Role of NEU3 Overexpression in the Prediction of Efficacy of EGFR-Targeted Therapies in Colon Cancer Cell Lines

 , and
, and
Abstract
:1. Introduction
2. Results
2.1. Mutational Status of Colorectal Cell Lines
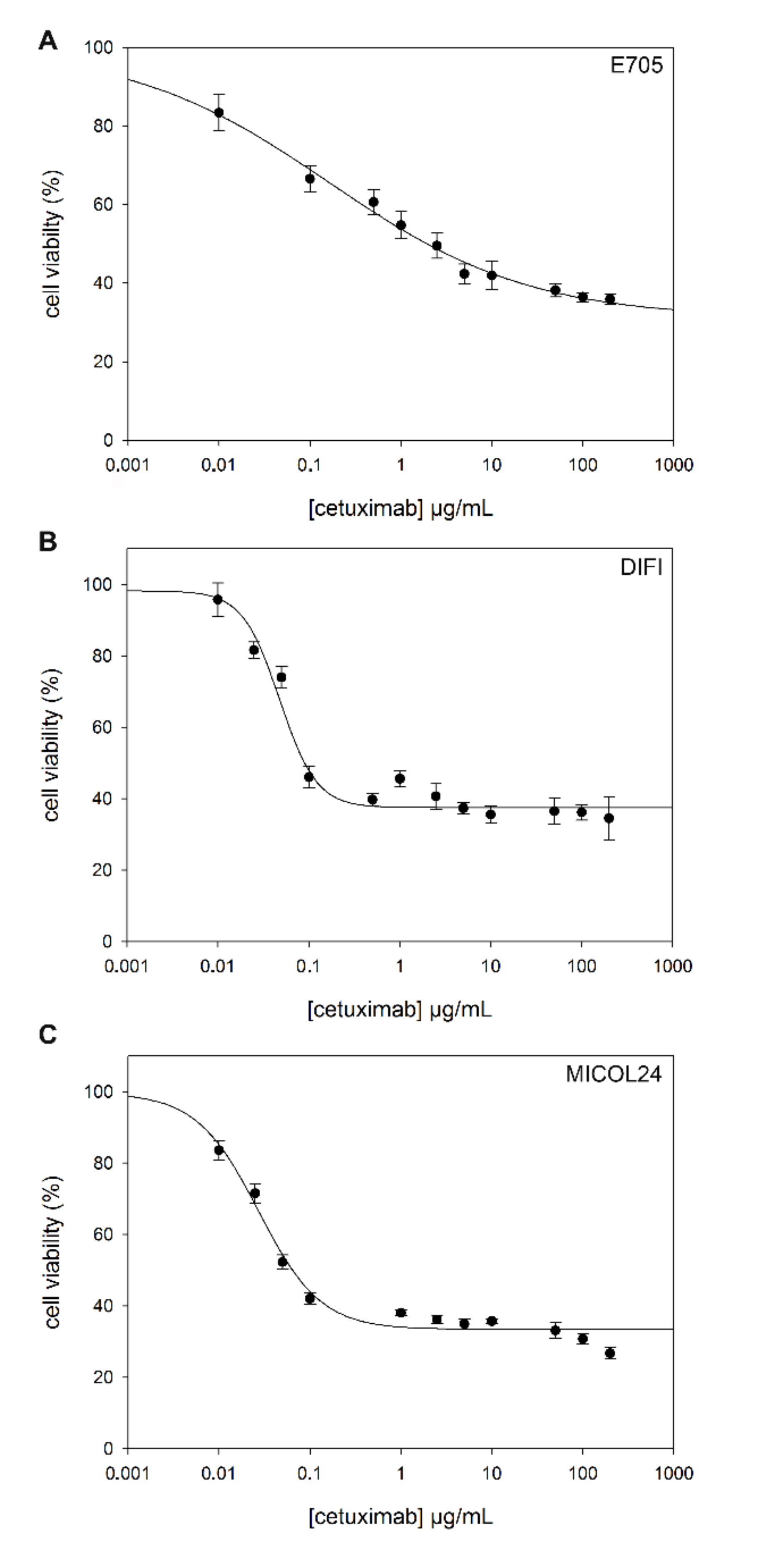
2.2. Sensitivity to Cetuximab
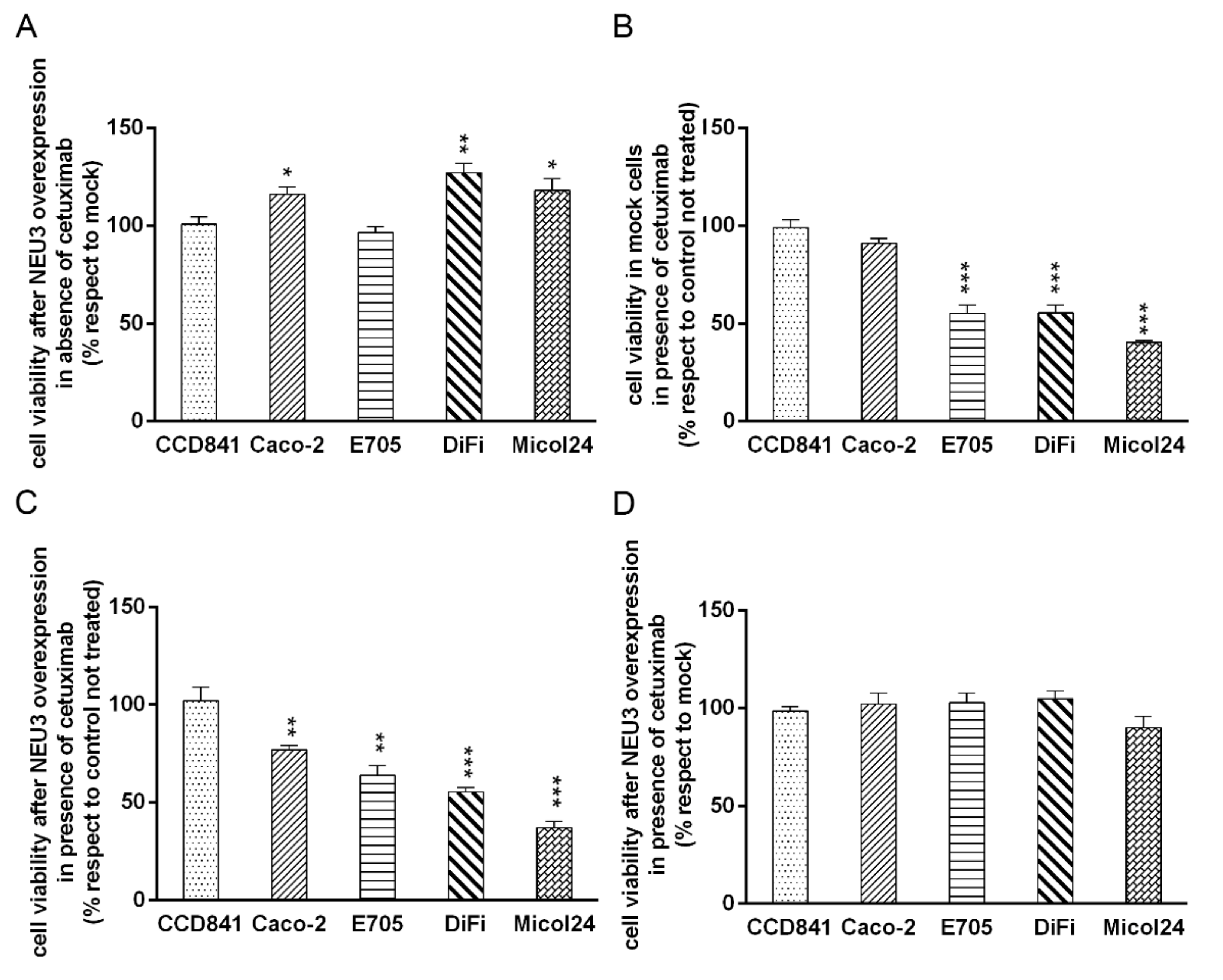
2.3. NEU3 Overexpression Effect on Cell Viability in the Presence or the Absence of Cetuximab
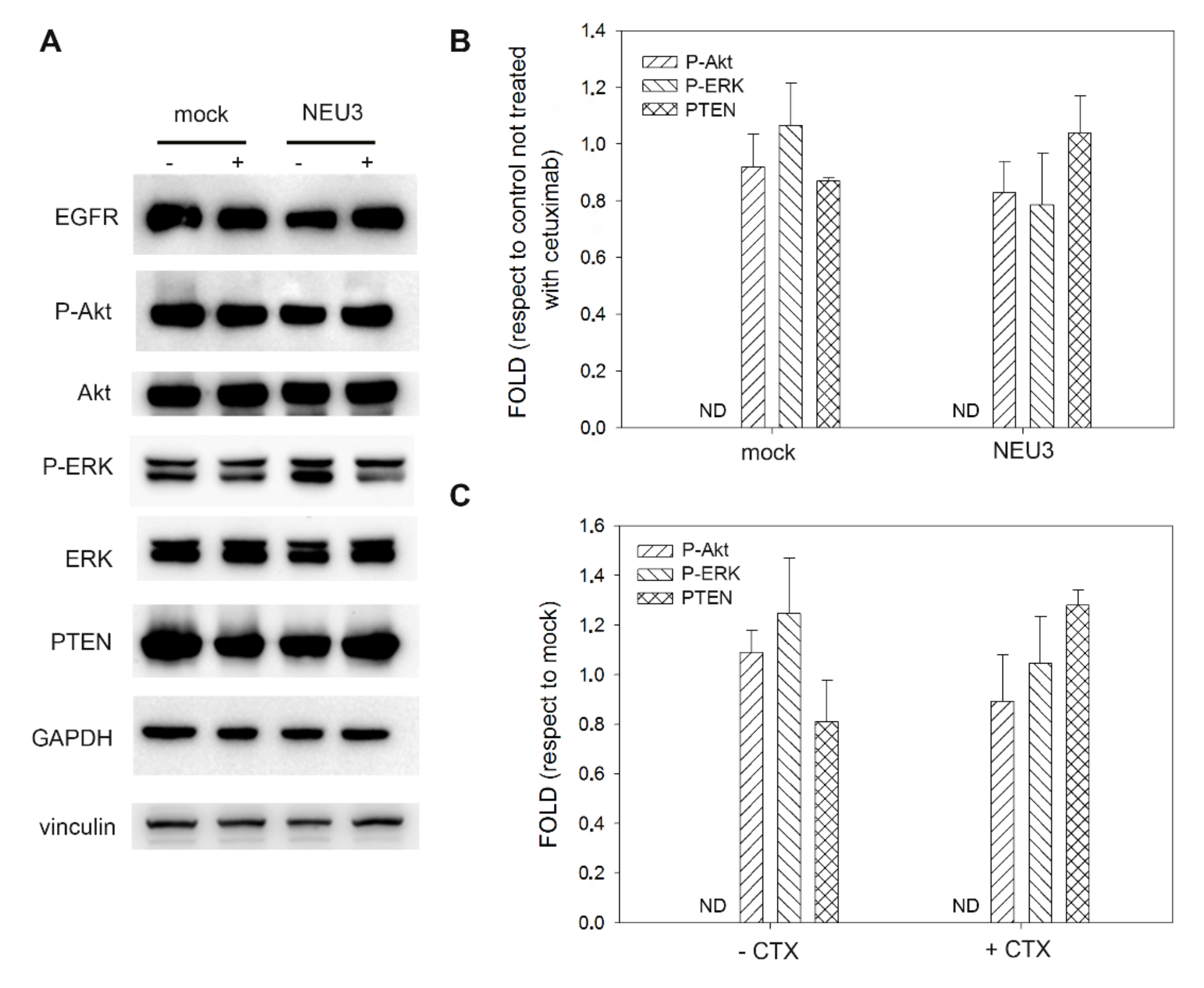
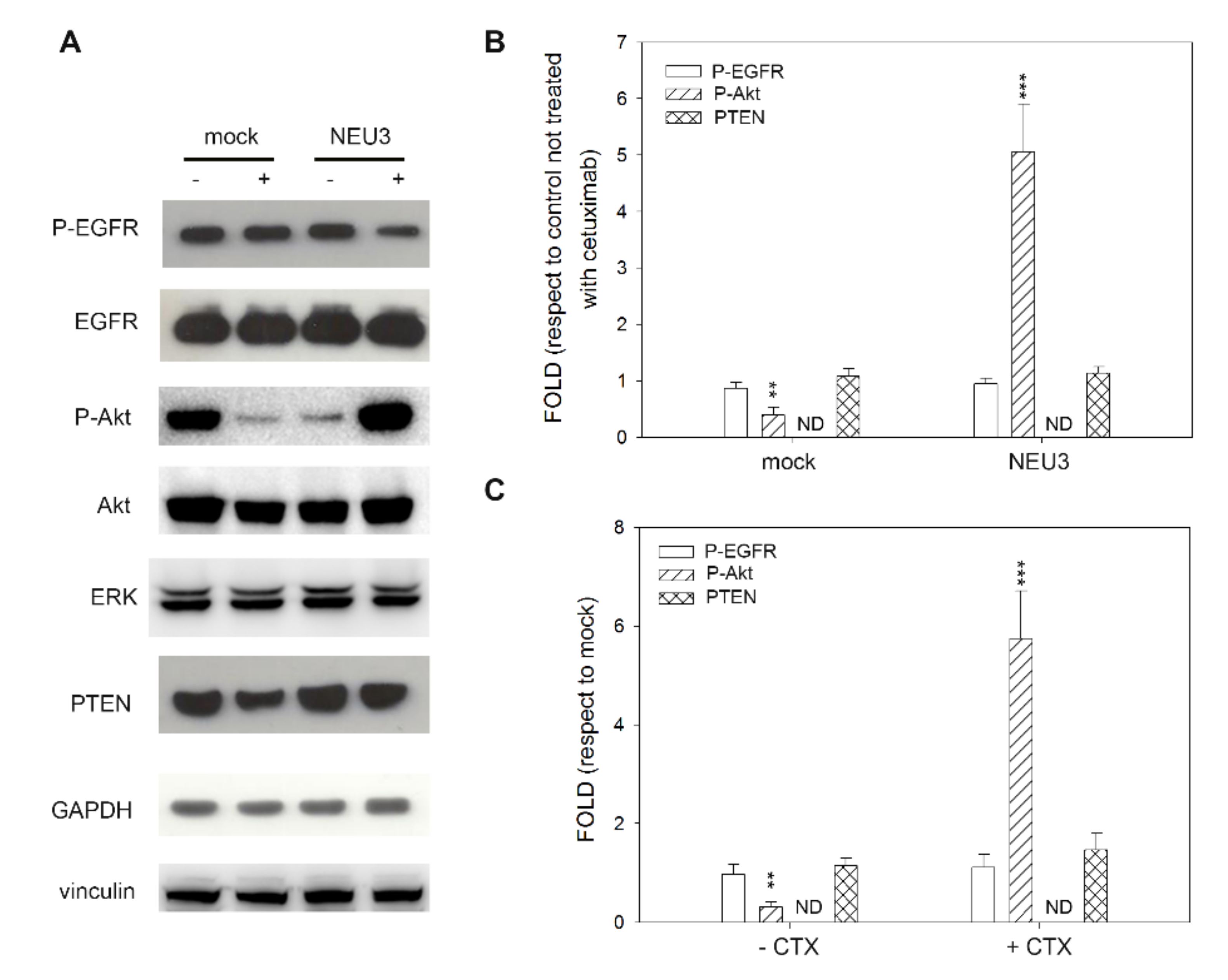
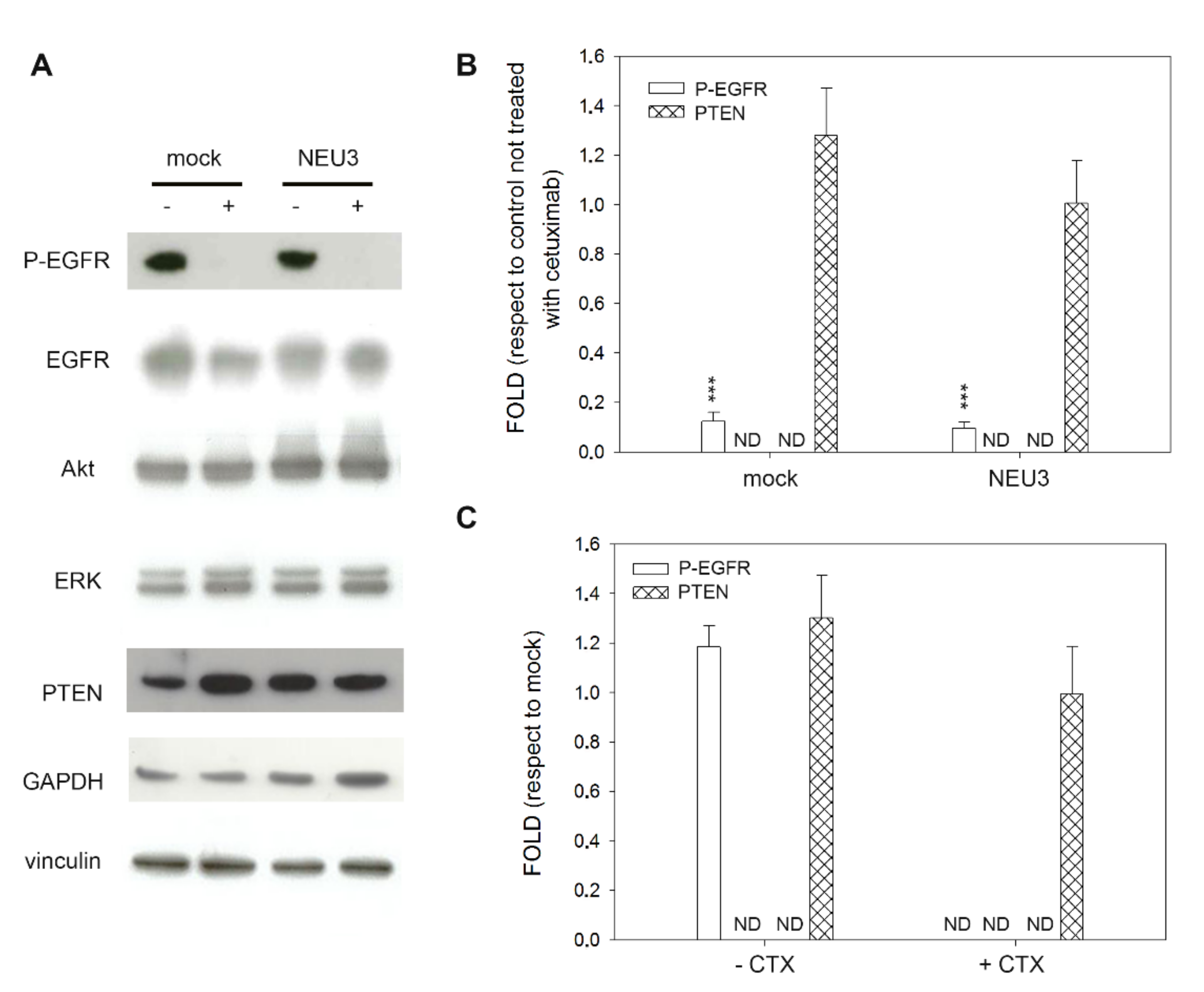
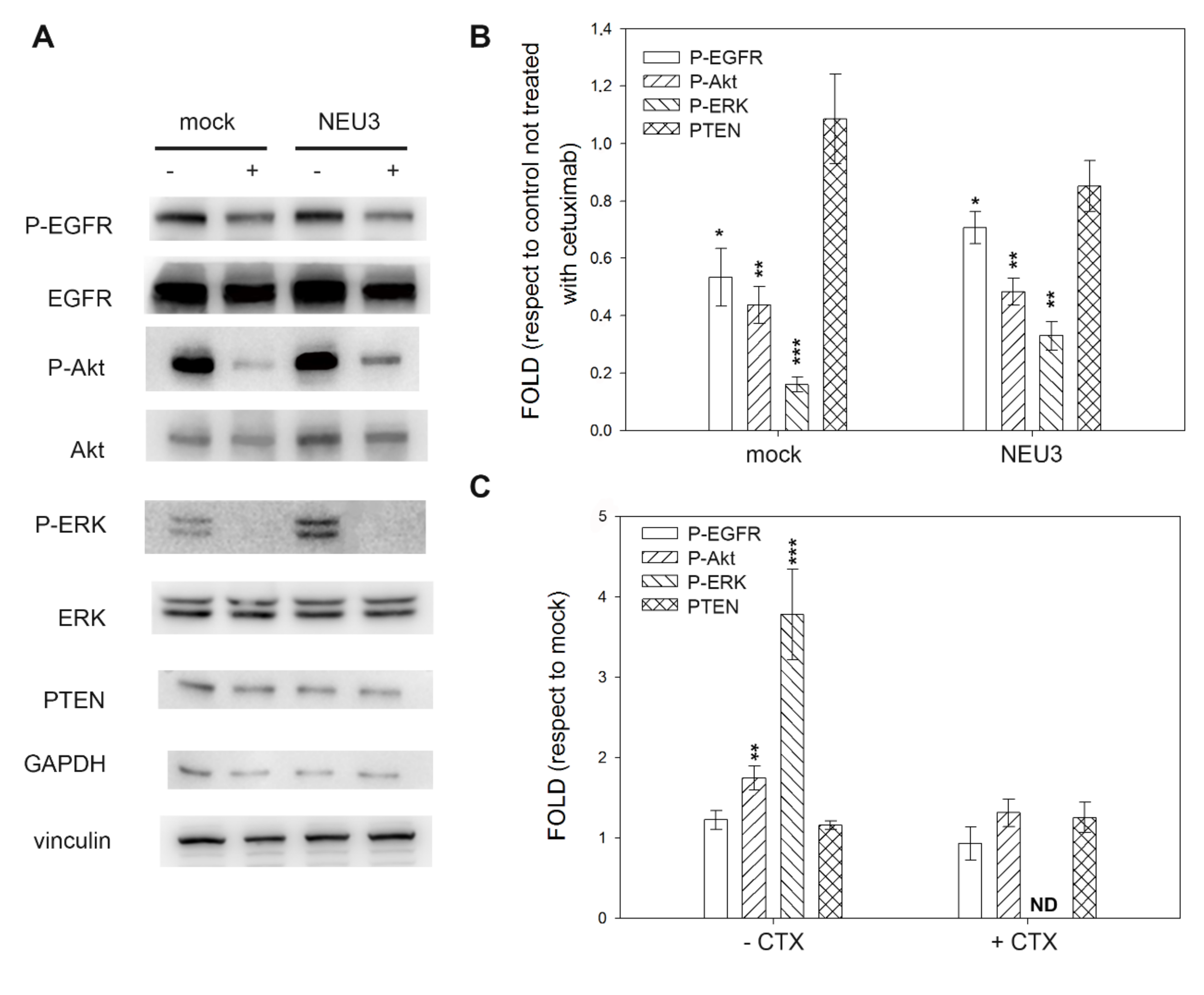
2.4. Effect of NEU3 Overexpression and Cetuximab on EGFR, Akt and ERK Phosphorylation
3. Discussion
4. Materials and Methods
4.1. Cell Cultures
4.2. Mutational Status of EGFR Pathway
4.3. Vector
4.4. Transfection
4.5. Viability Assay
4.6. SDS-PAGE and Western Blot
4.7. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Jorissen, R.N.; Walker, F.; Pouliot, N.; Garrett, T.P.; Ward, C.W.; Burgess, A.W. Epidermal growth factor receptor: Mechanisms of activation and signalling. Exp. Cell Res. 2003, 284, 31–53. [Google Scholar] [CrossRef]
- Yoshida, M.; Shimura, T.; Sato, M.; Ebi, M.; Nakazawa, T.; Takeyama, H.; Joh, T. A novel predictive strategy by immunohistochemical analysis of four EGFR ligands in metastatic colorectal cancer treated with anti-EGFR antibodies. J. Cancer Res. Clin. Oncol. 2013, 139, 367–378. [Google Scholar] [CrossRef] [PubMed]
- Price, T.J.; Tang, M.; Gibbs, P.; Haller, D.G.; Peeters, M.; Arnold, D.; Segelov, E.; Roy, A.; Tebbutt, N.; Pavlakis, N.; et al. Targeted therapy for metastatic colorectal cancer. Expert Rev. Anticancer Ther. 2018, 18, 991–1006. [Google Scholar] [CrossRef] [PubMed]
