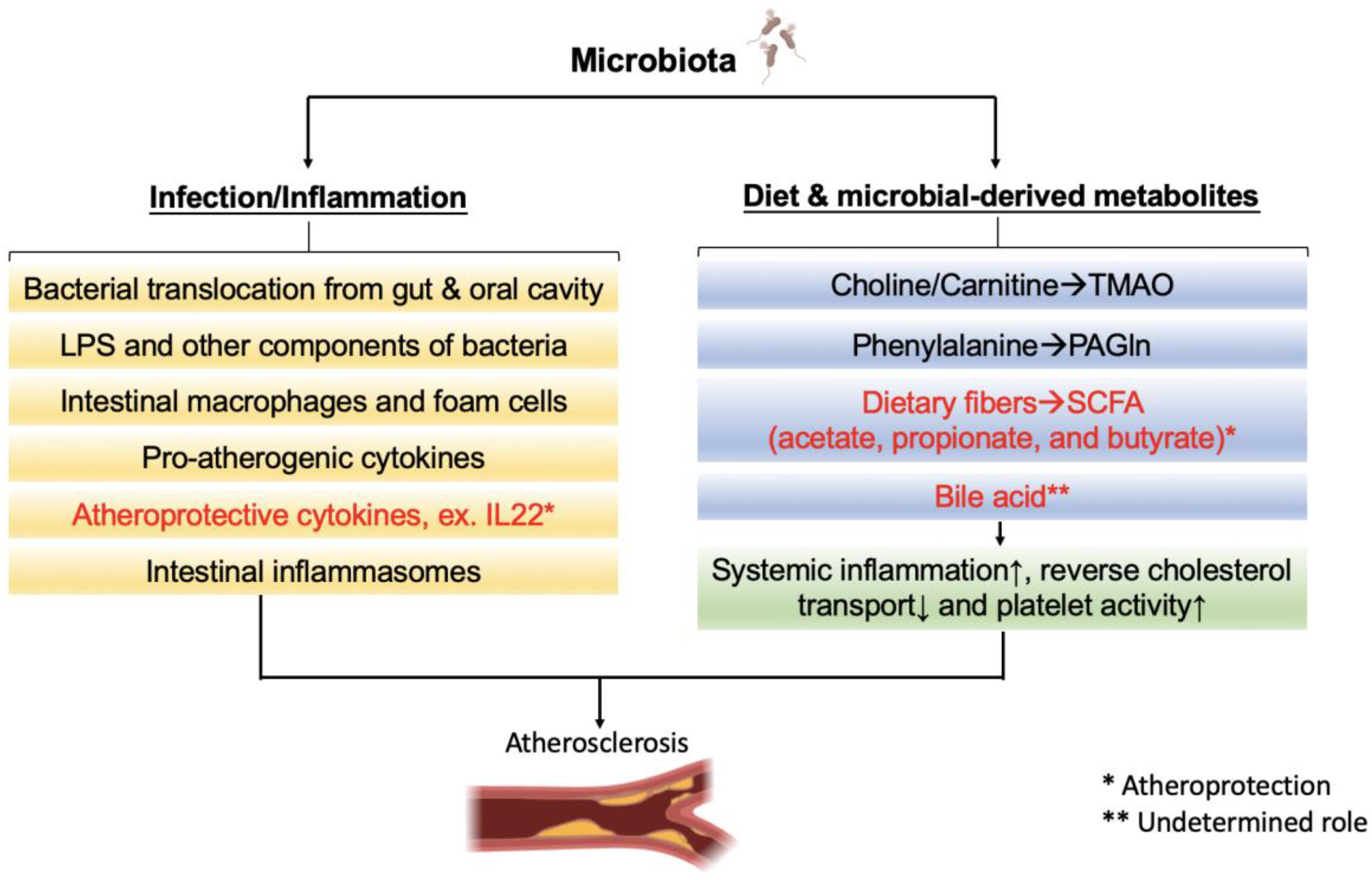
7. Microbial Metabolites and Atherosclerosis
7.1. Bile Acids (BAs)
7.2. Trimethylamine N-Oxide (TMAO)
7.3. Short-Chain Fatty Acid (SCFA)
7.4. Phenylacetylglutamine (PAGln)
8. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| ABCA1 | ATP Binding Cassette Subfamily A Member 1 |
| ABCG1 | ATP Binding Cassette Subfamily G Member 1 |
| ADR | adrenergic receptors |
| AhR | aryl hydrocarbon receptor |
| AMP | antimicrobial peptide |
| BA | bile acid |
| CAD | coronary artery diseases |
| CVD | cardiovascular diseases |
| DSS | dextran sodium sulfate |
| FMT | fecal microbial transplantation |
| FXR | farnesoid X receptor |
| GF | germ-free |
| GLP-1 | glucagon-like peptide-1 |
| HDL | high-density lipoprotein |
| HFD | high-fat diet |
| HIF | hypoxia-inducible factor |
| IgA | immunoglobulin A |
| IL-23R | IL-23 receptor |
| ILC | innate lymphoid cell |
| LPS | lipopolysaccharide |
| MACE | major adverse cardiovascular events |
| MerTK | Mer proto-oncogene tyrosine kinase |
| NLRP | NOD (nucleotide oligomerization domain)-, LRR (leucine-rich repeat)-, and PYD (pyrin domain)-containing protein |
| ox-LDL-C | oxidized low-density lipoprotein cholesterol |
| PAGln | phenylacetylglutamine |
| RCT | reverse cholesterol transport |
| SCFA | short-chain fatty acid |
| SPM | specialized pro-resolving mediator |
| TLR | toll-like receptor |
| TMAO | trimethylamine N-oxide |
| WT mice | wild-type mice |
References
- Moore, K.J.; Koplev, S.; Fisher, E.A.; Tabas, I.; Bjorkegren, J.L.M.; Doran, A.C.; Kovacic, J.C. Macrophage Trafficking, Inflammatory Resolution, and Genomics in Atherosclerosis: JACC Macrophage in CVD Series (Part 2). J. Am. Coll. Cardiol. 2018, 72, 2181–2197. [Google Scholar] [CrossRef] [PubMed]
- Chinetti-Gbaguidi, G.; Colin, S.; Staels, B. Macrophage subsets in atherosclerosis. Nat. Rev. Cardiol. 2015, 12, 10–17. [Google Scholar] [CrossRef] [PubMed]
- Chistiakov, D.A.; Bobryshev, Y.V.; Kozarov, E.; Sobenin, I.A.; Orekhov, A.N. Role of gut microbiota in the modulation of atherosclerosis-associated immune response. Front. Microbiol. 2015, 6, 671. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ponziani, F.R.; Scaldaferri, F.; Petito, V.; Paroni Sterbini, F.; Pecere, S.; Lopetuso, L.R.; Palladini, A.; Gerardi, V.; Masucci, L.; Pompili, M.; et al. The Role of Antibiotics in Gut Microbiota Modulation: The Eubiotic Effects of Rifaximin. Dig. Dis. 2016, 34, 269–278. [Google Scholar] [CrossRef] [PubMed]
- Clarke, G.; Stilling, R.M.; Kennedy, P.J.; Stanton, C.; Cryan, J.F.; Dinan, T.G. Minireview: Gut microbiota: The neglected endocrine organ. Mol. Endocrinol. 2014, 28, 1221–1238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brown, J.M.; Hazen, S.L. Microbial modulation of cardiovascular disease. Nat. Rev. Microbiol. 2018, 16, 171–181. [Google Scholar] [CrossRef] [PubMed]
- Tilg, H.; Zmora, N.; Adolph, T.E.; Elinav, E. The intestinal microbiota fuelling metabolic inflammation. Nat. Rev. Immunol. 2020, 20, 40–54. [Google Scholar] [CrossRef]
- Ma, J.; Li, H. The Role of Gut Microbiota in Atherosclerosis and Hypertension. Front. Pharmacol. 2018, 9, 1082. [Google Scholar] [CrossRef]
- Zheng, D.; Liwinski, T.; Elinav, E. Interaction between microbiota and immunity in health and disease. Cell Res. 2020, 30, 492–506. [Google Scholar] [CrossRef]
- Tang, W.H.W.; Li, D.Y.; Hazen, S.L. Dietary metabolism, the gut microbiome, and heart failure. Nat. Rev. Cardiol. 2019, 16, 137–154. [Google Scholar] [CrossRef]
- Fan, Y.; Pedersen, O. Gut microbiota in human metabolic health and disease. Nat. Rev. Microbiol. 2020, 1–17. [Google Scholar] [CrossRef]
- Backhed, F.; Ding, H.; Wang, T.; Hooper, L.V.; Koh, G.Y.; Nagy, A.; Semenkovich, C.F.; Gordon, J.I. The gut microbiota as an environmental factor that regulates fat storage. Proc. Natl. Acad. Sci. USA 2004, 101, 15718–15723. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koren, O.; Spor, A.; Felin, J.; Fak, F.; Stombaugh, J.; Tremaroli, V.; Behre, C.J.; Knight, R.; Fagerberg, B.; Ley, R.E.; et al. Human oral, gut, and plaque microbiota in patients with atherosclerosis. Proc. Natl. Acad. Sci. USA 2011, 108 (Suppl. 1), 4592–4598. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karlsson, F.H.; Fak, F.; Nookaew, I.; Tremaroli, V.; Fagerberg, B.; Petranovic, D.; Backhed, F.; Nielsen, J. Symptomatic atherosclerosis is associated with an altered gut metagenome. Nat. Commun. 2012, 3, 1245. [Google Scholar] [CrossRef] [Green Version]
- Emoto, T.; Yamashita, T.; Kobayashi, T.; Sasaki, N.; Hirota, Y.; Hayashi, T.; So, A.; Kasahara, K.; Yodoi, K.; Matsumoto, T.; et al. Characterization of gut microbiota profiles in coronary artery disease patients using data mining analysis of terminal restriction fragment length polymorphism: Gut microbiota could be a diagnostic marker of coronary artery disease. Heart Vessel. 2017, 32, 39–46. [Google Scholar] [CrossRef]
- Emoto, T.; Yamashita, T.; Sasaki, N.; Hirota, Y.; Hayashi, T.; So, A.; Kasahara, K.; Yodoi, K.; Matsumoto, T.; Mizoguchi, T.; et al. Analysis of Gut Microbiota in Coronary Artery Disease Patients: A Possible Link between Gut Microbiota and Coronary Artery Disease. J. Atheroscler. Thromb. 2016, 23, 908–921. [Google Scholar] [CrossRef] [Green Version]
- Jie, Z.; Xia, H.; Zhong, S.L.; Feng, Q.; Li, S.; Liang, S.; Zhong, H.; Liu, Z.; Gao, Y.; Zhao, H.; et al. The gut microbiome in atherosclerotic cardiovascular disease. Nat. Commun. 2017, 8, 845. [Google Scholar] [CrossRef] [Green Version]
- Kasahara, K.; Tanoue, T.; Yamashita, T.; Yodoi, K.; Matsumoto, T.; Emoto, T.; Mizoguchi, T.; Hayashi, T.; Kitano, N.; Sasaki, N.; et al. Commensal bacteria at the crossroad between cholesterol homeostasis and chronic inflammation in atherosclerosis. J. Lipid Res. 2017, 58, 519–528. [Google Scholar] [CrossRef] [Green Version]
- Jackel, S.; Kiouptsi, K.; Lillich, M.; Hendrikx, T.; Khandagale, A.; Kollar, B.; Hormann, N.; Reiss, C.; Subramaniam, S.; Wilms, E.; et al. Gut microbiota regulate hepatic von Willebrand factor synthesis and arterial thrombus formation via Toll-like receptor-2. Blood 2017, 130, 542–553. [Google Scholar] [CrossRef] [Green Version]
- Stepankova, R.; Tonar, Z.; Bartova, J.; Nedorost, L.; Rossman, P.; Poledne, R.; Schwarzer, M.; Tlaskalova-Hogenova, H. Absence of microbiota (germ-free conditions) accelerates the atherosclerosis in ApoE-deficient mice fed standard low cholesterol diet. J. Atheroscler. Thromb. 2010, 17, 796–804. [Google Scholar] [CrossRef] [Green Version]
- Lindskog Jonsson, A.; Caesar, R.; Akrami, R.; Reinhardt, C.; Fak Hallenius, F.; Boren, J.; Backhed, F. Impact of Gut Microbiota and Diet on the Development of Atherosclerosis in Apoe(-/-) Mice. Arterioscler. Thromb. Vasc. Biol. 2018, 38, 2318–2326. [Google Scholar] [CrossRef] [PubMed]
- Brown, J.M.; Hazen, S.L. The gut microbial endocrine organ: Bacterially derived signals driving cardiometabolic diseases. Annu. Rev. Med. 2015, 66, 343–359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hug, H.; Mohajeri, M.H.; La Fata, G. Toll-Like Receptors: Regulators of the Immune Response in the Human Gut. Nutrients 2018, 10, 203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Z.; Klipfell, E.; Bennett, B.J.; Koeth, R.; Levison, B.S.; Dugar, B.; Feldstein, A.E.; Britt, E.B.; Fu, X.; Chung, Y.M.; et al. Gut flora metabolism of phosphatidylcholine promotes cardiovascular disease. Nature 2011, 472, 57–63. [Google Scholar] [CrossRef] [Green Version]
- Koeth, R.A.; Wang, Z.; Levison, B.S.; Buffa, J.A.; Org, E.; Sheehy, B.T.; Britt, E.B.; Fu, X.; Wu, Y.; Li, L.; et al. Intestinal microbiota metabolism of L-carnitine, a nutrient in red meat, promotes atherosclerosis. Nat. Med. 2013, 19, 576–585. [Google Scholar] [CrossRef] [Green Version]
- Tang, W.H.; Wang, Z.; Levison, B.S.; Koeth, R.A.; Britt, E.B.; Fu, X.; Wu, Y.; Hazen, S.L. Intestinal microbial metabolism of phosphatidylcholine and cardiovascular risk. N. Engl. J. Med. 2013, 368, 1575–1584. [Google Scholar] [CrossRef] [Green Version]
