Role of iRhoms 1 and 2 in Endochondral Ossification


Abstract
:1. Introduction
2. Results
2.1. Expansion of the Zone of Hypertrophic Chondrocytes in the Femur Growth Plate of iR2−/−iR1∆Ch Mice
2.2. Long Bone Growth Defects in iR2−/−iR1∆Ch Mice
2.3. Selective Expansion of Von Kossa-Stained Mineralized Hypertrophic Chondrocytes in the Growth Plate of iR2−/−iR1∆Ch Mice
2.4. Analysis of TRAP+ Osteoclasts at the Chondro-Osseous Junction
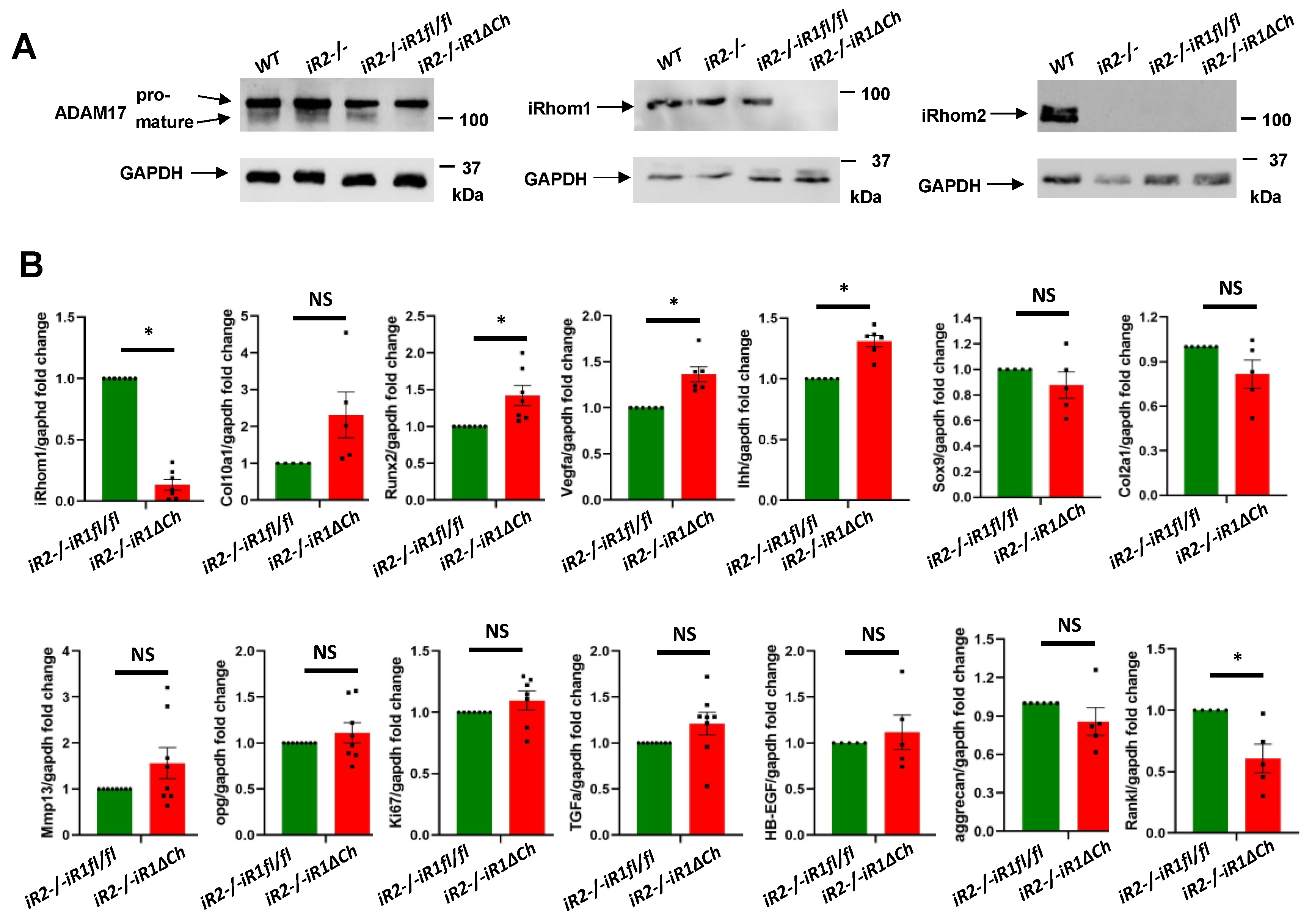
2.5. Western Blot and qPCR Analysis of Cultured Primary Chondrocytes from iR2−/−iR1∆Ch and Control Mice
2.6. Proteolytic Release of EGFR-Ligands from Cultured Primary Chondrocytes from iR2−/−iR1∆Ch and Control Mice
3. Discussion
4. Materials and Methods
4.1. Reagents and Antibodies
4.2. Mouse Strains
4.3. Sample Processing
4.4. Safranin O and Fast Green Staining
4.5. Von Kossa Staining
4.6. TRAP Staining
4.7. Immunohistochemistry
4.8. Faxitron Analysis
4.9. Cell Culture and Protein Ectodomain Shedding Assays
4.10. Western Blot Analysis
4.11. TGFα ELISA
4.12. qRT-PCR Analysis
4.13. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Schneider, M.R.; Sibilia, M.; Erben, R.G. The EGFR network in bone biology and pathology. Trends Endocrinol. Metab. 2009, 20, 517–524. [Google Scholar] [CrossRef] [PubMed]
- Karsenty, G.; Wagner, E.F. Reaching a genetic and molecular understanding of skeletal development. Dev. Cell 2002, 2, 389–406. [Google Scholar] [CrossRef] [Green Version]
- Kronenberg, H.M. Developmental regulation of the growth plate. Nature 2003, 423, 332–336. [Google Scholar] [CrossRef] [PubMed]
- Kozhemyakina, E.; Lassar, A.B.; Zelzer, E. A pathway to bone: Signaling molecules and transcription factors involved in chondrocyte development and maturation. Development 2015, 142, 817–831. [Google Scholar] [CrossRef] [Green Version]
- Harris, R.C.; Chung, E.; Coffey, R.J. EGF receptor ligands. Exp. Cell Res. 2003, 284, 2–13. [Google Scholar] [CrossRef]
- Blobel, C.P. ADAMs: Key players in EGFR-signaling, development and disease. Nat. Rev. Mol. Cell Biol. 2005, 6, 32–43. [Google Scholar] [CrossRef]
- Miettinen, P.J.; Berger, J.E.; Meneses, J.; Phung, Y.; Pedersen, R.A.; Werb, Z.; Derynck, R. Epithelial immaturity and multiorgan failure in mice lacking epidermal growth factor receptor. Nature 1995, 376, 337–341. [Google Scholar] [CrossRef]
- Peschon, J.J.; Slack, J.L.; Reddy, P.; Stocking, K.L.; Sunnarborg, S.W.; Lee, D.C.; Russel, W.E.; Castner, B.J.; Johnson, R.S.; Fitzner, J.N.; et al. An essential role for ectodomain shedding in mammalian development. Science 1998, 282, 1281–1284. [Google Scholar] [CrossRef]
- Sibilia, M.; Wagner, E.F. Strain-dependent epithelial defects in mice lacking the EGF receptor. Science 1995, 269, 234–238, Erratum in 1995, 269, 909. [Google Scholar] [CrossRef]
