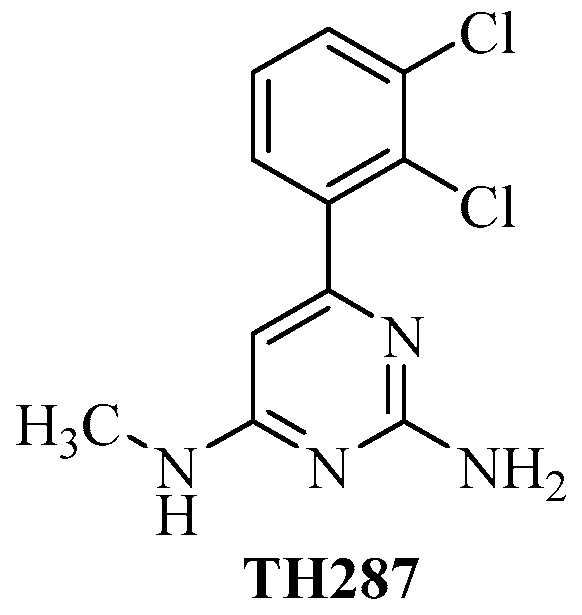
Radiolabeled 6-(2, 3-Dichlorophenyl)-N4-methylpyrimidine-2, 4-diamine (TH287): A Potential Radiotracer for Measuring and Imaging MTH1



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
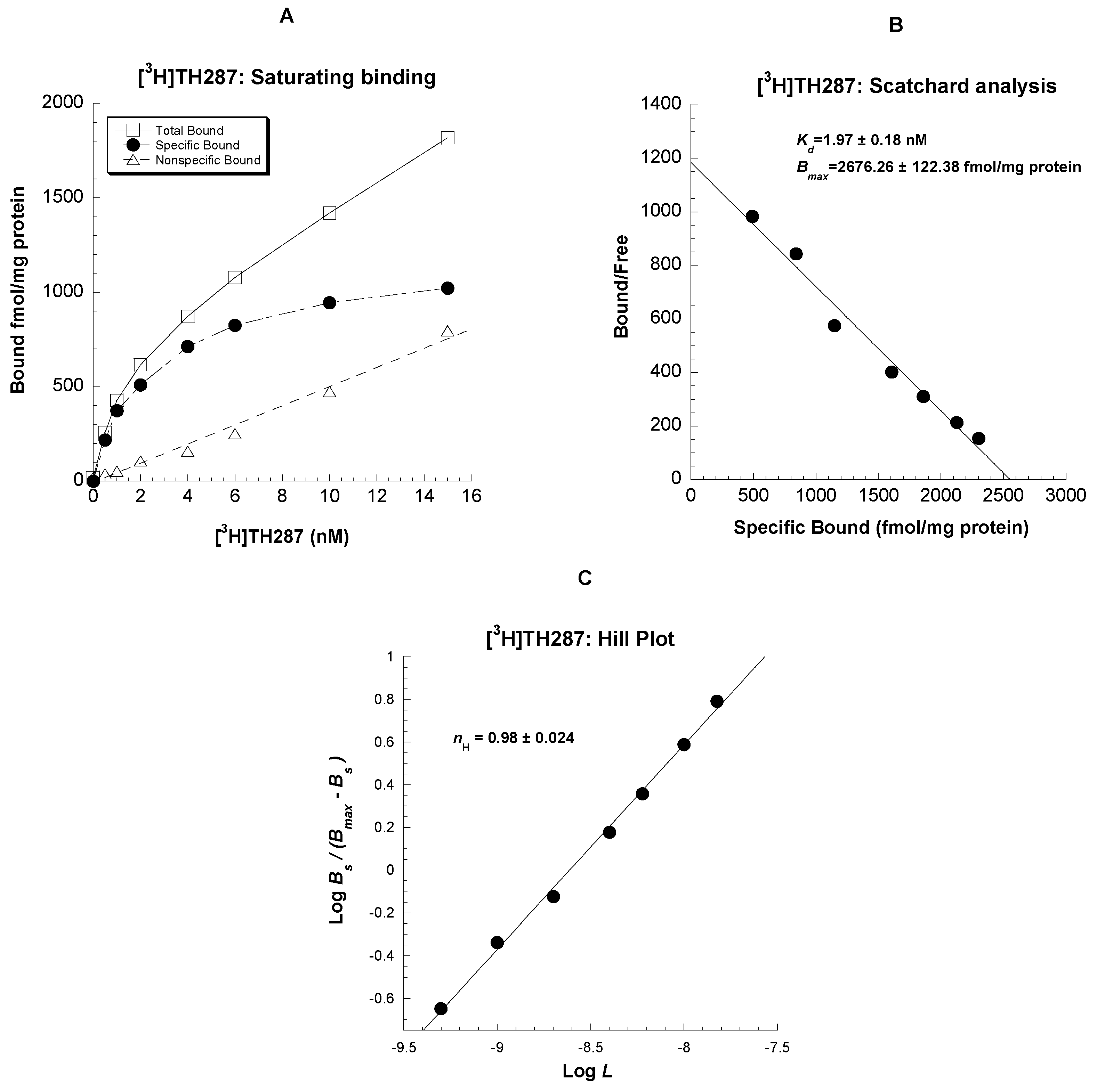
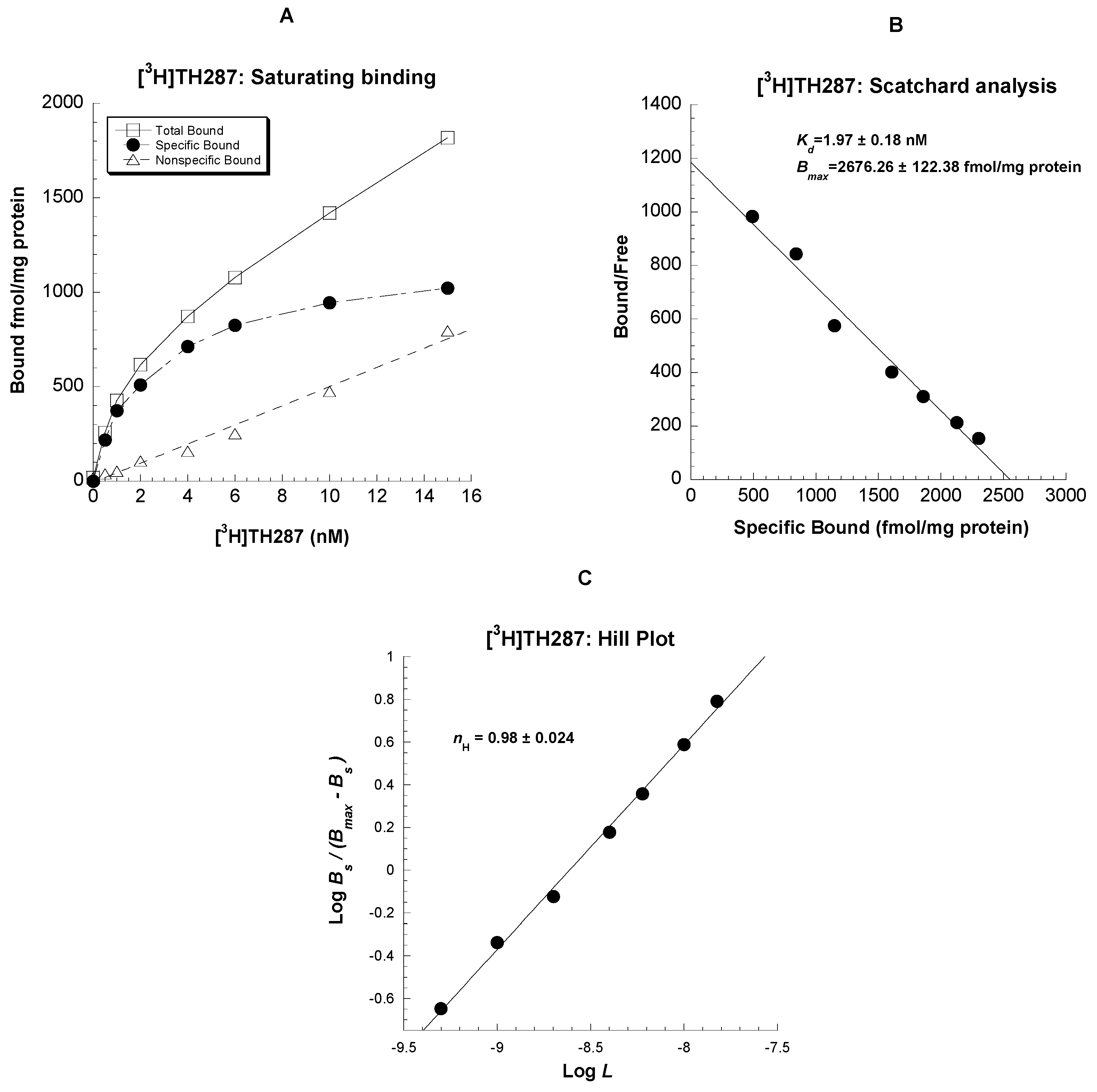
2.1. Saturation binding assays in U251MG cells
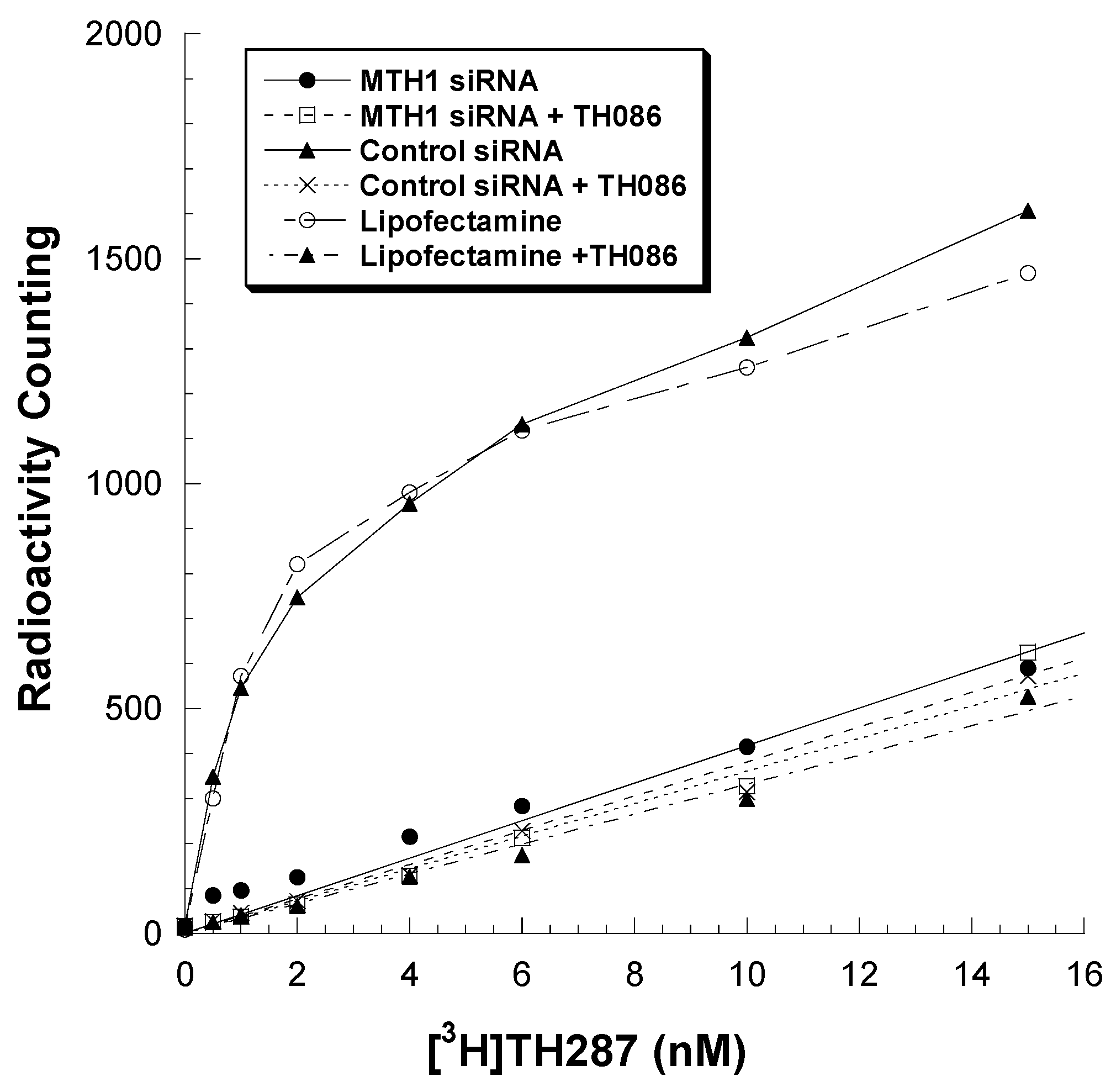
2.2. Knockdown MTH1 Expression through siRNA Abolishes [3H]TH287 Binding to U251MG Cells
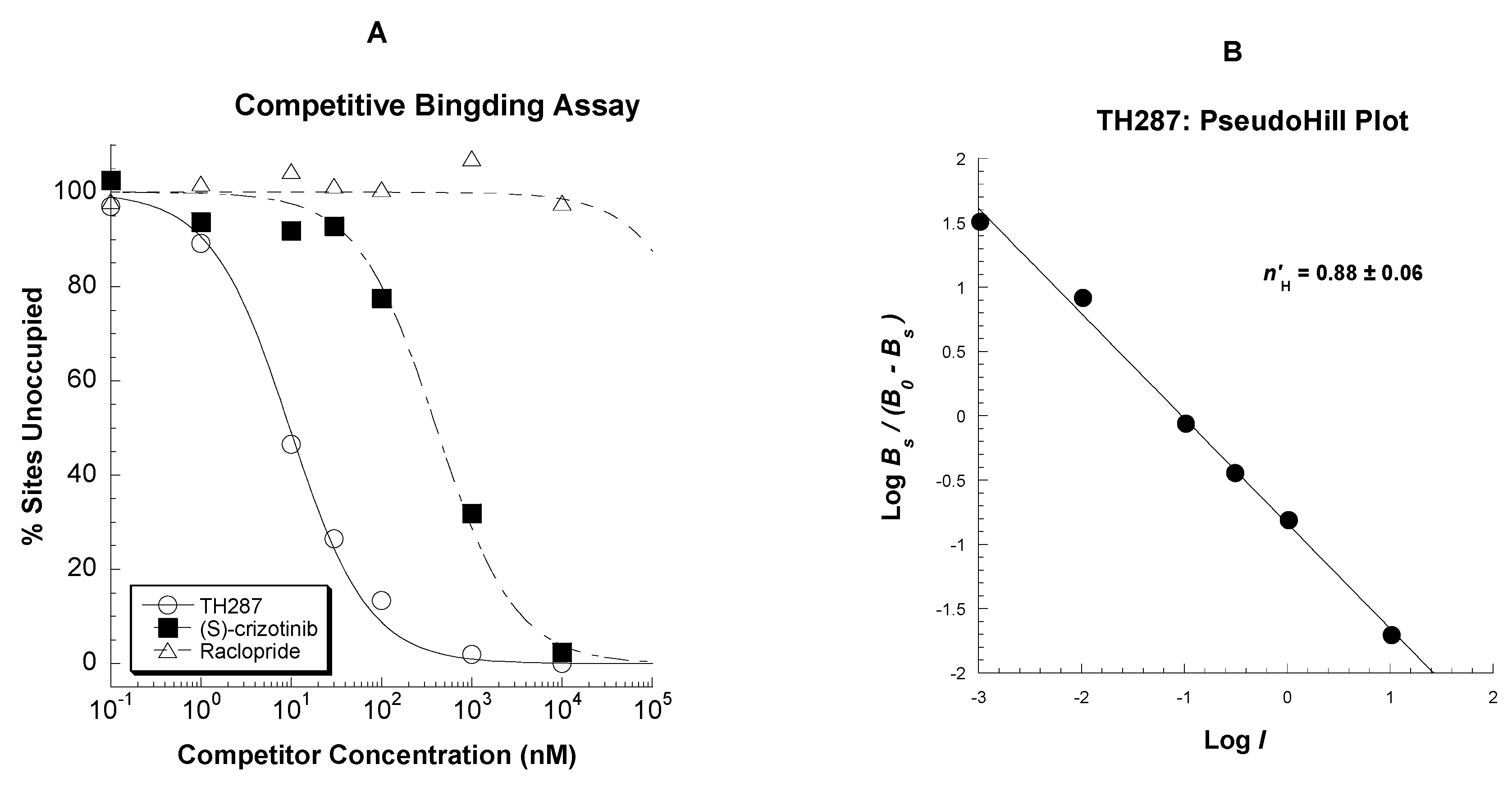
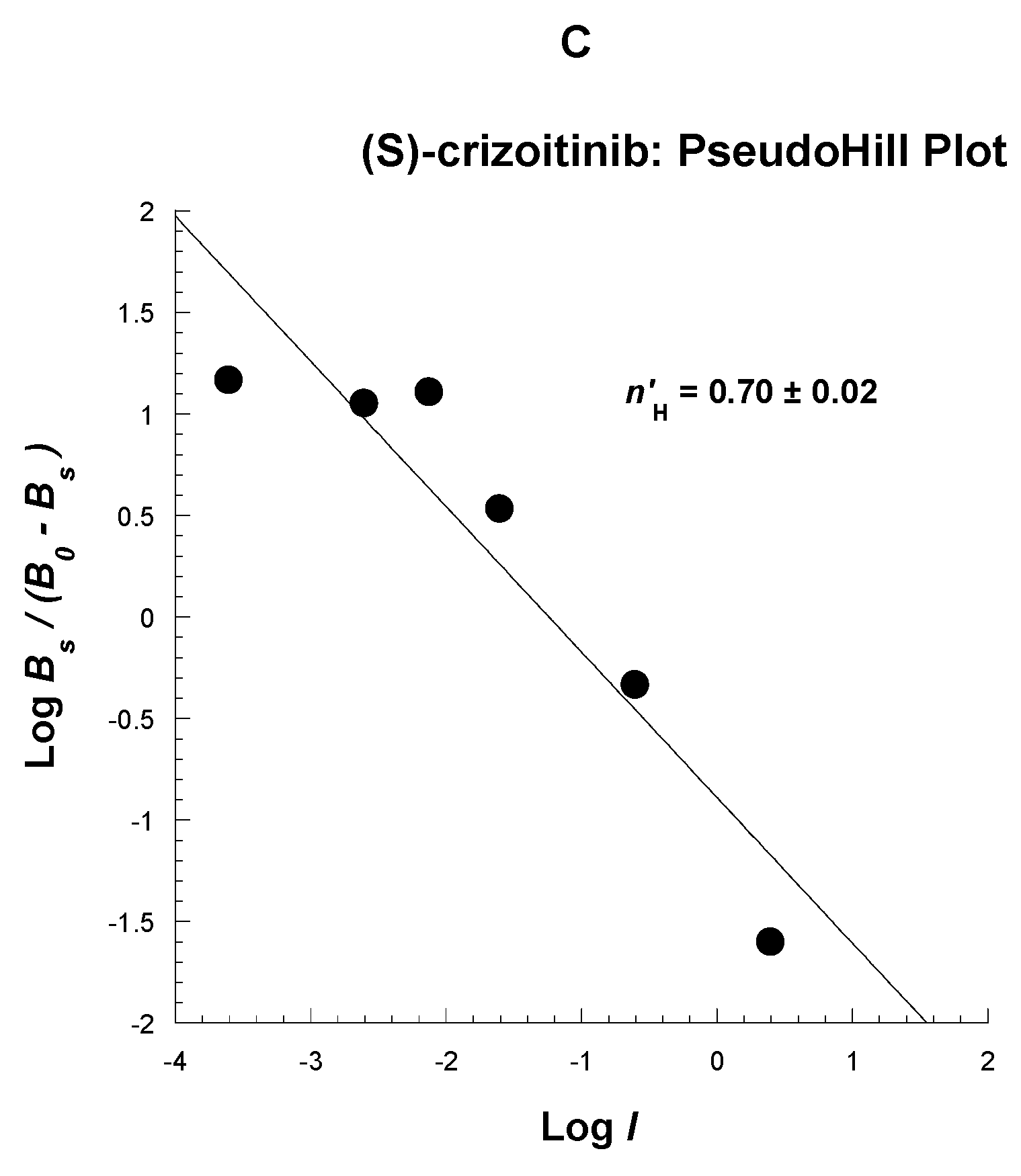
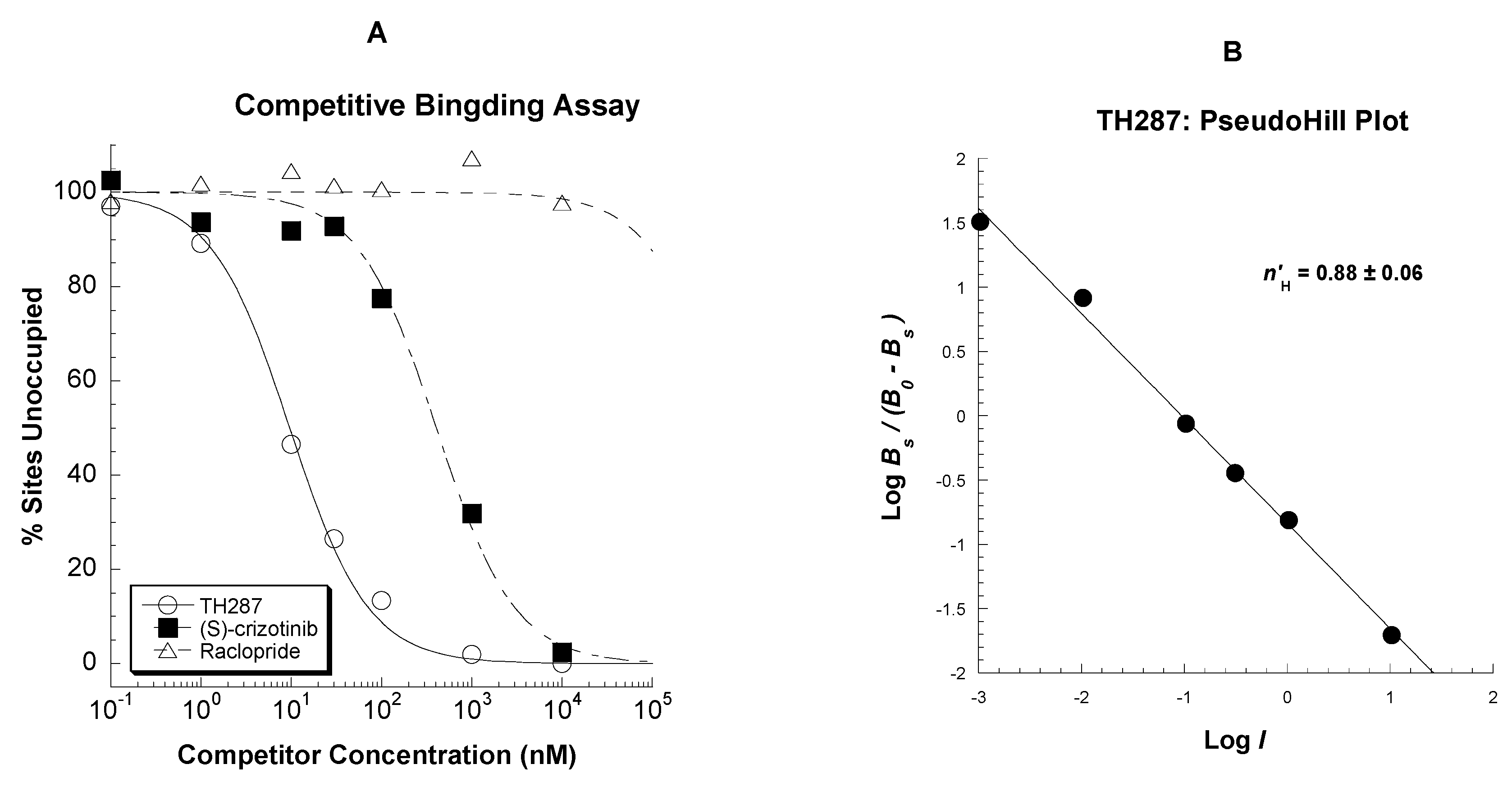
2.3. Competitive Profile of Standard and TH287 in U251MG Cells
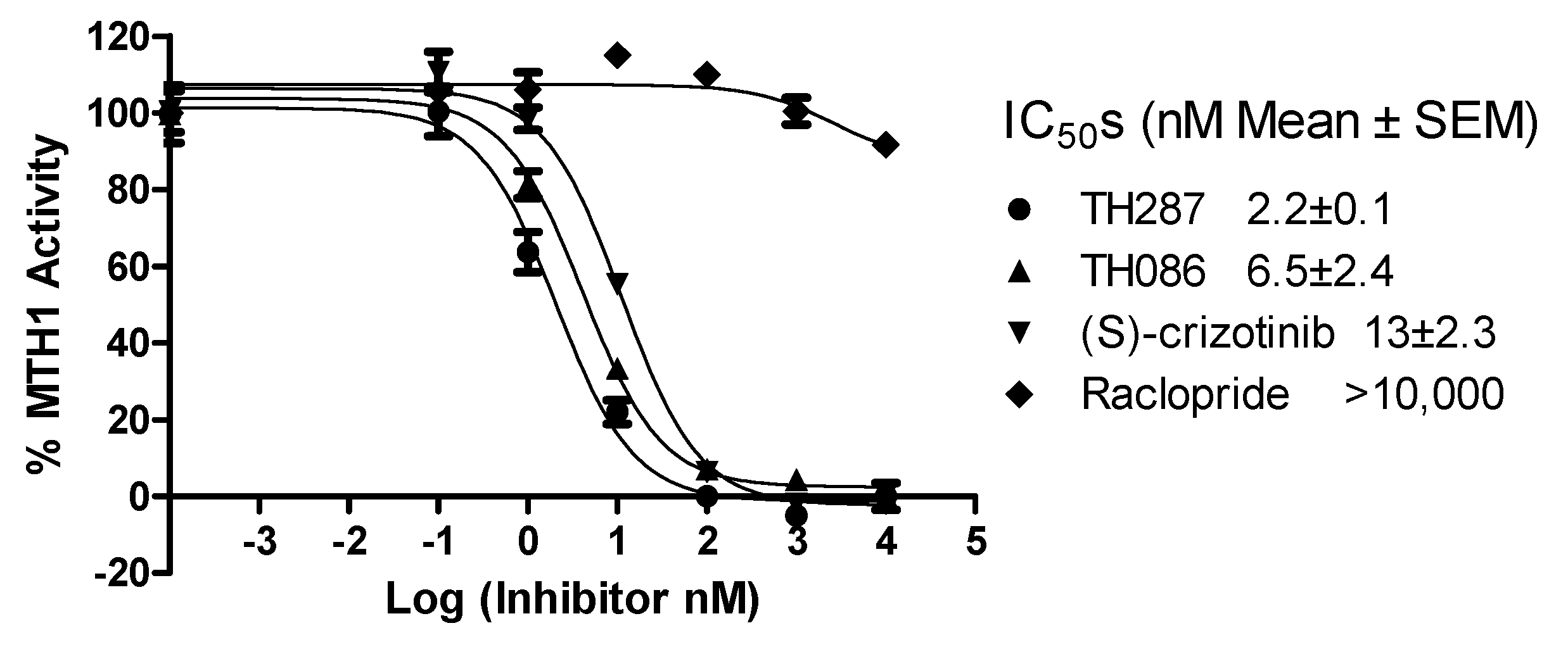
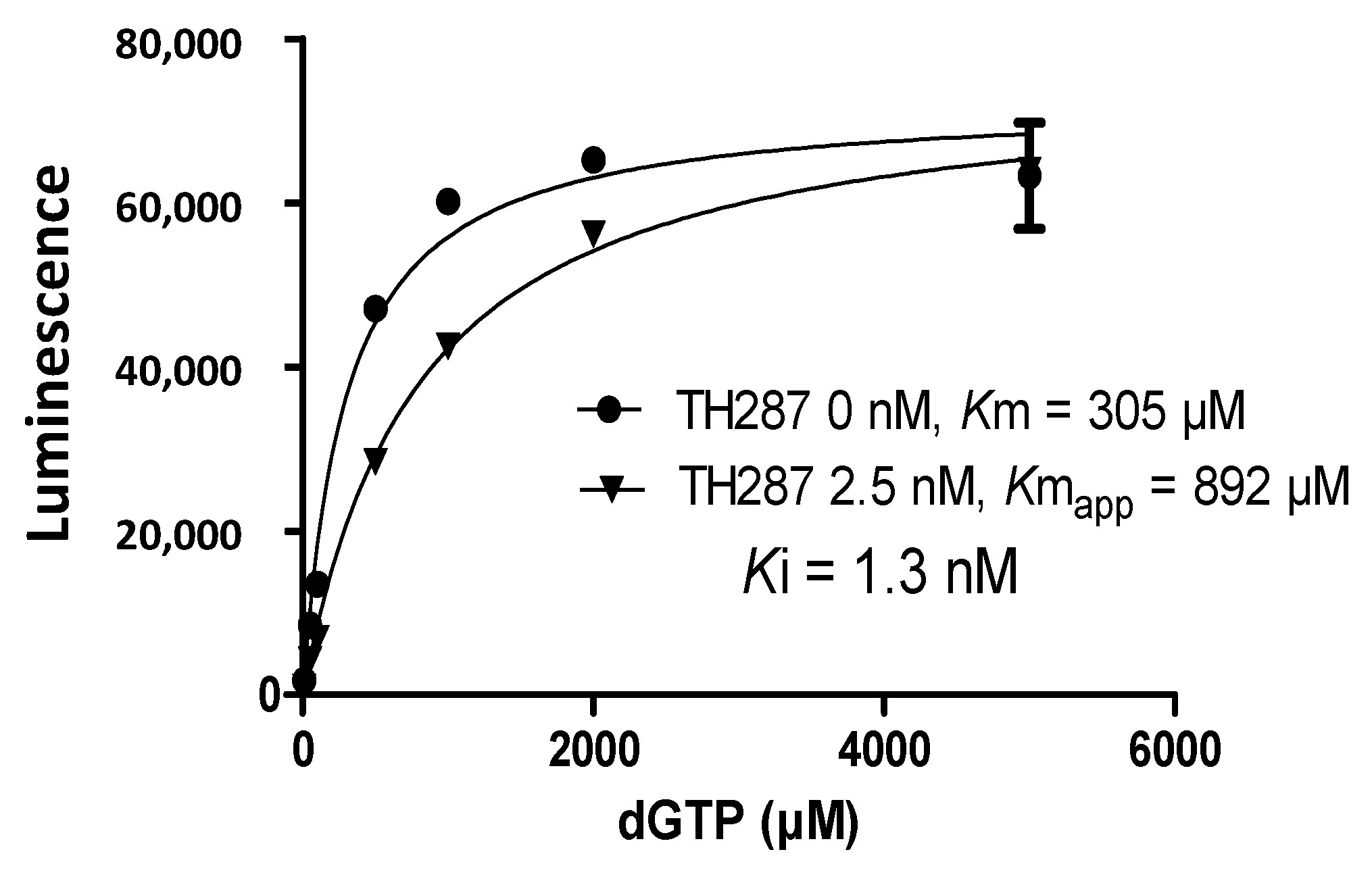
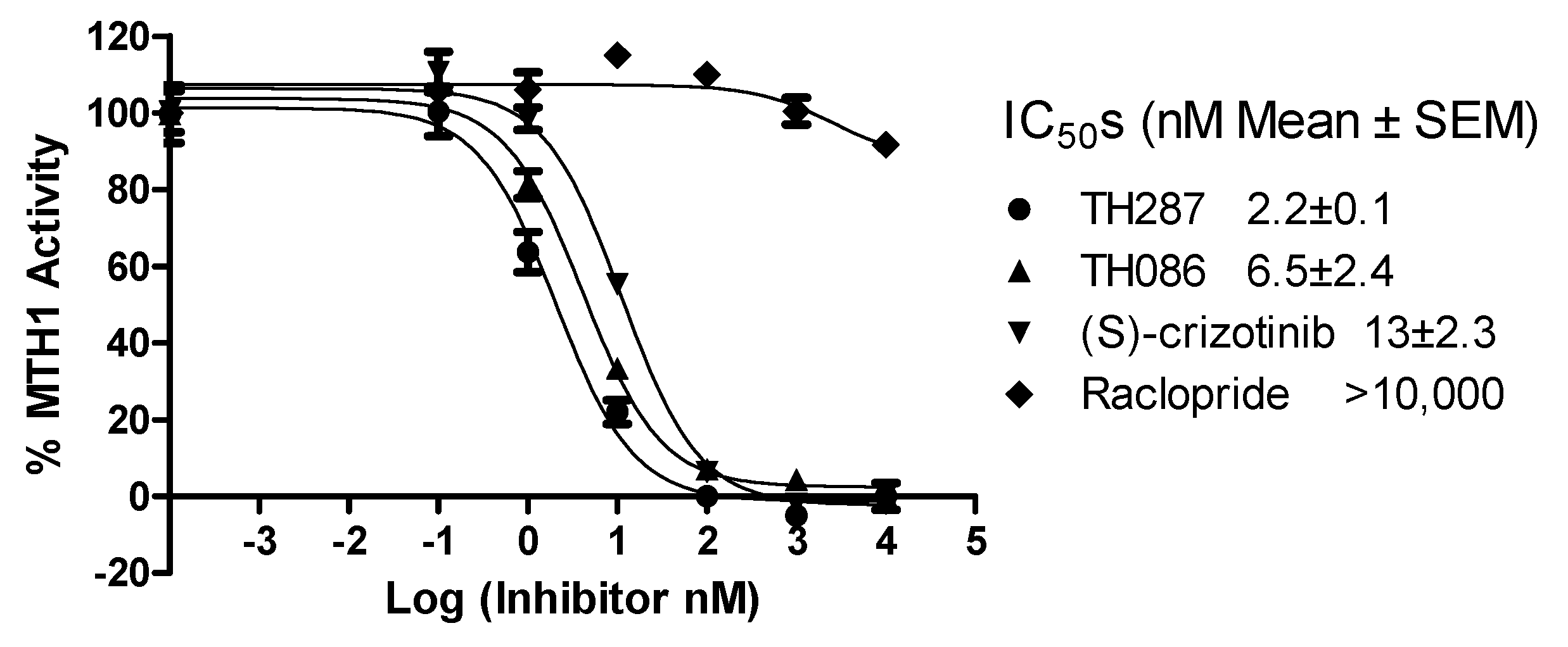
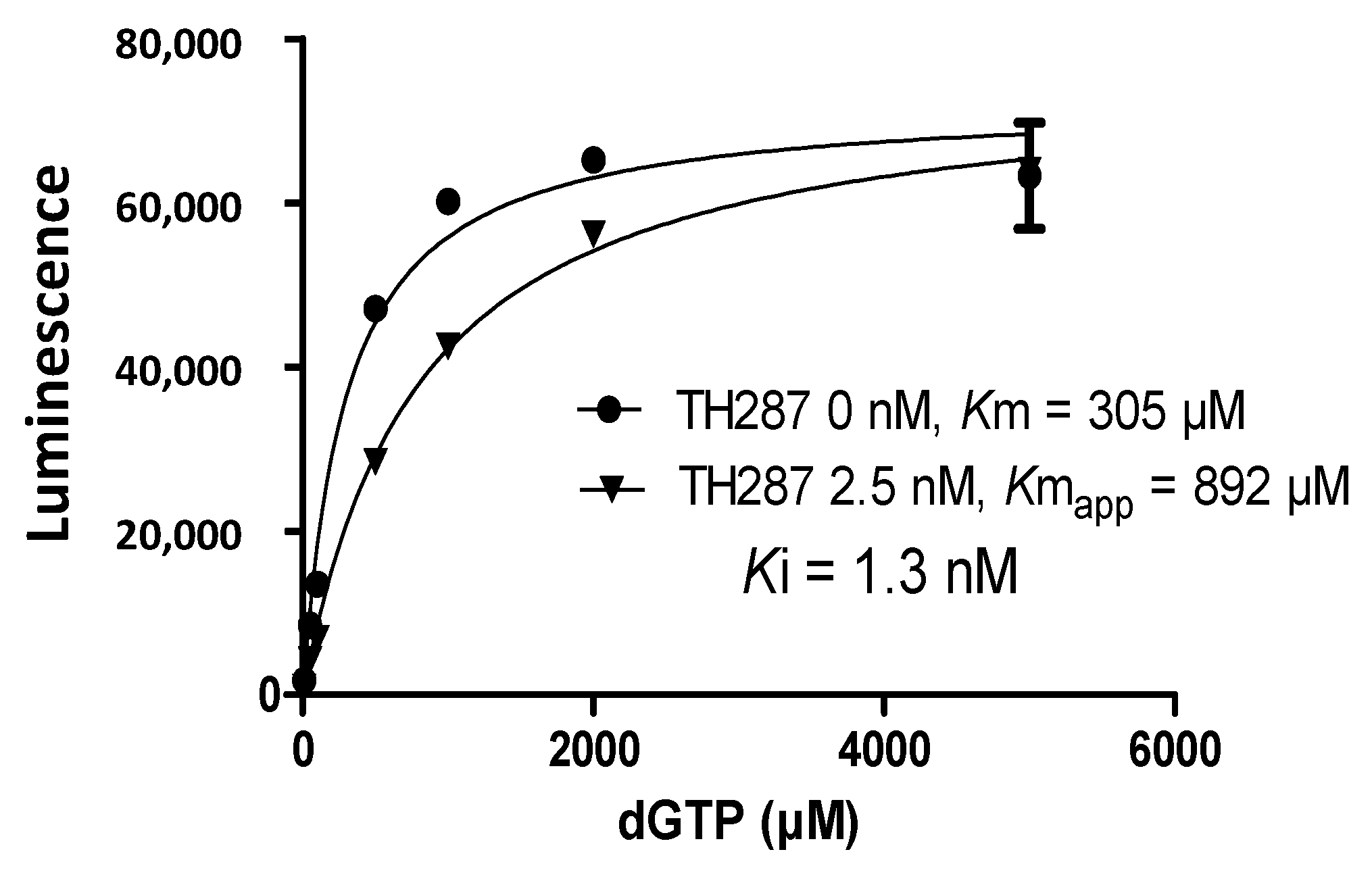
2.4. Inhibition Profile of Standard and TH287 on Enzyme Activity of MTH1
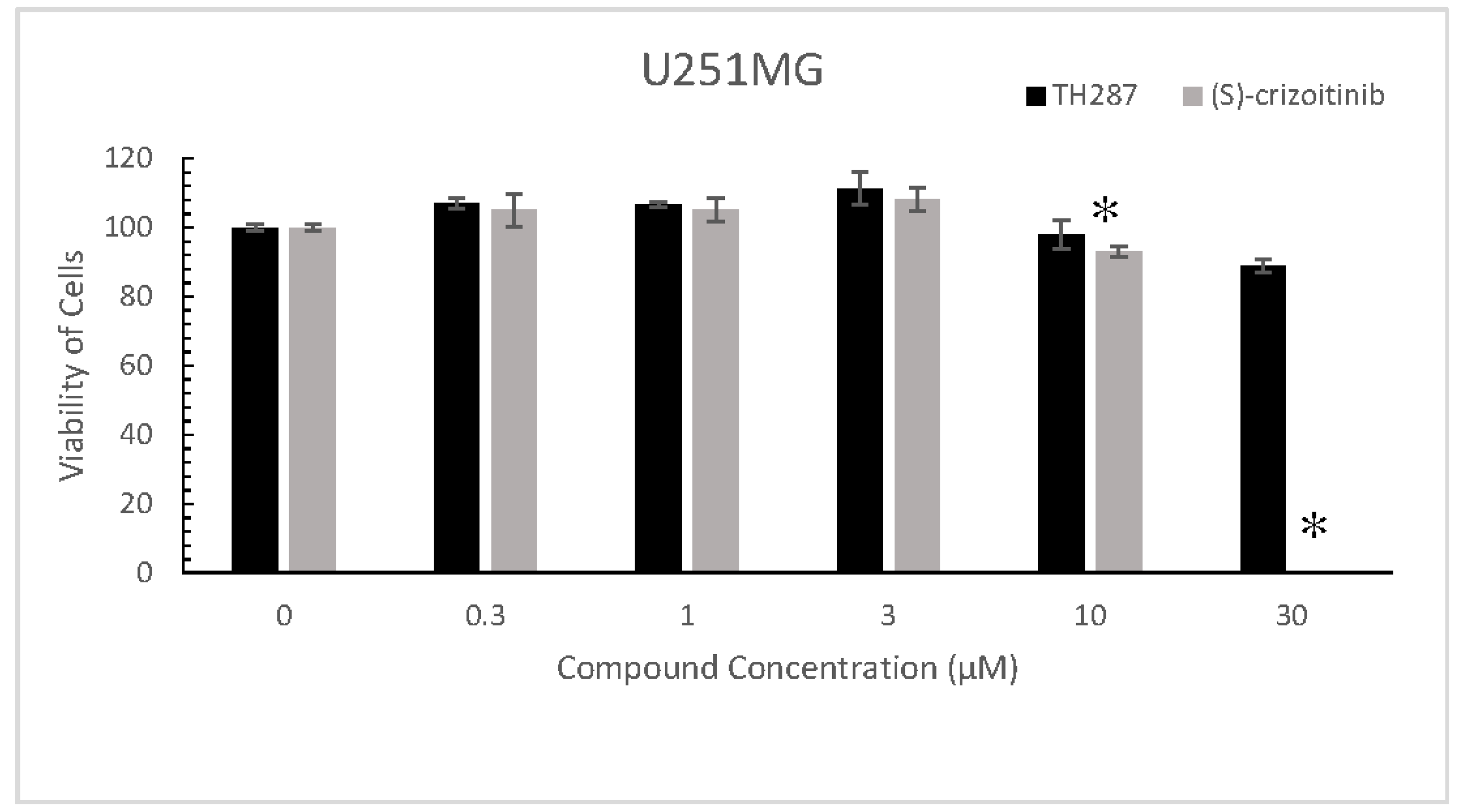
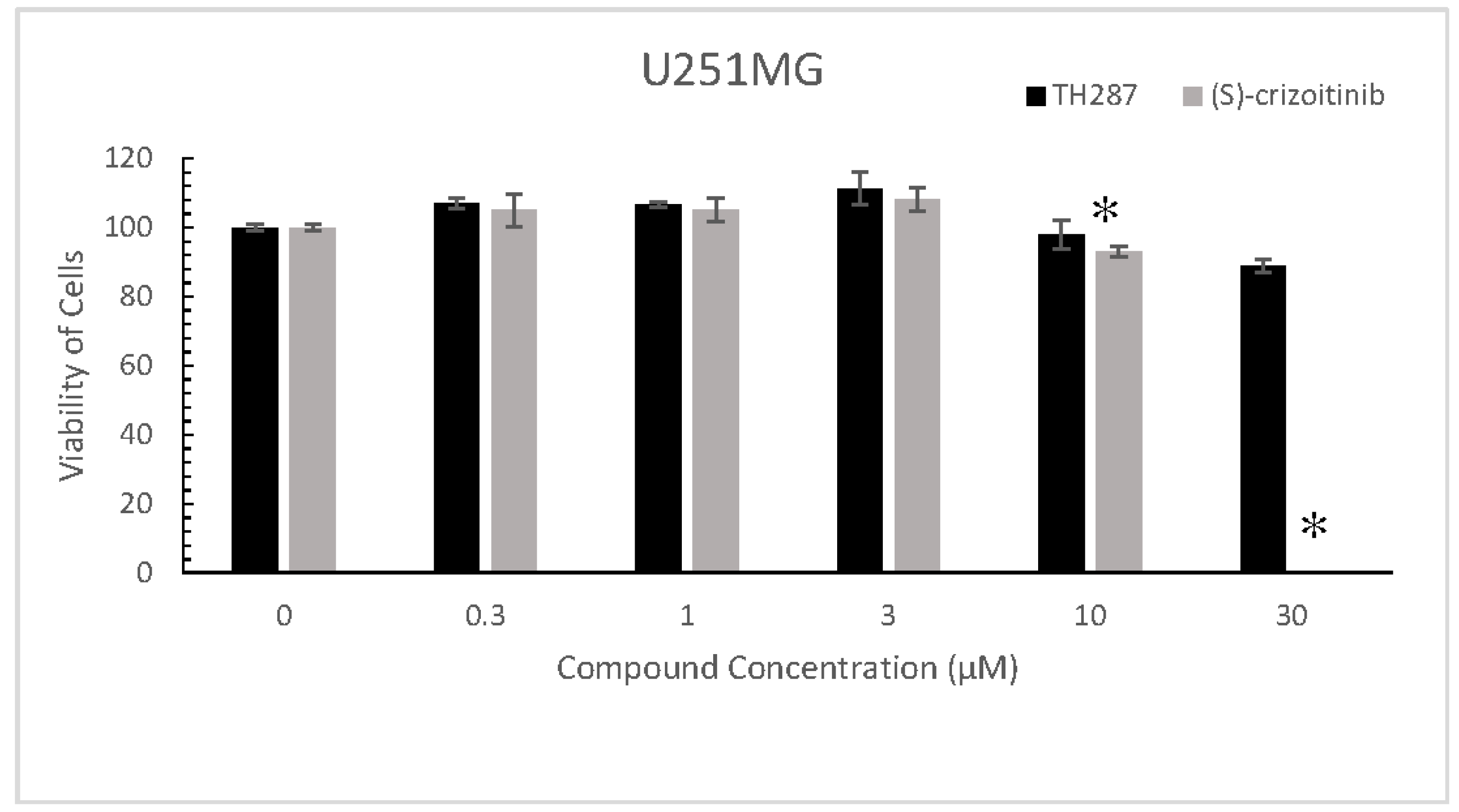
2.5. TH287 Exhibits no Killing Effect on Cells
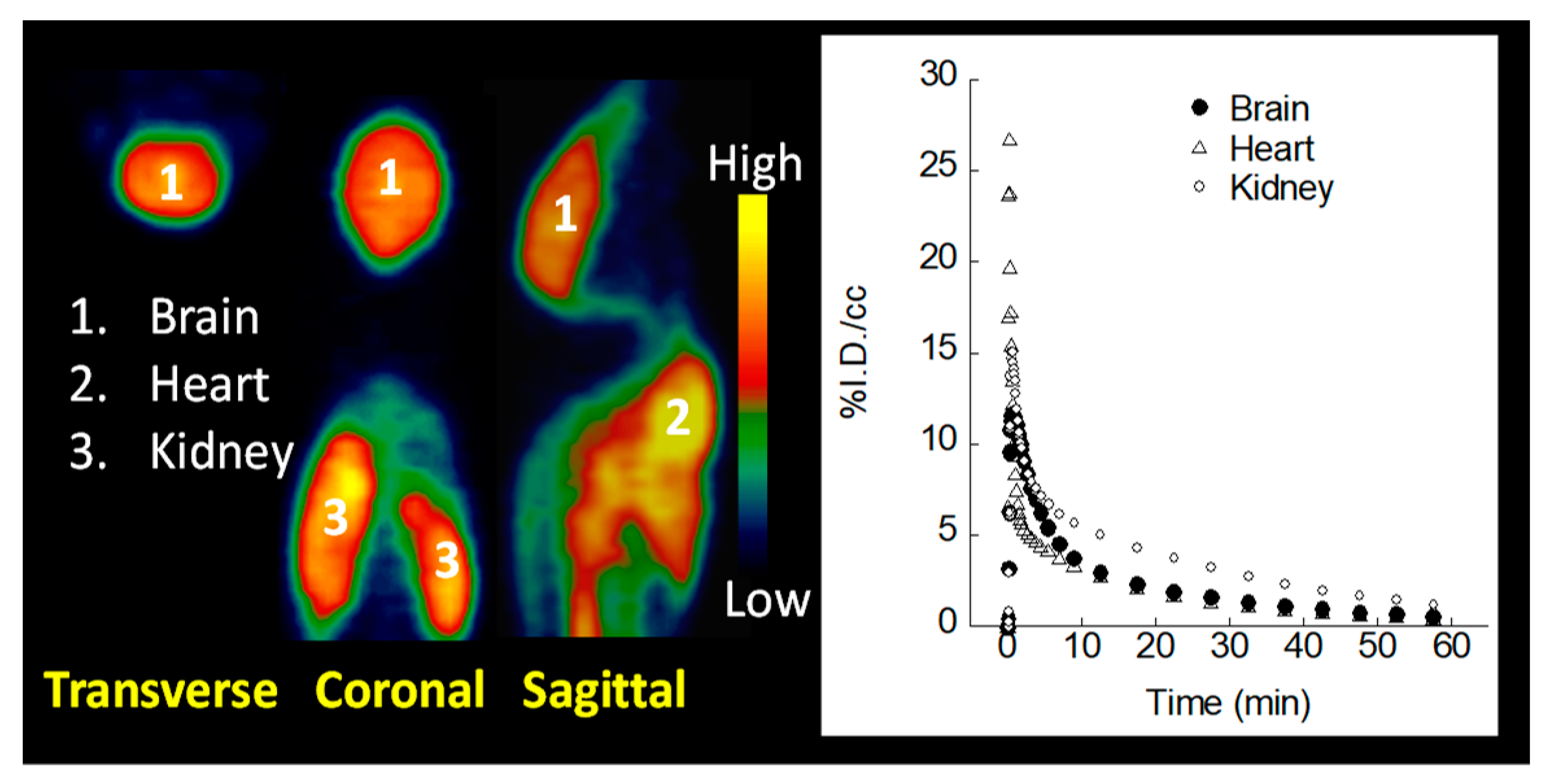
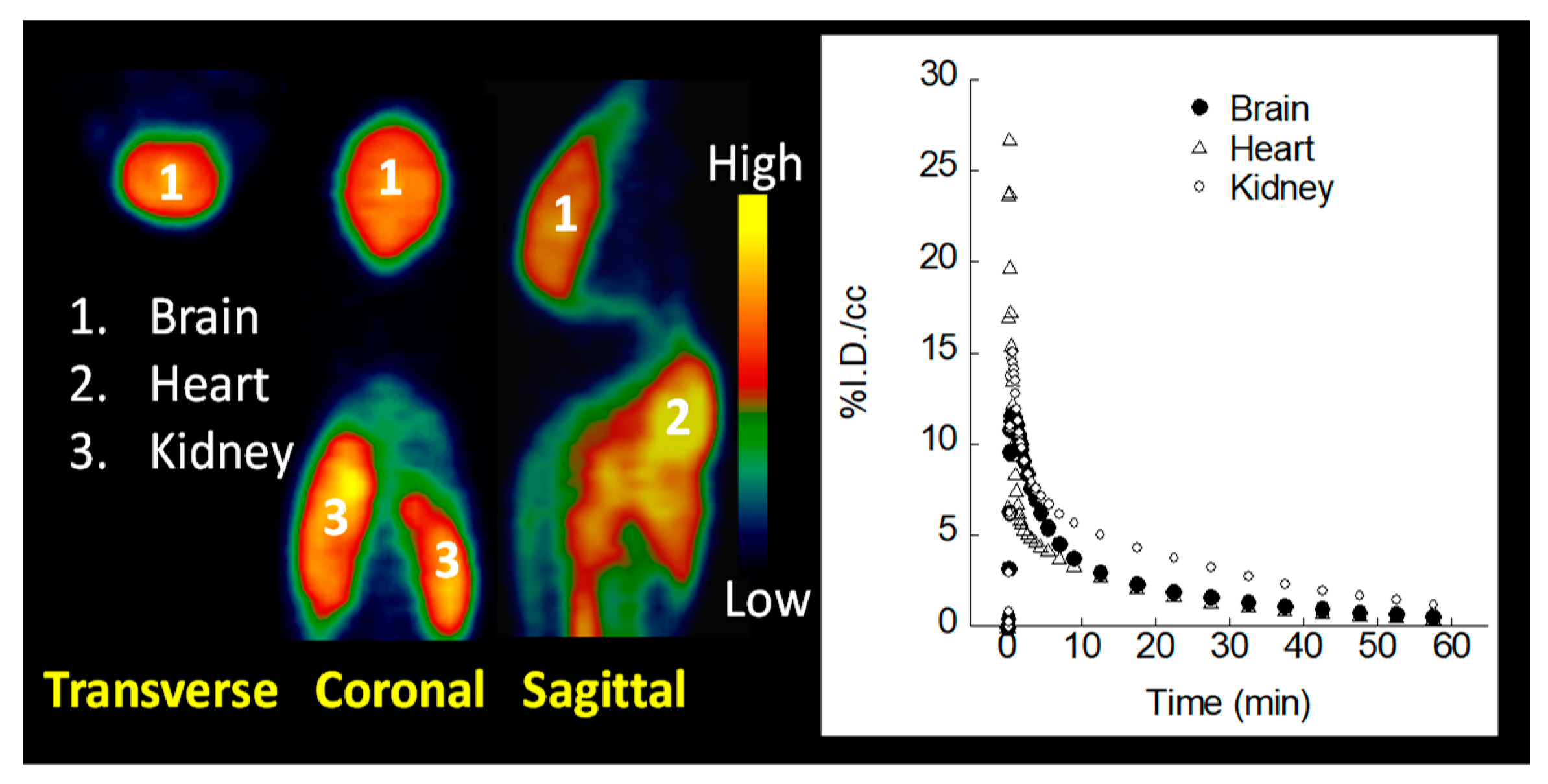
2.6. MicroPET Imaging
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Synthesis of Precursor and Radiosynthesis of [11C]TH287
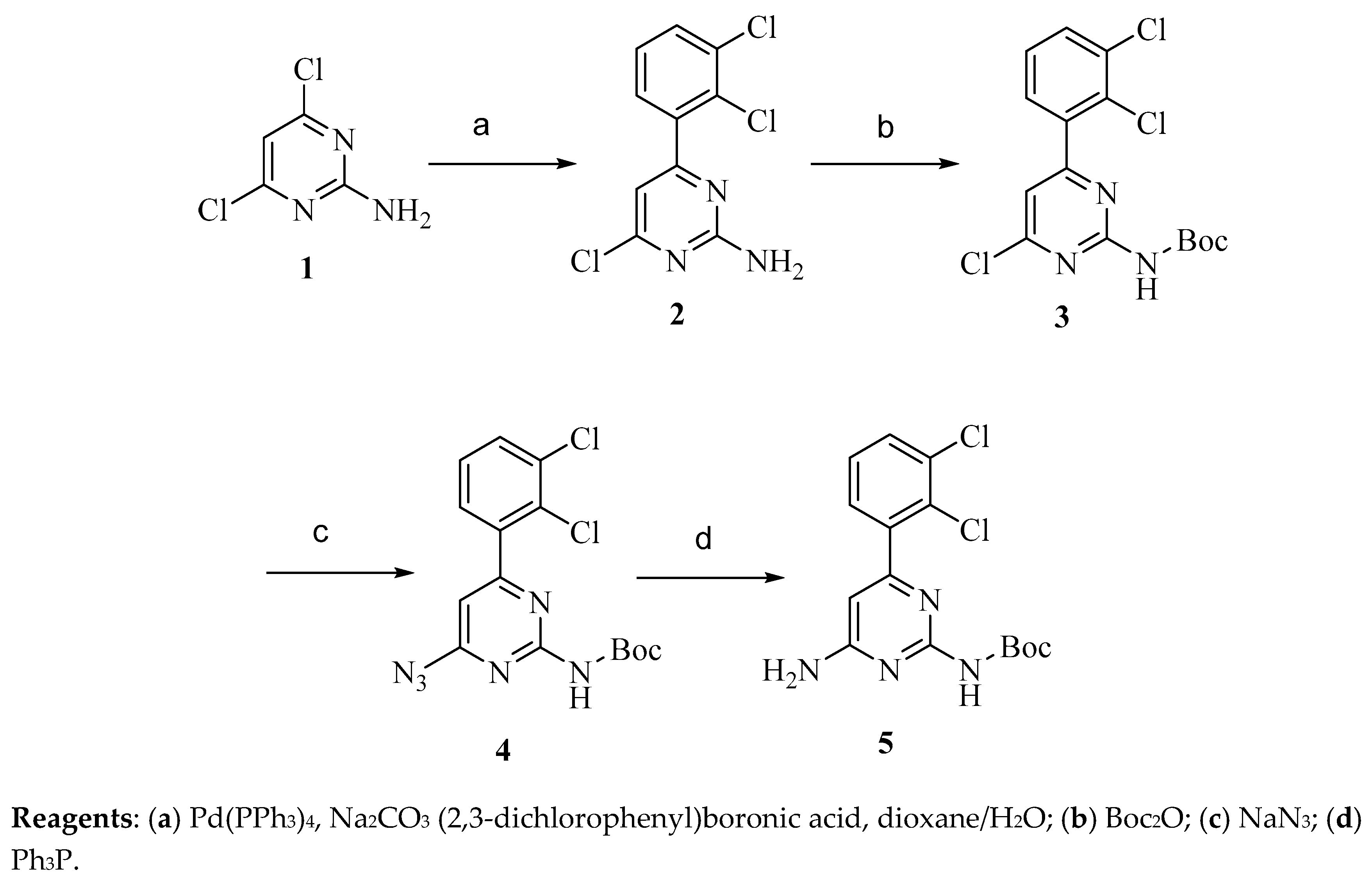
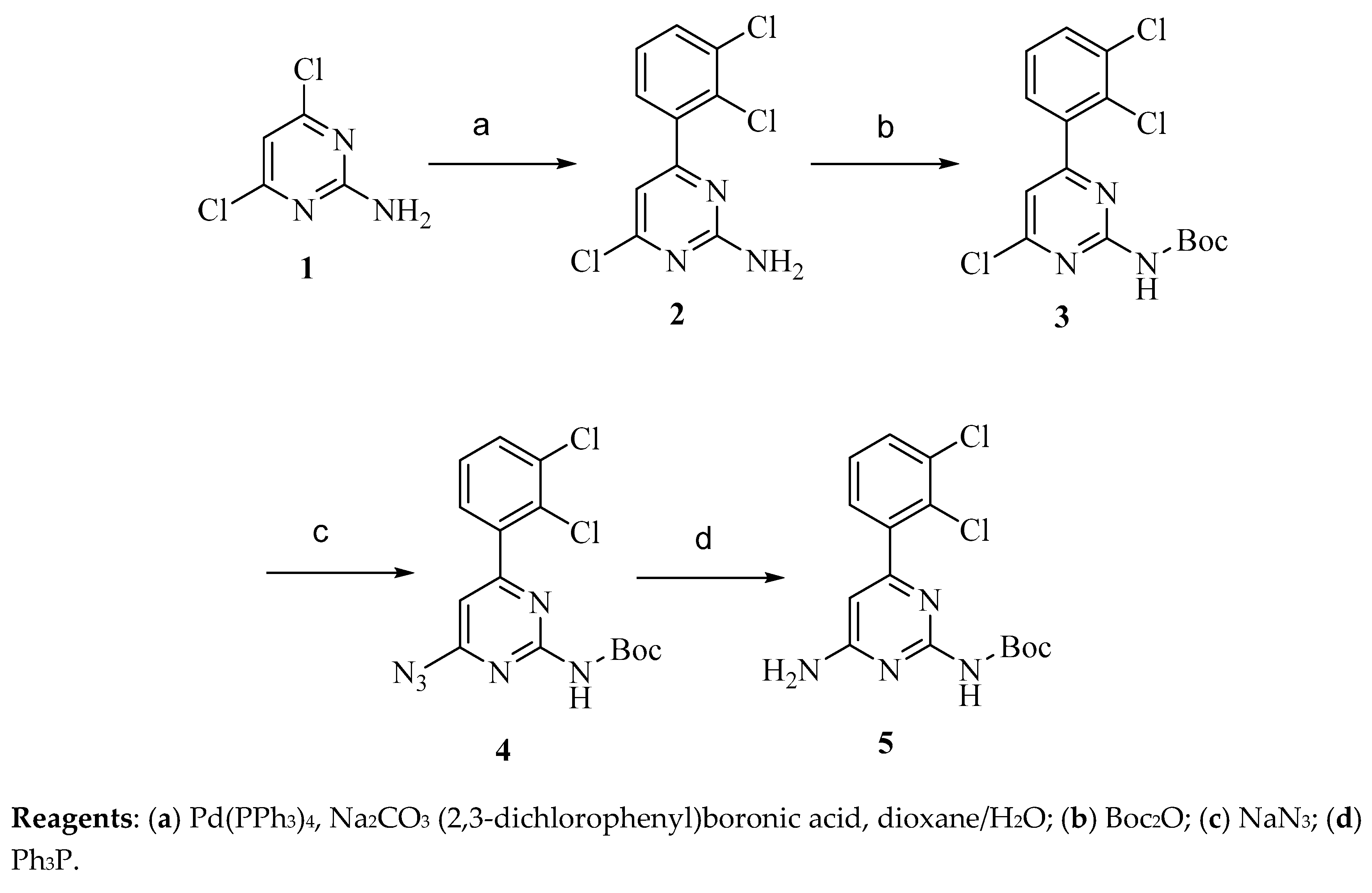
4.2.1. Synthesis of Precursor
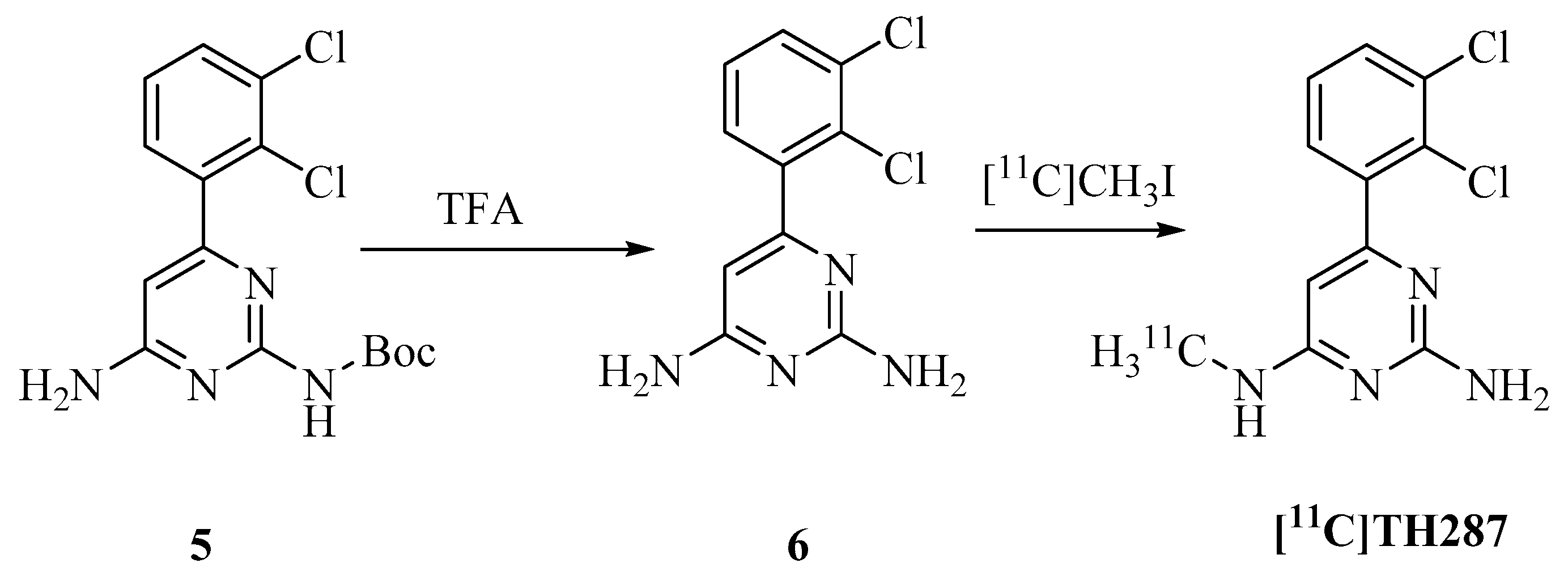
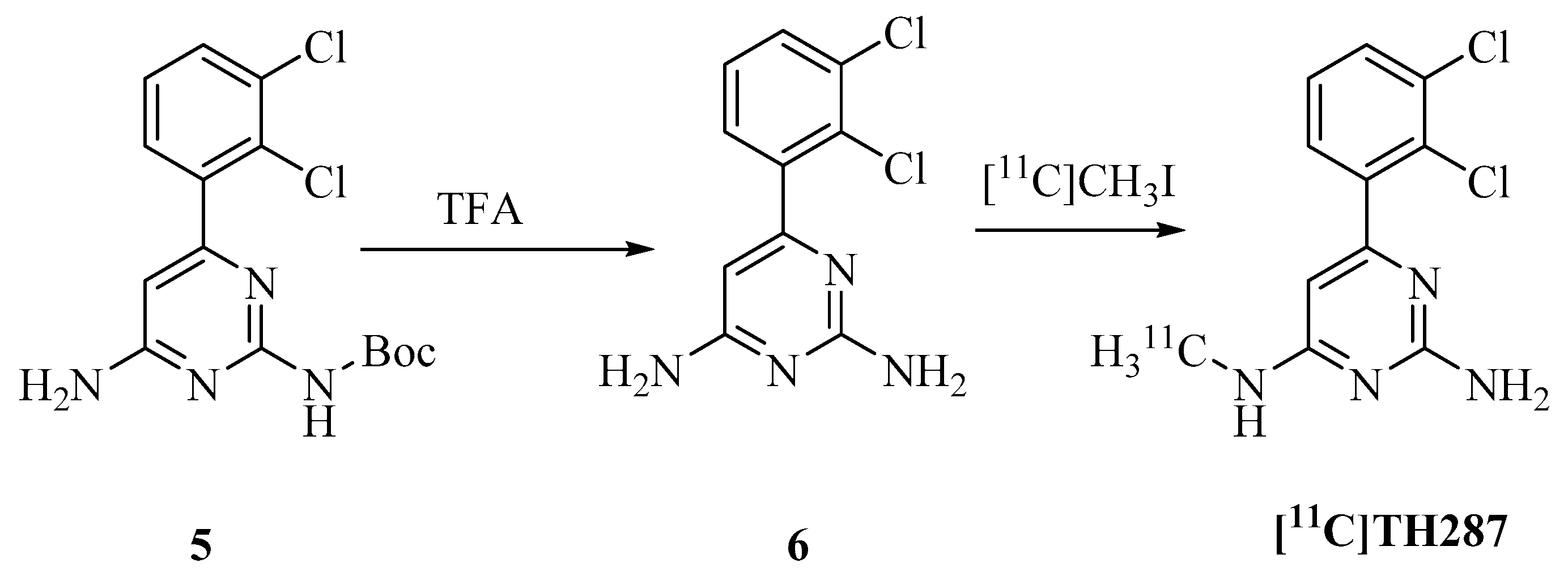
4.2.2. Radiosynthesis of [11C]TH287
4.3. Cell Culture
4.4. Knockdown MTH1 through siRNA Transfection
4.5. MTH1 Binding Assay
4.5.1. Saturation Binding and Scatchard Analysis
4.5.2. Competitive Binding
4.6. Enzyme Activity Assays
4.7. MTT Assays
4.8. MicroPET Imaging of Normal Mice with [11C]TH287
4.9. Data and Statistical Analysis
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| MTH1 | MutT homolog 1 |
