Effects of Ketone Bodies on Brain Metabolism and Function in Neurodegenerative Diseases


{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
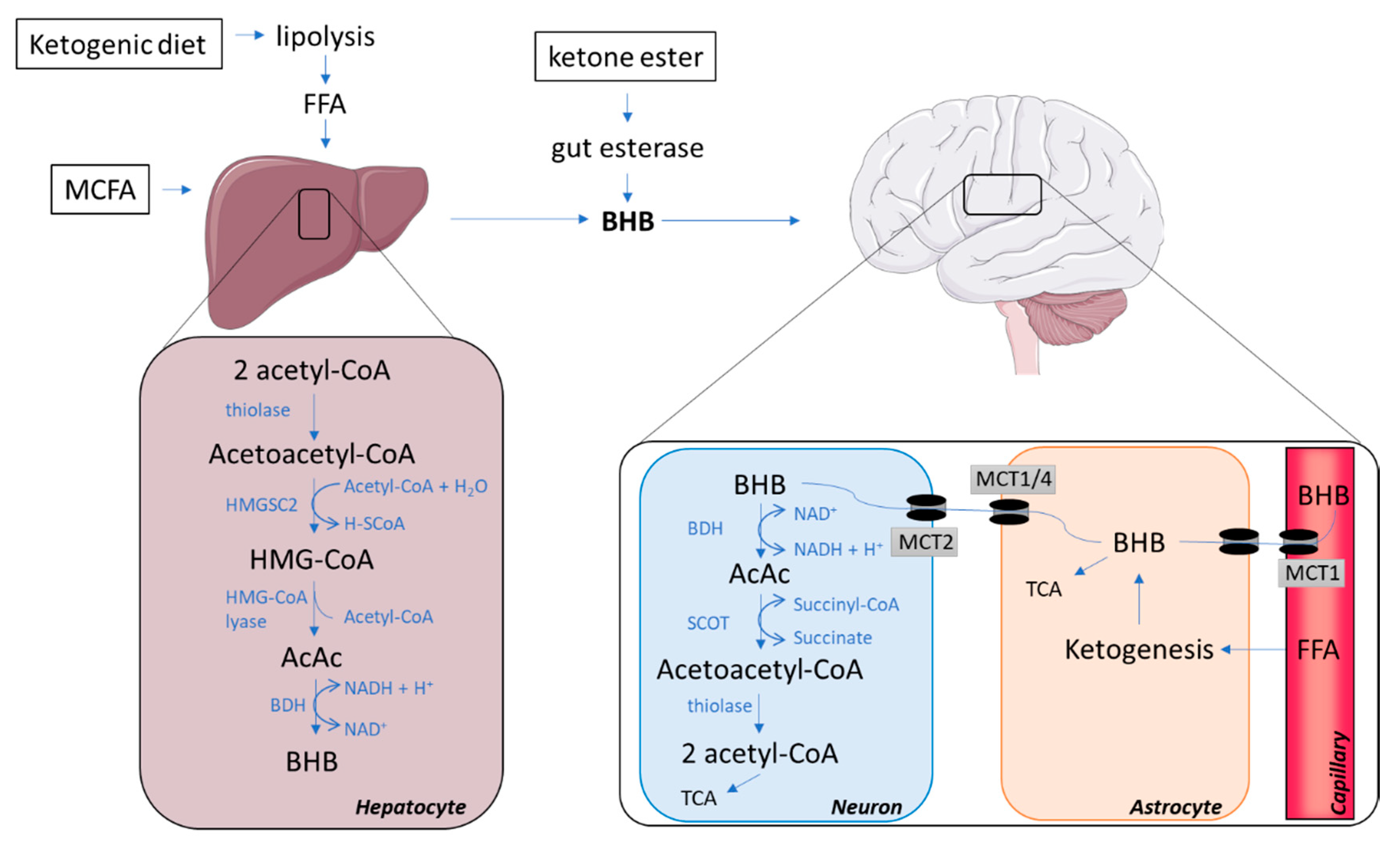
2. Ketone Bodies Reaching the Brain
2.1. The Synthesis of Ketone Bodies in the Liver
2.2. Ways of Increasing the Concentration of Circulating Ketone Bodies
2.3. Ketone Bodies Entering the Brain via Monocarboxylate Transporters
2.4. The Catabolism of Ketone Bodies in Neuronal and Glial Cells
2.5. Glial–Neural Compartment Mode for Ketone Body Supply to Neurones
3. The Effect of Ketone Bodies on Brain Metabolism
3.1. Ketone Bodies Affect Brain Metabolism in Healthy Individuals
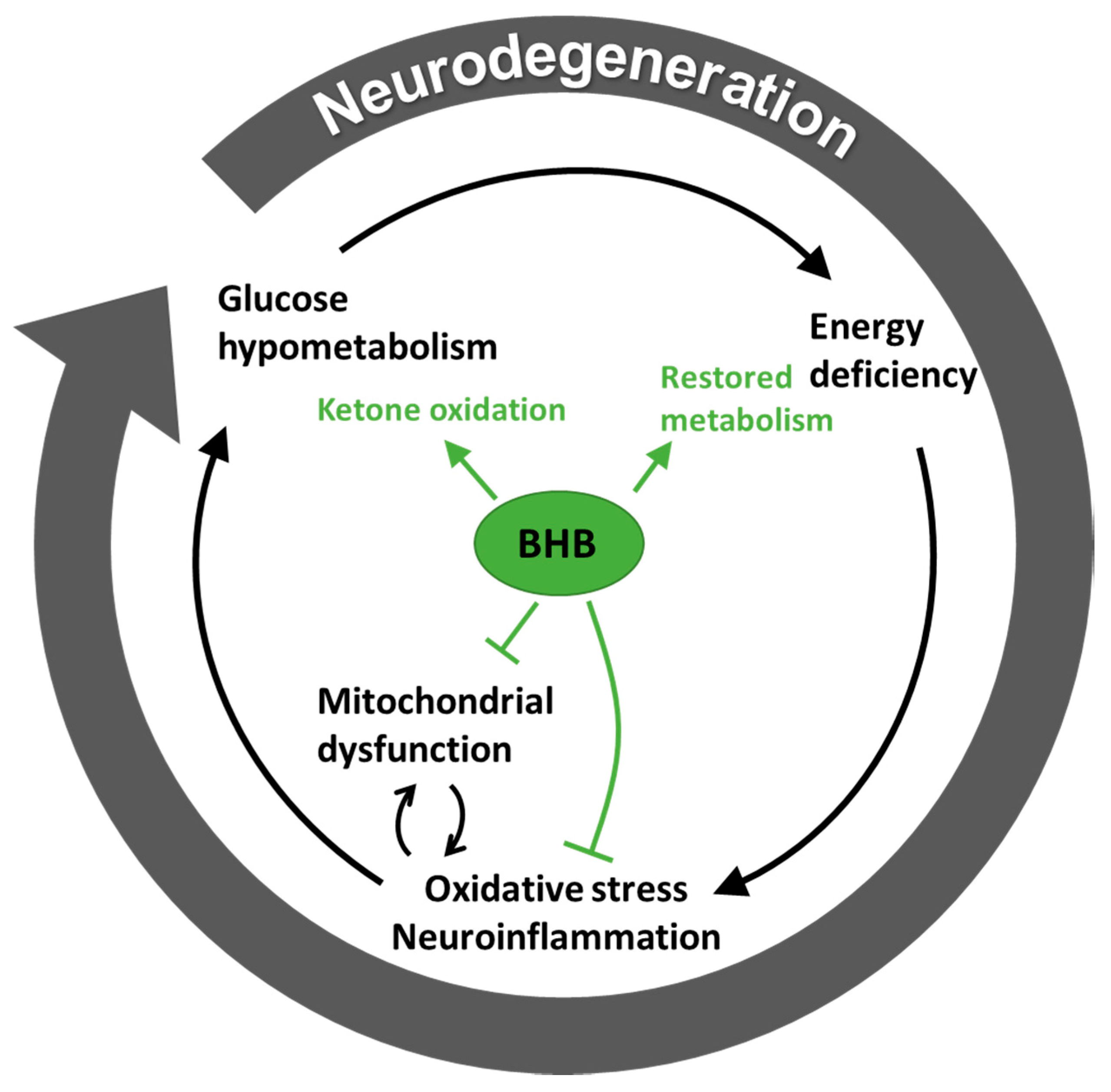
3.2. Hypometabolism in Neurodegenerative Disease is Ameliorated by Ketone Bodies
3.3. Ketone Bodies May Support Metabolism Besides Being a Substrate
4. The Therapeutic Role of Ketone Bodies in Neurodegeneration
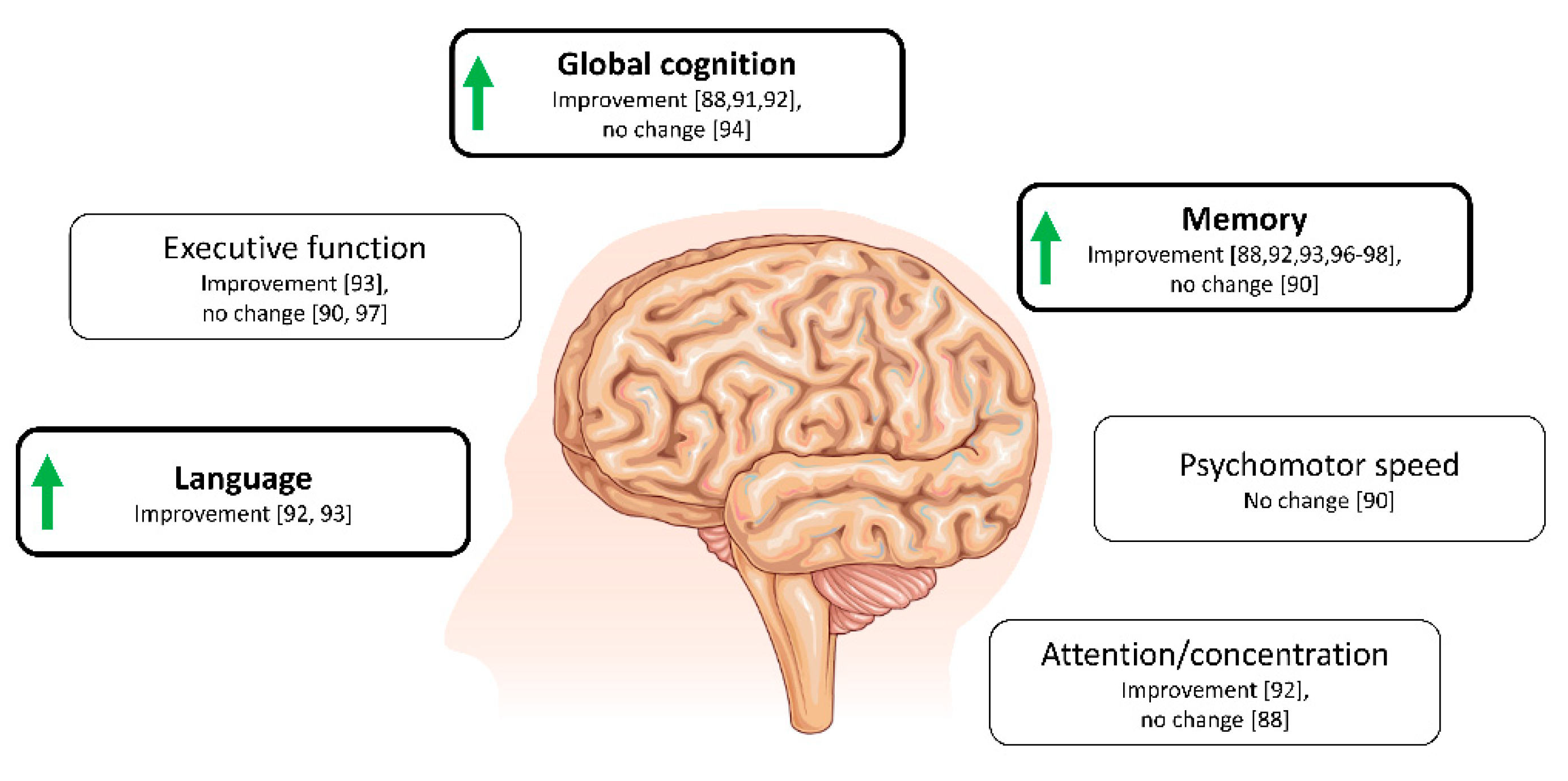
4.1. Ketone Bodies and Cognition in Alzheimer’s Disease and Related Conditions
4.2. Ketone Bodies in Other Neurodegenerative Diseases
5. Conclusions and Perspectives
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| Acetyl-CoA | acetyl coenzyme A |
| AD | Alzheimer’s disease |
| AICAR | 5-Aminoimidazole-4-carboxamide ribonucleotide |
| ALS | Amyotrophic lateral sclerosis |
| AMPK | AMP-activated protein kinase |
| BBB | Blood–brain barrier |
| BHB | beta-hydroxybutyrate |
| BHD | beta-hydroxybutyrate dehydrogenase |
| GLUT-1 | glucose transporter type 1 |
| HD | Huntington disease |
| HMG-CoA | 3-hydroxy-3-methylglutaryl-CoA |
| HMGCS2 | 3-Hydroxy-3-Methylglutaryl-CoA Synthase 2 |
| MCFA | medium-chain fatty acids |
| MCT | monocarboxylate transporter |
| PD | Parkinson’s disease |
| ROS | reactive oxygen species |
| SCOT | succinyl-CoA:3-ketoacid Coenzyme A transferase |
| SGLT2-i | sodium glucose cotransporter 2 inhibitors |
| TCA | tricarboxylic acid |
| UCP | uncoupling proteins |
References
- Rolfe, D.F.; Brown, G.C. Cellular energy utilization and molecular origin of standard metabolic rate in mammals. Physiol. Rev. 1997, 77, 731–758. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oyarzabal, A.; Marin-Valencia, I. Synaptic energy metabolism and neuronal excitability, in sickness and health. J. Inherit. Metab. Dis. 2019, 42, 220–236. [Google Scholar] [CrossRef] [PubMed]
- Harris, J.J.; Jolivet, R.; Attwell, D. Synaptic Energy Use and Supply. Neuron 2012, 75, 762–777. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bordone, M.P.; Salman, M.M.; Titus, H.E.; Amini, E.; Andersen, J.V.; Chakraborti, B.; Diuba, A.V.; Dubouskaya, T.G.; Ehrke, E.; Espindola De Freitas, A.; et al. The energetic brain–A review from students to students. J. Neurochem. 2019, 151, 139–165. [Google Scholar] [CrossRef] [PubMed]
- Bak, L.K.; Walls, A.B.; Schousboe, A.; Waagepetersen, H.S. Astrocytic glycogen metabolism in the healthy and diseased brain. J. Biol. Chem. 2018, 293, 7108–7116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pearson, T.S.; Akman, C.; Hinton, V.J.; Engelstad, K.; De Vivo, D.C. Phenotypic Spectrum of Glucose Transporter Type 1 Deficiency Syndrome (Glut1 DS). Curr. Neurol. Neurosci. Rep. 2013, 13. [Google Scholar] [CrossRef]
- Owen, O.E.; Morgan, A.P.; Kemp, H.G.; Sullivan, J.M.; Herrera, M.G.; Cahill, G.F. Brain Metabolism during Fasting. J. Clin. Investig. 1967, 46, 1589–1595. [Google Scholar] [CrossRef]
- Courchesne-Loyer, A.; Croteau, E.; Castellano, C.-A.; St-Pierre, V.; Hennebelle, M.; Cunnane, S.C. Inverse relationship between brain glucose and ketone metabolism in adults during short-term moderate dietary ketosis: A dual tracer quantitative positron emission tomography study. J. Cereb. Blood Flow Metab. 2017, 37, 2485–2493. [Google Scholar] [CrossRef] [Green Version]
- Pierre, K.; Pellerin, L. Monocarboxylate transporters in the central nervous system: Distribution, regulation and function. J. Neurochem. 2005, 94, 1–14. [Google Scholar] [CrossRef]
- Cunnane, S.C.; Courchesne-Loyer, A.; Vandenberghe, C.; St-Pierre, V.; Fortier, M.; Hennebelle, M.; Croteau, E.; Bocti, C.; Fulop, T.; Castellano, C.A. Can Ketones Help Rescue Brain Fuel Supply in Later Life? Implications for Cognitive Health during Aging and the Treatment of Alzheimer’s Disease. Front. Mol. Neurosci. 2016, 9, 53. [Google Scholar] [CrossRef]
