Phylostratic Shift of Whole-Genome Duplications in Normal Mammalian Tissues towards Unicellularity Is Driven by Developmental Bivalent Genes and Reveals a Link to Cancer

 , , and
, , and
Abstract
:
1. Introduction
2. Results
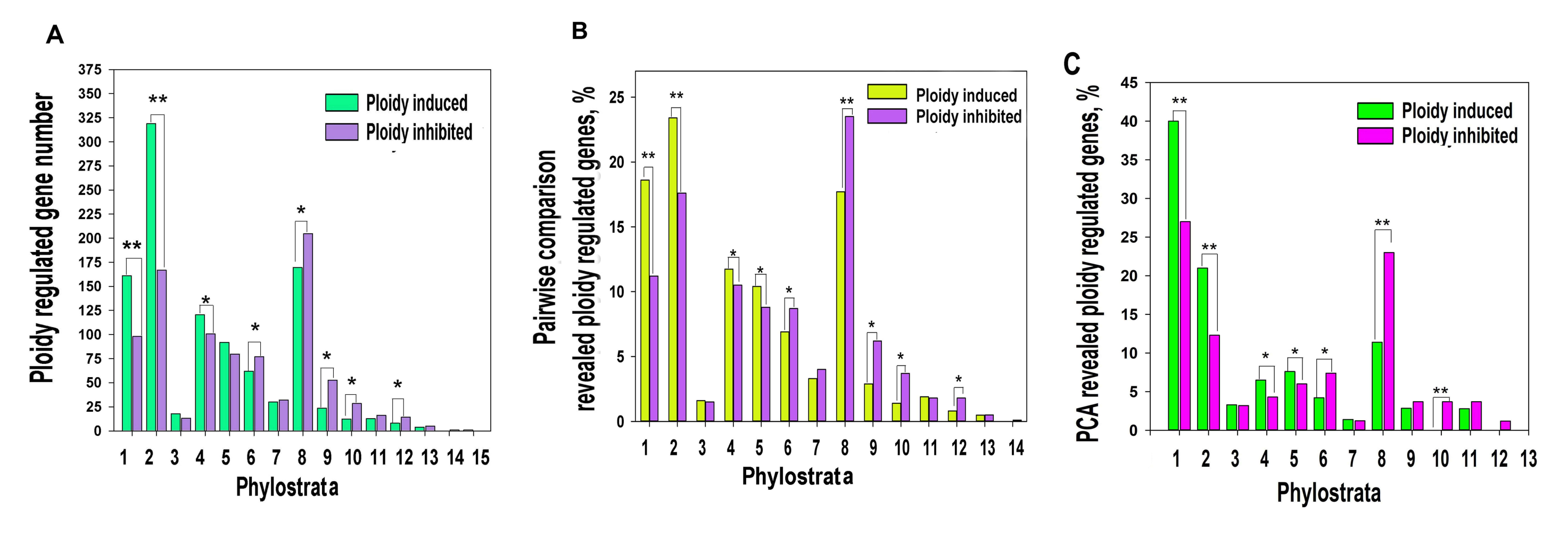
2.1. Polyploidy Causes Transition of Gene Phylostratic Balance towards Evolutionary Old Unicellular Phylostrata
2.2. Polyploidy Activates Recapitulation of Evolutionary Developmental Programs Associated with Carcinogenesis