| HCC | Hepatocellular carcinoma |
| NUDT1 | Nudix Hydrolase 1 |
| PET | Positron emission tomography |
| CT | Computed tomography |
| MRI | Magnetic resonance imaging |
| ROS | Reactive oxygen species |
| IC50 | The half-maximal inhibitory concentration |
References
- Cooke, M.S.; Evans, M.D.; Dizdaroglu, M.; Lunec, J. Oxidative DNA damage: Mechanisms, mutation, and disease. FASEB J.Off. Pub. Fed. Am. Soc. Exp. Biol. 2003, 17, 1195–1214. [Google Scholar]
- Roszkowski, K.; Olinski, R. Urinary 8-oxoguanine as a predictor of survival in patients undergoing radiotherapy. Cancer Epidemiol. Biomark. Prev. Publ. Am. Assoc. Cancer Res. Cosponsored Am. Soc. Prev. Oncol. 2012, 21, 629–634. [Google Scholar]
- Roszkowski, K.; Gackowski, D.; Rozalski, R.; Dziaman, T.; Siomek, A.; Guz, J.; Szpila, A.; Foksinski, M.; Olinski, R. Small field radiotherapy of head and neck cancer patients is responsible for oxidatively damaged DNA/oxidative stress on the level of a whole organism. Int. J. Cancer 2008, 123, 1964–1967. [Google Scholar] [PubMed]
- Vlassenko, A.G.; McConathy, J.; Couture, L.E.; Su, Y.; Massoumzadeh, P.; Leeds, H.S.; Chicoine, M.R.; Tran, D.D.; Huang, J.; Dahiya, S.; et al. Aerobic Glycolysis as a Marker of Tumor Aggressiveness: Preliminary Data in High Grade Human Brain Tumors. Dis. Markers 2015, 2015, 874904. [Google Scholar]
- Grossman, R.; Shimony, N.; Hadelsberg, U.; Soffer, D.; Sitt, R.; Strauss, N.; Corn, B.W.; Ram, Z. Impact of Resecting Radiation Necrosis and Pseudoprogression on Survival of Patients with Glioblastoma. World Neurosurg. 2016, 89, 37–41. [Google Scholar]
- Kang, D.; Takeshige, K.; Sekiguchi, M.; Singh, K.K. Introduction. In Mitochondrial DNA Mutations in Aging, Disease and Cancer; Singh, K.K., Ed.; Springer: New York, NY, USA, 1998; pp. 1–15. [Google Scholar]