- Kossoff, E.H.; Wang, H.S. Dietary therapies for epilepsy. Biomed. J. 2013, 36, 2–8. [Google Scholar] [CrossRef] [PubMed]
- Møller, N. Ketone Body, 3-Hydroxybutyrate: Minor Metabolite—Major Medical Manifestations. J. Clin. Endocrinol. Metab. 2020, 105. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.-M.; Ramprasath, T.; Zou, M.-H. β-hydroxybutyrate and its metabolic effects on age-associated pathology. Exp. Mol. Med. 2020, 52, 548–555. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vandoorne, T.; De Bock, K.; Van Den Bosch, L. Energy metabolism in ALS: An underappreciated opportunity? Acta Neuropathol. 2018, 135, 489–509. [Google Scholar] [CrossRef] [Green Version]
- Pagano, G.; Niccolini, F.; Politis, M. Current status of PET imaging in Huntington’s disease. Eur. J. Nucl. Med. Mol. Imaging 2016. [Google Scholar] [CrossRef] [Green Version]
- Zilberter, Y.; Zilberter, M. The vicious circle of hypometabolism in neurodegenerative diseases: Ways and mechanisms of metabolic correction. J. Neurosci. Res. 2017, 95, 2217–2235. [Google Scholar] [CrossRef] [Green Version]
- Cunnane, S.; Nugent, S.; Roy, M.; Courchesne-Loyer, A.; Croteau, E.; Tremblay, S.; Castellano, A.; Pifferi, F.; Bocti, C.; Paquet, N.; et al. Brain fuel metabolism, aging, and Alzheimer’s disease. Nutrition 2011, 27, 3–20. [Google Scholar] [CrossRef] [Green Version]
- Cunnane, S.C.; Trushina, E.; Morland, C.; Prigione, A.; Casadesus, G.; Andrews, Z.B.; Beal, M.F.; Bergersen, L.H.; Brinton, R.D.; de la Monte, S.; et al. Brain energy rescue: An emerging therapeutic concept for neurodegenerative disorders of ageing. Nat. Rev. Drug Discov. 2020, 19, 609–633. [Google Scholar] [CrossRef]
- Fukao, T.; Lopaschuk, G.D.; Mitchell, G.A. Pathways and control of ketone body metabolism: On the fringe of lipid biochemistry. Prostaglandins Leukot. Essent. Fat. Acids 2004, 70, 243–251. [Google Scholar] [CrossRef]
- Puchalska, P.; Crawford, P.A. Multi-dimensional Roles of Ketone Bodies in Fuel Metabolism, Signaling, and Therapeutics. Cell Metab. 2017, 25, 262–284. [Google Scholar] [CrossRef] [Green Version]
- Newman, J.C.; Verdin, E. β-Hydroxybutyrate: A Signaling Metabolite. Annu. Rev. Nutr. 2017, 37, 51–76. [Google Scholar] [CrossRef] [PubMed]
- Cullingford, T.E.; Eagles, D.A.; Sato, H. The ketogenic diet upregulates expression of the gene encoding the key ketogenic enzyme mitochondrial 3-hydroxy-3-methylglutaryl-CoA synthase in rat brain. Epilepsy Res. 2002, 49, 99–107. [Google Scholar] [CrossRef]
- Trimboli, P.; Castellana, M.; Bellido, D.; Casanueva, F.F. Confusion in the nomenclature of ketogenic diets blurs evidence. Rev. Endocr. Metab. Disord. 2020, 21, 1–3. [Google Scholar] [CrossRef]
- Włodarek, D. Role of Ketogenic Diets in Neurodegenerative Diseases (Alzheimer’s Disease and Parkinson’s Disease). Nutrients 2019, 11, 169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rusek, M.; Pluta, R.; Ułamek-Kozioł, M.; Czuczwar, S.J. Ketogenic Diet in Alzheimer’s Disease. Int. J. Mol. Sci. 2019, 20, 3892. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bach, A.C.; Babayan, V.K. Medium-chain triglycerides: An update. Am. J. Clin. Nutr. 1982, 36, 950–962. [Google Scholar] [CrossRef] [PubMed]
- Cunnane, S.C.; Courchesne-Loyer, A.; St-Pierre, V.; Vandenberghe, C.; Pierotti, T.; Fortier, M.; Croteau, E.; Castellano, C.A. Can ketones compensate for deteriorating brain glucose uptake during aging? Implications for the risk and treatment of Alzheimer’s disease. Ann. N. Y. Acad. Sci. 2016, 1367, 12–20. [Google Scholar] [CrossRef] [Green Version]
- Vandenberghe, C.; St-Pierre, V.; Pierotti, T.; Fortier, M.; Castellano, C.-A.; Cunnane, S.C. Tricaprylin Alone Increases Plasma Ketone Response More Than Coconut Oil or Other Medium-Chain Triglycerides: An Acute Crossover Study in Healthy Adults. Curr. Dev. Nutr. 2017, 1, e000257. [Google Scholar] [CrossRef] [Green Version]
- Courchesne-Loyer, A.; Lowry, C.-M.; St-Pierre, V.; Vandenberghe, C.; Fortier, M.; Castellano, C.-A.; Wagner, J.R.; Cunnane, S.C. Emulsification Increases the Acute Ketogenic Effect and Bioavailability of Medium-Chain Triglycerides in Humans. Curr. Dev. Nutr. 2017, 1, e000851. [Google Scholar] [CrossRef] [Green Version]
- Stubbs, B.J.; Cox, P.J.; Evans, R.D.; Santer, P.; Miller, J.J.; Faull, O.K.; Magor-Elliott, S.; Hiyama, S.; Stirling, M.; Clarke, K. On the Metabolism of Exogenous Ketones in Humans. Front. Physiol. 2017, 8. [Google Scholar] [CrossRef]