- Threadgill, D.W.; Dlugosz, A.A.; Hansen, L.A.; Tennenbaum, T.; Lichti, U.; Yee, D.; LaMantia, C.; Mourton, T.; Herrup, K.; Harris, R.C.; et al. Targeted disruption of mouse EGF receptor: Effect of genetic background on mutant phenotype. Science 1995, 269, 230–234. [Google Scholar] [CrossRef]
- Shi, W.; Chen, H.; Sun, J.; Buckley, S.; Zhao, J.; Anderson, K.D.; Williams, R.G.; Warburton, D. TACE is required for fetal murine cardiac development and modeling. Dev. Biol. 2003, 261, 371–380. [Google Scholar] [CrossRef] [Green Version]
- Zhao, J.; Chen, H.; Peschon, J.J.; Shi, W.; Zhang, Y.; Frank, S.J.; Warburton, D. Pulmonary hypoplasia in mice lacking tumor necrosis factor-alpha converting enzyme indicates an indispensable role for cell surface protein shedding during embryonic lung branching morphogenesis. Dev. Biol. 2001, 232, 204–218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Horiuchi, K.; Kimura, T.; Miyamoto, T.; Takaishi, H.; Okada, Y.; Toyama, Y.; Blobel, C.P. Cutting Edge: TNF-α-Converting Enzyme (TACE/ADAM17) Inactivation in Mouse Myeloid Cells Prevents Lethality from Endotoxin Shock. J. Immunol. 2007, 179, 2686–2689. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jackson, L.F.; Qiu, T.H.; Sunnarborg, S.W.; Chang, A.; Zhang, C.; Patterson, C.; Lee, D.C. Defective valvulogenesis in HB-EGF and TACE-null mice is associated with aberrant BMP signaling. EMBO J. 2003, 22, 2704–2716. [Google Scholar] [CrossRef] [Green Version]
- Hall, K.C.; Hill, D.; Otero, M.; Plumb, D.A.; Froemel, D.; Dragomir, C.L.; Maretzky, T.; Boskey, A.; Crawford, H.C.; Selleri, L.; et al. ADAM17 Controls Endochondral Ossification by Regulating Terminal Differentiation of Chondrocytes. Mol. Cell. Biol. 2013, 33, 3077–3090. [Google Scholar] [CrossRef] [Green Version]
- Saito, K.; Horiuchi, K.; Kimura, T.; Mizuno, S.; Yoda, M.; Morioka, H.; Akiyama, H.; Threadgill, D.; Okada, Y.; Toyama, Y.; et al. Conditional Inactivation of TNFalpha-Converting Enzyme in Chondrocytes Results in an Elongated Growth Plate and Shorter Long Bones. PLoS ONE 2013, 8, e54853. [Google Scholar] [CrossRef] [Green Version]
- Usmani, S.E.; Pest, M.A.; Kim, G.; Ohora, S.N.; Qin, L.; Beier, F. Transforming growth factor alpha controls the transition from hypertrophic cartilage to bone during endochondral bone growth. Bone 2012, 51, 131–141. [Google Scholar] [CrossRef]
- Wang, K.; Yamamoto, H.; Chin, J.R.; Werb, Z.; Vu, T.H. Epidermal growth factor receptor-deficient mice have delayed primary endochondral ossification because of defective osteoclast recruitment. J. Biol. Chem. 2004, 279, 53848–53856. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Siclari, V.A.; Lan, S.; Zhu, J.; Koyama, E.; Dupuis, H.L.; Enomoto-Iwamoto, M.; Beier, F.; Qin, L. The critical role of the epidermal growth factor receptor in endochondral ossification. J. Bone Miner. Res. 2011, 26, 2622–2633. [Google Scholar] [CrossRef] [Green Version]
- Qin, L.; Beier, F. EGFR Signaling: Friend or Foe for Cartilage? JBMR Plus 2019, 3, e10177. [Google Scholar] [CrossRef]
