The Genetic Basis of Primary Myelofibrosis and Its Clinical Relevance

 ,
,
Abstract
:1. Mutation and Disease Pathogenesis
2. Mutation and Clinical Phenotype
3. Mutation and Prognosis
4. Mutation and Treatment Response
4.1. Impact on Response to JAK Inhibitors or Interferon
4.2. Impact on Transplant Outcome
4.3. Minimal Residual Disease (MRD)
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Mora, B.; Siracusa, C.; Rumi, E.; Maffioli, M.; Casetti, I.C.; Barraco, D.; Merli, M.; Rossi, M.; Ubezio, M.; Accetta, R.; et al. Platelet count predicts driver mutations’ co-occurrence in low JAK2 mutated essential thrombocythemia and myelofibrosis. Leukemia 2020, 1–4. [Google Scholar] [CrossRef]
- Pikman, Y.; Lee, B.H.; Mercher, T.; McDowell, E.; Ebert, B.L.; Gozo, M.; Cuker, A.; Wernig, G.; Moore, S.; Galinsky, I.; et al. MPLW515L Is a Novel Somatic Activating Mutation in Myelofibrosis with Myeloid Metaplasia. PLoS Med. 2006, 3, e270. [Google Scholar] [CrossRef] [Green Version]
- Mullally, A.; Lane, S.W.; Ball, B.; Megerdichian, C.; Okabe, R.; Al-Shahrour, F.; Paktinat, M.; Haydu, J.E.; Housman, E.; Lord, A.M.; et al. Physiological Jak2V617F Expression Causes a Lethal Myeloproliferative Neoplasm with Differential Effects on Hematopoietic Stem and Progenitor Cells. Cancer Cell 2010, 17, 584–596. [Google Scholar] [CrossRef] [Green Version]
- Andrikovics, H.; Krahling, T.; Balassa, K.; Halm, G.; Bors, A.; Koszarska, M.; Batai, A.; Dolgos, J.; Csomor, J.; Egyed, M.; et al. Distinct clinical characteristics of myeloproliferative neoplasms with calreticulin mutations. Haematologica 2014, 99, 1184–1190. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Prins, D.; Park, H.J.; Grinfeld, J.; Gonzalez-Arias, C.; Loughran, S.; Dovey, O.M.; Klampfl, T.; Bennett, C.; Hamilton, T.L.; et al. Mutant calreticulin knockin mice develop thrombocytosis and myelofibrosis without a stem cell self-renewal advantage. Blood 2018, 131, 649–661. [Google Scholar] [CrossRef]
- Baxter, E.J.; Scott, L.M.; Campbell, P.J.; East, C.; Fourouclas, N.; Swanton, S.; Vassiliou, G.S.; Bench, A.J.; Boyd, E.M.; Curtin, N.; et al. Acquired mutation of the tyrosine kinase JAK2 in human myeloproliferative disorders. Lancet 2005, 365, 1054–1061. [Google Scholar] [CrossRef]
- James, C.; Ugo, V.; Le Couédic, J.P.; Staerk, J.; Delhommeau, F.; Lacout, C.; Garçon, L.; Raslova, H.; Berger, R.; Bennaceur-Griscelli, A.; et al. A unique clonal JAK2 mutation leading to constitutive signalling causes polycythaemia vera. Nature 2005, 43, 1144–1148. [Google Scholar] [CrossRef] [PubMed]
- Kralovics, R.; Passamonti, F.; Buser, A.S.; Teo, S.-S.; Tiedt, R.; Passweg, J.R.; Tichelli, A.; Cazzola, M.; Skoda, R.C. A Gain-of-Function Mutation ofJAK2in Myeloproliferative Disorders. N. Engl. J. Med. 2005, 352, 1779–1790. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levine, R.L.; Wadleigh, M.; Cools, J.; Ebert, B.L.; Wernig, G.; Huntly, B.J.; Boggon, T.J.; Wlodarska, I.; Clark, J.J.; Moore, S.; et al. Activating mutation in the tyrosine kinase JAK2 in polycythemia vera, essential thrombocythemia, and myeloid metaplasia with myelofibrosis. Cancer Cell 2005, 7, 387–397. [Google Scholar] [CrossRef] [Green Version]