- Soto-Mota, A.; Vansant, H.; Evans, R.D.; Clarke, K. Safety and tolerability of sustained exogenous ketosis using ketone monoester drinks for 28 days in healthy adults. Regul. Toxicol. Pharmacol. 2019, 109, 104506. [Google Scholar] [CrossRef] [PubMed]
- Augustin, K.; Khabbush, A.; Williams, S.; Eaton, S.; Orford, M.; Cross, J.H.; Heales, S.J.R.; Walker, M.C.; Williams, R.S.B. Mechanisms of action for the medium-chain triglyceride ketogenic diet in neurological and metabolic disorders. Lancet Neurol. 2018, 17, 84–93. [Google Scholar] [CrossRef]
- Achanta, L.B.; Rae, C.D. β-Hydroxybutyrate in the Brain: One Molecule, Multiple Mechanisms. Neurochem. Res. 2017, 42, 35–49. [Google Scholar] [CrossRef] [PubMed]
- Pierre, K.; Magistretti, P.J.; Pellerin, L. MCT2 is a Major Neuronal Monocarboxylate Transporter in the Adult Mouse Brain. J. Cereb. Blood Flow Metab. 2002, 22, 586–595. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiry, O.; Fishbein, W.N.; Merezhinskaya, N.; Clarke, S.; Galuske, R.; Magistretti, P.J.; Pellerin, L. Distribution of the monocarboxylate transporter MCT2 in human cerebral cortex: An immunohistochemical study. Brain Res. 2008, 1226, 61–69. [Google Scholar] [CrossRef] [PubMed]
- Chiry, O.; Pellerin, L.; Monnet-Tschudi, F.; Fishbein, W.N.; Merezhinskaya, N.; Magistretti, P.J.; Clarke, S. Expression of the monocarboxylate transporter MCT1 in the adult human brain cortex. Brain Res. 2006, 1070, 65–70. [Google Scholar] [CrossRef]
- Gjedde, A.; Crone, C. Induction processes in blood-brain transfer of ketone bodies during starvation. Am. J. Physiol. 1975, 229, 1165–1169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pan, J.W.; Telang, F.W.; Lee, J.H.; de Graaf, R.A.; Rothman, D.L.; Stein, D.T.; Hetherington, H.P. Measurement of beta-hydroxybutyrate in acute hyperketonemia in human brain. J. Neurochem. 2001, 79, 539–544. [Google Scholar] [CrossRef]
- Leino, R.L.; Gerhart, D.Z.; Duelli, R.; Enerson, B.E.; Drewes, L.R. Diet-induced ketosis increases monocarboxylate transporter (MCT1) levels in rat brain. Neurochem. Int. 2001, 38, 519–527. [Google Scholar] [CrossRef]
- Hargrave, S.L.; Davidson, T.L.; Lee, T.J.; Kinzig, K.P. Brain and behavioral perturbations in rats following Western diet access. Appetite 2015, 93, 35–43. [Google Scholar] [CrossRef]
- Takimoto, M.; Hamada, T. Acute exercise increases brain region-specific expression of MCT1, MCT2, MCT4, GLUT1, and COX IV proteins. J. Appl. Physiol. 1985 2014, 116, 1238–1250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fukao, T.; Song, X.-Q.; Mitchell, G.A.; Yamaguchi, S.; Sukegawa, K.; Or, T.; Kondo, N. Enzymes of Ketone Body Utilization in Human Tissues: Protein and Messenger RNA Levels of Succinyl-Coenzyme A (CoA):3-Ketoacid CoA Transferase and Mitochondrial and Cytosolic Acetoacetyl-CoA Thiolases. Pediatric Res. 1997, 42, 498–502. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koppel, S.J.; Swerdlow, R.H. Neuroketotherapeutics: A modern review of a century-old therapy. Neurochem. Int. 2018, 117, 114–125. [Google Scholar] [CrossRef]
- Edmond, J.; Robbins, R.A.; Bergstrom, J.D.; Cole, R.A.; De Vellis, J. Capacity for substrate utilization in oxidative metabolism by neurons, astrocytes, and oligodendrocytes from developing brain in primary culture. J. Neurosci. Res. 1987, 18, 551–561. [Google Scholar] [CrossRef]
- Aubert, A.; Costalat, R.; Magistretti, P.J.; Pellerin, L. Brain lactate kinetics: Modeling evidence for neuronal lactate uptake upon activation. Proc. Natl. Acad. Sci. USA 2005, 102, 16448–16453. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guzmán, M.; Blázquez, C. Is there an astrocyte–neuron ketone body shuttle? Trends Endocrinol. Metab. 2001, 12, 169–173. [Google Scholar] [CrossRef]
- Takahashi, S. Metabolic compartmentalization between astroglia and neurons in physiological and pathophysiological conditions of the neurovascular unit. Neuropathology 2020, 40, 121–137. [Google Scholar] [CrossRef] [Green Version]
- Thevenet, J.; De Marchi, U.; Domingo, J.S.; Christinat, N.; Bultot, L.; Lefebvre, G.; Sakamoto, K.; Descombes, P.; Masoodi, M.; Wiederkehr, A. Medium-chain fatty acids inhibit mitochondrial metabolism in astrocytes promoting astrocyte-neuron lactate and ketone body shuttle systems. FASEB J. 2016, 30, 1913–1926. [Google Scholar] [CrossRef] [Green Version]