- Sibilia, M.; Wagner, B.; Hoebertz, A.; Elliott, C.; Marino, S.; Jochum, W.; Wagner, E.F. Mice humanised for the EGF receptor display hypomorphic phenotypes in skin, bone and heart. Development 2003, 130, 4515–4525. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Linder, M.; Hecking, M.; Glitzner, E.; Zwerina, K.; Holcmann, M.; Bakiri, L.; Ruocco, M.G.; Tuckermann, J.; Schett, G.; Wagner, E.F.; et al. EGFR controls bone development by negatively regulating mTOR-signaling during osteoblast differentiation. Cell Death Differ. 2018, 25, 1094–1106. [Google Scholar] [CrossRef] [PubMed]
- Malemud, C.J. Matrix metalloproteinases: Role in skeletal development and growth plate disorders. Front. Biosci. 2006, 11, 1702–1715. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, J.; Shimizu, E.; Zhang, X.; Partridge, N.C.; Qin, L. EGFR signaling suppresses osteoblast differentiation and inhibits expression of master osteoblastic transcription factors Runx2 and Osterix. J. Cell. Biochem. 2011, 112, 1749–1760. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Tamasi, J.; Lu, X.; Zhu, J.; Chen, H.; Tian, X.; Lee, T.C.; Threadgill, D.W.; Kream, B.E.; Kang, Y.; et al. Epidermal growth factor receptor plays an anabolic role in bone metabolism in vivo. J. Bone Miner. Res. 2011, 26, 1022–1034. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adrain, C.; Zettl, M.; Christova, Y.; Taylor, N.; Freeman, M. Tumor necrosis factor signaling requires iRhom2 to promote trafficking and activation of TACE. Science 2012, 335, 225–228. [Google Scholar] [CrossRef] [Green Version]
- McIlwain, D.R.; Lang, P.A.; Maretzky, T.; Hamada, K.; Ohishi, K.; Maney, S.K.; Berger, T.; Murthy, A.; Duncan, G.; Xu, H.C.; et al. iRhom2 regulation of TACE controls TNF-mediated protection against Listeria and responses to LPS. Science 2012, 335, 229–232. [Google Scholar] [CrossRef] [Green Version]
- Maretzky, T.; McIlwain, D.R.; Issuree, P.D.; Li, X.; Malapeira, J.; Amin, S.; Lang, P.A.; Mak, T.W.; Blobel, C.P. iRhom2 controls the substrate selectivity of stimulated ADAM17-dependent ectodomain shedding. Proc. Natl. Acad. Sci. USA 2013, 110, 11433–11438. [Google Scholar] [CrossRef] [Green Version]
- Issuree, P.D.; Maretzky, T.; McIlwain, D.R.; Monette, S.; Qing, X.; Lang, P.A.; Swendeman, S.L.; Park-Min, K.H.; Binder, N.; Kalliolias, G.D.; et al. iRHOM2 is a critical pathogenic mediator of inflammatory arthritis. J. Clin. Investig. 2013, 123, 928–932. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Maretzky, T.; Weskamp, G.; Monette, S.; Qing, X.; Issuree, P.D.; Crawford, H.C.; McIlwain, D.R.; Mak, T.W.; Salmon, J.E.; et al. iRhoms 1 and 2 are essential upstream regulators of ADAM17-dependent EGFR signaling. Proc. Natl. Acad. Sci. USA 2015, 112, 6080–6085. [Google Scholar] [CrossRef] [Green Version]