- Koh, A.; De Vadder, F.; Kovatcheva-Datchary, P.; Backhed, F. From Dietary Fiber to Host Physiology: Short-Chain Fatty Acids as Key Bacterial Metabolites. Cell 2016, 165, 1332–1345. [Google Scholar] [CrossRef] [Green Version]
- Tang, T.W.H.; Chen, H.C.; Chen, C.Y.; Yen, C.Y.T.; Lin, C.J.; Prajnamitra, R.P.; Chen, L.L.; Ruan, S.C.; Lin, J.H.; Lin, P.J.; et al. Loss of Gut Microbiota Alters Immune System Composition and Cripples Postinfarction Cardiac Repair. Circulation 2019, 139, 647–659. [Google Scholar] [CrossRef]
- Nemet, I.; Saha, P.P.; Gupta, N.; Zhu, W.; Romano, K.A.; Skye, S.M.; Cajka, T.; Mohan, M.L.; Li, L.; Wu, Y.; et al. A Cardiovascular Disease-Linked Gut Microbial Metabolite Acts via Adrenergic Receptors. Cell 2020, 180, 862–877.e22. [Google Scholar] [CrossRef]
- Bogiatzi, C.; Gloor, G.; Allen-Vercoe, E.; Reid, G.; Wong, R.G.; Urquhart, B.L.; Dinculescu, V.; Ruetz, K.N.; Velenosi, T.J.; Pignanelli, M.; et al. Metabolic products of the intestinal microbiome and extremes of atherosclerosis. Atherosclerosis 2018, 273, 91–97. [Google Scholar] [CrossRef]
- Gryp, T.; Vanholder, R.; Vaneechoutte, M.; Glorieux, G. p-Cresyl Sulfate. Toxins 2017, 9, 52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sallee, M.; Dou, L.; Cerini, C.; Poitevin, S.; Brunet, P.; Burtey, S. The aryl hydrocarbon receptor-activating effect of uremic toxins from tryptophan metabolism: A new concept to understand cardiovascular complications of chronic kidney disease. Toxins 2014, 6, 934–949. [Google Scholar] [CrossRef] [PubMed]
- Barreto, F.C.; Barreto, D.V.; Liabeuf, S.; Meert, N.; Glorieux, G.; Temmar, M.; Choukroun, G.; Vanholder, R.; Massy, Z.A.; European Uraemic Toxin Work Group. Serum indoxyl sulfate is associated with vascular disease and mortality in chronic kidney disease patients. Clin. J. Am. Soc Nephrol. 2009, 4, 1551–1558. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liabeuf, S.; Barreto, D.V.; Barreto, F.C.; Meert, N.; Glorieux, G.; Schepers, E.; Temmar, M.; Choukroun, G.; Vanholder, R.; European Uraemic Toxin Work Group; et al. Free p-cresylsulphate is a predictor of mortality in patients at different stages of chronic kidney disease. Nephrol. Dial Transpl. 2010, 25, 1183–1191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Watanabe, H.; Miyamoto, Y.; Honda, D.; Tanaka, H.; Wu, Q.; Endo, M.; Noguchi, T.; Kadowaki, D.; Ishima, Y.; Kotani, S.; et al. p-Cresyl sulfate causes renal tubular cell damage by inducing oxidative stress by activation of NADPH oxidase. Kidney Int. 2013, 83, 582–592. [Google Scholar] [CrossRef] [Green Version]
- Pols, T.W.; Nomura, M.; Harach, T.; Lo Sasso, G.; Oosterveer, M.H.; Thomas, C.; Rizzo, G.; Gioiello, A.; Adorini, L.; Pellicciari, R.; et al. TGR5 activation inhibits atherosclerosis by reducing macrophage inflammation and lipid loading. Cell Metab. 2011, 14, 747–757. [Google Scholar] [CrossRef] [Green Version]
- Mencarelli, A.; Renga, B.; Distrutti, E.; Fiorucci, S. Antiatherosclerotic effect of farnesoid X receptor. Am. J. Physiol. Heart Circ. Physiol. 2009, 296, H272–H281. [Google Scholar] [CrossRef] [Green Version]
- Kuipers, F.; Bloks, V.W.; Groen, A.K. Beyond intestinal soap--bile acids in metabolic control. Nat. Rev. Endocrinol. 2014, 10, 488–498. [Google Scholar] [CrossRef]
- Humphrey, L.L.; Fu, R.; Buckley, D.I.; Freeman, M.; Helfand, M. Periodontal disease and coronary heart disease incidence: A systematic review and meta-analysis. J. Gen. Intern. Med. 2008, 23, 2079–2086. [Google Scholar] [CrossRef] [Green Version]
- Trevisan, M.; Dorn, J. The relationship between periodontal disease (pd) and cardiovascular disease (cvd). Mediterr J. Hematol. Infect. Dis. 2010, 2, e2010030. [Google Scholar] [CrossRef] [Green Version]
- Mattila, K.J.; Nieminen, M.S.; Valtonen, V.V.; Rasi, V.P.; Kesaniemi, Y.A.; Syrjala, S.L.; Jungell, P.S.; Isoluoma, M.; Hietaniemi, K.; Jokinen, M.J. Association between dental health and acute myocardial infarction. BMJ 1989, 298, 779–781. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Oliveira, C.; Watt, R.; Hamer, M. Toothbrushing, inflammation, and risk of cardiovascular disease: Results from Scottish Health Survey. BMJ 2010, 340, c2451. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Figuero, E.; Sanchez-Beltran, M.; Cuesta-Frechoso, S.; Tejerina, J.M.; del Castro, J.A.; Gutierrez, J.M.; Herrera, D.; Sanz, M. Detection of periodontal bacteria in atheromatous plaque by nested polymerase chain reaction. J. Periodontol. 2011, 82, 1469–1477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haraszthy, V.I.; Zambon, J.J.; Trevisan, M.; Zeid, M.; Genco, R.J. Identification of periodontal pathogens in atheromatous plaques. J. Periodontol. 2000, 71, 1554–1560. [Google Scholar] [CrossRef] [PubMed]
- Andraws, R.; Berger, J.S.; Brown, D.L. Effects of antibiotic therapy on outcomes of patients with coronary artery disease: A meta-analysis of randomized controlled trials. JAMA 2005, 293, 2641–2647. [Google Scholar] [CrossRef] [PubMed]
- Ross, R. Atherosclerosis—An inflammatory disease. N. Engl. J. Med. 1999, 340, 115–126. [Google Scholar] [CrossRef]
- Libby, P. Inflammation in atherosclerosis. Nature 2002, 420, 868–874. [Google Scholar] [CrossRef]
- Herrero-Fernandez, B.; Gomez-Bris, R.; Somovilla-Crespo, B.; Gonzalez-Granado, J.M. Immunobiology of Atherosclerosis: A Complex Net of Interactions. Int. J. Mol. Sci. 2019, 20, 5293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mantovani, A.; Sica, A.; Sozzani, S.; Allavena, P.; Vecchi, A.; Locati, M. The chemokine system in diverse forms of macrophage activation and polarization. Trends Immunol. 2004, 25, 677–686. [Google Scholar] [CrossRef]
- Williams, J.W.; Giannarelli, C.; Rahman, A.; Randolph, G.J.; Kovacic, J.C. Macrophage Biology, Classification, and Phenotype in Cardiovascular Disease: JACC Macrophage in CVD Series (Part 1). J. Am. Coll. Cardiol. 2018, 72, 2166–2180. [Google Scholar] [CrossRef] [PubMed]
- Moore, K.J.; Tabas, I. Macrophages in the pathogenesis of atherosclerosis. Cell 2011, 145, 341–355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robbins, C.S.; Hilgendorf, I.; Weber, G.F.; Theurl, I.; Iwamoto, Y.; Figueiredo, J.L.; Gorbatov, R.; Sukhova, G.K.; Gerhardt, L.M.; Smyth, D.; et al. Local proliferation dominates lesional macrophage accumulation in atherosclerosis. Nat. Med. 2013, 19, 1166–1172. [Google Scholar] [CrossRef] [PubMed]
- Tacke, F.; Alvarez, D.; Kaplan, T.J.; Jakubzick, C.; Spanbroek, R.; Llodra, J.; Garin, A.; Liu, J.; Mack, M.; van Rooijen, N.; et al. Monocyte subsets differentially employ CCR2, CCR5, and CX3CR1 to accumulate within atherosclerotic plaques. J. Clin. Investig. 2007, 117, 185–194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feig, J.E.; Parathath, S.; Rong, J.X.; Mick, S.L.; Vengrenyuk, Y.; Grauer, L.; Young, S.G.; Fisher, E.A. Reversal of hyperlipidemia with a genetic switch favorably affects the content and inflammatory state of macrophages in atherosclerotic plaques. Circulation 2011, 123, 989–998. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feig, J.E.; Rong, J.X.; Shamir, R.; Sanson, M.; Vengrenyuk, Y.; Liu, J.; Rayner, K.; Moore, K.; Garabedian, M.; Fisher, E.A. HDL promotes rapid atherosclerosis regression in mice and alters inflammatory properties of plaque monocyte-derived cells. Proc. Natl. Acad. Sci. USA 2011, 108, 7166–7171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van Gils, J.M.; Derby, M.C.; Fernandes, L.R.; Ramkhelawon, B.; Ray, T.D.; Rayner, K.J.; Parathath, S.; Distel, E.; Feig, J.L.; Alvarez-Leite, J.I.; et al. The neuroimmune guidance cue netrin-1 promotes atherosclerosis by inhibiting the emigration of macrophages from plaques. Nat. Immunol. 2012, 13, 136–143. [Google Scholar] [CrossRef] [Green Version]