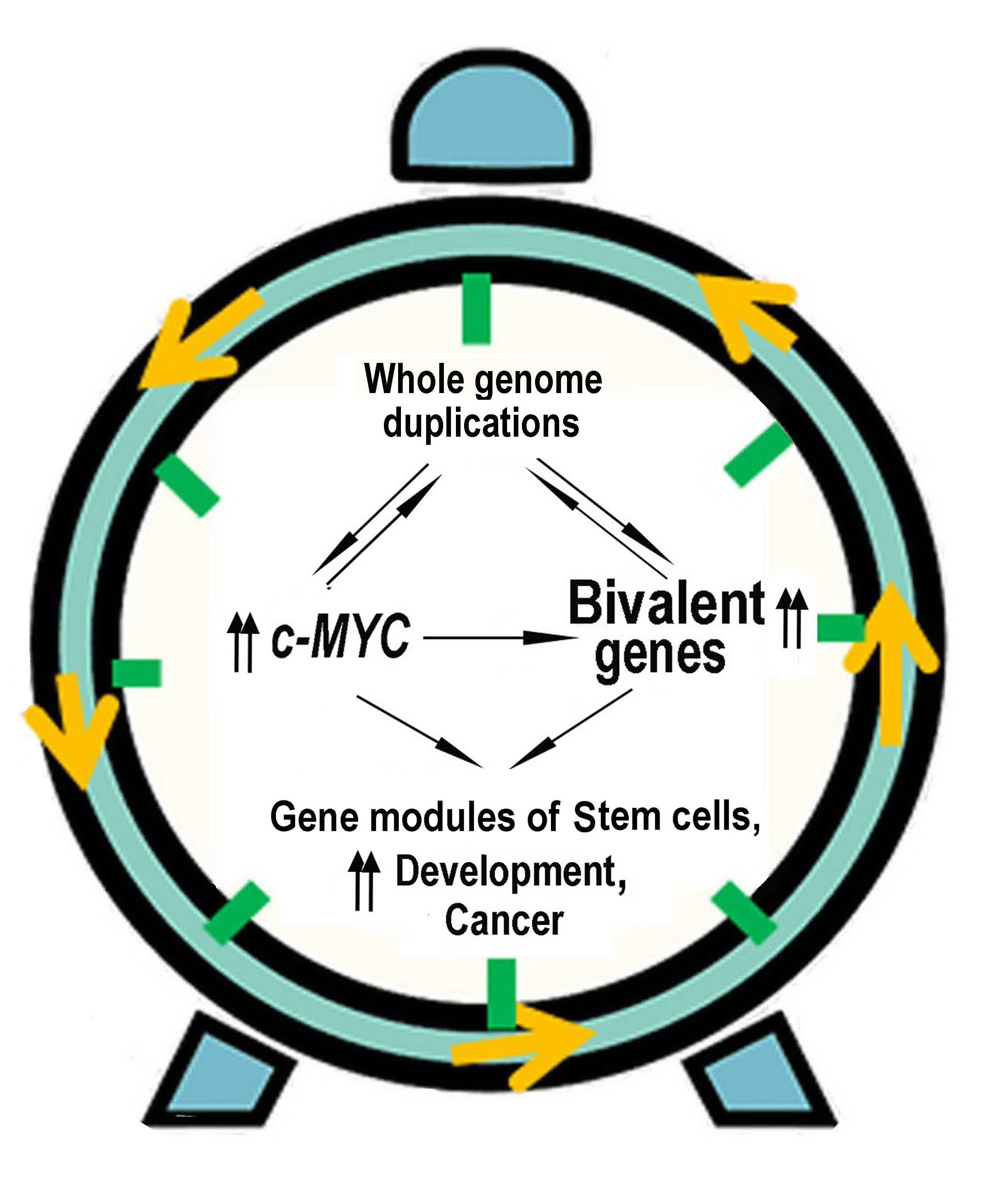
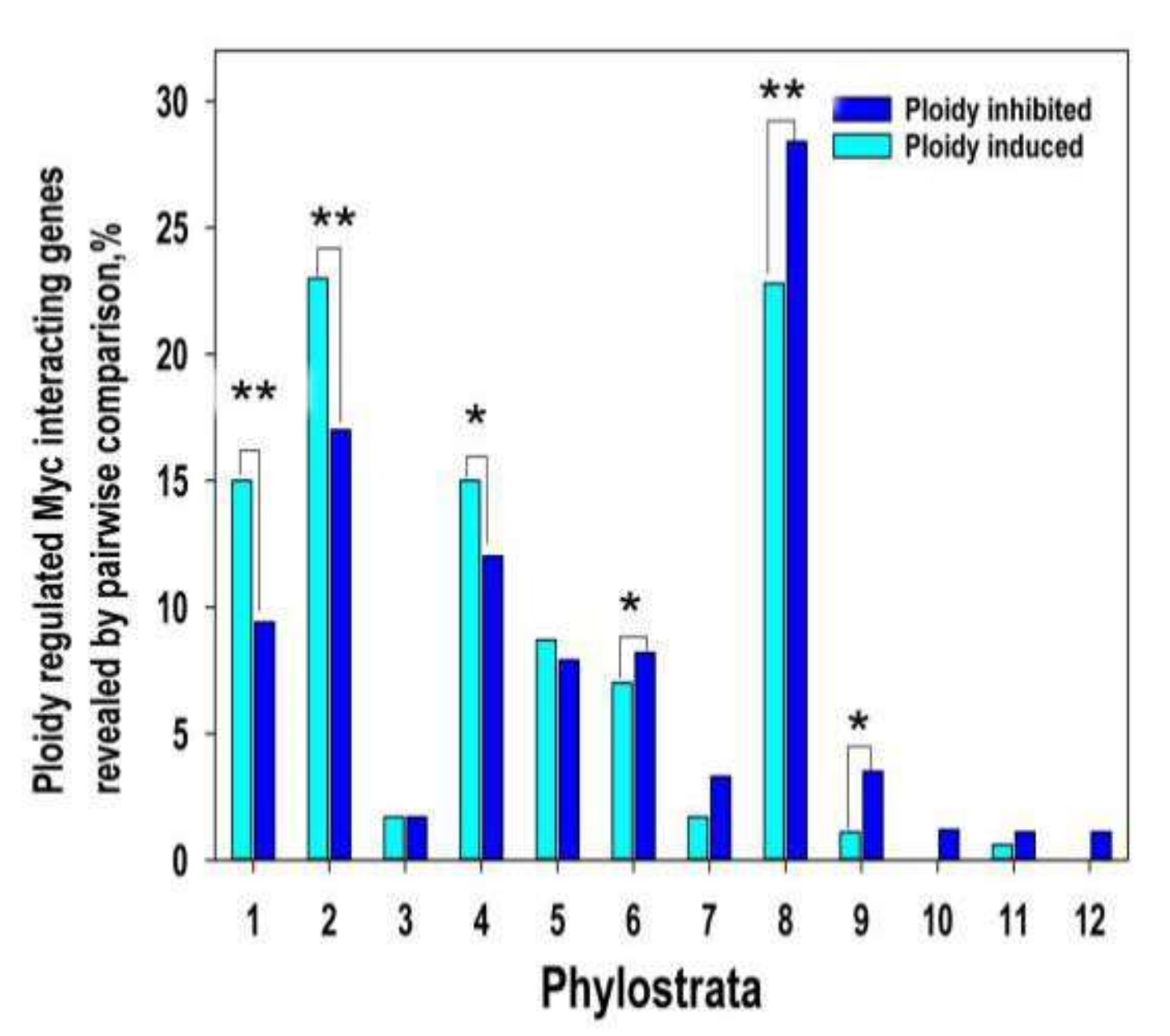
2.3. c-MYC Induction Drives Polyploidy-Associated Transcriptomic Changes towards Unicellularity
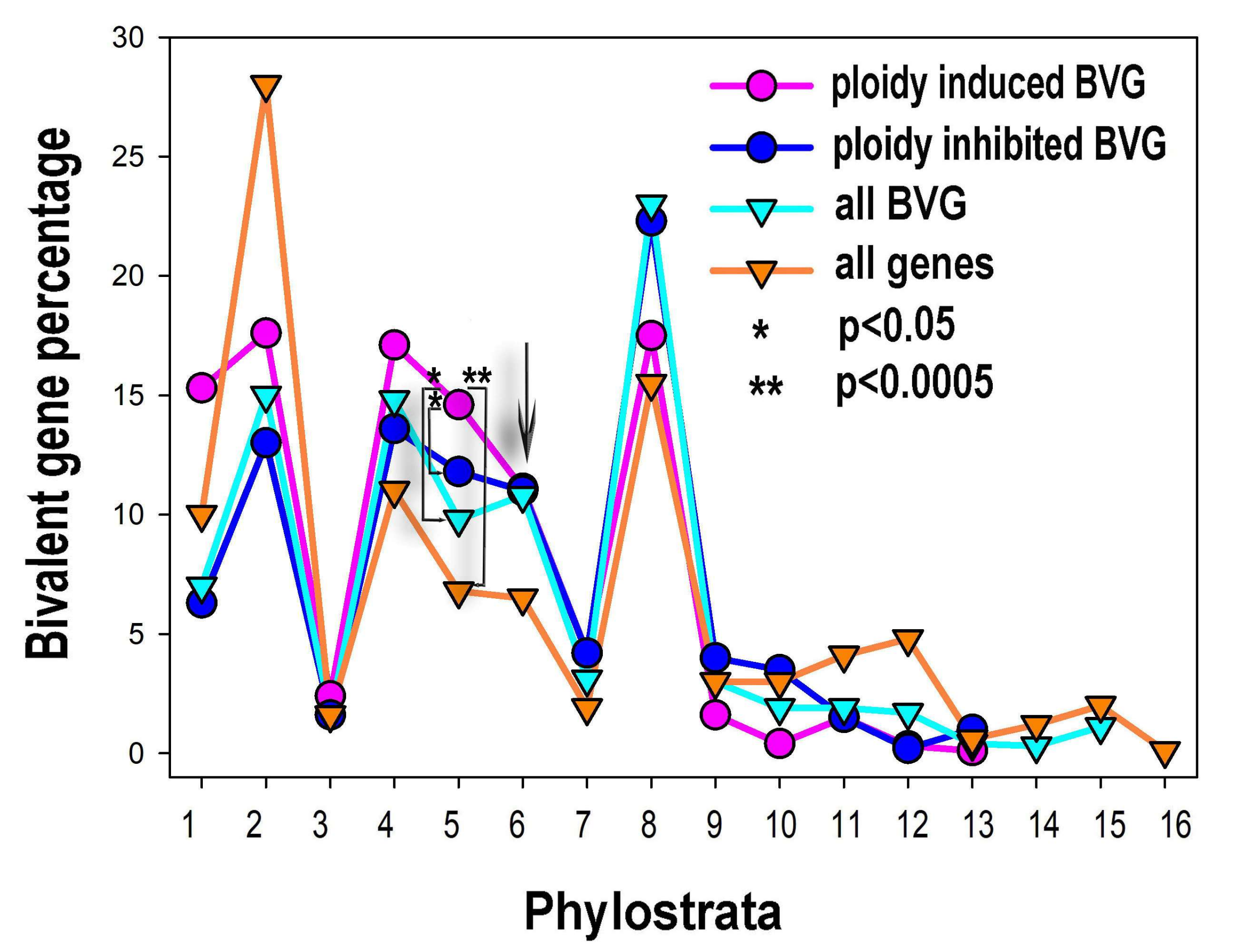
2.4. The Phylostratigraphic Polyploidy-Associated Effect of c-MYC Is Associated with the Regulation of Bivalent Genes
2.5. Ploidy-Associated Genes Are Enriched in Bivalent Genes of the Entire Genome, Prevailing among Unicellular and Eumetazoan Phylostrata.