- Zhao, B.; Wang, L.; Qiu, H.; Zhang, M.; Sun, L.; Peng, P.; Yu, Q.; Yuan, X. Mechanisms of resistance to anti-EGFR therapy in colorectal cancer. Oncotarget 2017, 8, 3980–4000. [Google Scholar] [CrossRef] [Green Version]
- Dienstmann, R.; Salazar, R.; Tabernero, J. Overcoming Resistance to Anti-EGFR Therapy in Colorectal Cancer. Am. Soc. Clin. Oncol. Educ. B 2015, e149–e156. [Google Scholar] [CrossRef] [Green Version]
- Gong, J.; Cho, M.; Fakih, M. RAS and BRAF in metastatic colorectal cancer management. J. Gastrointest. Oncol. 2016, 7, 687–704. [Google Scholar] [CrossRef] [Green Version]
- Van Emburgh, B.O.; Arena, S.; Siravegna, G.; Lazzari, L.; Crisafulli, G.; Corti, G.; Mussolin, B.; Baldi, F.; Buscarino, M.; Bartolini, A.; et al. Acquired RAS or EGFR mutations and duration of response to EGFR blockade in colorectal cancer. Nat. Commun. 2016, 7, 1–9. [Google Scholar] [CrossRef]
- Modest, D.P.; Pant, S.; Sartore-Bianchi, A. Treatment sequencing in metastatic colorectal cancer. Eur. J. Cancer 2019, 109, 70–83. [Google Scholar] [CrossRef]
- Ålgars, A.; Sundström, J.; Lintunen, M.; Jokilehto, T.; Kytölä, S.; Kaare, M.; Vainionpää, R.; Orpana, A.; Österlund, P.; Ristimäki, A.; et al. EGFR gene copy number predicts response to anti-EGFR treatment in RAS wild type and RAS/BRAF/PIK3CA wild type metastatic colorectal cancer. Int. J. Cancer 2017, 140, 922–929. [Google Scholar] [CrossRef] [Green Version]
- Pietrantonio, F.; Petrelli, F.; Coinu, A.; Di Bartolomeo, M.; Borgonovo, K.; Maggi, C.; Cabiddu, M.; Iacovelli, R.; Bossi, I.; Lonati, V.; et al. Predictive role of BRAF mutations in patients with advanced colorectal cancer receiving cetuximab and panitumumab: A meta-analysis. Eur. J. Cancer 2015, 51, 587–594. [Google Scholar] [CrossRef]
- Rossi, A.; Bria, E.; Maione, P.; Palazzolo, G.; Falanga, M.; Gridelli, C. The Role of Cetuximab and Other Epidermal Growth Factor Receptor Monoclonal Antibodies in the Treatment of Advanced Non-Small Cell Lung Cancer. Rev. Recent Clin. Trials 2008, 3, 217–227. [Google Scholar] [CrossRef] [PubMed]