- Trogan, E.; Choudhury, R.P.; Dansky, H.M.; Rong, J.X.; Breslow, J.L.; Fisher, E.A. Laser capture microdissection analysis of gene expression in macrophages from atherosclerotic lesions of apolipoprotein E-deficient mice. Proc. Natl. Acad. Sci. USA 2002, 99, 2234–2239. [Google Scholar] [CrossRef] [Green Version]
- Ouimet, M.; Barrett, T.J.; Fisher, E.A. HDL and Reverse Cholesterol Transport. Circ Res. 2019, 124, 1505–1518. [Google Scholar] [CrossRef]
- Tang, J.; Lobatto, M.E.; Hassing, L.; van der Staay, S.; van Rijs, S.M.; Calcagno, C.; Braza, M.S.; Baxter, S.; Fay, F.; Sanchez-Gaytan, B.L.; et al. Inhibiting macrophage proliferation suppresses atherosclerotic plaque inflammation. Sci. Adv. 2015, 1, e1400223. [Google Scholar] [CrossRef] [Green Version]
- Fredman, G.; Tabas, I. Boosting Inflammation Resolution in Atherosclerosis: The Next Frontier for Therapy. Am. J. Pathol. 2017, 187, 1211–1221. [Google Scholar] [CrossRef] [Green Version]
- Russell, D.G.; Huang, L.; VanderVen, B.C. Immunometabolism at the interface between macrophages and pathogens. Nat. Rev. Immunol. 2019, 19, 291–304. [Google Scholar] [CrossRef] [PubMed]
- Bobryshev, Y.V.; Ivanova, E.A.; Chistiakov, D.A.; Nikiforov, N.G.; Orekhov, A.N. Macrophages and Their Role in Atherosclerosis: Pathophysiology and Transcriptome Analysis. BioMed Res. Int. 2016, 2016, 9582430. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colin, S.; Chinetti-Gbaguidi, G.; Staels, B. Macrophage phenotypes in atherosclerosis. Immunol. Rev. 2014, 262, 153–166. [Google Scholar] [CrossRef] [PubMed]
- Williams, K.J.; Tabas, I. The response-to-retention hypothesis of early atherogenesis. Arterioscler. Thromb. Vasc. Biol. 1995, 15, 551–561. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tabas, I.; Williams, K.J.; Boren, J. Subendothelial lipoprotein retention as the initiating process in atherosclerosis: Update and therapeutic implications. Circulation 2007, 116, 1832–1844. [Google Scholar] [CrossRef]
- Alciato, F.; Sainaghi, P.P.; Sola, D.; Castello, L.; Avanzi, G.C. TNF-alpha, IL-6, and IL-1 expression is inhibited by GAS6 in monocytes/macrophages. J. Leukoc. Biol. 2010, 87, 869–875. [Google Scholar] [CrossRef]
- Doran, A.C.; Yurdagul, A., Jr.; Tabas, I. Efferocytosis in health and disease. Nat. Rev. Immunol. 2020, 20, 254–267. [Google Scholar] [CrossRef]
- Dalli, J.; Serhan, C.N. Specific lipid mediator signatures of human phagocytes: Microparticles stimulate macrophage efferocytosis and pro-resolving mediators. Blood 2012, 120, e60–e72. [Google Scholar] [CrossRef] [Green Version]
- Cai, B.; Thorp, E.B.; Doran, A.C.; Subramanian, M.; Sansbury, B.E.; Lin, C.S.; Spite, M.; Fredman, G.; Tabas, I. MerTK cleavage limits proresolving mediator biosynthesis and exacerbates tissue inflammation. Proc. Natl. Acad. Sci. USA 2016, 113, 6526–6531. [Google Scholar] [CrossRef] [Green Version]
- Cai, B.; Thorp, E.B.; Doran, A.C.; Sansbury, B.E.; Daemen, M.J.; Dorweiler, B.; Spite, M.; Fredman, G.; Tabas, I. MerTK receptor cleavage promotes plaque necrosis and defective resolution in atherosclerosis. J. Clin. Investig. 2017, 127, 564–568. [Google Scholar] [CrossRef]
- Ridker, P.M.; Everett, B.M.; Thuren, T.; MacFadyen, J.G.; Chang, W.H.; Ballantyne, C.; Fonseca, F.; Nicolau, J.; Koenig, W.; Anker, S.D.; et al. Antiinflammatory Therapy with Canakinumab for Atherosclerotic Disease. N. Engl. J. Med. 2017, 377, 1119–1131. [Google Scholar] [CrossRef] [PubMed]
- Tardif, J.C.; Kouz, S.; Waters, D.D.; Bertrand, O.F.; Diaz, R.; Maggioni, A.P.; Pinto, F.J.; Ibrahim, R.; Gamra, H.; Kiwan, G.S.; et al. Efficacy and Safety of Low-Dose Colchicine after Myocardial Infarction. N. Engl. J. Med. 2019, 381, 2497–2505. [Google Scholar] [CrossRef] [PubMed]
- Nidorf, S.M.; Fiolet, A.T.L.; Mosterd, A.; Eikelboom, J.W.; Schut, A.; Opstal, T.S.J.; The, S.H.K.; Xu, X.F.; Ireland, M.A.; Lenderink, T.; et al. Colchicine in Patients with Chronic Coronary Disease. N. Engl. J. Med. 2020. [Google Scholar] [CrossRef] [PubMed]
- Ridker, P.M.; Everett, B.M.; Pradhan, A.; MacFadyen, J.G.; Solomon, D.H.; Zaharris, E.; Mam, V.; Hasan, A.; Rosenberg, Y.; Iturriaga, E.; et al. Low-Dose Methotrexate for the Prevention of Atherosclerotic Events. N. Engl. J. Med. 2019, 380, 752–762. [Google Scholar] [CrossRef]
- Dabke, K.; Hendrick, G.; Devkota, S. The gut microbiome and metabolic syndrome. J. Clin. Investig. 2019, 129, 4050–4057. [Google Scholar] [CrossRef]
- Garrett, W.S.; Gordon, J.I.; Glimcher, L.H. Homeostasis and inflammation in the intestine. Cell 2010, 140, 859–870. [Google Scholar] [CrossRef] [Green Version]
- Allaire, J.M.; Morampudi, V.; Crowley, S.M.; Stahl, M.; Yu, H.; Bhullar, K.; Knodler, L.A.; Bressler, B.; Jacobson, K.; Vallance, B.A. Frontline defenders: Goblet cell mediators dictate host-microbe interactions in the intestinal tract during health and disease. Am. J. Physiol. Gastrointest Liver Physiol. 2018, 314, G360–G377. [Google Scholar] [CrossRef]
- Brown, E.M.; Sadarangani, M.; Finlay, B.B. The role of the immune system in governing host-microbe interactions in the intestine. Nat. Immunol. 2013, 14, 660–667. [Google Scholar] [CrossRef]
- Vaishnava, S.; Yamamoto, M.; Severson, K.M.; Ruhn, K.A.; Yu, X.; Koren, O.; Ley, R.; Wakeland, E.K.; Hooper, L.V. The antibacterial lectin RegIIIgamma promotes the spatial segregation of microbiota and host in the intestine. Science 2011, 334, 255–258. [Google Scholar] [CrossRef] [Green Version]
- Selsted, M.E.; Ouellette, A.J. Mammalian defensins in the antimicrobial immune response. Nat. Immunol. 2005, 6, 551–557. [Google Scholar] [CrossRef]
- Vijay-Kumar, M.; Aitken, J.D.; Carvalho, F.A.; Cullender, T.C.; Mwangi, S.; Srinivasan, S.; Sitaraman, S.V.; Knight, R.; Ley, R.E.; Gewirtz, A.T. Metabolic syndrome and altered gut microbiota in mice lacking Toll-like receptor 5. Science 2010, 328, 228–231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vijay-Kumar, M.; Sanders, C.J.; Taylor, R.T.; Kumar, A.; Aitken, J.D.; Sitaraman, S.V.; Neish, A.S.; Uematsu, S.; Akira, S.; Williams, I.R.; et al. Deletion of TLR5 results in spontaneous colitis in mice. J. Clin. Investig. 2007, 117, 3909–3921. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spits, H.; Di Santo, J.P. The expanding family of innate lymphoid cells: Regulators and effectors of immunity and tissue remodeling. Nat. Immunol. 2011, 12, 21–27. [Google Scholar] [CrossRef] [PubMed]
- Panda, S.K.; Colonna, M. Innate Lymphoid Cells in Mucosal Immunity. Front. Immunol. 2019, 10, 861. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mjosberg, J.; Spits, H. Human innate lymphoid cells. J. Allergy Clin. Immunol. 2016, 138, 1265–1276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ebbo, M.; Crinier, A.; Vely, F.; Vivier, E. Innate lymphoid cells: Major players in inflammatory diseases. Nat. Rev. Immunol. 2017, 17, 665–678. [Google Scholar] [CrossRef]
- Spits, H.; Cupedo, T. Innate lymphoid cells: Emerging insights in development, lineage relationships, and function. Annu. Rev. Immunol. 2012, 30, 647–675. [Google Scholar] [CrossRef]
- Qiu, J.; Heller, J.J.; Guo, X.; Chen, Z.M.; Fish, K.; Fu, Y.X.; Zhou, L. The aryl hydrocarbon receptor regulates gut immunity through modulation of innate lymphoid cells. Immunity 2012, 36, 92–104. [Google Scholar] [CrossRef] [Green Version]
- Aujla, S.J.; Chan, Y.R.; Zheng, M.; Fei, M.; Askew, D.J.; Pociask, D.A.; Reinhart, T.A.; McAllister, F.; Edeal, J.; Gaus, K.; et al. IL-22 mediates mucosal host defense against Gram-negative bacterial pneumonia. Nat. Med. 2008, 14, 275–281. [Google Scholar] [CrossRef]
- Zheng, Y.; Valdez, P.A.; Danilenko, D.M.; Hu, Y.; Sa, S.M.; Gong, Q.; Abbas, A.R.; Modrusan, Z.; Ghilardi, N.; de Sauvage, F.J.; et al. Interleukin-22 mediates early host defense against attaching and effacing bacterial pathogens. Nat. Med. 2008, 14, 282–289. [Google Scholar] [CrossRef]