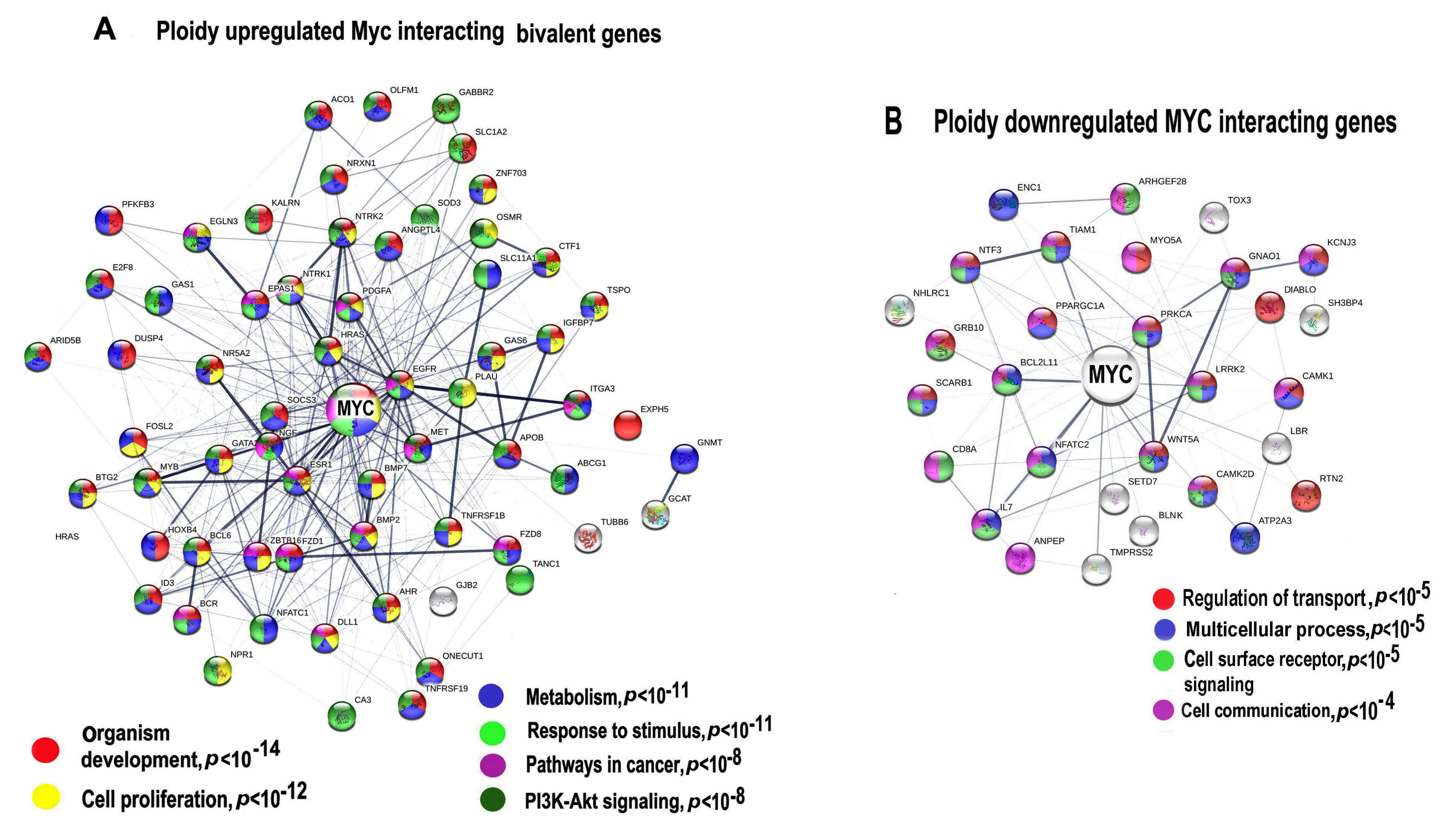
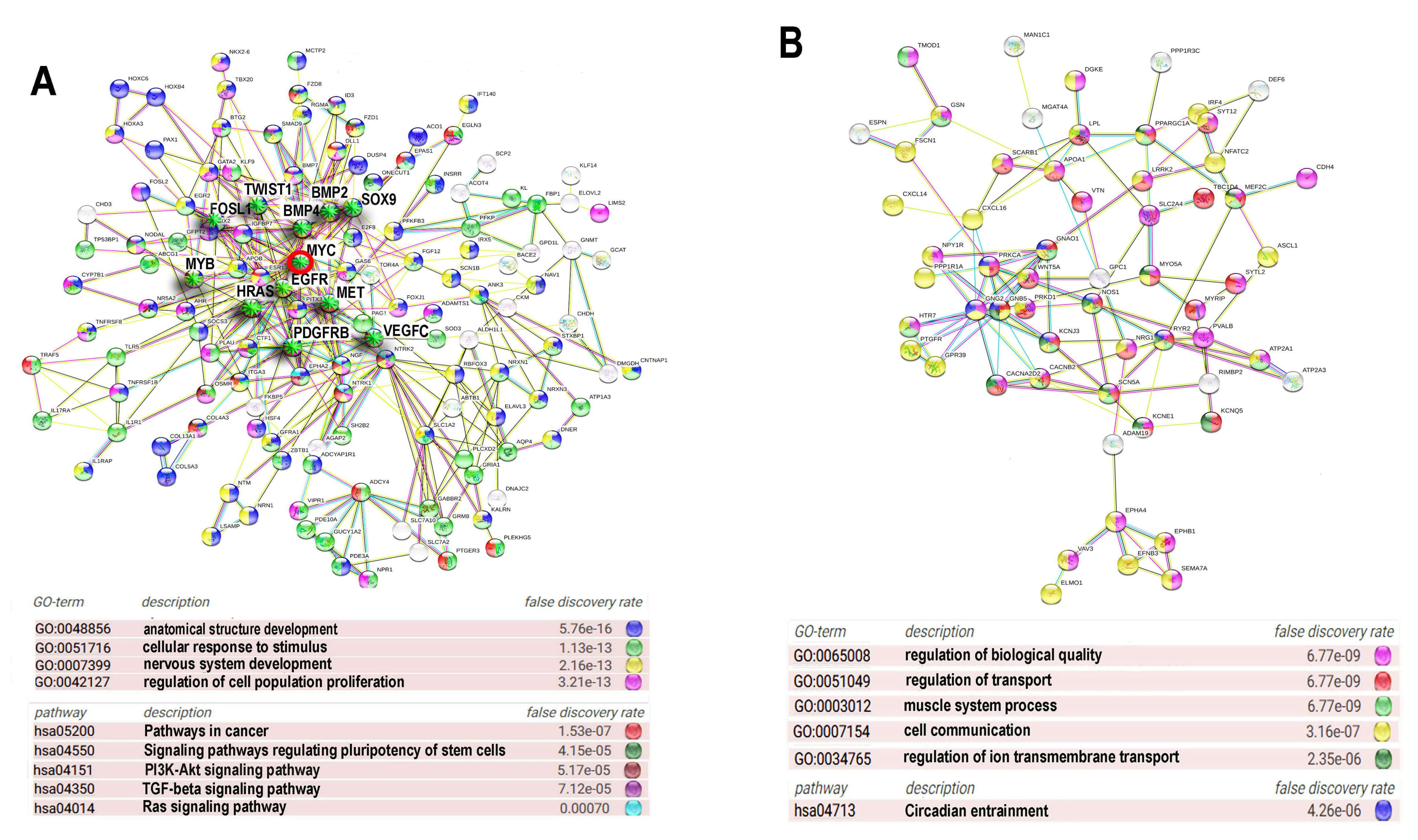
2.6. Protein Interaction Networks for Ploidy-Associated Bivalent Genes Are Involved in the Upregulation of the Developmental and Carcinogenesis Genes and the Downregulation of the Networks Related to Differentiation Biological Quality and Circadian Clock
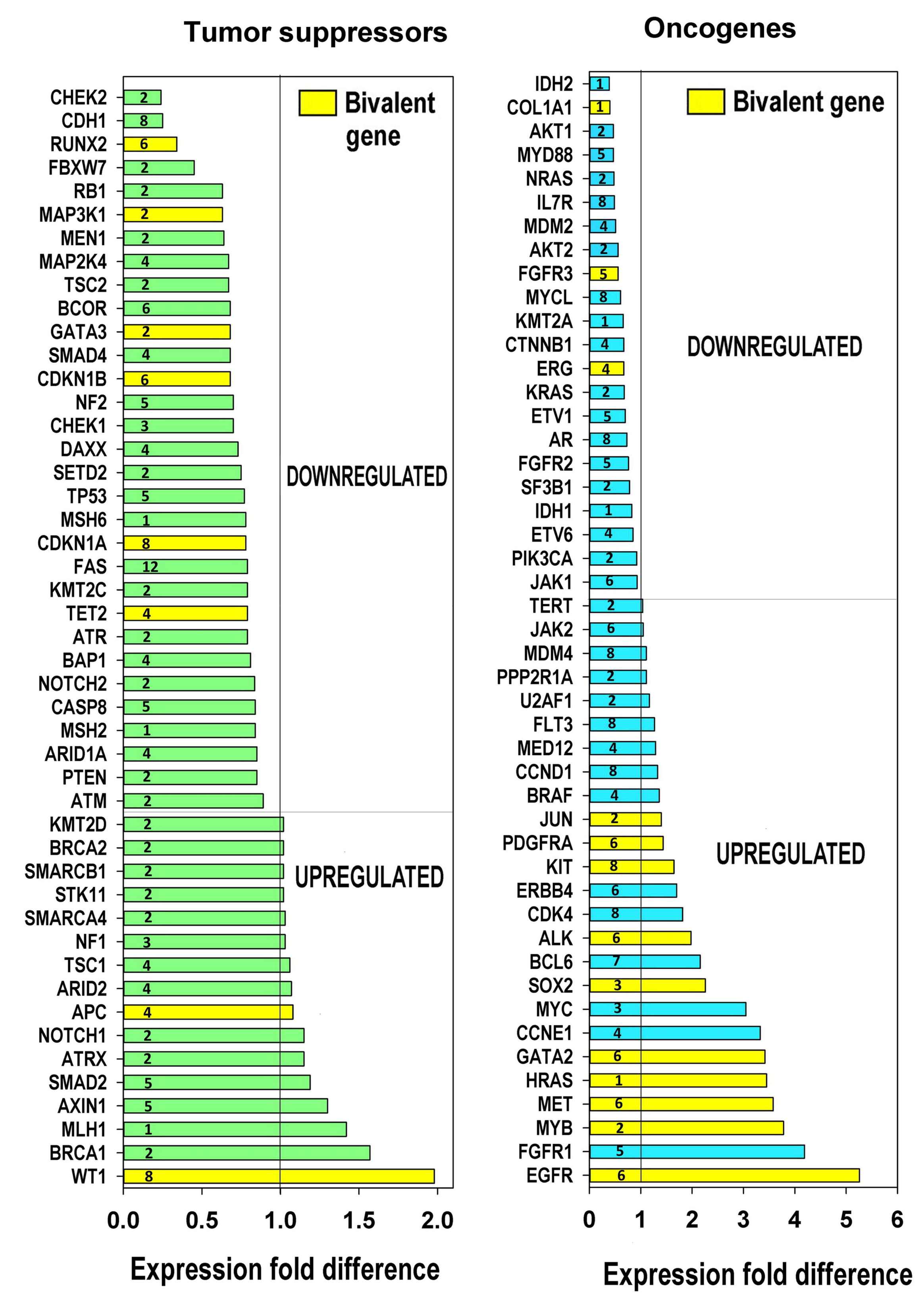
2.7. The Relation of Cancer Driver Genes to Polyploidy, Bivalency and Their Phylostratigraphic Origin
3. Discussion
4. Methods
4.1. Data Sources and Comparative Criss-Cross Analysis
4.2. Principal Component Analysis
4.3. Analysis of Gene Modules
4.4. Protein-Protein Interaction Network
4.5. Phylostratigraphic Analysis of Ploidy Associated Genes
4.6. Identification of Ploidy Associated Bivalent Gene
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Comai, L. The advantages and disadvantages of being polyploid. Nat. Rev. Genet. 2005, 6, 836–846. [Google Scholar] [CrossRef] [PubMed]
- Fox, D.T.; Soltis, D.E.; Soltis, P.S.; Ashman, T.-L.; Van de Peer, Y. Polyploidy: A Biological Force from Cells to Ecosystems. Trends Cell Biol. 2020, 30, 688–694. [Google Scholar] [CrossRef] [PubMed]
- Avdeyev, P.; Alexeev, N.; Rong, Y.; Alekseyev, M.A. A unified ILP framework for core ancestral genome reconstruction problems. Bioinformatics 2020, 36, 2993–3003. [Google Scholar] [CrossRef] [PubMed]
- Anatskaya, O.V.; Vinogradov, A.E. Myocyte ploidy in heart chambers of birds with different locomotor activity. J. Exp. Zool. 2002, 293, 427–441. [Google Scholar] [CrossRef] [PubMed]
- Anatskaya, O.V.; Vinogradov, A.E. Heart and liver as developmental bottlenecks of mammal design: Evidence from cell polyploidization. Biol. J. Linn. Soc. 2004, 83, 175–186. [Google Scholar] [CrossRef]
- Sher, N.; Von Stetina, J.R.; Bell, G.W.; Matsuura, S.; Ravid, K.; Orr-Weaver, T.L. Fundamental differences in endoreplication in mammals and Drosophila revealed by analysis of endocycling and endomitotic cells. Proc. Natl. Acad. Sci. USA 2013, 110, 9368–9373. [Google Scholar] [CrossRef] [Green Version]
- Orr-Weaver, T.L. When bigger is better: The role of polyploidy in organogenesis. Trends Genet. 2015, 31, 307–315. [Google Scholar] [CrossRef] [Green Version]
- Lazzeri, E.; Angelotti, M.L.; Conte, C.; Anders, H.-J.; Romagnani, P. Surviving Acute Organ Failure: Cell Polyploidization and Progenitor Proliferation. Trends Mol. Med. 2019, 25, 366–381. [Google Scholar] [CrossRef] [Green Version]
- Derks, W.; Bergmann, O. Polyploidy in Cardiomyocytes: Roadblock to Heart Regeneration? Circ. Res. 2020, 126, 552–565. [Google Scholar] [CrossRef]
- Gan, P.; Patterson, M.; Sucov, H.M. Cardiomyocyte Polyploidy and Implications for Heart Regeneration. Annu. Rev. Physiol. 2020, 82, 45–61. [Google Scholar] [CrossRef] [Green Version]
- Anatskaya, O.V.; Vinogradov, A.E.; Kudryavtsev, B.N. Cardiomyocyte ploidy levels in birds with different growth rates. J. Exp. Zool. 2001, 289, 48–58. [Google Scholar] [CrossRef]
- Anatskaya, O.V.; Vinogradov, A.E. Genome multiplication as adaptation to tissue survival: Evidence from gene expression in mammalian heart and liver. Genomics 2007, 89, 70–80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Silva, I.S.; Ghiraldini, F.G.; Veronezi, G.M.B.; Mello, M.L.S. Polyploidy and nuclear phenotype characteristics of cardiomyocytes from diabetic adult and normoglycemic aged mice. Acta Histochem. 2018, 120, 84–94. [Google Scholar] [CrossRef] [PubMed]
- Bensley, J.G.; Stacy, V.K.; De Matteo, R.; Harding, R.; Black, M.J. Cardiac remodelling as a result of pre-term birth: Implications for future cardiovascular disease. Eur. Heart J. 2010, 31, 2058–2066. [Google Scholar] [CrossRef]
- Gjelsvik, K.J.; Besen-McNally, R.; Losick, V.P. Solving the Polyploid Mystery in Health and Disease. Trends Genet. 2019, 35, 6–14. [Google Scholar] [CrossRef]
- Donne, R.; Saroul-Aïnama, M.; Cordier, P.; Celton-Morizur, S.; Desdouets, C. Polyploidy in liver development, homeostasis and disease. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 391–405. [Google Scholar] [CrossRef]
- Storchova, Z.; Pellman, D. From polyploidy to aneuploidy, genome instability and cancer. Nat. Rev. Mol. Cell Biol. 2004, 5, 45–54. [Google Scholar] [CrossRef]
- Coward, J.; Harding, A. Size Does Matter: Why Polyploid Tumor Cells are Critical Drug Targets in the War on Cancer. Front. Oncol. 2014, 4, 123. [Google Scholar] [CrossRef]
- Ben-Porath, I.; Thomson, M.W.; Carey, V.J.; Ge, R.; Bell, G.W.; Regev, A.; Weinberg, R.A. An embryonic stem cell-like gene expression signature in poorly differentiated aggressive human tumors. Nat. Genet. 2008, 40, 499–507. [Google Scholar] [CrossRef]
- Salmina, K.; Jankevics, E.; Huna, A.; Perminov, D.; Radovica, I.; Klymenko, T.; Ivanov, A.; Jascenko, E.; Scherthan, H.; Cragg, M.; et al. Up-regulation of the embryonic self-renewal network through reversible polyploidy in irradiated p53-mutant tumour cells. Exp. Cell Res. 2010, 316, 2099–2112. [Google Scholar] [CrossRef]
- Nguyen, H.G.; Ravid, K. Polyploidy: Mechanisms and Cancer Promotion in Hematopoietic and Other Cells. In Polyploidization and Cancer; Poon, R.Y.C., Ed.; Advances in Experimental Medicine and Biology; Springer: New York, NY, USA, 2010; Volume 676, pp. 105–122. ISBN 978-1-4419-6198-3. [Google Scholar]