- Blázquez, C.; Woods, A.; de Ceballos, M.L.; Carling, D.; Guzmán, M. The AMP-activated protein kinase is involved in the regulation of ketone body production by astrocytes. J. Neurochem. 1999, 73, 1674–1682. [Google Scholar] [CrossRef] [Green Version]
- Westhaus, A.; Blumrich, E.M.; Dringen, R. The Antidiabetic Drug Metformin Stimulates Glycolytic Lactate Production in Cultured Primary Rat Astrocytes. Neurochem. Res. 2017, 42, 294–305. [Google Scholar] [CrossRef]
- Svart, M.; Gormsen, L.C.; Hansen, J.; Zeidler, D.; Gejl, M.; Vang, K.; Aanerud, J.; Moeller, N. Regional cerebral effects of ketone body infusion with 3-hydroxybutyrate in humans: Reduced glucose uptake, unchanged oxygen consumption and increased blood flow by positron emission tomography. A randomized, controlled trial. PLoS ONE 2018, 13, e0190556. [Google Scholar] [CrossRef] [PubMed]
- Hasselbalch, S.G.; Madsen, P.L.; Hageman, L.P.; Olsen, K.S.; Justesen, N.; Holm, S.; Paulson, O.B. Changes in cerebral blood flow and carbohydrate metabolism during acute hyperketonemia. Am. J. Physiol. Endocrinol. Metab. 1996, 270, E746–E751. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mikkelsen, K.H.; Seifert, T.; Secher, N.H.; Grøndal, T.; van Hall, G. Systemic, cerebral and skeletal muscle ketone body and energy metabolism during acute hyper-D-β-hydroxybutyratemia in post-absorptive healthy males. J. Clin. Endocrinol. Metab. 2015, 100, 636–643. [Google Scholar] [CrossRef] [Green Version]
- Cahill, G.F., Jr. Fuel metabolism in starvation. Annu. Rev. Nutr. 2006, 26, 1–22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blomqvist, G.; Alvarsson, M.; Grill, V.; Von Heijne, G.; Ingvar, M.; Thorell, J.O.; Stone-Elander, S.; Widén, L.; Ekberg, K. Effect of acute hyperketonemia on the cerebral uptake of ketone bodies in nondiabetic subjects and IDDM patients. Am. J. Physiol. Endocrinol. Metab. 2002, 283, E20–E28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morris, A.A. Cerebral ketone body metabolism. J. Inherit. Metab. Dis. 2005, 28, 109–121. [Google Scholar] [CrossRef]
- Xin, L.; Ipek, Ö.; Beaumont, M.; Shevlyakova, M.; Christinat, N.; Masoodi, M.; Greenberg, N.; Gruetter, R.; Cuenoud, B. Nutritional Ketosis Increases NAD(+)/NADH Ratio in Healthy Human Brain: An in Vivo Study by (31)P-MRS. Front. Nutr. 2018, 5, 62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bough, K.J.; Wetherington, J.; Hassel, B.; Pare, J.F.; Gawryluk, J.W.; Greene, J.G.; Shaw, R.; Smith, Y.; Geiger, J.D.; Dingledine, R.J. Mitochondrial biogenesis in the anticonvulsant mechanism of the ketogenic diet. Ann. Neurol. 2006, 60, 223–235. [Google Scholar] [CrossRef]
- Page, K.A.; Williamson, A.; Yu, N.; McNay, E.C.; Dzuira, J.; McCrimmon, R.J.; Sherwin, R.S. Medium-Chain Fatty Acids Improve Cognitive Function in Intensively Treated Type 1 Diabetic Patients and Support In Vitro Synaptic Transmission During Acute Hypoglycemia. Diabetes 2009, 58, 1237–1244. [Google Scholar] [CrossRef] [Green Version]
- Veneman, T.; Mitrakou, A.; Mokan, M.; Cryer, P.; Gerich, J. Effect of Hyperketonemia and Hyperlacticacidemia on Symptoms, Cognitive Dysfunction, and Counterregulatory Hormone Responses During Hypoglycemia in Normal Humans. Diabetes 1994, 43, 1311–1317. [Google Scholar] [CrossRef]
- Muddapu, V.R.; Dharshini, S.A.P.; Chakravarthy, V.S.; Gromiha, M.M. Neurodegenerative Diseases—Is Metabolic Deficiency the Root Cause? Front. Neurosci. 2020, 14. [Google Scholar] [CrossRef] [PubMed]
- Bakshi, R.; Miletich, R.S.; Kinkel, P.R.; Emmet, M.L.; Kinkel, W.R. High-Resolution Fluorodeoxyglucose Positron Emission Tomography Shows Both Global and Regional Cerebral Hypometabolism in Multiple Sclerosis. J. Neuroimaging 1998, 8, 228–234. [Google Scholar] [CrossRef] [PubMed]
- Henderson, S.T. Ketone bodies as a therapeutic for Alzheimer’s disease. Neurotherapeutics 2008, 5, 470–480. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gejl, M.; Brock, B.; Egefjord, L.; Vang, K.; Rungby, J.; Gjedde, A. Blood-Brain Glucose Transfer in Alzheimer’s disease: Effect of GLP-1 Analog Treatment. Sci. Rep. 2017, 7, 17490. [Google Scholar] [CrossRef] [Green Version]