- Ames, B.N.; Shigenaga, M.K.; Hagen, T.M. Oxidants, antioxidants, and the degenerative diseases of aging. Proc. Natl. Acad. Sci. USA 1993, 90, 7915–7922. [Google Scholar]
- Kasai, H.; Nishimura, S. Hydroxylation of deoxyguanosine at the C-8 position by ascorbic acid and other reducing agents. Nucleic Acids Res. 1984, 12, 2137–2145. [Google Scholar]
- De Souza-Pinto, N.C.; Eide, L.; Hogue, B.A.; Thybo, T.; Stevnsner, T.; Seeberg, E.; Klungland, A.; Bohr, V.A. Repair of 8-oxodeoxyguanosine lesions in mitochondrial dna depends on the oxoguanine dna glycosylase (OGG1) gene and 8-oxoguanine accumulates in the mitochondrial dna of OGG1-defective mice. Cancer Res. 2001, 61, 5378–5381. [Google Scholar]
- Kang, D.; Nishida, J.; Iyama, A.; Nakabeppu, Y.; Furuichi, M.; Fujiwara, T.; Sekiguchi, M.; Takeshige, K. Intracellular localization of 8-oxo-dGTPase in human cells, with special reference to the role of the enzyme in mitochondria. J. Biol. Chem. 1995, 270, 14659–14665. [Google Scholar]
- Tsuzuki, T.; Egashira, A.; Igarashi, H.; Iwakuma, T.; Nakatsuru, Y.; Tominaga, Y.; Kawate, H.; Nakao, K.; Nakamura, K.; Ide, F.; et al. Spontaneous tumorigenesis in mice defective in the MTH1 gene encoding 8-oxo-dGTPase. Proc. Natl. Acad. Sci. USA 2001, 98, 11456–11461. [Google Scholar]
- Fujikawa, K.; Kamiya, H.; Yakushiji, H.; Nakabeppu, Y.; Kasai, H. Human MTH1 protein hydrolyzes the oxidized ribonucleotide, 2-hydroxy-ATP. Nucleic Acids Res. 2001, 29, 449–454. [Google Scholar] [PubMed] [Green Version]
- Nakabeppu, Y. Regulation of intracellular localization of human MTH1, OGG1, and MYH proteins for repair of oxidative DNA damage. Prog. Nucleic Acid Res. Mol. Biol. 2001, 68, 75–94. [Google Scholar]
- Nakabeppu, Y.; Tsuchimoto, D.; Yamaguchi, H.; Sakumi, K. Oxidative damage in nucleic acids and Parkinson’s disease. J. Neurosci. Res. 2007, 85, 919–934. [Google Scholar] [PubMed]