- Sugimoto, K.; Ogawa, A.; Mizoguchi, E.; Shimomura, Y.; Andoh, A.; Bhan, A.K.; Blumberg, R.S.; Xavier, R.J.; Mizoguchi, A. IL-22 ameliorates intestinal inflammation in a mouse model of ulcerative colitis. J. Clin. Investig. 2008, 118, 534–544. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reboldi, A.; Arnon, T.I.; Rodda, L.B.; Atakilit, A.; Sheppard, D.; Cyster, J.G. IgA production requires B cell interaction with subepithelial dendritic cells in Peyer’s patches. Science 2016, 352, aaf4822. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lochner, M.; Ohnmacht, C.; Presley, L.; Bruhns, P.; Si-Tahar, M.; Sawa, S.; Eberl, G. Microbiota-induced tertiary lymphoid tissues aggravate inflammatory disease in the absence of RORgamma t and LTi cells. J. Exp. Med. 2011, 208, 125–134. [Google Scholar] [CrossRef] [PubMed]
- Sonnenberg, G.F.; Monticelli, L.A.; Alenghat, T.; Fung, T.C.; Hutnick, N.A.; Kunisawa, J.; Shibata, N.; Grunberg, S.; Sinha, R.; Zahm, A.M.; et al. Innate lymphoid cells promote anatomical containment of lymphoid-resident commensal bacteria. Science 2012, 336, 1321–1325. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ding, S.; Chi, M.M.; Scull, B.P.; Rigby, R.; Schwerbrock, N.M.; Magness, S.; Jobin, C.; Lund, P.K. High-fat diet: Bacteria interactions promote intestinal inflammation which precedes and correlates with obesity and insulin resistance in mouse. PLoS ONE 2010, 5, e12191. [Google Scholar] [CrossRef] [Green Version]
- Thaiss, C.A.; Levy, M.; Grosheva, I.; Zheng, D.; Soffer, E.; Blacher, E.; Braverman, S.; Tengeler, A.C.; Barak, O.; Elazar, M.; et al. Hyperglycemia drives intestinal barrier dysfunction and risk for enteric infection. Science 2018, 359, 1376–1383. [Google Scholar] [CrossRef] [Green Version]
- Cani, P.D.; Bibiloni, R.; Knauf, C.; Waget, A.; Neyrinck, A.M.; Delzenne, N.M.; Burcelin, R. Changes in gut microbiota control metabolic endotoxemia-induced inflammation in high-fat diet-induced obesity and diabetes in mice. Diabetes 2008, 57, 1470–1481. [Google Scholar] [CrossRef] [Green Version]
- Guo, S.; Nighot, M.; Al-Sadi, R.; Alhmoud, T.; Nighot, P.; Ma, T.Y. Lipopolysaccharide Regulation of Intestinal Tight Junction Permeability Is Mediated by TLR4 Signal Transduction Pathway Activation of FAK and MyD88. J. Immunol. 2015, 195, 4999–5010. [Google Scholar] [CrossRef]
- Cani, P.D.; Amar, J.; Iglesias, M.A.; Poggi, M.; Knauf, C.; Bastelica, D.; Neyrinck, A.M.; Fava, F.; Tuohy, K.M.; Chabo, C.; et al. Metabolic endotoxemia initiates obesity and insulin resistance. Diabetes 2007, 56, 1761–1772. [Google Scholar] [CrossRef] [Green Version]
- Stenman, L.K.; Holma, R.; Korpela, R. High-fat-induced intestinal permeability dysfunction associated with altered fecal bile acids. World J. Gastroenterol. 2012, 18, 923–929. [Google Scholar] [CrossRef]
- Horioka, K.; Tanaka, H.; Isozaki, S.; Konishi, H.; Fujiya, M.; Okuda, K.; Asari, M.; Shiono, H.; Ogawa, K.; Shimizu, K. Acute Colchicine Poisoning Causes Endotoxemia via the Destruction of Intestinal Barrier Function: The Curative Effect of Endotoxin Prevention in a Murine Model. Dig. Dis. Sci. 2020, 65, 132–140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hansson, G.K.; Hermansson, A. The immune system in atherosclerosis. Nat. Immunol. 2011, 12, 204–212. [Google Scholar] [CrossRef] [PubMed]
- Tang, C.; Chen, S.; Qian, H.; Huang, W. Interleukin-23: As a drug target for autoimmune inflammatory diseases. Immunology 2012, 135, 112–124. [Google Scholar] [CrossRef] [PubMed]
- Wiekowski, M.T.; Leach, M.W.; Evans, E.W.; Sullivan, L.; Chen, S.C.; Vassileva, G.; Bazan, J.F.; Gorman, D.M.; Kastelein, R.A.; Narula, S.; et al. Ubiquitous transgenic expression of the IL-23 subunit p19 induces multiorgan inflammation, runting, infertility, and premature death. J. Immunol. 2001, 166, 7563–7570. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Engelbertsen, D.; Depuydt, M.A.C.; Verwilligen, R.A.F.; Rattik, S.; Levinsohn, E.; Edsfeldt, A.; Kuperwaser, F.; Jarolim, P.; Lichtman, A.H. IL-23R Deficiency Does Not Impact Atherosclerotic Plaque Development in Mice. J. Am. Heart Assoc. 2018, 7, 8. [Google Scholar] [CrossRef]
- Fatkhullina, A.R.; Peshkova, I.O.; Dzutsev, A.; Aghayev, T.; McCulloch, J.A.; Thovarai, V.; Badger, J.H.; Vats, R.; Sundd, P.; Tang, H.Y.; et al. An Interleukin-23-Interleukin-22 Axis Regulates Intestinal Microbial Homeostasis to Protect from Diet-Induced Atherosclerosis. Immunity 2018, 49, 943–957.e9. [Google Scholar] [CrossRef] [Green Version]
- Lok, Z.S.Y.; Lyle, A.N. Osteopontin in Vascular Disease. Arterioscler. Thromb. Vasc. Biol. 2019, 39, 613–622. [Google Scholar] [CrossRef] [Green Version]
- Langley, R.G.; Papp, K.; Gottlieb, A.B.; Krueger, G.G.; Gordon, K.B.; Williams, D.; Valdes, J.; Setze, C.; Strober, B. Safety results from a pooled analysis of randomized, controlled phase II and III clinical trials and interim data from an open-label extension trial of the interleukin-12/23 monoclonal antibody, briakinumab, in moderate to severe psoriasis. J. Eur. Acad. Dermatol. Venereol. 2013, 27, 1252–1261. [Google Scholar] [CrossRef]
- Ryan, C.; Leonardi, C.L.; Krueger, J.G.; Kimball, A.B.; Strober, B.E.; Gordon, K.B.; Langley, R.G.; de Lemos, J.A.; Daoud, Y.; Blankenship, D.; et al. Association between biologic therapies for chronic plaque psoriasis and cardiovascular events: A meta-analysis of randomized controlled trials. JAMA 2011, 306, 864–871. [Google Scholar] [CrossRef]
- Tzellos, T.; Kyrgidis, A.; Trigoni, A.; Zouboulis, C.C. Association of anti-IL-12/23 biologic agents ustekinumab and briakinumab with major adverse cardiovascular events. J. Eur. Acad. Dermatol. Venereol. 2013, 27, 1586–1587. [Google Scholar] [CrossRef]
- Al-Janabi, A.; Jabbar-Lopez, Z.K.; Griffiths, C.E.M.; Yiu, Z.Z.N. Risankizumab vs. ustekinumab for plaque psoriasis: A critical appraisal. Br. J. Dermatol. 2019, 180, 1348–1351. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Broz, P.; Dixit, V.M. Inflammasomes: Mechanism of assembly, regulation and signalling. Nat. Rev. Immunol. 2016, 16, 407–420. [Google Scholar] [CrossRef] [PubMed]
- Grebe, A.; Hoss, F.; Latz, E. NLRP3 Inflammasome and the IL-1 Pathway in Atherosclerosis. Circ. Res. 2018, 122, 1722–1740. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.; Zhao, Y.; Wang, Y.; Gao, W.; Ding, J.; Li, P.; Hu, L.; Shao, F. Inflammatory caspases are innate immune receptors for intracellular LPS. Nature 2014, 514, 187–192. [Google Scholar] [CrossRef]
- Zanoni, I.; Tan, Y.; Di Gioia, M.; Broggi, A.; Ruan, J.; Shi, J.; Donado, C.A.; Shao, F.; Wu, H.; Springstead, J.R.; et al. An endogenous caspase-11 ligand elicits interleukin-1 release from living dendritic cells. Science 2016, 352, 1232–1236. [Google Scholar] [CrossRef] [Green Version]
- Kayagaki, N.; Wong, M.T.; Stowe, I.B.; Ramani, S.R.; Gonzalez, L.C.; Akashi-Takamura, S.; Miyake, K.; Zhang, J.; Lee, W.P.; Muszynski, A.; et al. Noncanonical inflammasome activation by intracellular LPS independent of TLR4. Science 2013, 341, 1246–1249. [Google Scholar] [CrossRef]
- Latz, E.; Xiao, T.S.; Stutz, A. Activation and regulation of the inflammasomes. Nat. Rev. Immunol. 2013, 13, 397–411. [Google Scholar] [CrossRef]
- Lu, A.; Wu, H. Structural mechanisms of inflammasome assembly. FEBS J. 2015, 282, 435–444. [Google Scholar] [CrossRef]
- Zhen, Y.; Zhang, H. NLRP3 Inflammasome and Inflammatory Bowel Disease. Front. Immunol. 2019, 10, 276. [Google Scholar] [CrossRef] [Green Version]
- Wlodarska, M.; Thaiss, C.A.; Nowarski, R.; Henao-Mejia, J.; Zhang, J.P.; Brown, E.M.; Frankel, G.; Levy, M.; Katz, M.N.; Philbrick, W.M.; et al. NLRP6 inflammasome orchestrates the colonic host-microbial interface by regulating goblet cell mucus secretion. Cell 2014, 156, 1045–1059. [Google Scholar] [CrossRef] [Green Version]