- Small, G.W.; Ercoli, L.M.; Silverman, D.H.S.; Huang, S.C.; Komo, S.; Bookheimer, S.Y.; Lavretsky, H.; Miller, K.; Siddarth, P.; Rasgon, N.L.; et al. Cerebral metabolic and cognitive decline in persons at genetic risk for Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2000, 97, 6037–6042. [Google Scholar] [CrossRef] [Green Version]
- Reiman, E.M.; Chen, K.; Alexander, G.E.; Caselli, R.J.; Bandy, D.; Osborne, D.; Saunders, A.M.; Hardy, J. Functional brain abnormalities in young adults at genetic risk for late-onset Alzheimer’s dementia. Proc. Natl. Acad. Sci. USA 2004, 101, 284–289. [Google Scholar] [CrossRef] [Green Version]
- Hwang, J.J.; Jiang, L.; Hamza, M.; Sanchez Rangel, E.; Dai, F.; Belfort-Deaguiar, R.; Parikh, L.; Koo, B.B.; Rothman, D.L.; Mason, G.; et al. Blunted rise in brain glucose levels during hyperglycemia in adults with obesity and T2DM. JCI Insight 2017, 2. [Google Scholar] [CrossRef] [Green Version]
- Kullmann, S.; Heni, M.; Hallschmid, M.; Fritsche, A.; Preissl, H.; Häring, H.-U. Brain Insulin Resistance at the Crossroads of Metabolic and Cognitive Disorders in Humans. Physiol. Rev. 2016, 96, 1169–1209. [Google Scholar] [CrossRef] [Green Version]
- Baker, L.D.; Cross, D.J.; Minoshima, S.; Belongia, D.; Watson, G.S.; Craft, S. Insulin Resistance and Alzheimer-like Reductions in Regional Cerebral Glucose Metabolism for Cognitively Normal Adults With Prediabetes or Early Type 2 Diabetes. Arch. Neurol. 2011, 68. [Google Scholar] [CrossRef]
- Castellano, C.A.; Nugent, S.; Paquet, N.; Tremblay, S.; Bocti, C.; Lacombe, G.; Imbeault, H.; Turcotte, É.; Fulop, T.; Cunnane, S.C. Lower brain 18F-fluorodeoxyglucose uptake but normal 11C-acetoacetate metabolism in mild Alzheimer’s disease dementia. J. Alzheimers Dis. 2015, 43, 1343–1353. [Google Scholar] [CrossRef]
- Croteau, E.; Castellano, C.A.; Fortier, M.; Bocti, C.; Fulop, T.; Paquet, N.; Cunnane, S.C. A cross-sectional comparison of brain glucose and ketone metabolism in cognitively healthy older adults, mild cognitive impairment and early Alzheimer’s disease. Exp. Gerontol. 2018, 107, 18–26. [Google Scholar] [CrossRef] [PubMed]
- Morris, G.; Maes, M.; Berk, M.; Carvalho, A.F.; Puri, B.K. Nutritional ketosis as an intervention to relieve astrogliosis: Possible therapeutic applications in the treatment of neurodegenerative and neuroprogressive disorders. Eur. Psychiatry 2020, 63, e8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fortier, M.; Castellano, C.A.; Croteau, E.; Langlois, F.; Bocti, C.; St-Pierre, V.; Vandenberghe, C.; Bernier, M.; Roy, M.; Descoteaux, M.; et al. A ketogenic drink improves brain energy and some measures of cognition in mild cognitive impairment. Alzheimers Dement. 2019, 15, 625–634. [Google Scholar] [CrossRef] [PubMed]
- Croteau, E.; Castellano, C.A.; Richard, M.A.; Fortier, M.; Nugent, S.; Lepage, M.; Duchesne, S.; Whittingstall, K.; Turcotte, É.E.; Bocti, C.; et al. Ketogenic Medium Chain Triglycerides Increase Brain Energy Metabolism in Alzheimer’s Disease. J. Alzheimers Dis. 2018, 64, 551–561. [Google Scholar] [CrossRef] [PubMed]
- Lacourt, T.E.; Vichaya, E.G.; Chiu, G.S.; Dantzer, R.; Heijnen, C.J. The High Costs of Low-Grade Inflammation: Persistent Fatigue as a Consequence of Reduced Cellular-Energy Availability and Non-adaptive Energy Expenditure. Front. Behav. Neurosci. 2018, 12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rojas-Morales, P.; Pedraza-Chaverri, J.; Tapia, E. Ketone bodies, stress response, and redox homeostasis. Redox Biol. 2020, 29, 101395. [Google Scholar] [CrossRef]
- Haces, M.L.; Hernández-Fonseca, K.; Medina-Campos, O.N.; Montiel, T.; Pedraza-Chaverri, J.; Massieu, L. Antioxidant capacity contributes to protection of ketone bodies against oxidative damage induced during hypoglycemic conditions. Exp. Neurol. 2008, 211, 85–96. [Google Scholar] [CrossRef]
- Maalouf, M.; Sullivan, P.G.; Davis, L.; Kim, D.Y.; Rho, J.M. Ketones inhibit mitochondrial production of reactive oxygen species production following glutamate excitotoxicity by increasing NADH oxidation. Neuroscience 2007, 145, 256–264. [Google Scholar] [CrossRef] [Green Version]
- Pinto, A.; Bonucci, A.; Maggi, E.; Corsi, M.; Businaro, R. Anti-Oxidant and Anti-Inflammatory Activity of Ketogenic Diet: New Perspectives for Neuroprotection in Alzheimer’s Disease. Antioxidants 2018, 7, 63. [Google Scholar] [CrossRef] [Green Version]