- Lagadec, C.; Vlashi, E.; Della Donna, L.; Dekmezian, C.; Pajonk, F. Radiation-Induced Reprogramming of Breast Cancer Cells: Radiation-Induced Cancer Stem Cells. Stem Cells 2012, 30, 833–844. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Erenpreisa, J.; Cragg, M.S. Cancer: A matter of life cycle? Cell Biol. Int. 2007, 31, 1507–1510. [Google Scholar] [CrossRef] [PubMed]
- Erenpreisa, J.; Cragg, M.S. Life-Cycle Features of Tumour Cells. In Evolutionary Biology from Concept to Application; Pontarotti, P., Ed.; Springer: Berlin/Heidelberg, Germany, 2008; pp. 61–71. ISBN 978-3-540-78992-5. [Google Scholar]
- Mirzayans, R.; Andrais, B.; Scott, A.; Wang, Y.; Kumar, P.; Murray, D. Multinucleated Giant Cancer Cells Produced in Response to Ionizing Radiation Retain Viability and Replicate Their Genome. Int. J. Mol. Sci. 2017, 18, 360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, J. The dualistic origin of human tumors. Semin. Cancer Biol. 2018, 53, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Salmina, K.; Bojko, A.; Inashkina, I.; Staniak, K.; Dudkowska, M.; Podlesniy, P.; Rumnieks, F.; Vainshelbaum, N.M.; Pjanova, D.; Sikora, E.; et al. “Mitotic Slippage” and Extranuclear DNA in Cancer Chemoresistance: A Focus on Telomeres. Int. J. Mol. Sci. 2020, 21, 2779. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mirzayans, R.; Andrais, B.; Murray, D. Roles of Polyploid/Multinucleated Giant Cancer Cells in Metastasis and Disease Relapse Following Anticancer Treatment. Cancers 2018, 10, 118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vincent, M.D. Cancer: Beyond Speciation. In Advances in Cancer Research; Elsevier: Amsterdam, The Netherlands, 2011; Volume 112, pp. 283–350. ISBN 978-0-12-387688-1. [Google Scholar]
- Davies, P.C.W.; Lineweaver, C.H. Cancer tumors as Metazoa 1.0: Tapping genes of ancient ancestors. Phys. Biol. 2011, 8, 015001. [Google Scholar] [CrossRef] [PubMed]
- Davies, P. Exposing cancer’s deep evolutionary roots. Phys. Cancer Phys. World 2013, 26, 37–40. [Google Scholar] [CrossRef]
- Aktipis, C.A.; Boddy, A.M.; Jansen, G.; Hibner, U.; Hochberg, M.E.; Maley, C.C.; Wilkinson, G.S. Cancer across the tree of life: Cooperation and cheating in multicellularity. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2015, 370. [Google Scholar] [CrossRef] [Green Version]
- Niculescu, V.F. Developmental and Non Developmental Polyploidy in Xenic and Axenic Cultured Stem Cell Lines of Entamoeba invadens and E. histolytica. Insights Stem Cells 2016, 2, 1–9. [Google Scholar]
- Vinogradov, A.E. Human transcriptome nexuses: Basic-eukaryotic and metazoan. Genomics 2010, 95, 345–354. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vinogradov, A.E.; Anatskaya, O.V. Evolutionary framework of the human interactome: Unicellular and multicellular giant clusters. Biosystems 2019, 181, 82–87. [Google Scholar] [CrossRef] [PubMed]
- Domazet-Lošo, T.; Tautz, D. Phylostratigraphic tracking of cancer genes suggests a link to the emergence of multicellularity in metazoa. BMC Biol. 2010, 8, 66. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gu, X.; Zou, Z.; Yang, J. Tracing Evolutionary Ages of Cancer-Driving Sites by Cancer Somatic Mutations. bioRxiv 2020. [Google Scholar] [CrossRef] [Green Version]
- Trigos, A.S.; Pearson, R.B.; Papenfuss, A.T.; Goode, D.L. Altered interactions between unicellular and multicellular genes drive hallmarks of transformation in a diverse range of solid tumors. Proc. Natl. Acad. Sci. USA 2017, 114, 6406–6411. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bussey, K.J.; Cisneros, L.H.; Lineweaver, C.H.; Davies, P.C.W. Ancestral gene regulatory networks drive cancer. Proc. Natl. Acad. Sci. USA 2017, 114, 6160–6162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trigos, A.S.; Pearson, R.B.; Papenfuss, A.T.; Goode, D.L. How the evolution of multicellularity set the stage for cancer. Br. J. Cancer 2018, 118, 145–152. [Google Scholar] [CrossRef] [PubMed]
- Vinogradov, A.E.; Anatskaya, O.V. Cell-cycle dependence of transcriptome gene modules: Comparison of regression lines. FEBS J. 2020, 287, 4427–4439. [Google Scholar] [CrossRef]
- Bizzarri, M.; Giuliani, A.; Cucina, A.; D’Anselmi, F.; Soto, A.M.; Sonnenschein, C. Fractal analysis in a systems biology approach to cancer. Semin. Cancer Biol. 2011, 21, 175–182. [Google Scholar] [CrossRef] [Green Version]
- Bizzarri, M.; Giuliani, A.; Minini, M.; Monti, N.; Cucina, A. Constraints Shape Cell Function and Morphology by Canalizing the Developmental Path along the Waddington’s Landscape. Bioessays 2020, 42, e1900108. [Google Scholar] [CrossRef]
- Erenpreisa, J.; Cragg, M.S. Three steps to the immortality of cancer cells: Senescence, polyploidy and self-renewal. Cancer Cell Int. 2013, 13, 92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van de Peer, Y.; Mizrachi, E.; Marchal, K. The evolutionary significance of polyploidy. Nat. Rev. Genet. 2017, 18, 411–424. [Google Scholar] [CrossRef] [PubMed]
- Moein, S.; Adibi, R.; da Silva Meirelles, L.; Nardi, N.B.; Gheisari, Y. Cancer regeneration: Polyploid cells are the key drivers of tumor progression. Biochim. Biophys. Acta (BBA) Rev. Cancer 2020, 188408. [Google Scholar] [CrossRef] [PubMed]