- Sakai, Y.; Furuichi, M.; Takahashi, M.; Mishima, M.; Iwai, S.; Shirakawa, M.; Nakabeppu, Y. A molecular basis for the selective recognition of 2-hydroxy-dATP and 8-oxo-dGTP by human MTH1. J. Biol. Chem. 2002, 277, 8579–8587. [Google Scholar]
- Sakai, Y.; Oda, H.; Yoshimura, D.; Furuichi, M.; Kang, D.; Iwai, S.; Hara, T.; Nakabeppu, Y. The GT to GC single nucleotide polymorphism at the beginning of an alternative exon 2C of human MTH1 gene confers an amino terminal extension that functions as a mitochondrial targeting signal. J. Mol. Med. (Berl) 2006, 84, 660–670. [Google Scholar] [PubMed]
- Iida, T.; Furuta, A.; Kawashima, M.; Nishida, J.; Nakabeppu, Y.; Iwaki, T. Accumulation of 8-oxo-2’-deoxyguanosine and increased expression of hMTH1 protein in brain tumors. Neuro Oncol. 2001, 3, 73–81. [Google Scholar] [PubMed]
- Tu, Y.; Wang, Z.; Wang, X.; Yang, H.; Zhang, P.; Johnson, M.; Liu, N.; Liu, H.; Jin, W.; Zhang, Y.; et al. Birth of MTH1 as a therapeutic target for glioblastoma: MTH1 is indispensable for gliomatumorigenesis. Am. J. Transl. Res. 2016, 8, 2803–2811. [Google Scholar] [PubMed]
- Kennedy, C.H.; Pass, H.I.; Mitchell, J.B. Expression of human MutT homologue (hMTH1) protein in primary non-small-cell lung carcinomas and histologically normal surrounding tissue. Free Radic. Biol. Med. 2003, 34, 1447–1457. [Google Scholar]
- Fujishita, T.; Okamoto, T.; Akamine, T.; Takamori, S.; Takada, K.; Katsura, M.; Toyokawa, G.; Shoji, F.; Shimokawa, M.; Oda, Y.; et al. Association of MTH1 expression with the tumor malignant potential and poor prognosis in patients with resected lung cancer. Lung Cancer 2017, 109, 52–57. [Google Scholar]
- Zhang, X.; Song, W.; Zhou, Y.; Mao, F.; Lin, Y.; Guan, J.; Sun, Q. Expression and function of MutT homolog 1 in distinct subtypes of breast cancer. Oncol. Lett. 2017, 13, 2161–2168. [Google Scholar]