- Elamin, M.; Ruskin, D.N.; Masino, S.A.; Sacchetti, P. Ketone-Based Metabolic Therapy: Is Increased NAD(+) a Primary Mechanism? Front. Mol. Neurosci. 2017, 10, 377. [Google Scholar] [CrossRef]
- Tieu, K.; Perier, C.; Caspersen, C.; Teismann, P.; Wu, D.-C.; Yan, S.-D.; Naini, A.; Vila, M.; Jackson-Lewis, V.; Ramasamy, R.; et al. D-β-Hydroxybutyrate rescues mitochondrial respiration and mitigates features of Parkinson disease. J. Clin. Investig. 2003, 112, 892–901. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kashiwaya, Y.; Pawlosky, R.; Markis, W.; King, M.T.; Bergman, C.; Srivastava, S.; Murray, A.; Clarke, K.; Veech, R.L. A Ketone Ester Diet Increases Brain Malonyl-CoA and Uncoupling Proteins 4 and 5 while Decreasing Food Intake in the Normal Wistar Rat. J. Biol. Chem. 2010, 285, 25950–25956. [Google Scholar] [CrossRef] [Green Version]
- Sullivan, P.G.; Rippy, N.A.; Dorenbos, K.; Concepcion, R.C.; Agarwal, A.K.; Rho, J.M. The ketogenic diet increases mitochondrial uncoupling protein levels and activity. Ann. Neurol. 2004, 55, 576–580. [Google Scholar] [CrossRef] [PubMed]
- Andrews, Z.B.; Diano, S.; Horvath, T.L. Mitochondrial uncoupling proteins in the cns: In support of function and survival. Nat. Rev. Neurosci. 2005, 6, 829–840. [Google Scholar] [CrossRef] [PubMed]
- Fu, S.P.; Wang, J.F.; Xue, W.J.; Liu, H.M.; Liu, B.R.; Zeng, Y.L.; Li, S.N.; Huang, B.X.; Lv, Q.K.; Wang, W.; et al. Anti-inflammatory effects of BHBA in both in vivo and in vitro Parkinson’s disease models are mediated by GPR109A-dependent mechanisms. J. Neuroinflamm. 2015, 12, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, Y.; Gong, Y.; Luan, Y.; Li, Y.; Liu, J.; Yue, Z.; Yuan, B.; Sun, J.; Xie, C.; Li, L.; et al. BHBA treatment improves cognitive function by targeting pleiotropic mechanisms in transgenic mouse model of Alzheimer’s disease. FASEB J. 2020, 34, 1412–1429. [Google Scholar] [CrossRef] [Green Version]
- Jensen, N.J.; Nilsson, M.; Ingerslev, J.S.; Olsen, D.A.; Fenger, M.; Svart, M.; Møller, N.; Zander, M.; Miskowiak, K.W.; Rungby, J. Effects of β-hydroxybutyrate on cognition in patients with type 2 diabetes. Eur. J. Endocrinol. 2020, 182, 233–242. [Google Scholar] [CrossRef]
- Reger, M.A.; Henderson, S.T.; Hale, C.; Cholerton, B.; Baker, L.D.; Watson, G.S.; Hyde, K.; Chapman, D.; Craft, S. Effects of beta-hydroxybutyrate on cognition in memory-impaired adults. Neurobiol. Aging 2004, 25, 311–314. [Google Scholar] [CrossRef]
- Ota, M.; Matsuo, J.; Ishida, I.; Hattori, K.; Teraishi, T.; Tonouchi, H.; Ashida, K.; Takahashi, T.; Kunugi, H. Effect of a ketogenic meal on cognitive function in elderly adults: Potential for cognitive enhancement. Psychopharmacology 2016, 233, 3797–3802. [Google Scholar] [CrossRef]
- Ota, M.; Matsuo, J.; Ishida, I.; Takano, H.; Yokoi, Y.; Hori, H.; Yoshida, S.; Ashida, K.; Nakamura, K.; Takahashi, T.; et al. Effects of a medium-chain triglyceride-based ketogenic formula on cognitive function in patients with mild-to-moderate Alzheimer’s disease. Neurosci. Lett. 2019, 690, 232–236. [Google Scholar] [CrossRef]
- Henderson, S.T.; Vogel, J.L.; Barr, L.J.; Garvin, F.; Jones, J.J.; Costantini, L.C. Study of the ketogenic agent AC-1202 in mild to moderate Alzheimer’s disease: A randomized, double-blind, placebo-controlled, multicenter trial. Nutr. Metab. 2009, 6, 31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, Q.; Zhang, Y.; Zhang, X.; Liu, L.; Zhou, B.; Mo, R.; Li, Y.; Li, H.; Li, F.; Tao, Y.; et al. Medium-chain triglycerides improved cognition and lipid metabolomics in mild to moderate Alzheimer’s disease patients with APOE4(-/-): A double-blind, randomized, placebo-controlled crossover trial. Clin. Nutr. 2020, 39, 2092–2105. [Google Scholar] [CrossRef] [PubMed]
- Fortier, M.; Castellano, C.A.; St-Pierre, V.; Myette-Côté, É.; Langlois, F.; Roy, M.; Morin, M.C.; Bocti, C.; Fulop, T.; Godin, J.P.; et al. A ketogenic drink improves cognition in mild cognitive impairment: Results of a 6-month RCT. Alzheimer’s Dement. 2020. [Google Scholar] [CrossRef] [PubMed]
- Henderson, S.T.; Morimoto, B.H.; Cummings, J.L.; Farlow, M.R.; Walker, J. A Placebo-Controlled, Parallel-Group, Randomized Clinical Trial of AC-1204 in Mild-to-Moderate Alzheimer’s Disease. J. Alzheimers Dis. 2020, 75, 547–557. [Google Scholar] [CrossRef]
- Abe, S.; Ezaki, O.; Suzuki, M. Medium-Chain Triglycerides (8:0 and 10:0) Increase Mini-Mental State Examination (MMSE) Score in Frail Elderly Adults in a Randomized Controlled Trial. J. Nutr. 2020, 150, 2383–2390. [Google Scholar] [CrossRef]