- Christova, Y.; Adrain, C.; Bambrough, P.; Ibrahim, A.; Freeman, M. Mammalian iRhoms have distinct physiological functions including an essential role in TACE regulation. EMBO Rep. 2013, 14, 884–890. [Google Scholar] [CrossRef] [PubMed]
- Ovchinnikov, D.A.; Deng, J.M.; Ogunrinu, G.; Behringer, R.R. Col2a1-directed expression of Cre recombinase in differentiating chondrocytes in transgenic mice. Genesis 2000, 26, 145–146. [Google Scholar] [CrossRef]
- Li, X.; Maretzky, T.; Perez-Aguilar, J.M.; Monette, S.; Weskamp, G.; Le Gall, S.; Beutler, B.; Weinstein, H.; Blobel, C.P. Structural modeling defines transmembrane residues in ADAM17 that are crucial for Rhbdf2/ADAM17-dependent proteolysis. J. Cell Sci. 2017, 130, 868–878. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sunnarborg, S.W.; Hinkle, C.L.; Stevenson, M.; Russell, W.E.; Raska, C.S.; Peschon, J.J.; Castner, B.J.; Gerhart, M.J.; Paxton, R.J.; Black, R.A.; et al. Tumor necrosis factor-alpha converting enzyme (TACE) regulates epidermal growth factor receptor ligand availability. J. Biol. Chem. 2002, 277, 12838–12845. [Google Scholar] [CrossRef] [Green Version]
- Sahin, U.; Weskamp, G.; Zhou, H.M.; Higashiyama, S.; Peschon, J.J.; Hartmann, D.; Saftig, P.; Blobel, C.P. Distinct roles for ADAM10 and ADAM17 in ectodomain shedding of six EGFR-ligands. J. Cell Biol. 2004, 164, 769–779. [Google Scholar] [CrossRef] [Green Version]
- Sahin, U.; Blobel, C.P. Ectodomain shedding of the EGF-receptor ligand epigen is mediated by ADAM17. FEBS Lett. 2007, 581, 41–44. [Google Scholar] [CrossRef] [Green Version]
- Le Gall, S.; Bobe, P.; Reiss, K.; Horiuchi, K.; Niu, X.-D.; Lundell, D.; Gibb, D.; Conrad, D.; Saftig, P.; Blobel, C. ADAMs 10 and 17 represent differentially regulated components of a general shedding machinery for membrane proteins such as TGFα, L-Selectin and TNFα. Mol. Biol. Cell 2009, 20, 1785–1794. [Google Scholar] [CrossRef] [Green Version]
- Hall, K.; Blobel, C.P. Interleukin-1 Stimulates ADAM17 Through a Mechanism Independent of its Cytoplasmic Domain or Phosphorylation at Threonine 735. PLoS ONE 2012, 7, e31600. [Google Scholar] [CrossRef] [Green Version]
- Usmani, S.E.; Appleton, C.T.; Beier, F. Transforming growth factor-alpha induces endothelin receptor A expression in osteoarthritis. J. Orthop. Res. 2012, 30, 1391–1397. [Google Scholar] [CrossRef]
- Levy, R.; Levet, C.; Cohen, K.; Freeman, M.; Mott, R.; Iraqi, F.; Gabet, Y. A genome-wide association study in mice reveals a role for Rhbdf2 in skeletal homeostasis. Sci. Rep. 2020, 10, 3286. [Google Scholar] [CrossRef]