- Mukohara, T.; Engelman, J.A.; Hanna, N.H.; Yeap, B.Y.; Kobayashi, S.; Lindeman, N.; Halmos, B.; Pearlberg, J.; Tsuchihashi, Z.; Cantley, L.C.; et al. Differential Effects of Gefitinib and Cetuximab on Non–small-cell Lung Cancers Bearing Epidermal Growth Factor Receptor Mutations. J. Natl. Cancer Inst. 2005, 97, 1185–1194. [Google Scholar] [CrossRef] [PubMed]
- Schauer, R. Sialic acids as regulators of molecular and cellular interactions. Curr. Opin. Struct. Biol. 2009, 19, 507–514. [Google Scholar] [CrossRef] [PubMed]
- Buschiazzo, A.; Alzari, P.M. Structural insights into sialic acid enzymology. Curr. Opin. Chem. Biol. 2008, 12, 565–572. [Google Scholar] [CrossRef]
- Miljan, E.A.; Meuillet, E.J.; Mania-Farnell, B.; George, D.; Yamamoto, H.; Simon, H.G.; Bremer, E.G. Interaction of the extracellular domain of the epidermal growth factor receptor with gangliosides. J. Biol. Chem. 2002, 277, 10108–10113. [Google Scholar] [CrossRef] [Green Version]
- Zhen, Y.; Caprioli, R.M.; Staros, J. V Characterization of glycosylation sites of the epidermal growth factor receptor. Biochemistry 2003, 42, 5478–5492. [Google Scholar] [CrossRef] [Green Version]
- Yoon, S.J.; Nakayama, K.; Hikita, T.; Handa, K.; Hakomori, S.I. Epidermal growth factor receptor tyrosine kinase is modulated by GM3 interaction with N-linked GlcNAc termini of the receptor. Proc. Natl. Acad. Sci. USA 2006, 103, 18987–18991. [Google Scholar] [CrossRef] [Green Version]
- Anastasia, L.; Papini, N.; Colazzo, F.; Palazzolo, G.; Tringali, C.; Dileo, L.; Piccoli, M.; Conforti, E.; Sitzia, C.; Monti, E.; et al. NEU3 Sialidase Strictly Modulates GM3 Levels in Skeletal Myoblasts C2C12 Thus Favoring Their Differentiation and Protecting Them from Apoptosis. J. Biol. Chem. 2008, 283, 36265–36271. [Google Scholar] [CrossRef] [Green Version]