- de la Rubia Ortí, J.E.; García-Pardo, M.P.; Drehmer, E.; Sancho Cantus, D.; Julián Rochina, M.; Aguilar, M.A.; Hu Yang, I. Improvement of Main Cognitive Functions in Patients with Alzheimer’s Disease after Treatment with Coconut Oil Enriched Mediterranean Diet: A Pilot Study. J. Alzheimers Dis. 2018, 65, 577–587. [Google Scholar] [CrossRef]
- Krikorian, R.; Shidler, M.D.; Dangelo, K.; Couch, S.C.; Benoit, S.C.; Clegg, D.J. Dietary ketosis enhances memory in mild cognitive impairment. Neurobiol. Aging 2012, 33, 425.e419–425.e427. [Google Scholar] [CrossRef] [Green Version]
- Brandt, J.; Buchholz, A.; Henry-Barron, B.; Vizthum, D.; Avramopoulos, D.; Cervenka, M.C. Preliminary Report on the Feasibility and Efficacy of the Modified Atkins Diet for Treatment of Mild Cognitive Impairment and Early Alzheimer’s Disease. J. Alzheimers Dis. 2019, 68, 969–981. [Google Scholar] [CrossRef]
- Taylor, M.K.; Sullivan, D.K.; Mahnken, J.D.; Burns, J.M.; Swerdlow, R.H. Feasibility and efficacy data from a ketogenic diet intervention in Alzheimer’s disease. Alzheimers Dement. 2018, 4, 28–36. [Google Scholar] [CrossRef]
- Zhang, J.; Chen, C.; Hua, S.; Liao, H.; Wang, M.; Xiong, Y.; Cao, F. An updated meta-analysis of cohort studies: Diabetes and risk of Alzheimer’s disease. Diabetes Res. Clin. Pract. 2017, 124, 41–47. [Google Scholar] [CrossRef]
- Wium-Andersen, I.K.; Rungby, J.; Jørgensen, M.B.; Sandbæk, A.; Osler, M.; Wium-Andersen, M.K. Risk of dementia and cognitive dysfunction in individuals with diabetes or elevated blood glucose. Epidemiol. Psychiatr. Sci. 2020, 29, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castellano, C.-A.; Hudon, C.; Croteau, E.; Fortier, M.; St-Pierre, V.; Vandenberghe, C.; Nugent, S.; Tremblay, S.; Paquet, N.; Lepage, M.; et al. Links Between Metabolic and Structural Changes in the Brain of Cognitively Normal Older Adults: A 4-Year Longitudinal Follow-Up. Front. Aging Neurosci. 2019, 11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Newport, M.T.; VanItallie, T.B.; Kashiwaya, Y.; King, M.T.; Veech, R.L. A new way to produce hyperketonemia: Use of ketone ester in a case of Alzheimer’s disease. Alzheimers Dement. 2015, 11, 99–103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lassen, N.A.; Feinberg, I.; Lane, M.H. Bilateral studies of cerebral oxygen uptake in young and aged normal subjects and in patients with organic dementia. J. Clin. Investig. 1960, 39, 491–500. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lying-Tunell, U.; Lindblad, B.S.; Malmlund, H.O.; Persson, B. Cerebral blood flow and metabolic rate of oxygen, glucose, lactate, pyruvate, ketone bodies and amino acids. Acta Neurol. Scand. 1981, 63, 337–350. [Google Scholar] [CrossRef]
- Neth, B.J.; Mintz, A.; Whitlow, C.; Jung, Y.; Solingapuram Sai, K.; Register, T.C.; Kellar, D.; Lockhart, S.N.; Hoscheidt, S.; Maldjian, J.; et al. Modified ketogenic diet is associated with improved cerebrospinal fluid biomarker profile, cerebral perfusion, and cerebral ketone body uptake in older adults at risk for Alzheimer’s disease: A pilot study. Neurobiol. Aging 2020, 86, 54–63. [Google Scholar] [CrossRef]
- Nagpal, R.; Neth, B.J.; Wang, S.; Craft, S.; Yadav, H. Modified Mediterranean-ketogenic diet modulates gut microbiome and short-chain fatty acids in association with Alzheimer’s disease markers in subjects with mild cognitive impairment. EBioMedicine 2019, 47, 529–542. [Google Scholar] [CrossRef] [Green Version]
- Torosyan, N.; Sethanandha, C.; Grill, J.D.; Dilley, M.L.; Lee, J.; Cummings, J.L.; Ossinalde, C.; Silverman, D.H. Changes in regional cerebral blood flow associated with a 45day course of the ketogenic agent, caprylidene, in patients with mild to moderate Alzheimer’s disease: Results of a randomized, double-blinded, pilot study. Exp. Gerontol. 2018, 111, 118–121. [Google Scholar] [CrossRef]
- Yang, X.; Cheng, B. Neuroprotective and anti-inflammatory activities of ketogenic diet on MPTP-induced neurotoxicity. J. Mol. Neurosci. 2010, 42, 145–153. [Google Scholar] [CrossRef]
- Cheng, B.; Yang, X.; An, L.; Gao, B.; Liu, X.; Liu, S. Ketogenic diet protects dopaminergic neurons against 6-OHDA neurotoxicity via up-regulating glutathione in a rat model of Parkinson’s disease. Brain Res. 2009, 1286, 25–31. [Google Scholar] [CrossRef]