- Coskun, E.; Jaruga, P.; Jemth, A.S.; Loseva, O.; Scanlan, L.D.; Tona, A.; Lowenthal, M.S.; Helleday, T.; Dizdaroglu, M. Addiction to MTH1 protein results in intense expression in human breast cancer tissue as measured by liquid chromatography-isotope-dilution tandem mass spectrometry. DNA Repair (Amst) 2015, 33, 101–110. [Google Scholar] [PubMed]
- Hua, X.; Sanjiv, K.; Gad, H.; Pham, T.; Gokturk, C.; Rasti, A.; Zhao, Z.; He, K.; Feng, M.; Zang, Y.; et al. Karonudib is a promising anticancer therapy in hepatocellular carcinoma. Ther. Adv. Med. Oncol. 2019, 11, 1758835919866960. [Google Scholar] [PubMed] [Green Version]
- Okamoto, K.; Toyokuni, S.; Kim, W.J.; Ogawa, O.; Kakehi, Y.; Arao, S.; Hiai, H.; Yoshida, O. Overexpression of human mutT homologue gene messenger RNA in renal-cell carcinoma: Evidence of persistent oxidative stress in cancer. Int. J. Cancer 1996, 65, 437–441. [Google Scholar] [PubMed]
- Koketsu, S.; Watanabe, T.; Nagawa, H. Expression of DNA repair protein: MYH, NTH1, and MTH1 in colorectal cancer. Hepatogastroenterology 2004, 51, 638–642. [Google Scholar] [PubMed]
- Akiyama, S.; Saeki, H.; Nakashima, Y.; Iimori, M.; Kitao, H.; Oki, E.; Oda, Y.; Nakabeppu, Y.; Kakeji, Y.; Maehara, Y. Prognostic impact of MutT homolog-1 expression on esophageal squamous cell carcinoma. Cancer Med. 2017, 6, 258–266. [Google Scholar] [PubMed]
- Zhou, W.; Ma, L.; Yang, J.; Qiao, H.; Li, L.; Guo, Q.; Ma, J.; Zhao, L.; Wang, J.; Jiang, G.; et al. Potent and specific MTH1 inhibitors targeting gastric cancer. Cell Death Dis. 2019, 10, 434. [Google Scholar]
- Zhou, H.; Jian, Y.; Leng, Y.; Liu, N.; Tian, Y.; Wang, G.; Gao, W.; Yang, G.; Chen, W. Human MutT homologue 1 mRNA overexpression correlates to poor response of multiple myeloma. Int. J. Hematol. 2017, 105, 318–325. [Google Scholar]