- Miyagi, T.; Takahashi, K.; Moriya, S.; Hata, K.; Yamamoto, K.; Wada, T.; Yamaguchi, K.; Shiozaki, K. Altered expression of sialidases in human cancer. Adv. Exp. Med. Biol. 2012, 749, 257–267. [Google Scholar] [CrossRef]
- Liu, Y.C.; Yen, H.Y.; Chen, C.Y.; Chen, C.H.; Cheng, P.F.; Juan, Y.H.; Chen, C.H.; Khoo, K.H.; Yu, C.J.; Yang, P.C.; et al. Sialylation and fucosylation of epidermal growth factor receptor suppress its dimerization and activation in lung cancer cells. Proc. Natl. Acad. Sci. USA 2011, 108, 11332–11337. [Google Scholar] [CrossRef] [Green Version]
- Mozzi, A.; Forcella, M.; Riva, A.; Difrancesco, C.; Molinari, F.; Martin, V.; Papini, N.; Bernasconi, B.; Nonnis, S.; Tedeschi, G.; et al. NEU3 activity enhances EGFR activation without affecting EGFR expression and acts on its sialylation levels. Glycobiology 2015, 25, 855–868. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Forcella, M.; Oldani, M.; Epistolio, S.; Freguia, S.; Monti, E.; Fusi, P.; Frattini, M. Non-small cell lung cancer (NSCLC), EGFR downstream pathway activation and TKI targeted therapies sensitivity: Effect of the plasma membrane-associated NEU3. PLoS ONE 2017, 12, e0187289. [Google Scholar] [CrossRef] [PubMed]
- Van Cutsem, E.; Cervantes, A.; Adam, R.; Sobrero, A.; Van Krieken, J.H.; Aderka, D.; Aranda Aguilar, E.; Bardelli, A.; Benson, A.; Bodoky, G.; et al. ESMO consensus guidelines for the management of patients with metastatic colorectal cancer. Ann. Oncol. 2016, 27, 1386–1422. [Google Scholar] [CrossRef] [PubMed]