- Pardanani, A.D.; Levine, R.L.; Lasho, T.; Pikman, Y.; Mesa, R.; Wadleigh, M.; Steensma, D.P.; Elliott, M.; Wolanskyj, A.P.; Hogan, W.J.; et al. MPL515 mutations in myeloproliferative and other myeloid disorders: A study of 1182 patients. Blood 2006, 108, 3472–3476. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klampfl, T.; Gisslinger, H.; Harutyunyan, A.S.; Nivarthi, H.; Rumi, E.; Milosevic, J.D.; Them, N.C.C.; Berg, T.; Gisslinger, B.; Pietra, D.; et al. Somatic Mutations of Calreticulin in Myeloproliferative Neoplasms. N. Engl. J. Med. 2013, 369, 2379–2390. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nangalia, J.; Massie, C.; Baxter, E.; Nice, F.; Gundem, G.; Wedge, D.; Avezov, E.; Li, J.; Kollmann, K.; Kent, D.; et al. SomaticCALRMutations in Myeloproliferative Neoplasms with NonmutatedJAK2. N. Engl. J. Med. 2013, 369, 2391–2405. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Araki, M.; Yang, Y.; Masubuchi, N.; Hironaka, Y.; Takei, H.; Morishita, S.; Mizukami, Y.; Kan, S.; Shirane, S.; Edahiro, Y.; et al. Activation of the thrombopoietin receptor by mutant calreticulin in CALR-mutant myeloproliferative neoplasms. Blood 2016, 127, 1307–1316. [Google Scholar] [CrossRef] [PubMed]
- Chachoua, I.; Pecquet, C.; El-Khoury, M.; Nivarthi, H.; Albu, R.-I.; Marty, C.; Gryshkova, V.; Defour, J.-P.; Vertenoeil, G.; Ngo, A.; et al. Thrombopoietin receptor activation by myeloproliferative neoplasm associated calreticulin mutants. Blood 2016, 127, 1325–1335. [Google Scholar] [CrossRef]
- Marty, C.; Pecquet, C.; Nivarthi, H.; El-Khoury, M.; Chachoua, I.; Tulliez, M.; Villeval, J.-L.; Raslova, H.; Kralovics, R.; Constantinescu, S.N.; et al. Calreticulin mutants in mice induce an MPL-dependent thrombocytosis with frequent progression to myelofibrosis. Blood 2016, 127, 1317–1324. [Google Scholar] [CrossRef] [Green Version]
- Elf, S.; Abdelfattah, N.S.; Chen, E.; Perales-Patón, J.; Rosen, E.A.; Ko, A.; Peisker, F.; Florescu, N.; Giannini, S.; Wolach, O.; et al. Mutant Calreticulin Requires Both Its Mutant C-terminus and the Thrombopoietin Receptor for Oncogenic Transformation. Cancer Discov. 2016, 6, 368–381. [Google Scholar] [CrossRef] [Green Version]
- Pietra, D.; Rumi, E.; Ferretti, V.V.; Di Buduo, C.A.; Milanes, C.; Cavalloni, C.; Sant’Antonio, E.; Abbonante, V.; Moccia, F.; Casetti, I.C.; et al. Differential clinical effects of different mutation subtypes in CALR-mutant myeloproliferative neoplasms. Leukemia 2016, 30, 431–438. [Google Scholar] [CrossRef] [Green Version]
- Cabagnols, X.; Favale, F.; Pasquier, F.; Messaoudi, K.; Defour, J.P.; Ianotto, J.-C.; Marzac, C.; Le Couédic, J.P.; Droin, N.; Chachoua, I.; et al. Presence of atypical thrombopoietin receptor (MPL) mutations in triple-negative essential thrombocythemia patients. Blood 2016, 127, 333–342. [Google Scholar] [CrossRef] [Green Version]
- Feenstra, J.D.M.; Nivarthi, H.; Gisslinger, H.; Leroy, E.; Rumi, E.; Chachoua, I.; Bagienski, K.; Kubesova, B.; Pietra, D.; Gisslinger, B.; et al. Whole-exome sequencing identifies novel MPL and JAK2 mutations in triple-negative myeloproliferative neoplasms. Blood 2016, 127, 325–332. [Google Scholar] [CrossRef]