- Haxaire, C.; Hakobyan, N.; Pannellini, T.; Carballo, C.; McIlwain, D.; Mak, T.W.; Rodeo, S.; Acharya, S.; Li, D.; Szymonifka, J.; et al. Blood-induced bone loss in murine hemophilic arthropathy is prevented by blocking the iRhom2/ADAM17/TNF-alpha pathway. Blood 2018, 132, 1064–1074. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ono, N.; Ono, W.; Nagasawa, T.; Kronenberg, H.M. A subset of chondrogenic cells provides early mesenchymal progenitors in growing bones. Nat. Cell Biol. 2014, 16, 1157–1167. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Tsang, K.Y.; Tang, H.C.; Chan, D.; Cheah, K.S. Hypertrophic chondrocytes can become osteoblasts and osteocytes in endochondral bone formation. Proc. Natl. Acad. Sci. USA 2014, 111, 12097–12102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, X.; von der Mark, K.; Henry, S.; Norton, W.; Adams, H.; de Crombrugghe, B. Chondrocytes transdifferentiate into osteoblasts in endochondral bone during development, postnatal growth and fracture healing in mice. PLoS Genet. 2014, 10, e1004820. [Google Scholar] [CrossRef] [Green Version]
- Schlöndorff, J.; Becherer, J.D.; Blobel, C.P. Intracellular maturation and localization of the tumour necrosis factor alpha convertase (TACE). Biochem. J. 2000, 347 Pt 1, 131–138. [Google Scholar] [CrossRef]
- Weskamp, G.; Tushaus, J.; Li, D.; Feederle, R.; Maretzky, T.; Swendeman, S.; Falck-Pedersen, E.; McIlwain, D.R.; Mak, T.W.; Salmon, J.E.; et al. ADAM17 stabilizes its interacting partner inactive Rhomboid 2 (iRhom2) but not inactive Rhomboid 1 (iRhom1). J. Biol. Chem. 2020, 295, 4350–4358. [Google Scholar] [CrossRef] [Green Version]
- Gosset, M.; Berenbaum, F.; Thirion, S.; Jacques, C. Primary culture and phenotyping of murine chondrocytes. Nat. Protoc. 2008, 3, 1253–1260. [Google Scholar] [CrossRef]
- Olivotto, E.; Otero, M.; Astolfi, A.; Platano, D.; Facchini, A.; Pagani, S.; Flamigni, F.; Facchini, A.; Goldring, M.B.; Borzi, R.M.; et al. IKKalpha/CHUK regulates extracellular matrix remodeling independent of its kinase activity to facilitate articular chondrocyte differentiation. PLoS ONE 2013, 8, e73024. [Google Scholar] [CrossRef]
- Singh, P.; Lessard, S.G.; Mukherjee, P.; Rourke, B.; Otero, M. Changes in DNA methylation accompany changes in gene expression during chondrocyte hypertrophic differentiation in vitro. Ann. N. Y. Acad. Sci. 2020. [Google Scholar] [CrossRef]
- Sahin, U.; Weskamp, G.; Zheng, Y.; Chesneau, V.; Horiuchi, K.; Blobel, C.P. A sensitive method to monitor ectodomain shedding of ligands of the epidermal growth factor receptor. In Epidermal Growth Factor: Methods and Protocols; Patel, T.B., Bertics, P.J., Eds.; Humana Press Inc.: Totowa, NJ, USA, 2006; Volume 327, pp. 99–113. [Google Scholar]
- Horiuchi, K.; Le Gall, S.; Schulte, M.; Yamaguchi, T.; Reiss, K.; Murphy, G.; Toyama, Y.; Hartmann, D.; Saftig, P.; Blobel, C. Substrate Selectivity of EGF-Receptor Ligand Sheddases and Their Regulation by Phorbol Esters and Calcium Influx. Mol. Biol. Cell 2007, 18, 176–188. [Google Scholar] [CrossRef] [Green Version]