- Shaafi, S.; Najmi, S.; Aliasgharpour, H.; Mahmoudi, J.; Sadigh-Etemad, S.; Farhoudi, M.; Baniasadi, N. The efficacy of the ketogenic diet on motor functions in Parkinson’s disease: A rat model. Iran. J. Neurol. 2016, 15, 63–69. [Google Scholar] [PubMed]
- Vanitallie, T.B.; Nonas, C.; Di Rocco, A.; Boyar, K.; Hyams, K.; Heymsfield, S.B. Treatment of Parkinson disease with diet-induced hyperketonemia: A feasibility study. Neurology 2005, 64, 728–730. [Google Scholar] [CrossRef]
- Koyuncu, H.; Fidan, V.; Toktas, H.; Binay, O.; Celik, H. Effect of ketogenic diet versus regular diet on voice quality of patients with Parkinson’s disease. Acta Neurol. Belg. 2020. [Google Scholar] [CrossRef] [PubMed]
- Phillips, M.C.L.; Murtagh, D.K.J.; Gilbertson, L.J.; Asztely, F.J.S.; Lynch, C.D.P. Low-fat versus ketogenic diet in Parkinson’s disease: A pilot randomized controlled trial. Mov. Disord. 2018, 33, 1306–1314. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.; Lange, D.J.; Voustianiouk, A.; MacGrogan, D.; Ho, L.; Suh, J.; Humala, N.; Thiyagarajan, M.; Wang, J.; Pasinetti, G.M. A ketogenic diet as a potential novel therapeutic intervention in amyotrophic lateral sclerosis. BMC Neurosci. 2006, 7, 29. [Google Scholar] [CrossRef] [Green Version]
- Zhao, W.; Varghese, M.; Vempati, P.; Dzhun, A.; Cheng, A.; Wang, J.; Lange, D.; Bilski, A.; Faravelli, I.; Pasinetti, G.M. Caprylic triglyceride as a novel therapeutic approach to effectively improve the performance and attenuate the symptoms due to the motor neuron loss in ALS disease. PLoS ONE 2012, 7, e49191. [Google Scholar] [CrossRef]
- Santa-Cruz, L.D.; Tapia, R. Role of energy metabolic deficits and oxidative stress in excitotoxic spinal motor neuron degeneration in vivo. ASN Neuro 2014, 6. [Google Scholar] [CrossRef]
- Netzahualcoyotzi, C.; Tapia, R. Degeneration of spinal motor neurons by chronic AMPA-induced excitotoxicity in vivo and protection by energy substrates. Acta Neuropathol. Commun. 2015, 3, 27. [Google Scholar] [CrossRef] [Green Version]
- Lim, S.; Chesser, A.S.; Grima, J.C.; Rappold, P.M.; Blum, D.; Przedborski, S.; Tieu, K. D-β-hydroxybutyrate is protective in mouse models of Huntington’s disease. PLoS ONE 2011, 6, e24620. [Google Scholar] [CrossRef] [Green Version]
- Ruskin, D.N.; Ross, J.L.; Kawamura, M., Jr.; Ruiz, T.L.; Geiger, J.D.; Masino, S.A. A ketogenic diet delays weight loss and does not impair working memory or motor function in the R6/2 1J mouse model of Huntington’s disease. Physiol. Behav. 2011, 103, 501–507. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.Y.; Tran, C.; Hwang, L.; Deng, G.; Jung, M.E.; Faull, K.F.; Levine, M.S.; Cepeda, C. Partial Amelioration of Peripheral and Central Symptoms of Huntington’s Disease via Modulation of Lipid Metabolism. J. Huntingt. Dis. 2016, 5, 65–81. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferrannini, E.; Baldi, S.; Frascerra, S.; Astiarraga, B.; Barsotti, E.; Clerico, A.; Muscelli, E. Renal Handling of Ketones in Response to Sodium–Glucose Cotransporter 2 Inhibition in Patients With Type 2 Diabetes. Diabetes Care 2017. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wium-Andersen, I.K.; Osler, M.; Jørgensen, M.B.; Rungby, J.; Wium-Andersen, M.K. Antidiabetic medication and risk of dementia in patients with type 2 diabetes: A nested case–control study. Eur. J. Endocrinol. 2019, 181, 499–507. [Google Scholar] [CrossRef] [PubMed]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jensen, N.J.; Wodschow, H.Z.; Nilsson, M.; Rungby, J. Effects of Ketone Bodies on Brain Metabolism and Function in Neurodegenerative Diseases. Int. J. Mol. Sci. 2020, 21, 8767. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21228767
Jensen NJ, Wodschow HZ, Nilsson M, Rungby J. Effects of Ketone Bodies on Brain Metabolism and Function in Neurodegenerative Diseases. International Journal of Molecular Sciences. 2020; 21(22):8767. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21228767
Chicago/Turabian StyleJensen, Nicole Jacqueline, Helena Zander Wodschow, Malin Nilsson, and Jørgen Rungby. 2020. "Effects of Ketone Bodies on Brain Metabolism and Function in Neurodegenerative Diseases" International Journal of Molecular Sciences 21, no. 22: 8767. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21228767