- Papaemmanuil, E.; Döhner, H.; Campbell, P.J. Genomic Classification in Acute Myeloid Leukemia. N. Engl. J. Med. 2016, 374, 2209–2221. [Google Scholar] [CrossRef]
- Grinfeld, J.; Nangalia, J.; Baxter, E.J.; Wedge, D.C.; Angelopoulos, N.; Cantrill, R.; Godfrey, A.L.; Papaemmanuil, E.; Gundem, G.; MacLean, C.; et al. Classification and Personalized Prognosis in Myeloproliferative Neoplasms. N. Engl. J. Med. 2018, 379, 1416–1430. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Liu, Y.; Rappaport, A.R.; Kitzing, T.; Schultz, N.; Zhao, Z.; Shroff, A.S.; Dickins, R.A.; Vakoc, C.R.; Bradner, J.E.; et al. MLL3 Is a Haploinsufficient 7q Tumor Suppressor in Acute Myeloid Leukemia. Cancer Cell 2014, 25, 652–665. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arber, D.A.; Orazi, A.; Hasserjian, R.; Thiele, J.; Borowitz, M.J.; Le Beau, M.M.; Bloomfield, C.D.; Cazzola, M.; Vardiman, J.W. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood 2016, 127, 2391–2405. [Google Scholar] [CrossRef] [PubMed]
- Genovese, G.; Kähler, A.K.; Handsaker, R.E.; Lindberg, J.; Rose, S.A.; Bakhoum, S.F.; Chambert, K.; Mick, E.; Neale, B.M.; Fromer, M.; et al. Clonal Hematopoiesis and Blood-Cancer Risk Inferred from Blood DNA Sequence. N. Engl. J. Med. 2014, 371, 2477–2487. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mertz, P.; Chalayer, E.; Amoura, Z.; Cathebras, P.; Chiche, L.; Coestedoat, N.; Deroux, A.; Diot, E.; Durupt, S.; Forestier, E.; et al. Clinical spectrum and therapeutic management of auto-immune myelofibrosis: A nation-wide study of 30 cases. Haematologica 2020. [Google Scholar] [CrossRef]
- Vannucchi, A.M.; Antonioli, E.; Guglielmelli, P.; Pardanani, A.; Tefferi, A. Clinical correlates of JAK2V617F presence or allele burden in myeloproliferative neoplasms: A critical reappraisal. Leukemia 2008, 22, 1299–1307. [Google Scholar] [CrossRef]
- Vannucchi, A.M.; Lasho, T.L.; Guglielmelli, P.; Am, V.; Pardanani, A.; Pereira, A.; Finke, C.M.; Score, J.; Gangat, N.; Mannarelli, C.; et al. Mutations and prognosis in primary myelofibrosis. Leukemia 2013, 27, 1861–1869. [Google Scholar] [CrossRef]
- Rumi, E.; Pietra, D.; Pascutto, C.; Guglielmelli, P.; Martínez-Trillos, A.; Casetti, I.; Colomer, D.; Pieri, L.; Pratcorona, M.; Rotunno, G.; et al. Clinical effect of driver mutations of JAK2, CALR, or MPL in primary myelofibrosis. Blood 2014, 124, 1062–1069. [Google Scholar] [CrossRef] [Green Version]
- Tefferi, A.; Lasho, T.L.; Finke, C.; Belachew, A.A.; Wassie, E.A.; Ketterling, R.P.; Hanson, C.A.; Pardani, A. Type 1 vs. type 2 calreticulin mutations in primary myelofibrosis: Differences in phenotype and prognostic impact. Leukemia 2014, 28, 1568–1570. [Google Scholar] [CrossRef]
- Tefferi, A.; Lasho, T.L.; Tischer, A.; Wassie, E.A.; Finke, C.M.; Belachew, A.A.; Ketterling, R.P.; Hanson, C.A.; Pardanani, A.D. The prognostic advantage of calreticulin mutations in myelofibrosis might be confined to type 1 or type 1-like CALR variants. Blood 2014, 124, 2465–2466. [Google Scholar] [CrossRef] [Green Version]
- Finazzi, M.C.; Carobbio, A.; Cervantes, F.; Isola, I.M.; Vannucchi, A.M.; Guglielmelli, P.; Rambaldi, A.; Finazzi, G.; Barosi, G.; Barbui, T. CALR mutation, MPL mutation and triple negativity identify patients with the lowest vascular risk in primary myelofibrosis. Leukemia 2014, 29, 1209–1210. [Google Scholar] [CrossRef] [PubMed]