- Mirzayans, R.; Murray, D. Intratumor Heterogeneity and Therapy Resistance: Contributions of Dormancy, Apoptosis Reversal (Anastasis) and Cell Fusion to Disease Recurrence. Int. J. Mol. Sci. 2020, 21, 1308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bernstein, B.E.; Mikkelsen, T.S.; Xie, X.; Kamal, M.; Huebert, D.J.; Cuff, J.; Fry, B.; Meissner, A.; Wernig, M.; Plath, K.; et al. A bivalent chromatin structure marks key developmental genes in embryonic stem cells. Cell 2006, 125, 315–326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mikkelsen, T.S.; Ku, M.; Jaffe, D.B.; Issac, B.; Lieberman, E.; Giannoukos, G.; Alvarez, P.; Brockman, W.; Kim, T.-K.; Koche, R.P.; et al. Genome-wide maps of chromatin state in pluripotent and lineage-committed cells. Nature 2007, 448, 553–560. [Google Scholar] [CrossRef]
- Widschwendter, M.; Fiegl, H.; Egle, D.; Mueller-Holzner, E.; Spizzo, G.; Marth, C.; Weisenberger, D.J.; Campan, M.; Young, J.; Jacobs, I.; et al. Epigenetic stem cell signature in cancer. Nat. Genet. 2007, 39, 157–158. [Google Scholar] [CrossRef]
- Balch, C.; Nephew, K.P.; Huang, T.H.-M.; Bapat, S.A. Epigenetic “bivalently marked” process of cancer stem cell-driven tumorigenesis. Bioessays 2007, 29, 842–845. [Google Scholar] [CrossRef]
- Blanco, E.; González-Ramírez, M.; Alcaine-Colet, A.; Aranda, S.; Di Croce, L. The Bivalent Genome: Characterization, Structure, and Regulation. Trends Genet. 2020, 36, 118–131. [Google Scholar] [CrossRef]
- Bernhart, S.H.; Kretzmer, H.; Holdt, L.M.; Jühling, F.; Ammerpohl, O.; Bergmann, A.K.; Northoff, B.H.; Doose, G.; Siebert, R.; Stadler, P.F.; et al. Changes of bivalent chromatin coincide with increased expression of developmental genes in cancer. Sci. Rep. 2016, 6, 37393. [Google Scholar] [CrossRef]
- Court, F.; Arnaud, P. An annotated list of bivalent chromatin regions in human ES cells: A new tool for cancer epigenetic research. Oncotarget 2017, 8, 4110–4124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuroda, M.I.; Kang, H.; De, S.; Kassis, J.A. Dynamic Competition of Polycomb and Trithorax in Transcriptional Programming. Annu. Rev. Biochem. 2020, 89, 235–253. [Google Scholar] [CrossRef] [PubMed]
- Sonnenschein, C.; Soto, A.M. Cancer Metastases: So Close and So Far. JNCI J. Natl. Cancer Inst. 2015, 107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Q.; Dang, C.V. Myc Overexpression Uncouples DNA Replication from Mitosis. Mol. Cell. Biol. 1999, 19, 5339–5351. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grendler, J.; Lowgren, S.; Mills, M.; Losick, V.P. Wound-induced polyploidization is driven by Myc and supports tissue repair in the presence of DNA damage. Development 2019, 146, dev173005. [Google Scholar] [CrossRef] [Green Version]
- Soucek, L.; Whitfield, J.; Martins, C.P.; Finch, A.J.; Murphy, D.J.; Sodir, N.M.; Karnezis, A.N.; Swigart, L.B.; Nasi, S.; Evan, G.I. Modelling Myc inhibition as a cancer therapy. Nature 2008, 455, 679–683. [Google Scholar] [CrossRef] [Green Version]
- Evan, G. Taking a Back Door to Target Myc. Science 2012, 335, 293–294. [Google Scholar] [CrossRef]
- Kortlever, R.M.; Sodir, N.M.; Wilson, C.H.; Burkhart, D.L.; Pellegrinet, L.; Brown Swigart, L.; Littlewood, T.D.; Evan, G.I. Myc Cooperates with Ras by Programming Inflammation and Immune Suppression. Cell 2017, 171, 1301–1315. [Google Scholar] [CrossRef]
- Sui, Y.; Gu, R.; Janknecht, R. Crucial Functions of the JMJD1/KDM3 Epigenetic Regulators in Cancer. Mol. Cancer Res. 2020. [Google Scholar] [CrossRef]
- Neri, F.; Zippo, A.; Krepelova, A.; Cherubini, A.; Rocchigiani, M.; Oliviero, S. Myc Regulates the Transcription of the PRC2 Gene to Control the Expression of Developmental Genes in Embryonic Stem Cells. Mol. Cell. Biol. 2012, 32, 840–851. [Google Scholar] [CrossRef] [Green Version]
- Ullius, A.; Luscher-Firzlaff, J.; Costa, I.G.; Walsemann, G.; Forst, A.H.; Gusmao, E.G.; Kapelle, K.; Kleine, H.; Kremmer, E.; Vervoorts, J.; et al. The interaction of MYC with the trithorax protein ASH2L promotes gene transcription by regulating H3K27 modification. Nucleic Acids Res. 2014, 42, 6901–6920. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vinogradov, A.E.; Shilina, M.A.; Anatskaya, O.V.; Alekseenko, L.L.; Fridlyanskaya, I.I.; Krasnenko, A.; Kim, A.; Korostin, D.; Ilynsky, V.; Elmuratov, A.; et al. Molecular Genetic Analysis of Human Endometrial Mesenchymal Stem Cells That Survived Sublethal Heat Shock. Stem Cells Int 2017, 17, 2362630. [Google Scholar] [CrossRef] [Green Version]
- Kallioniemi, A. Bone morphogenetic protein 4—A fascinating regulator of cancer cell behavior. Cancer Genet. 2012, 205, 267–277. [Google Scholar] [CrossRef] [PubMed]
- Bach, D.-H.; Park, H.J.; Lee, S.K. The Dual Role of Bone Morphogenetic Proteins in Cancer. Mol. Ther. Oncolytics 2018, 8, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, H.; Liu, H.; Qing, G. Targeting oncogenic Myc as a strategy for cancer treatment. Signal Transduct. Target. Ther. 2018, 3, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tovar, E.A.; Graveel, C.R. MET in human cancer: Germline and somatic mutations. Ann. Transl. Med. 2017, 5, 205. [Google Scholar] [CrossRef] [Green Version]
- Mitra, P. Transcription regulation of MYB: A potential and novel therapeutic target in cancer. Ann. Transl. Med. 2018, 6, 443. [Google Scholar] [CrossRef]
- Erenpreiss, J.O. Current Concepts of Malignant Growth; Zinâtne Publ.: Riga, Latvia, 1993. [Google Scholar]
- Moore, A.R.; Rosenberg, S.C.; McCormick, F.; Malek, S. RAS-targeted therapies: Is the undruggable drugged? Nat. Rev. Drug Discov. 2020, 19, 533–552. [Google Scholar] [CrossRef]
- Sigismund, S.; Avanzato, D.; Lanzetti, L. Emerging functions of the EGFR in cancer. Mol. Oncol. 2018, 12, 3–20. [Google Scholar] [CrossRef]