- Elinav, E.; Strowig, T.; Kau, A.L.; Henao-Mejia, J.; Thaiss, C.A.; Booth, C.J.; Peaper, D.R.; Bertin, J.; Eisenbarth, S.C.; Gordon, J.I.; et al. NLRP6 inflammasome regulates colonic microbial ecology and risk for colitis. Cell 2011, 145, 745–757. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gremel, G.; Wanders, A.; Cedernaes, J.; Fagerberg, L.; Hallstrom, B.; Edlund, K.; Sjostedt, E.; Uhlen, M.; Ponten, F. The human gastrointestinal tract-specific transcriptome and proteome as defined by RNA sequencing and antibody-based profiling. J. Gastroenterol. 2015, 50, 46–57. [Google Scholar] [CrossRef] [PubMed]
- Levy, M.; Thaiss, C.A.; Zeevi, D.; Dohnalova, L.; Zilberman-Schapira, G.; Mahdi, J.A.; David, E.; Savidor, A.; Korem, T.; Herzig, Y.; et al. Microbiota-Modulated Metabolites Shape the Intestinal Microenvironment by Regulating NLRP6 Inflammasome Signaling. Cell 2015, 163, 1428–1443. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hirota, S.A.; Ng, J.; Lueng, A.; Khajah, M.; Parhar, K.; Li, Y.; Lam, V.; Potentier, M.S.; Ng, K.; Bawa, M.; et al. NLRP3 inflammasome plays a key role in the regulation of intestinal homeostasis. Inflamm. Bowel Dis. 2011, 17, 1359–1372. [Google Scholar] [CrossRef] [PubMed]
- Bauer, C.; Duewell, P.; Mayer, C.; Lehr, H.A.; Fitzgerald, K.A.; Dauer, M.; Tschopp, J.; Endres, S.; Latz, E.; Schnurr, M. Colitis induced in mice with dextran sulfate sodium (DSS) is mediated by the NLRP3 inflammasome. Gut 2010, 59, 1192–1199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zaki, M.H.; Boyd, K.L.; Vogel, P.; Kastan, M.B.; Lamkanfi, M.; Kanneganti, T.D. The NLRP3 inflammasome protects against loss of epithelial integrity and mortality during experimental colitis. Immunity 2010, 32, 379–391. [Google Scholar] [CrossRef] [Green Version]
- Itani, S.; Watanabe, T.; Nadatani, Y.; Sugimura, N.; Shimada, S.; Takeda, S.; Otani, K.; Hosomi, S.; Nagami, Y.; Tanaka, F.; et al. NLRP3 inflammasome has a protective effect against oxazolone-induced colitis: A possible role in ulcerative colitis. Sci. Rep. 2016, 6, 39075. [Google Scholar] [CrossRef]
- Song-Zhao, G.X.; Srinivasan, N.; Pott, J.; Baban, D.; Frankel, G.; Maloy, K.J. Nlrp3 activation in the intestinal epithelium protects against a mucosal pathogen. Mucosal Immunol. 2014, 7, 763–774. [Google Scholar] [CrossRef] [Green Version]
- Shi, X.; Xie, W.L.; Kong, W.W.; Chen, D.; Qu, P. Expression of the NLRP3 Inflammasome in Carotid Atherosclerosis. J. Stroke Cerebrovasc. Dis. 2015, 24, 2455–2466. [Google Scholar] [CrossRef]
- Paramel, V.G.; Folkersen, L.; Strawbridge, R.J.; Halvorsen, B.; Yndestad, A.; Ranheim, T.; Krohg-Sorensen, K.; Skjelland, M.; Espevik, T.; Aukrust, P.; et al. NLRP3 Inflammasome Expression and Activation in Human Atherosclerosis. J. Am. Heart Assoc. 2016, 5, e003031. [Google Scholar] [CrossRef] [Green Version]
- Westerterp, M.; Fotakis, P.; Ouimet, M.; Bochem, A.E.; Zhang, H.; Molusky, M.M.; Wang, W.; Abramowicz, S.; la Bastide-van Gemert, S.; Wang, N.; et al. Cholesterol Efflux Pathways Suppress Inflammasome Activation, NETosis, and Atherogenesis. Circulation 2018, 138, 898–912. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duewell, P.; Kono, H.; Rayner, K.J.; Sirois, C.M.; Vladimer, G.; Bauernfeind, F.G.; Abela, G.S.; Franchi, L.; Nunez, G.; Schnurr, M.; et al. NLRP3 inflammasomes are required for atherogenesis and activated by cholesterol crystals. Nature 2010, 464, 1357–1361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hendrikx, T.; Jeurissen, M.L.; van Gorp, P.J.; Gijbels, M.J.; Walenbergh, S.M.; Houben, T.; van Gorp, R.; Pottgens, C.C.; Stienstra, R.; Netea, M.G.; et al. Bone marrow-specific caspase-1/11 deficiency inhibits atherosclerosis development in Ldlr(-/-) mice. FEBS J. 2015, 282, 2327–2338. [Google Scholar] [CrossRef] [PubMed]
- Brandsma, E.; Kloosterhuis, N.J.; Koster, M.; Dekker, D.C.; Gijbels, M.J.J.; van der Velden, S.; Rios-Morales, M.; van Faassen, M.J.R.; Loreti, M.G.; de Bruin, A.; et al. A Proinflammatory Gut Microbiota Increases Systemic Inflammation and Accelerates Atherosclerosis. Circ. Res. 2019, 124, 94–100. [Google Scholar] [CrossRef]
- Birchenough, G.M.; Nystrom, E.E.; Johansson, M.E.; Hansson, G.C. A sentinel goblet cell guards the colonic crypt by triggering Nlrp6-dependent Muc2 secretion. Science 2016, 352, 1535–1542. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fritz, J.V.; Desai, M.S.; Shah, P.; Schneider, J.G.; Wilmes, P. From meta-omics to causality: Experimental models for human microbiome research. Microbiome 2013, 1, 14. [Google Scholar] [CrossRef] [Green Version]
- Chistiakov, D.A.; Bobryshev, Y.V.; Kozarov, E.; Sobenin, I.A.; Orekhov, A.N. Intestinal mucosal tolerance and impact of gut microbiota to mucosal tolerance. Front. Microbiol. 2014, 5, 781. [Google Scholar] [CrossRef]
- Belizario, J.E.; Faintuch, J.; Garay-Malpartida, M. Gut Microbiome Dysbiosis and Immunometabolism: New Frontiers for Treatment of Metabolic Diseases. Mediat. Inflamm. 2018, 2018, 2037838. [Google Scholar] [CrossRef]
- Kim, D.; Zeng, M.Y.; Nunez, G. The interplay between host immune cells and gut microbiota in chronic inflammatory diseases. Exp. Mol. Med. 2017, 49, e339. [Google Scholar] [CrossRef] [Green Version]
- Feng, Y.; Huang, Y.; Wang, Y.; Wang, P.; Song, H.; Wang, F. Antibiotics induced intestinal tight junction barrier dysfunction is associated with microbiota dysbiosis, activated NLRP3 inflammasome and autophagy. PLoS ONE 2019, 14, e0218384. [Google Scholar] [CrossRef]
- Yoshida, N.; Emoto, T.; Yamashita, T.; Watanabe, H.; Hayashi, T.; Tabata, T.; Hoshi, N.; Hatano, N.; Ozawa, G.; Sasaki, N.; et al. Bacteroides vulgatus and Bacteroides dorei Reduce Gut Microbial Lipopolysaccharide Production and Inhibit Atherosclerosis. Circulation 2018, 138, 2486–2498. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Lin, S.; Vanhoutte, P.M.; Woo, C.W.; Xu, A. Akkermansia Muciniphila Protects Against Atherosclerosis by Preventing Metabolic Endotoxemia-Induced Inflammation in Apoe-/- Mice. Circulation 2016, 133, 2434–2446. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, K.; Yuan, Y.; Yu, H.; Dai, X.; Wang, S.; Sun, Z.; Wang, F.; Fei, H.; Lin, Q.; Jiang, H.; et al. The gut microbial metabolite trimethylamine N-oxide aggravates GVHD by inducing M1 macrophage polarization in mice. Blood 2020, 136, 501–515. [Google Scholar] [CrossRef] [PubMed]
- Schulthess, J.; Pandey, S.; Capitani, M.; Rue-Albrecht, K.C.; Arnold, I.; Franchini, F.; Chomka, A.; Ilott, N.E.; Johnston, D.G.W.; Pires, E.; et al. The Short Chain Fatty Acid Butyrate Imprints an Antimicrobial Program in Macrophages. Immunity 2019, 50, 432–445.e7. [Google Scholar] [CrossRef] [Green Version]
- Varol, C.; Landsman, L.; Fogg, D.K.; Greenshtein, L.; Gildor, B.; Margalit, R.; Kalchenko, V.; Geissmann, F.; Jung, S. Monocytes give rise to mucosal, but not splenic, conventional dendritic cells. J. Exp. Med. 2007, 204, 171–180. [Google Scholar] [CrossRef]
- Bain, C.C.; Bravo-Blas, A.; Scott, C.L.; Perdiguero, E.G.; Geissmann, F.; Henri, S.; Malissen, B.; Osborne, L.C.; Artis, D.; Mowat, A.M. Constant replenishment from circulating monocytes maintains the macrophage pool in the intestine of adult mice. Nat. Immunol. 2014, 15, 929–937. [Google Scholar] [CrossRef] [Green Version]
- Kang, B.; Alvarado, L.J.; Kim, T.; Lehmann, M.L.; Cho, H.; He, J.; Li, P.; Kim, B.H.; Larochelle, A.; Kelsall, B.L. Commensal microbiota drive the functional diversification of colon macrophages. Mucosal Immunol. 2020, 13, 216–229. [Google Scholar] [CrossRef] [Green Version]
- Suh, S.H.; Choe, K.; Hong, S.P.; Jeong, S.H.; Makinen, T.; Kim, K.S.; Alitalo, K.; Surh, C.D.; Koh, G.Y.; Song, J.H. Gut microbiota regulates lacteal integrity by inducing VEGF-C in intestinal villus macrophages. EMBO Rep. 2019, 20, 4. [Google Scholar] [CrossRef]
- Bernier-Latmani, J.; Petrova, T.V. Intestinal lymphatic vasculature: Structure, mechanisms and functions. Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 510–526. [Google Scholar] [CrossRef]
- Cifarelli, V.; Eichmann, A. The Intestinal Lymphatic System: Functions and Metabolic Implications. Cell Mol. Gastroenterol. Hepatol. 2019, 7, 503–513. [Google Scholar] [CrossRef] [Green Version]