- Martin, V.; Botta, F.; Zanellato, E.; Molinari, F.; Crippa, S.; Mazzucchelli, L.; Frattini, M. Molecular characterization of EGFR and EGFR-downstream pathways in triple negative breast carcinomas with basal like features. Histol. Histopathol. 2012, 27, 785–792. [Google Scholar] [CrossRef]
- Sartore-Bianchi, A.; Fieuws, S.; Veronese, S.; Moroni, M.; Personeni, N.; Frattini, M.; Torri, V.; Cappuzzo, F.; Vander Borght, S.; Martin, V.; et al. Standardisation of EGFR FISH in colorectal cancer: Results of an international interlaboratory reproducibility ring study. J. Clin. Pathol. 2012, 65, 218–223. [Google Scholar] [CrossRef] [Green Version]
- Howlader, M.A.; Guo, T.; Chakraberty, R.; Cairo, C.W. Isoenzyme-Selective Inhibitors of Human Neuraminidases Reveal Distinct Effects on Cell Migration. ACS Chem. Biol. 2020, 15, 1328–1339. [Google Scholar] [CrossRef]
- Iioka, H.; Saito, K.; Kondo, E. Crumbs3 regulates the expression of glycosphingolipids on the plasma membrane to promote colon cancer cell migration. Biochem. Biophys. Res. Commun. 2019, 519, 287–293. [Google Scholar] [CrossRef]
- Park, J.J.; Yi, J.Y.; Jin, Y.B.; Lee, Y.J.; Lee, J.S.; Lee, Y.S.; Ko, Y.G.; Lee, M. Sialylation of epidermal growth factor receptor regulates receptor activity and chemosensitivity to gefitinib in colon cancer cells. Biochem. Pharmacol. 2012, 83, 849–857. [Google Scholar] [CrossRef]
- Uhlyarik, A.; Piurko, V.; Vizkeleti, L.; Pápai, Z.; Rásó, E.; Lahm, E.; Kiss, E.; Sikter, M.; Vachaja, J.; Kenessey, I.; et al. EGFR Protein Expression of KRAS Wild-Type Colorectal Cancer: Predictive Value of the Sidedness for Efficacy of Anti-EGFR Therapy. Pathol. Oncol. Res. 2020, 26, 1429–1434. [Google Scholar] [CrossRef]
- Arnold, D.; Lueza, B.; Douillard, J.Y.; Peeters, M.; Lenz, H.J.; Venook, A.; Heinemann, V.; Van Cutsem, E.; Pignon, J.P.; Tabernero, J.; et al. Prognostic and predictive value of primary tumour side in patients with RAS wild-type metastatic colorectal cancer treated with chemotherapy and EGFR directed antibodies in six randomized trials. Ann. Oncol. 2017, 28, 1713–1729. [Google Scholar] [CrossRef]