- Hosaka, K.; Yang, Y.; Seki, T.; Du, Q.; Jing, X.; He, X.; Wu, J.; Zhang, Y.; Morikawa, H.; Nakamura, M.; et al. Therapeutic paradigm of dual targeting VEGF and PDGF for effectively treating FGF-2 off-target tumors. Nat. Commun. 2020, 11, 3704. [Google Scholar] [CrossRef]
- Khurana, N.; Sikka, S.C. Interplay between SOX9, Wnt/β-Catenin and Androgen Receptor Signaling in Castration-Resistant Prostate Cancer. Int. J. Mol. Sci. 2019, 20, 2066. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qin, Q.; Xu, Y.; He, T.; Qin, C.; Xu, J. Normal and disease-related biological functions of Twist1 and underlying molecular mechanisms. Cell Res. 2012, 22, 90–106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petrizzo, A.; Caruso, F.P.; Tagliamonte, M.; Tornesello, M.L.; Ceccarelli, M.; Costa, V.; Aprile, M.; Esposito, R.; Ciliberto, G.; Buonaguro, F.M.; et al. Identification and Validation of HCC-specific Gene Transcriptional Signature for Tumor Antigen Discovery. Sci. Rep. 2016, 6, 29258. [Google Scholar] [CrossRef] [PubMed]
- Davoli, T.; Xu, A.W.; Mengwasser, K.E.; Sack, L.M.; Yoon, J.C.; Park, P.J.; Elledge, S.J. Cumulative Haploinsufficiency and Triplosensitivity Drive Aneuploidy Patterns and Shape the Cancer Genome. Cell 2013, 155, 948–962. [Google Scholar] [CrossRef] [Green Version]
- Justice, J.; Malhotra, S.; Ruano, M.; Li, Y.; Zavala, G.; Lee, N.; Morgan, R.; Beemon, K. The MET Gene Is a Common Integration Target in Avian Leukosis Virus Subgroup J-Induced Chicken Hemangiomas. J. Virol. 2015, 89, 4712–4719. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pienta, K.J.; Hammarlund, E.U.; Axelrod, R.; Amend, S.R.; Brown, J.S. Convergent Evolution, Evolving Evolvability, and the Origins of Lethal Cancer. Mol. Cancer Res. 2020, 18, 801–810. [Google Scholar] [CrossRef] [Green Version]
- Walther, V.; Hiley, C.T.; Shibata, D.; Swanton, C.; Turner, P.E.; Maley, C.C. Can oncology recapitulate paleontology? Lessons from species extinctions. Nat. Rev. Clin. Oncol. 2015, 12, 273–285. [Google Scholar] [CrossRef] [Green Version]
- Versteeg, R. Tumours outside the mutation box. Nature 2014, 506, 438–439. [Google Scholar] [CrossRef]
- Gatenby, R.A. Is the Genetic Paradigm of Cancer Complete? Radiology 2017, 284, 1–3. [Google Scholar] [CrossRef]
- Ganem, N.J.; Storchova, Z.; Pellman, D. Tetraploidy, aneuploidy and cancer. Curr. Opin. Genet. Dev. 2007, 17, 157–162. [Google Scholar] [CrossRef]
- López, S.; Lim, E.L.; Horswell, S.; Haase, K.; Huebner, A.; Dietzen, M.; Mourikis, T.P.; Watkins, T.B.K.; Rowan, A.; Dewhurst, S.M.; et al. Interplay between whole-genome doubling and the accumulation of deleterious alterations in cancer evolution. Nat. Genet. 2020, 52, 283–293. [Google Scholar] [CrossRef] [PubMed]
- Beach, R.R.; Ricci-Tam, C.; Brennan, C.M.; Moomau, C.A.; Hsu, P.-H.; Hua, B.; Silberman, R.E.; Springer, M.; Amon, A. Aneuploidy Causes Non-genetic Individuality. Cell 2017, 169, 229–242.e21. [Google Scholar] [CrossRef] [PubMed]
- Salmina, K.; Huna, A.; Kalejs, M.; Pjanova, D.; Scherthan, H.; Cragg, M.S.; Erenpreisa, J. The Cancer Aneuploidy Paradox: In the Light of Evolution. Genes 2019, 10, 83. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mojtahedi, M.; Skupin, A.; Zhou, J.; Castaño, I.G.; Leong-Quong, R.Y.Y.; Chang, H.; Trachana, K.; Giuliani, A.; Huang, S. Cell Fate Decision as High-Dimensional Critical State Transition. PLoS Biol. 2016, 14, e2000640. [Google Scholar] [CrossRef] [Green Version]
- Cavalli, G. Chromosome kissing. Curr. Opin. Genet. Dev. 2007, 17, 443–450. [Google Scholar] [CrossRef]
- Lieberman-Aiden, E.; van Berkum, N.L.; Williams, L.; Imakaev, M.; Ragoczy, T.; Telling, A.; Amit, I.; Lajoie, B.R.; Sabo, P.J.; Dorschner, M.O.; et al. Comprehensive Mapping of Long-Range Interactions Reveals Folding Principles of the Human Genome. Science 2009, 326, 289–293. [Google Scholar] [CrossRef] [Green Version]
- Solovei, I.; Thanisch, K.; Feodorova, Y. How to rule the nucleus: Divide et impera. Curr. Opin. Cell Biol. 2016, 40, 47–59. [Google Scholar] [CrossRef] [Green Version]
- Bakhmet, E.I.; Nazarov, I.B.; Gazizova, A.R.; Vorobyeva, N.E.; Kuzmin, A.A.; Gordeev, M.N.; Sinenko, S.A.; Aksenov, N.D.; Artamonova, T.O.; Khodorkovskii, M.A.; et al. hnRNP-K Targets Open Chromatin in Mouse Embryonic Stem Cells in Concert with Multiple Regulators: hnRNP-K Targets Open Chromatin in Mouse ESCs. Stem Cells 2019, 37, 1018–1029. [Google Scholar] [CrossRef] [Green Version]
- Nazarov, I.B.; Bakhmet, E.I.; Tomilin, A.N. KH-Domain Poly(C)-Binding Proteins as Versatile Regulators of Multiple Biological Processes. Biochem. Mosc. 2019, 84, 205–219. [Google Scholar] [CrossRef]
- Kauffman, S.A. The Origins of Order: Self-Organization and Selection in Evolution; Series on Directions in Condensed Matter Physics; World Scientific: Singapore, 1992; Volume 6, pp. 61–100. ISBN 978-9971-5-0537-0. [Google Scholar]
- Conant, G.C. Rapid reorganization of the transcriptional regulatory network after genome duplication in yeast. Proc. Biol. Sci. 2010, 277, 869–876. [Google Scholar] [CrossRef] [Green Version]
- Conant, G.C. The lasting after-effects of an ancient polyploidy on the genomes of teleosts. PLoS ONE 2020, 15, e0231356. [Google Scholar] [CrossRef] [PubMed]
- Cherstvy, A.G.; Teif, V.B. Structure-driven homology pairing of chromatin fibers: The role of electrostatics and protein-induced bridging. J. Biol. Phys. 2013, 39, 363–385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jerković, I.; Szabo, Q.; Bantignies, F.; Cavalli, G. Higher-Order Chromosomal Structures Mediate Genome Function. J. Mol. Biol. 2020, 432, 676–681. [Google Scholar] [CrossRef]