- Gad, H.; Koolmeister, T.; Jemth, A.S.; Eshtad, S.; Jacques, S.A.; Strom, C.E.; Svensson, L.M.; Schultz, N.; Lundback, T.; Einarsdottir, B.O.; et al. MTH1 inhibition eradicates cancer by preventing sanitation of the dNTP pool. Nature 2014, 508, 215–221. [Google Scholar]
- Farand, J.; Kropf, J.E.; Blomgren, P.; Xu, J.; Schmitt, A.C.; Newby, Z.E.; Wang, T.; Murakami, E.; Barauskas, O.; Sudhamsu, J.; et al. Discovery of Potent and Selective MTH1 Inhibitors for Oncology: Enabling Rapid Target (In)Validation. ACS Med. Chem. Lett. 2019, 11, 358–364. [Google Scholar]
- Ellermann, M.; Eheim, A.; Rahm, F.; Viklund, J.; Guenther, J.; Andersson, M.; Ericsson, U.; Forsblom, R.; Ginman, T.; Lindstrom, J.; et al. Novel Class of Potent and Cellularly Active Inhibitors Devalidates MTH1 as Broad-Spectrum Cancer Target. ACS Chem. Biol. 2017, 12, 1986–1992. [Google Scholar]
- Kettle, J.G.; Alwan, H.; Bista, M.; Breed, J.; Davies, N.L.; Eckersley, K.; Fillery, S.; Foote, K.M.; Goodwin, L.; Jones, D.R.; et al. Potent and Selective Inhibitors of MTH1 Probe Its Role in Cancer Cell Survival. J. Med. Chem. 2016, 59, 2346–2361. [Google Scholar] [PubMed]
- Huber, K.V.; Salah, E.; Radic, B.; Gridling, M.; Elkins, J.M.; Stukalov, A.; Jemth, A.S.; Gokturk, C.; Sanjiv, K.; Stromberg, K.; et al. Stereospecific targeting of MTH1 by (S)-crizotinib as an anticancer strategy. Nature 2014, 508, 222–227. [Google Scholar]
- Kawamura, T.; Kawatani, M.; Muroi, M.; Kondoh, Y.; Futamura, Y.; Aono, H.; Tanaka, M.; Honda, K.; Osada, H. Proteomic profiling of small-molecule inhibitors reveals dispensability of MTH1 for cancer cell survival. Sci. Rep. 2016, 6, 26521. [Google Scholar] [PubMed] [Green Version]
- Warpman Berglund, U.; Sanjiv, K.; Gad, H.; Kalderen, C.; Koolmeister, T.; Pham, T.; Gokturk, C.; Jafari, R.; Maddalo, G.; Seashore-Ludlow, B.; et al. Validation and development of MTH1 inhibitors for treatment of cancer. Ann. Oncol. 2016, 27, 2275–2283. [Google Scholar] [PubMed]