- Franzke, C.W.; Cobzaru, C.; Triantafyllopoulou, A.; Loffek, S.; Horiuchi, K.; Threadgill, D.W.; Kurz, T.; van Rooijen, N.; Bruckner-Tuderman, L.; Blobel, C.P. Epidermal ADAM17 maintains the skin barrier by regulating EGFR ligand-dependent terminal keratinocyte differentiation. J. Exp. Med. 2012, 209, 1105–1119. [Google Scholar] [CrossRef] [PubMed] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Primers Sequence for qPCR Amplification | Direction | NT |
|---|---|---|---|
| iRhom1 | 5′-TCC ACG TAC CCA GAT GAG GTG-3′ | Forward | 21 |
| 5′-GCC TTC CTT CTG CTT TCT CCA-3′ | Reverse | 21 | |
| Col10a1 | 5′-ACG CAT CTC CCA GCA CCA GAA TC-3′ | Forward | 23 |
| 5′-GGG GCT AGC AAG TGG GCC CT-3′ | Reverse | 20 | |
| Runx2 | 5′-TCC CCG GGA ACC AAG AAG GCA-3′ | Forward | 21 |
| 5′-AGG GAG GGC CGT GGG TTC TG-3′ | Reverse | 20 | |
| Vegfα | 5′-CTC GCA GTC CGA GCC GGA GA-3′ | Forward | 20 |
| 5′-GCA GCC TGG GAC CAC TTG GC-3′ | Reverse | 20 | |
| Ihh | 5′-GTC AAG TCT GAG CAT TCG GC-3′ | Forward | 20 |
| 5′-CAT CAC TGA AGG TGG GGG TC-3′ | Reverse | 20 | |
| Mmp13 | 5′-ATG GTC CAG GCG ATG AAG ACC CC-3′ | Forward | 23 |
| 5′-GTG CAG GCG CCA GAA GAA TCT GT-3′ | Reverse | 23 | |
| Tnfrsf11b | 5′-ACA GTT TGC CTG GGA CCA AA-3′ | Forward | 20 |
| (OPG) | 5′-TCA CAG AGG TCA ATG TCT TGG A-3′ | Reverse | 22 |
| Rankl | 5′-CGA GCG CAG ATG GAT CCT AA-3′ | Forward | 20 |
| 5′-CCC CCT GAA AGG CTT GTT TC-3′ | Reverse | 20 | |
| TGFα | 5′-CTC TGG GTA CGT GGG TGT TC-3 | Forward | 20 |
| 5′-CTG ACA GCA GTG GAT CAG CA-3′ | Reverse | 20 | |
| Hb-egf | 5′-CCT CTT GCA AAT GCC TCC CT-3′ | Forward | 20 |
| 5′-ACA AGA AGA CAG ACG GAC GAC-3′ | Reverse | 21 | |
| Gapdh | 5′-GGG CTC ATG ACC ACA GTC CAT GC-3′ | Forward | 23 |
| 5′-CCT TGC CCA CAG CCT TGG CA-3′ | Reverse | 20 | |
| Sox9 | 5′-AAG CTC TGG AGG CTG CTG AAC GAG-3′ | Forward | 24 |
| 5′-CGG CCT CCG CTT GTC CGT TCT-3′ | Reverse | 21 | |
| Col2a1 | 5′-CGA TCA CAG AAG ACC TCC CG-3′ | Forward | 20 |
| 5′-GCG GTT GCA AAG TGT TTG GC-3′ | Reverse | 20 | |
| Acan | 5′-GGT CAC TGT TAC CGC CAC TT-3′ | Forward | 20 |
| 5′-CCC CTT CGA TAG TCC TGT CA-3′ | Reverse | 20 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fang, R.; Haxaire, C.; Otero, M.; Lessard, S.; Weskamp, G.; McIlwain, D.R.; Mak, T.W.; Lichtenthaler, S.F.; Blobel, C.P. Role of iRhoms 1 and 2 in Endochondral Ossification. Int. J. Mol. Sci. 2020, 21, 8732. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21228732
Fang R, Haxaire C, Otero M, Lessard S, Weskamp G, McIlwain DR, Mak TW, Lichtenthaler SF, Blobel CP. Role of iRhoms 1 and 2 in Endochondral Ossification. International Journal of Molecular Sciences. 2020; 21(22):8732. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21228732
Chicago/Turabian StyleFang, Renpeng, Coline Haxaire, Miguel Otero, Samantha Lessard, Gisela Weskamp, David R. McIlwain, Tak W. Mak, Stefan F. Lichtenthaler, and Carl P. Blobel. 2020. "Role of iRhoms 1 and 2 in Endochondral Ossification" International Journal of Molecular Sciences 21, no. 22: 8732. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21228732