- Vinogradov, A.E. Buffering: A Possible Passive-homeostasis Role for Redundant DNA. J. Theor. Biol. 1998, 193, 197–199. [Google Scholar] [CrossRef] [PubMed]
- Vinogradov, A.E. Genome size and chromatin condensation in vertebrates. Chromosoma 2005, 113, 362–369. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Sato, Y.; Furumichi, M.; Morishima, K.; Tanabe, M. New approach for understanding genome variations in KEGG. Nucleic Acids Res. 2019, 47, D590–D595. [Google Scholar] [CrossRef] [Green Version]
- Land, H.; Parada, L.F.; Weinberg, R.A. Tumorigenic conversion of primary embryo fibroblasts requires at least two cooperating oncogenes. Nature 1983, 304, 596–602. [Google Scholar] [CrossRef]
- Knudson, A.G. Two genetic hits (more or less) to cancer. Nat. Rev. Cancer 2001, 1, 157–162. [Google Scholar] [CrossRef]
- Wang, C.; Lisanti, M.P.; Liao, D.J. Reviewing once more the c-myc and Ras collaboration: Converging at the cyclin D1-CDK4 complex and challenging basic concepts of cancer biology. Cell Cycle 2011, 10, 57–67. [Google Scholar] [CrossRef]
- Huang, S.; Ernberg, I.; Kauffman, S. Cancer attractors: A systems view of tumors from a gene network dynamics and developmental perspective. Semin. Cell Dev. Biol. 2009, 20, 869–876. [Google Scholar] [CrossRef] [Green Version]
- Huang, S.; Kauffman, S. How to escape the cancer attractor: Rationale and limitations of multi-target drugs. Semin. Cancer Biol. 2013, 23, 270–278. [Google Scholar] [CrossRef] [PubMed]
- Erenpreisa, J.; Roach, H.I. Epigenetic selection as a possible component of transdifferentiation. Further study of the commitment of hypertrophic chondrocytes to become osteocytes. Mech. Ageing Dev. 1996, 87, 165–182. [Google Scholar] [CrossRef]
- Erenpreisa, J.; Salmiņa, K.; Belyayev, A.; Inashkina, I.; Cragg, M.S. Survival at the Brink. In Autophagy: Cancer, Other Pathologies, Inflammation, Immunity, Infection, and Aging; Elsevier: Amsterdam, The Netherlands, 2017; pp. 275–294. ISBN 978-0-12-812146-7. [Google Scholar]
- Domazet-Lošo, T.; Klimovich, A.; Anokhin, B.; Anton-Erxleben, F.; Hamm, M.J.; Lange, C.; Bosch, T.C.G. Naturally occurring tumours in the basal metazoan Hydra. Nat. Commun. 2014, 5, 4222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arthur, W. Evolution: A Developmental Approach; Wiley-Blackwell: Chichester, UK, 2011; ISBN 978-1-4051-8658-2. [Google Scholar]
- Erenpreisa, J.; Salmina, K.; Huna, A.; Jackson, T.R.; Vazquez-Martin, A.; Cragg, M.S. The “virgin birth”, polyploidy, and the origin of cancer. Oncoscience 2014, 2, 3–14. [Google Scholar] [CrossRef]
- Niu, N.; Mercado-Uribe, I.; Liu, J. Dedifferentiation into blastomere-like cancer stem cells via formation of polyploid giant cancer cells. Oncogene 2017, 36, 4887–4900. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fais, S.; Overholtzer, M. Cell-in-cell phenomena, cannibalism, and autophagy: Is there a relationship? Cell Death Dis. 2018, 9, 95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goodkov, A.V.; Berdieva, M.A.; Podlipaeva, Y.I.; Demin, S.Y. The Chromatin Extrusion Phenomenon in Amoeba proteus Cell Cycle. J. Eukaryot. Microbiol. 2020, 67, 203–208. [Google Scholar] [CrossRef]
- Arguello, F. Atavistic Metamorphosis: A New and Logical Explanation for the Origin and Biological Nature of Cancer: With a Discussion on a Novel Approach to Treat Cancer. CreateSpace: Scotts Valley, CA, USA, 2011; ISBN 978-1-4609-6899-4. [Google Scholar]
- Lineweaver, C.H.; Davies, P.C.W.; Vincent, M.D. Targeting cancer’s weaknesses (not its strengths): Therapeutic strategies suggested by the atavistic model: Insights & Perspectives. BioEssays 2014, 36, 827–835. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raikov, I.B. Nuclear apparatus of Mesokaryotic Protozoa. In The Protozoan Nucleus, Morphology and Evolution; Cell Biology Monographs; Springer: New York, NY, USA, 1982; pp. 1–174. [Google Scholar]
- Demin, S.Y.; Berdieva, M.A.; Podlipaeva, Y.I.; Goodkov, A.V. Karyotypic instability of endoprophase and mitotic cells of Amoeba sp. strain Cont from the “proteus-type” group (Amoebozoa, Euamoebida, Amoebidae). Eur. J. Protistol. 2020, 74, 125691. [Google Scholar] [CrossRef]
- Niculescu, V.F. The reproductive life cycle of cancer: Hypotheses of cell of origin, TP53 drivers and stem cell conversions in the light of the atavistic cancer cell theory. Med Hypotheses 2019, 123, 19–23. [Google Scholar] [CrossRef]
- Nanjundiah, V. Cellular slime mold development as a paradigm for the transition from unicellular to multicellular life. In Multicellularity: Origins and Evolution; MIT Press: Cambridge, MA, USA, 2016. [Google Scholar]
- Andreani, T.S.; Itoh, T.Q.; Yildirim, E.; Hwangbo, D.-S.; Allada, R. Genetics of Circadian Rhythms. Sleep Med. Clin. 2015, 10, 413–421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Camponeschi, I.; Damasco, A.; Uversky, V.N.; Giuliani, A.; Bianchi, M.M. Phenotypic suppression caused by resonance with light-dark cycles indicates the presence of a 24-hours oscillator in yeast and suggests a new role of intrinsically disordered protein regions as internal mediators. J. Biomol. Struct. Dyn. 2020. [Google Scholar] [CrossRef] [PubMed]