- Sanjiv, K.; Gad, H.; Rudd, S.G.; Mortusewicz, O.; Stolz, A.; Amaral, N.; Brautigham, L.; Pudelko, L.; Kalderen, C.; Jemth, A.S.; et al. MTH1 promotes mitotic progression to avoid oxidative DNA damage in cancer cells. Cancer Res. 2019, 79. [Google Scholar]
- Vlassenko, A.G.; Gordon, B.A.; Goyal, M.S.; Su, Y.; Blazey, T.M.; Durbin, T.J.; Couture, L.E.; Christensen, J.J.; Jafri, H.; Morris, J.C.; et al. Aerobic glycolysis and tau deposition in preclinical Alzheimer’s disease. Neurobiol. Aging 2018, 67, 95–98. [Google Scholar]
- Lovell, M.A.; Markesbery, W.R. Oxidative DNA damage in mild cognitive impairment and late-stage Alzheimer’s disease. Nucleic Acids Res. 2007, 35, 7497–7504. [Google Scholar]
- Zhou, D.; Chu, W.; Chen, D.L.; Wang, Q.; Reichert, D.E.; Rothfuss, J.; D’Avignon, A.; Welch, M.J.; Mach, R.H. [18F]- and [11C]-labeled N-benzyl-isatin sulfonamide analogues as PET tracers for apoptosis: Synthesis, radiolabeling mechanism, and in vivo imaging study of apoptosis in Fas-treated mice using [11C]WC-98. Org. Biomol. Chem. 2009, 7, 1337–1348. [Google Scholar]
- Scatchard, G. The Attractions of Proteins for Small Molecules and Ions. Ann. N. Y. Acad. Sci. 1949, 51, 660–672. [Google Scholar]
- Xu, J.; Hassanzadeh, B.; Chu, W.; Tu, Z.; Jones, L.A.; Luedtke, R.R.; Perlmutter, J.S.; Mintun, M.A.; Mach, R.H. [3H]4-(dimethylamino)-N-(4-(4-(2-methoxyphenyl)piperazin-1-yl) butyl)benzamide: A selective radioligand for dopamine D(3) receptors. II. Quantitative analysis of dopamine D(3) and D(2) receptor density ratio in the caudate-putamen. Synapse 2010, 64, 449–459. [Google Scholar]
- Sun, J.; Cairns, N.J.; Perlmutter, J.S.; Mach, R.H.; Xu, J. Regulation of dopamine D(3) receptor in the striatal regions and substantia nigra in diffuse Lewy body disease. Neuroscience 2013, 248, 112–126. [Google Scholar] [PubMed] [Green Version]