- Martel, C.; Li, W.; Fulp, B.; Platt, A.M.; Gautier, E.L.; Westerterp, M.; Bittman, R.; Tall, A.R.; Chen, S.H.; Thomas, M.J.; et al. Lymphatic vasculature mediates macrophage reverse cholesterol transport in mice. J. Clin. Investig. 2013, 123, 1571–1579. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aspelund, A.; Robciuc, M.R.; Karaman, S.; Makinen, T.; Alitalo, K. Lymphatic System in Cardiovascular Medicine. Circ. Res. 2016, 118, 515–530. [Google Scholar] [CrossRef] [PubMed]
- Schuijt, T.J.; Lankelma, J.M.; Scicluna, B.P.; de Sousa e Melo, F.; Roelofs, J.J.; de Boer, J.D.; Hoogendijk, A.J.; de Beer, R.; de Vos, A.; Belzer, C.; et al. The gut microbiota plays a protective role in the host defence against pneumococcal pneumonia. Gut 2016, 65, 575–583. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caesar, R.; Reigstad, C.S.; Backhed, H.K.; Reinhardt, C.; Ketonen, M.; Lunden, G.O.; Cani, P.D.; Backhed, F. Gut-derived lipopolysaccharide augments adipose macrophage accumulation but is not essential for impaired glucose or insulin tolerance in mice. Gut 2012, 61, 1701–1707. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luche, E.; Cousin, B.; Garidou, L.; Serino, M.; Waget, A.; Barreau, C.; Andre, M.; Valet, P.; Courtney, M.; Casteilla, L.; et al. Metabolic endotoxemia directly increases the proliferation of adipocyte precursors at the onset of metabolic diseases through a CD14-dependent mechanism. Mol. Metab. 2013, 2, 281–291. [Google Scholar] [CrossRef]
- Saita, D.; Ferrarese, R.; Foglieni, C.; Esposito, A.; Canu, T.; Perani, L.; Ceresola, E.R.; Visconti, L.; Burioni, R.; Clementi, M.; et al. Adaptive immunity against gut microbiota enhances apoE-mediated immune regulation and reduces atherosclerosis and western-diet-related inflammation. Sci. Rep. 2016, 6, 29353. [Google Scholar] [CrossRef] [Green Version]
- Huber, S.; Gagliani, N.; Zenewicz, L.A.; Huber, F.J.; Bosurgi, L.; Hu, B.; Hedl, M.; Zhang, W.; O’Connor, W., Jr.; Murphy, A.J.; et al. IL-22BP is regulated by the inflammasome and modulates tumorigenesis in the intestine. Nature 2012, 491, 259–263. [Google Scholar] [CrossRef] [Green Version]
- Komada, T.; Muruve, D.A. The role of inflammasomes in kidney disease. Nat. Rev. Nephrol. 2019, 15, 501–520. [Google Scholar] [CrossRef]
- Belkaid, Y.; Hand, T.W. Role of the microbiota in immunity and inflammation. Cell 2014, 157, 121–141. [Google Scholar] [CrossRef] [Green Version]
- Kamada, N.; Seo, S.U.; Chen, G.Y.; Nunez, G. Role of the gut microbiota in immunity and inflammatory disease. Nat. Rev. Immunol. 2013, 13, 321–335. [Google Scholar] [CrossRef]
- Kazemian, N.; Mahmoudi, M.; Halperin, F.; Wu, J.C.; Pakpour, S. Gut microbiota and cardiovascular disease: Opportunities and challenges. Microbiome 2020, 8, 36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wahlstrom, A.; Sayin, S.I.; Marschall, H.U.; Backhed, F. Intestinal Crosstalk between Bile Acids and Microbiota and Its Impact on Host Metabolism. Cell Metab. 2016, 24, 41–50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Foley, M.H.; O’Flaherty, S.; Barrangou, R.; Theriot, C.M. Bile salt hydrolases: Gatekeepers of bile acid metabolism and host-microbiome crosstalk in the gastrointestinal tract. PLoS Pathog. 2019, 15, e1007581. [Google Scholar] [CrossRef]
- Lefebvre, P.; Cariou, B.; Lien, F.; Kuipers, F.; Staels, B. Role of bile acids and bile acid receptors in metabolic regulation. Physiol. Rev. 2009, 89, 147–191. [Google Scholar] [CrossRef] [Green Version]
- Choucair, I.; Nemet, I.; Li, L.; Cole, M.A.; Skye, S.M.; Kirsop, J.D.; Fischbach, M.A.; Gogonea, V.; Brown, J.M.; Tang, W.H.W.; et al. Quantification of bile acids: A mass spectrometry platform for studying gut microbe connection to metabolic diseases. J. Lipid Res. 2020, 61, 159–177. [Google Scholar] [CrossRef] [PubMed]
- Haeusler, R.A.; Astiarraga, B.; Camastra, S.; Accili, D.; Ferrannini, E. Human insulin resistance is associated with increased plasma levels of 12alpha-hydroxylated bile acids. Diabetes 2013, 62, 4184–4191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schoenfield, L.J.; Lachin, J.M. Chenodiol (chenodeoxycholic acid) for dissolution of gallstones: The National Cooperative Gallstone Study. A controlled trial of efficacy and safety. Ann. Intern. Med. 1981, 95, 257–282. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, M.; Houten, S.M.; Wang, L.; Moschetta, A.; Mangelsdorf, D.J.; Heyman, R.A.; Moore, D.D.; Auwerx, J. Bile acids lower triglyceride levels via a pathway involving FXR, SHP, and SREBP-1c. J. Clin. Investig. 2004, 113, 1408–1418. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fiorucci, S.; Rizzo, G.; Donini, A.; Distrutti, E.; Santucci, L. Targeting farnesoid X receptor for liver and metabolic disorders. Trends Mol. Med. 2007, 13, 298–309. [Google Scholar] [CrossRef] [PubMed]
- Witkowski, M.; Weeks, T.L.; Hazen, S.L. Gut Microbiota and Cardiovascular Disease. Circ. Res. 2020, 127, 553–570. [Google Scholar] [CrossRef]
- Randrianarisoa, E.; Lehn-Stefan, A.; Wang, X.; Hoene, M.; Peter, A.; Heinzmann, S.S.; Zhao, X.; Konigsrainer, I.; Konigsrainer, A.; Balletshofer, B.; et al. Relationship of Serum Trimethylamine N-Oxide (TMAO) Levels with early Atherosclerosis in Humans. Sci. Rep 2016, 6, 26745. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mente, A.; Chalcraft, K.; Ak, H.; Davis, A.D.; Lonn, E.; Miller, R.; Potter, M.A.; Yusuf, S.; Anand, S.S.; McQueen, M.J. The Relationship Between Trimethylamine-N-Oxide and Prevalent Cardiovascular Disease in a Multiethnic Population Living in Canada. Can. J. Cardiol. 2015, 31, 1189–1194. [Google Scholar] [CrossRef] [PubMed]
- Zhu, W.; Gregory, J.C.; Org, E.; Buffa, J.A.; Gupta, N.; Wang, Z.; Li, L.; Fu, X.; Wu, Y.; Mehrabian, M.; et al. Gut Microbial Metabolite TMAO Enhances Platelet Hyperreactivity and Thrombosis Risk. Cell 2016, 165, 111–124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- den Besten, G.; van Eunen, K.; Groen, A.K.; Venema, K.; Reijngoud, D.J.; Bakker, B.M. The role of short-chain fatty acids in the interplay between diet, gut microbiota, and host energy metabolism. J. Lipid Res. 2013, 54, 2325–2340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alex, S.; Lange, K.; Amolo, T.; Grinstead, J.S.; Haakonsson, A.K.; Szalowska, E.; Koppen, A.; Mudde, K.; Haenen, D.; Al-Lahham, S.; et al. Short-chain fatty acids stimulate angiopoietin-like 4 synthesis in human colon adenocarcinoma cells by activating peroxisome proliferator-activated receptor gamma. Mol. Cell Biol. 2013, 33, 1303–1316. [Google Scholar] [CrossRef] [Green Version]
- Pluznick, J.L.; Protzko, R.J.; Gevorgyan, H.; Peterlin, Z.; Sipos, A.; Han, J.; Brunet, I.; Wan, L.X.; Rey, F.; Wang, T.; et al. Olfactory receptor responding to gut microbiota-derived signals plays a role in renin secretion and blood pressure regulation. Proc. Natl. Acad. Sci. USA 2013, 110, 4410–4415. [Google Scholar] [CrossRef] [Green Version]
- Macia, L.; Tan, J.; Vieira, A.T.; Leach, K.; Stanley, D.; Luong, S.; Maruya, M.; Ian McKenzie, C.; Hijikata, A.; Wong, C.; et al. Metabolite-sensing receptors GPR43 and GPR109A facilitate dietary fibre-induced gut homeostasis through regulation of the inflammasome. Nat. Commun. 2015, 6, 6734. [Google Scholar] [CrossRef] [Green Version]
- Kasahara, K.; Krautkramer, K.A.; Org, E.; Romano, K.A.; Kerby, R.L.; Vivas, E.I.; Mehrabian, M.; Denu, J.M.; Backhed, F.; Lusis, A.J.; et al. Interactions between Roseburia intestinalis and diet modulate atherogenesis in a murine model. Nat. Microbiol. 2018, 3, 1461–1471. [Google Scholar] [CrossRef]
- Bazzano, L.A.; He, J.; Ogden, L.G.; Loria, C.M.; Whelton, P.K. Dietary fiber intake and reduced risk of coronary heart disease in US men and women: The National Health and Nutrition Examination Survey I Epidemiologic Follow-up Study. Arch. Intern. Med. 2003, 163, 1897–1904. [Google Scholar] [CrossRef] [Green Version]