- Chen, D.; Zhang, X.; Gao, G.; Shen, L.; Xie, J.; Qian, X.; Wang, H. Should anti-EGFR mAbs be discontinued for conversion surgery in untreated right-sided metastatic colorectal cancer? A systematic review and meta-analysis 11 Medical and Health Sciences 1112 Oncology and Carcinogenesis. World J. Surg. Oncol. 2018, 16, 200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Forcella, M.; Mozzi, A.; Stefanini, F.M.; Riva, A.; Epistolio, S.; Molinari, F.; Merlo, E.; Monti, E.; Fusi, P.; Frattini, M. Deregulation of sialidases in human normal and tumor tissues. Cancer Biomark. 2018, 21, 591–601. [Google Scholar] [CrossRef] [PubMed]
- Frattini, M.; Saletti, P.; Romagnani, E.; Martin, V.; Molinari, F.; Ghisletta, M.; Camponovo, A.; Etienne, L.L.; Cavalli, F.; Mazzucchelli, L. PTEN loss of expression predicts cetuximab efficacy in metastatic colorectal cancer patients. Br. J. Cancer 2007, 97, 1139–1145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Nicolantonio, F.; Martini, M.; Molinari, F.; Sartore-Bianchi, A.; Arena, S.; Saletti, P.; De Dosso, S.; Mazzucchelli, L.; Frattini, M.; Siena, S.; et al. Wild-type BRAF is required for response to panitumumab or cetuximab in metastatic colorectal cancer. J. Clin. Oncol. 2008, 26, 5705–5712. [Google Scholar] [CrossRef] [PubMed]
- Sartore-Bianchi, A.; Martini, M.; Molinari, F.; Veronese, S.; Nichelatti, M.; Artale, S.; Di Nicolantonio, F.; Saletti, P.; De Dosso, S.; Mazzucchelli, L.; et al. PIK3CA mutations in colorectal cancer are associated with clinical resistance to EGFR-targeted monoclonal antibodies. Cancer Res. 2009, 69, 1851–1857. [Google Scholar] [CrossRef] [Green Version]