- Øvrebø, J.I.; Edgar, B.A. Polyploidy in tissue homeostasis and regeneration. Development 2018, 145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vazquez-Martin, A.; Anatskaya, O.V.; Giuliani, A.; Erenpreisa, J.; Huang, S.; Salmina, K.; Inashkina, I.; Huna, A.; Nikolsky, N.N.; Vinogradov, A.E. Somatic polyploidy is associated with the upregulation of c-MYC interacting genes and EMT-like signature. Oncotarget 2016, 7, 75235–75260. [Google Scholar] [CrossRef] [PubMed]
- Whitehead, A.; Crawford, D.L. Variation within and among species in gene expression: Raw material for evolution: Review of gene expression variation. Mol. Ecol. 2006, 15, 1197–1211. [Google Scholar] [CrossRef]
- Otto, S.P. The Evolutionary Consequences of Polyploidy. Cell 2007, 131, 452–462. [Google Scholar] [CrossRef] [Green Version]
- Brawand, D.; Soumillon, M.; Necsulea, A.; Julien, P.; Csárdi, G.; Harrigan, P.; Weier, M.; Liechti, A.; Aximu-Petri, A.; Kircher, M.; et al. The evolution of gene expression levels in mammalian organs. Nature 2011, 478, 343–348. [Google Scholar] [CrossRef]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef]
- Vinogradov, A.E.; Anatskaya, O.V. Systemic evolutionary changes in mammalian gene expression. Biosystems 2020, 198, 104256. [Google Scholar] [CrossRef]
- Sayers, E.W.; Beck, J.; Brister, J.R.; Bolton, E.E.; Canese, K.; Comeau, D.C.; Funk, K.; Ketter, A.; Kim, S.; Kimchi, A.; et al. Database resources of the National Center for Biotechnology Information. Nucleic Acids Res. 2020, 48, D9–D16. [Google Scholar] [CrossRef] [Green Version]
- Seyednasrollah, F.; Laiho, A.; Elo, L.L. Comparison of software packages for detecting differential expression in RNA-seq studies. Brief. Bioinform. 2015, 16, 59–70. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giuliani, A. The application of principal component analysis to drug discovery and biomedical data. Drug Discov. Today 2017, 22, 1069–1076. [Google Scholar] [CrossRef] [PubMed]
- Roden, J.C.; King, B.W.; Trout, D.; Mortazavi, A.; Wold, B.J.; Hart, C.E. Mining gene expression data by interpreting principal components. BMC Bioinform. 2006, 7, 194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vinogradov, A.E. ’Genome design’ model and multicellular complexity: Golden middle. Nucleic Acids Res. 2006, 34, 5906–5914. [Google Scholar] [CrossRef] [Green Version]
- Vinogradov, A.E.; Anatskaya, O.V. DNA helix: The importance of being AT-rich. Mamm. Genome 2017, 28, 455–464. [Google Scholar] [CrossRef]
- The Gene Ontology Consortium. The Gene Ontology Resource: 20 years and still GOing strong. Nucleic Acids Res. 2019, 47, D330–D338. [Google Scholar] [CrossRef] [Green Version]
- Storey, J.D.; Tibshirani, R. Statistical significance for genomewide studies. Proc. Natl. Acad. Sci. USA 2003, 100, 9440–9445. [Google Scholar] [CrossRef] [Green Version]
- Szklarczyk, D.; Gable, A.L.; Lyon, D.; Junge, A.; Wyder, S.; Huerta-Cepas, J.; Simonovic, M.; Doncheva, N.T.; Morris, J.H.; Bork, P.; et al. STRING v11: Protein–protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2019, 47, D607–D613. [Google Scholar] [CrossRef] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene/ Protein | Folds Up-Regulation | Number of Connections | Phylo Stratum | Function Involvement in Cancer | References |
|---|---|---|---|---|---|
| BMP4 | 3.24 | 26 | 4 | Development, cell motility, oncogene | [66] |
| BMP2 | 3.84 | 14 | 5 | Development, cell motility, oncogene | [67] |
| c-MYC | 3.2 | 29 | 3 | Stemness, proliferation, apoptosis, polyploidy induction, driver oncogene | [68] |
| MET | 3.7 | 15 | 6 | Embryonic development, proliferation; when mutationally activated is involved in multiple cancers | [69] |
| MYB | 3.8 | 8 | 2 | Development of colon epithelial progenitors, high in gastric, colon, and breast cancer | [70] |
| HRAS | 3.6 | 19 | 1 | Proliferation, senescence, germ development, driver oncogene | [71,72] |
| EGFR | 4.94 | 34 | 6 | Epithelium development, driver oncogene | [73] |
| PDGFRB | 2.23 | 15 | 6 | Development, cancer-related angiogenesis | [74] |
| VEGFC (C) | 2.88 | 12 | 5 | Blood vessels development, cancer-related angiogenesis | [74] |
| SOX9 | 3.86 | 15 | 4 | Germ (male) and skeleton development | [75] |
| TWIST1 | 2.53 | 11 | 2 | Mesoderm development, motility, metastatic cancer | [76] |
| FOSL1 | 2.19 | 11 | 5 | Early stress response, reinforces Myc, oncogenesis | [77] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Anatskaya, O.V.; Vinogradov, A.E.; Vainshelbaum, N.M.; Giuliani, A.; Erenpreisa, J. Phylostratic Shift of Whole-Genome Duplications in Normal Mammalian Tissues towards Unicellularity Is Driven by Developmental Bivalent Genes and Reveals a Link to Cancer. Int. J. Mol. Sci. 2020, 21, 8759. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21228759
Anatskaya OV, Vinogradov AE, Vainshelbaum NM, Giuliani A, Erenpreisa J. Phylostratic Shift of Whole-Genome Duplications in Normal Mammalian Tissues towards Unicellularity Is Driven by Developmental Bivalent Genes and Reveals a Link to Cancer. International Journal of Molecular Sciences. 2020; 21(22):8759. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21228759
Chicago/Turabian StyleAnatskaya, Olga V., Alexander E. Vinogradov, Ninel M. Vainshelbaum, Alessandro Giuliani, and Jekaterina Erenpreisa. 2020. "Phylostratic Shift of Whole-Genome Duplications in Normal Mammalian Tissues towards Unicellularity Is Driven by Developmental Bivalent Genes and Reveals a Link to Cancer" International Journal of Molecular Sciences 21, no. 22: 8759. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21228759