- Chambers, E.S.; Viardot, A.; Psichas, A.; Morrison, D.J.; Murphy, K.G.; Zac-Varghese, S.E.; MacDougall, K.; Preston, T.; Tedford, C.; Finlayson, G.S.; et al. Effects of targeted delivery of propionate to the human colon on appetite regulation, body weight maintenance and adiposity in overweight adults. Gut 2015, 64, 1744–1754. [Google Scholar] [CrossRef] [Green Version]
- den Besten, G.; Lange, K.; Havinga, R.; van Dijk, T.H.; Gerding, A.; van Eunen, K.; Muller, M.; Groen, A.K.; Hooiveld, G.J.; Bakker, B.M.; et al. Gut-derived short-chain fatty acids are vividly assimilated into host carbohydrates and lipids. Am. J. Physiol. Gastrointest Liver Physiol. 2013, 305, G900–G910. [Google Scholar] [CrossRef] [PubMed]
- Tolhurst, G.; Heffron, H.; Lam, Y.S.; Parker, H.E.; Habib, A.M.; Diakogiannaki, E.; Cameron, J.; Grosse, J.; Reimann, F.; Gribble, F.M. Short-chain fatty acids stimulate glucagon-like peptide-1 secretion via the G-protein-coupled receptor FFAR2. Diabetes 2012, 61, 364–371. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Hou, Y.; Wang, G.; Zheng, X.; Hao, H. Gut Microbial Metabolites of Aromatic Amino Acids as Signals in Host-Microbe Interplay. Trends Endocrinol. Metab. 2020, 31, 818–834. [Google Scholar] [CrossRef] [PubMed]
- Dodd, D.; Spitzer, M.H.; Van Treuren, W.; Merrill, B.D.; Hryckowian, A.J.; Higginbottom, S.K.; Le, A.; Cowan, T.M.; Nolan, G.P.; Fischbach, M.A.; et al. A gut bacterial pathway metabolizes aromatic amino acids into nine circulating metabolites. Nature 2017, 551, 648–652. [Google Scholar] [CrossRef] [PubMed]
- Grant, J.A.; Scrutton, M.C. Novel alpha2-adrenoreceptors primarily responsible for inducing human platelet aggregation. Nature 1979, 277, 659–661. [Google Scholar] [CrossRef]
- Rodriguez-Morato, J.; Matthan, N.R. Nutrition and Gastrointestinal Microbiota, Microbial-Derived Secondary Bile Acids, and Cardiovascular Disease. Curr. Atheroscler. Rep. 2020, 22, 47. [Google Scholar] [CrossRef] [PubMed]
- Hofmann, A.F.; Hagey, L.R. Bile acids: Chemistry, pathochemistry, biology, pathobiology, and therapeutics. Cell. Mol. Life Sci. 2008, 65, 2461–2483. [Google Scholar] [CrossRef] [PubMed]
- Begley, M.; Gahan, C.G.; Hill, C. The interaction between bacteria and bile. FEMS Microbiol. Rev. 2005, 29, 625–651. [Google Scholar] [CrossRef] [Green Version]
- Inagaki, T.; Moschetta, A.; Lee, Y.K.; Peng, L.; Zhao, G.; Downes, M.; Yu, R.T.; Shelton, J.M.; Richardson, J.A.; Repa, J.J.; et al. Regulation of antibacterial defense in the small intestine by the nuclear bile acid receptor. Proc. Natl. Acad. Sci. USA 2006, 103, 3920–3925. [Google Scholar] [CrossRef] [Green Version]
- Islam, K.B.; Fukiya, S.; Hagio, M.; Fujii, N.; Ishizuka, S.; Ooka, T.; Ogura, Y.; Hayashi, T.; Yokota, A. Bile acid is a host factor that regulates the composition of the cecal microbiota in rats. Gastroenterology 2011, 141, 1773–1781. [Google Scholar] [CrossRef]
- Ridlon, J.M.; Alves, J.M.; Hylemon, P.B.; Bajaj, J.S. Cirrhosis, bile acids and gut microbiota: Unraveling a complex relationship. Gut Microbes 2013, 4, 382–387. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parseus, A.; Sommer, N.; Sommer, F.; Caesar, R.; Molinaro, A.; Stahlman, M.; Greiner, T.U.; Perkins, R.; Backhed, F. Microbiota-induced obesity requires farnesoid X receptor. Gut 2017, 66, 429–437. [Google Scholar] [CrossRef] [Green Version]
- Hanniman, E.A.; Lambert, G.; McCarthy, T.C.; Sinal, C.J. Loss of functional farnesoid X receptor increases atherosclerotic lesions in apolipoprotein E-deficient mice. J. Lipid Res. 2005, 46, 2595–2604. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Wang, X.; Vales, C.; Lee, F.Y.; Lee, H.; Lusis, A.J.; Edwards, P.A. FXR deficiency causes reduced atherosclerosis in Ldlr-/- mice. Arterioscler. Thromb. Vasc. Biol. 2006, 26, 2316–2321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, G.L.; Santamarina-Fojo, S.; Akiyama, T.E.; Amar, M.J.; Paigen, B.J.; Brewer, B., Jr.; Gonzalez, F.J. Effects of FXR in foam-cell formation and atherosclerosis development. Biochim. Biophys. Acta 2006, 1761, 1401–1409. [Google Scholar] [CrossRef] [Green Version]
- Fang, S.; Suh, J.M.; Reilly, S.M.; Yu, E.; Osborn, O.; Lackey, D.; Yoshihara, E.; Perino, A.; Jacinto, S.; Lukasheva, Y.; et al. Intestinal FXR agonism promotes adipose tissue browning and reduces obesity and insulin resistance. Nat. Med. 2015, 21, 159–165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, K.; Saha, P.K.; Chan, L.; Moore, D.D. Farnesoid X receptor is essential for normal glucose homeostasis. J. Clin. Investig. 2006, 116, 1102–1109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maruyama, T.; Miyamoto, Y.; Nakamura, T.; Tamai, Y.; Okada, H.; Sugiyama, E.; Nakamura, T.; Itadani, H.; Tanaka, K. Identification of membrane-type receptor for bile acids (M-BAR). Biochem. Biophys. Res. Commun. 2002, 298, 714–719. [Google Scholar] [CrossRef]
- Watanabe, M.; Houten, S.M.; Mataki, C.; Christoffolete, M.A.; Kim, B.W.; Sato, H.; Messaddeq, N.; Harney, J.W.; Ezaki, O.; Kodama, T.; et al. Bile acids induce energy expenditure by promoting intracellular thyroid hormone activation. Nature 2006, 439, 484–489. [Google Scholar] [CrossRef]
- Thomas, C.; Gioiello, A.; Noriega, L.; Strehle, A.; Oury, J.; Rizzo, G.; Macchiarulo, A.; Yamamoto, H.; Mataki, C.; Pruzanski, M.; et al. TGR5-mediated bile acid sensing controls glucose homeostasis. Cell Metab. 2009, 10, 167–177. [Google Scholar] [CrossRef] [Green Version]
- Guo, C.; Xie, S.; Chi, Z.; Zhang, J.; Liu, Y.; Zhang, L.; Zheng, M.; Zhang, X.; Xia, D.; Ke, Y.; et al. Bile Acids Control Inflammation and Metabolic Disorder through Inhibition of NLRP3 Inflammasome. Immunity 2016, 45, 802–816. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, W.H.; Wang, Z.; Fan, Y.; Levison, B.; Hazen, J.E.; Donahue, L.M.; Wu, Y.; Hazen, S.L. Prognostic value of elevated levels of intestinal microbe-generated metabolite trimethylamine-N-oxide in patients with heart failure: Refining the gut hypothesis. J. Am. Coll. Cardiol. 2014, 64, 1908–1914. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, W.H.; Wang, Z.; Shrestha, K.; Borowski, A.G.; Wu, Y.; Troughton, R.W.; Klein, A.L.; Hazen, S.L. Intestinal microbiota-dependent phosphatidylcholine metabolites, diastolic dysfunction, and adverse clinical outcomes in chronic systolic heart failure. J. Card. Fail. 2015, 21, 91–96. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bennett, B.J.; Davis, R.C.; Civelek, M.; Orozco, L.; Wu, J.; Qi, H.; Pan, C.; Packard, R.R.; Eskin, E.; Yan, M.; et al. Genetic Architecture of Atherosclerosis in Mice: A Systems Genetics Analysis of Common Inbred Strains. PLoS Genet. 2015, 11, e1005711. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gregory, J.C.; Buffa, J.A.; Org, E.; Wang, Z.; Levison, B.S.; Zhu, W.; Wagner, M.A.; Bennett, B.J.; Li, L.; DiDonato, J.A.; et al. Transmission of atherosclerosis susceptibility with gut microbial transplantation. J. Biol. Chem. 2015, 290, 5647–5660. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seldin, M.M.; Meng, Y.; Qi, H.; Zhu, W.; Wang, Z.; Hazen, S.L.; Lusis, A.J.; Shih, D.M. Trimethylamine N-Oxide Promotes Vascular Inflammation Through Signaling of Mitogen-Activated Protein Kinase and Nuclear Factor-kappaB. J. Am. Heart Assoc. 2016, 5, 2. [Google Scholar] [CrossRef] [Green Version]
- Gao, Z.; Yin, J.; Zhang, J.; Ward, R.E.; Martin, R.J.; Lefevre, M.; Cefalu, W.T.; Ye, J. Butyrate improves insulin sensitivity and increases energy expenditure in mice. Diabetes 2009, 58, 1509–1517. [Google Scholar] [CrossRef] [Green Version]
- Whelton, S.P.; Hyre, A.D.; Pedersen, B.; Yi, Y.; Whelton, P.K.; He, J. Effect of dietary fiber intake on blood pressure: A meta-analysis of randomized, controlled clinical trials. J. Hypertens 2005, 23, 475–481. [Google Scholar] [CrossRef]