- Monti, E.; Bassi, M.T.; Papini, N.; Riboni, M.; Manzoni, M.; Venerando, B.; Croci, G.; Preti, A.; Ballabio, A.; Tettamanti, G.; et al. Identification and expression of NEU3, a novel human sialidase associated to the plasma membrane. Biochem. J. 2000, 349, 343–351. [Google Scholar] [CrossRef]
- Smith, P.K.; Krohn, R.I.; Hermanson, G.T.; Mallia, A.K.; Gartner, F.H.; Provenzano, M.D.; Fujimoto, E.K.; Goeke, N.M.; Olson, B.J.; Klenk, D.C. Measurement of protein using bicinchoninic acid. Anal. Biochem. 1985, 150, 76–85. [Google Scholar] [CrossRef]
- Laemmli, U.K. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 1970, 227, 680–685. [Google Scholar] [CrossRef]
- Pitts, T.M.; Newton, T.P.; Bradshaw-Pierce, E.L.; Addison, R.; Arcaroli, J.J.; Klauck, P.J.; Bagby, S.M.; Hyatt, S.L.; Purkey, A.; Tentler, J.J.; et al. Dual Pharmacological Targeting of the MAP Kinase and PI3K/mTOR Pathway in Preclinical Models of Colorectal Cancer. PLoS ONE 2014, 9, e113037. [Google Scholar] [CrossRef] [Green Version]
- Bahrami, A.; Khazaei, M.; Hasanzadeh, M.; ShahidSales, S.; Joudi Mashhad, M.; Farazestanian, M.; Sadeghnia, H.R.; Rezayi, M.; Maftouh, M.; Hassanian, S.M.; et al. Therapeutic Potential of Targeting PI3K/AKT Pathway in Treatment of Colorectal Cancer: Rational and Progress. J. Cell. Biochem. 2018, 119, 2460–2469. [Google Scholar] [CrossRef]
- Narayanankutty, A. PI3K/ Akt/ mTOR Pathway as a Therapeutic Target for Colorectal Cancer: A Review of Preclinical and Clinical Evidence. Curr. Drug Targets 2019, 20, 1217–1226. [Google Scholar] [CrossRef] [PubMed]
- Miyagi, T.; Yamaguchi, K. Mammalian sialidases: Physiological and pathological roles in cellular functions. Glycobiology 2012, 22, 880–896. [Google Scholar] [CrossRef] [PubMed] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell Line | KRAS | NRAS | BRAF | PIK3CA | EGFR |