- Cheng, Y.; Prusoff, W.H. Relationship between the inhibition constant (K1) and the concentration of inhibitor which causes 50 per cent inhibition (I50) of an enzymatic reaction. Biochem. Pharmacol. 1973, 22, 3099–3108. [Google Scholar] [PubMed]
- Curtis, M.J.; Alexander, S.; Cirino, G.; Docherty, J.R.; George, C.H.; Giembycz, M.A.; Hoyer, D.; Insel, P.A.; Izzo, A.A.; Ji, Y.; et al. Experimental design and analysis and their reporting II: Updated and simplified guidance for authors and peer reviewers. Br. J. Pharmacol. 2018, 175, 987–993. [Google Scholar] [PubMed] [Green Version]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, H.; Afrin, S.; Guo, Y.; Chu, W.; Benzinger, T.L.S.; Rogers, B.E.; Garbow, J.R.; Perlmutter, J.S.; Zhou, D.; Xu, J. Radiolabeled 6-(2, 3-Dichlorophenyl)-N4-methylpyrimidine-2, 4-diamine (TH287): A Potential Radiotracer for Measuring and Imaging MTH1. Int. J. Mol. Sci. 2020, 21, 8860. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21228860
Chen H, Afrin S, Guo Y, Chu W, Benzinger TLS, Rogers BE, Garbow JR, Perlmutter JS, Zhou D, Xu J. Radiolabeled 6-(2, 3-Dichlorophenyl)-N4-methylpyrimidine-2, 4-diamine (TH287): A Potential Radiotracer for Measuring and Imaging MTH1. International Journal of Molecular Sciences. 2020; 21(22):8860. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21228860
Chicago/Turabian StyleChen, Huaping, Sadia Afrin, Yingqiu Guo, Wenhua Chu, Tammie L.S. Benzinger, Buck E. Rogers, Joel R. Garbow, Joel S. Perlmutter, Dong Zhou, and Jinbin Xu. 2020. "Radiolabeled 6-(2, 3-Dichlorophenyl)-N4-methylpyrimidine-2, 4-diamine (TH287): A Potential Radiotracer for Measuring and Imaging MTH1" International Journal of Molecular Sciences 21, no. 22: 8860. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21228860