- Li, J.; Zhao, F.; Wang, Y.; Chen, J.; Tao, J.; Tian, G.; Wu, S.; Liu, W.; Cui, Q.; Geng, B.; et al. Gut microbiota dysbiosis contributes to the development of hypertension. Microbiome 2017, 5, 14. [Google Scholar] [CrossRef] [Green Version]
- Tye, H.; Yu, C.H.; Simms, L.A.; de Zoete, M.R.; Kim, M.L.; Zakrzewski, M.; Penington, J.S.; Harapas, C.R.; Souza-Fonseca-Guimaraes, F.; Wockner, L.F.; et al. NLRP1 restricts butyrate producing commensals to exacerbate inflammatory bowel disease. Nat. Commun. 2018, 9, 3728. [Google Scholar] [CrossRef] [Green Version]
- Yang, W.; Yu, T.; Huang, X.; Bilotta, A.J.; Xu, L.; Lu, Y.; Sun, J.; Pan, F.; Zhou, J.; Zhang, W.; et al. Intestinal microbiota-derived short-chain fatty acids regulation of immune cell IL-22 production and gut immunity. Nat. Commun. 2020, 11, 4457. [Google Scholar] [CrossRef] [PubMed]
- Woo, A.Y.; Xiao, R.P. beta-Adrenergic receptor subtype signaling in heart: From bench to bedside. Acta Pharmacol. Sin. 2012, 33, 335–341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Gareri, C.; Rockman, H.A. G-Protein-Coupled Receptors in Heart Disease. Circ. Res. 2018, 123, 716–735. [Google Scholar] [CrossRef] [PubMed]
- Ilardi, F.; Gargiulo, G.; Schiattarella, G.G.; Giugliano, G.; Paolillo, R.; Menafra, G.; De Angelis, E.; Scudiero, L.; Franzone, A.; Stabile, E.; et al. Effects of Carvedilol Versus Metoprolol on Platelet Aggregation in Patients With Acute Coronary Syndrome: The PLATE-BLOCK Study. Am. J. Cardiol. 2018, 122, 6–11. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Metabolite | Precursor | Gut Microbial Metabolism | Site of Action | Atherosclerosis Effect | Physiological Action | Mechanism in Atherosclerosis |
|---|---|---|---|---|---|---|
| Bile acid | Primary bile acids [162,163] | Dehydroxylation, deconjugation, dehydrogenation, epimerization, [162,163] | FXR [164] | Undetermined | Increase insulin resistance Increase hepatic and serum triglyceride and cholesterol levels [165,166] | FXR-dependent [167] |
| TGR5 [164] | Improve insulin resistance Increase (extra)hepatic metabolism of VLDL and fatty acid [168,169] | |||||
| TMAO | Choline L-carnitine [24] | TMA-lyase (CutC/D and others) [24,170] | Proatherogenic [26,171,172] | Induce vascular inflammation [24] Induce macrophage foam cell formation [24] Platelet activation [24,173] | Upregulate multiple macrophage scavenger receptors [24] Inhibit macrophage reverse cholesterol transport [25] | |
| SCFA | Dietary fiber [27,174] | Wood-Ljungdahl pathway [27,175] Succinate pathway, propanediol pathway, acrylate pathway [27,175] | Olfr78 [176] Gpr43 [177] | Atheroprotective [178,179] | Decrease blood pressure [176] Body weight reduction [180] Decrease appetite [180] Improve liver steatosis [181] | Induce GLP-1 [180,182] Reduce migration of macrophages into plaques [178] |
| PAGln | Phenylalanine [183] | Pyruvate ferredoxin, oxidoreductase A (PorA) [183,184] | Adrenergic receptors [29] | Proatherogenic [29,30] | Increase platelet reactivity [29,185] | Induce thrombosis [29,185] |
| Reference | Model | Aim of Study | Experiment Design | Main Finding |
|---|---|---|---|---|
| [24] | Human Mouse | Gut flora-dependent metabolism of dietary phosphatidylcholine on CVD pathogenesis | Metabolomics approach in human cohort Choline isotope tracer feeding in mice | Dietary supplementation with choline or TMAO promoted upregulation of macrophage scavenger receptors linked to atherosclerosis, and aggravated atherosclerosis |
| [25] | Human Mouse | Role of gut microbiota on TMAO production from dietary L-carnitine and relationship of TMAO and CVD risk | Metabolomics approach Human/mouse microbiota analyses Isotopic L-carnitine feeding in mice | L-carnitine supplementation significantly altered cecal microbial composition, markedly enhanced synthesis of TMA/TMAO, and increased atherosclerosis |
| [29] | Human Mouse Microorganism | Identifying novel pathways linked to CVD | Metabolomics approach in CVD vs. non-CVD patients In vivo FeCl3-induced thrombosis model | PAGln represents a new CVD-promoting gut microbiota-dependent metabolite that signals via adrenergic receptors |
| [55] | Mouse | Mechanism of HDL promoting regression of atherosclerosis | Aortic transplantation Lipid and Lipoprotein Analyses | HDL as a regulator of the migration and inflammation of monocyte-derived cells in murine atherosclerotic plaques |
| [59] | Mouse | Effect of simvastatin on macrophages and plaque regression | Nanoparticle-based delivery of simvastatin in mice with advanced atherosclerotic plaques | Pharmacologically inhibiting local macrophage proliferation can effectively treat inflammation in atherosclerosis |
| [71] | Human | Feasibility of reducing inflammation to decrease the risk of CVD clinically | Canakinumab 150mg every 3 months, randomized controlled and double blind trial | Antiinflammatory therapy targeting the IL-1β led to a significantly lower rate of recurrent cardiovascular events |
| [79] | Mouse | Role of RegIIIγ on the bacterial colonization of the mucosal surface | RegIIIγ−/− and Myd88−/− vs. wild-type littermate FISH analysis for spatial relationships between the microbiota and the host mucosal surface | RegIIIγ is a fundamental immune mechanism that promotes host-bacterial mutualism by regulating the spatial relationships between microbiota and host |
| [106] | Mouse | Role of IL-23 on atherosclerosis | Bone marrow deletion of IL-23 FMT | The IL-23-IL-22 signaling as a regulator of atherosclerosis that restrains expansion of pro-atherogenic microbiota |
| [134] | Mouse | Impact of microbiota from Casp1−/− mice on atherogenesis in Ldlr−/− mice | FMT from Casp1−/− mice to Ldlr−/− mice following antibiotics treatment | FMT of proinflammatory Casp1−/− microbiota into Ldlr−/− mice enhances systemic inflammation and accelerates atherogenesis |
| [142] | Mouse | Role of Akkermansia muciniphila in the pathogenesis of atherosclerosis | ApoE−/− mice treated with A. muciniphila by daily oral gavage | A. muciniphila attenuates atherosclerotic lesions by ameliorating metabolic endotoxemia-induced inflammation through restoration of the gut barrier |
| [173] | Human Mouse | Role of TMAO on platelet activity and thrombosis | In vivo FeCl3-induced thrombosis model Metagenomic analyses by sequencing 16S ribosomal RNA in cecal microbiota FMT Platelet activity from human samples | Gut microbes, via generation of TMAO, can directly modulate platelet hyperresponsiveness and clot formation rate in vivo |
| [178] | Mouse | Effect of butyrate-producing bacteria on atherosclerosis | FMT of either low or high butyrate-producing human microbiota to GF mice | Colonization with butyrate producing R. intestinalis decreases levels of inflammatory markers and atherosclerosis in a diet-dependent manner |
| [205] | Mouse | Effect of gut microbial transplantation from high to low TMAO-producing mice on atherosclerosis susceptibility | FMT from high to low TMAO-producing mice | Atherosclerosis susceptibility may be transmitted via transplantation of gut microbiota |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yeh, C.-F.; Chen, Y.-H.; Liu, S.-F.; Kao, H.-L.; Wu, M.-S.; Yang, K.-C.; Wu, W.-K. Mutual Interplay of Host Immune System and Gut Microbiota in the Immunopathology of Atherosclerosis. Int. J. Mol. Sci. 2020, 21, 8729. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21228729
Yeh C-F, Chen Y-H, Liu S-F, Kao H-L, Wu M-S, Yang K-C, Wu W-K. Mutual Interplay of Host Immune System and Gut Microbiota in the Immunopathology of Atherosclerosis. International Journal of Molecular Sciences. 2020; 21(22):8729. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21228729
Chicago/Turabian StyleYeh, Chih-Fan, Ying-Hsien Chen, Sheng-Fu Liu, Hsien-Li Kao, Ming-Shiang Wu, Kai-Chien Yang, and Wei-Kai Wu. 2020. "Mutual Interplay of Host Immune System and Gut Microbiota in the Immunopathology of Atherosclerosis" International Journal of Molecular Sciences 21, no. 22: 8729. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21228729