|---|---|---|---|---|---|
| SW48 | WT | WT | WT | WT | G719S |
| CO115 | WT | WT | V600E | WT | WT |
| SW403 | G12V | WT | WT | WT | WT |
| SW1116 | G12A | WT | WT | WT | WT |
| SW480 | G12V | WT | WT | WT | WT |
| SW1463 | G12C | WT | WT | WT | WT |
| E705 | WT | WT | WT | H1047H | WT |
| MICOL29 | G12D | WT | WT | WT | WT |
| DIFI | WT | WT | WT | WT | WT |
| MICOL24 | WT | WT | WT | WT | WT |
| HT-29 | WT | WT | V600E | WT | WT |
| SW620 | G12V | WT | WT | WT | WT |
| CACO-2 | WT | WT | WT | WT | WT |
| CCD 841 | WT | WT | WT | WT | WT |
| Cell Lines | IC50 (µg/mL, Mean ± SEM) |
|---|---|
| CCD 841 | >200 |
| CACO-2 | >200 |
| E705 | 0.165 ± 0.047 |
| DIFI | 0.048 ± 0.005 |
| MICOL24 | 0.027 ± 0.004 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bovio, F.; Epistolio, S.; Mozzi, A.; Monti, E.; Fusi, P.; Forcella, M.; Frattini, M. Role of NEU3 Overexpression in the Prediction of Efficacy of EGFR-Targeted Therapies in Colon Cancer Cell Lines. Int. J. Mol. Sci. 2020, 21, 8805. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21228805
Bovio F, Epistolio S, Mozzi A, Monti E, Fusi P, Forcella M, Frattini M. Role of NEU3 Overexpression in the Prediction of Efficacy of EGFR-Targeted Therapies in Colon Cancer Cell Lines. International Journal of Molecular Sciences. 2020; 21(22):8805. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21228805
Chicago/Turabian StyleBovio, Federica, Samantha Epistolio, Alessandra Mozzi, Eugenio Monti, Paola Fusi, Matilde Forcella, and Milo Frattini. 2020. "Role of NEU3 Overexpression in the Prediction of Efficacy of EGFR-Targeted Therapies in Colon Cancer Cell Lines" International Journal of Molecular Sciences 21, no. 22: 8805. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21228805