- Tefferi, A.; Lasho, T.L.; Huang, J.; Finke, C.M.; Mesa, R.; Li, C.Y.; Wu, W.; Hanson, C.; Pardanani, A. Low JAK2V617F allele burden in primary myelofibrosis, compared to either a higher allele burden or unmutated status, is associated with inferior overall and leukemia-free survival. Leukemia 2008, 22, 756–761. [Google Scholar] [CrossRef] [PubMed]
- Guglielmelli, P.; Barosi, G.; Specchia, G.; Rambaldi, A.; Lo-Coco, F.; Antonioli, E.; Pieri, L.; Pancrazzi, A.; Ponziani, V.; Delaini, F.; et al. Identification of patients with poorer survival in primary myelofibrosis based on the burden of JAK2V617F mutated allele. Blood 2009, 114, 1477–1483. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barosi, G.; Massa, M.; Campanelli, R.; Fois, G.; Catarsi, P.; Viarengo, G.; Villani, L.; Poletto, V.; Bosoni, T.; Magrini, U.; et al. Primary myelofibrosis: Older age and high JAK2 V617F allele burden are associated with elevated plasma high-sensitivity C-reactive protein levels and a phenotype of progressive disease. Leuk. Res. 2017, 60, 18–23. [Google Scholar] [CrossRef]
- Barosi, G.; Klersy, C.; Villani, L.; Bonetti, E.; Catarsi, P.; Poletto, V.; Campanelli, R.; Impera, S.; Latagliata, R.; Viarengo, G.; et al. JAK2(V617F) allele burden ≥50% is associated with response to ruxolitinib in persons with MPN-associated myelofibrosis and splenomegaly requiring therapy. Leukemia 2016, 30, 1772–1775. [Google Scholar] [CrossRef]
- Cabagnols, X.; Defour, J.P.; Ugo, V.; Ianotto, J.C.; Mossuz, P.; Mondet, J.; Girodon, F.; Alexandre, J.H.; Mansier, O.; Viallard, J.F.; et al. Differential association of calreticulin type 1 and type 2 mutations with myelofibrosis and essential thrombocytemia: Relevance for disease evolution. Leukemia 2015, 29, 249–252. [Google Scholar] [CrossRef]
- Rumi, E.; Pietra, D.; Ferretti, V.; Klampfl, T.; Harutyunyan, A.S.; Milosevic, J.D.; Them, N.C.C.; Berg, T.; Elena, C.; Casetti, I.C.; et al. JAK2 or CALR mutation status defines subtypes of essential thrombocythemia with substantially different clinical course and outcomes. Blood 2014, 123, 1544–1551. [Google Scholar] [CrossRef]
- Godfrey, A.L.; Chen, E.; Pagano, F.; Ortmann, C.A.; Silber, Y.; Bellosillo, B.; Guglielmelli, P.; Harrison, C.N.; Reilly, J.T.; Stegelmann, F.; et al. JAK2V617F homozygosity arises commonly and recurrently in PV and ET, but PV is characterized by expansion of a dominant homozygous subclone. Blood 2012, 120, 2704–2707. [Google Scholar] [CrossRef]
- Rumi, E.; Pietra, D.; Guglielmelli, P.; Bordoni, R.; Casetti, I.; Milanesi, C.; Sant’Antonio, E.; Ferretti, V.V.; Pancrazzi, A.; Rotunno, G.; et al. Acquired copy-neutral loss of heterozygosity of chromosome 1p as a molecular event associated with marrow fibrosis in MPL-mutated myeloproliferative neoplasms. Blood 2013, 121, 4388–4395. [Google Scholar] [CrossRef] [Green Version]
- Cavalloni, C.; Rumi, E.; Ferretti, V.V.; Pietra, D.; Roncoroni, E.; Bellini, M.; Ciboddo, M.; Casetti, I.C.; Landini, B.; Fugazza, E.; et al. Sequential evaluation of CALR mutant burden in patients with myeloproliferative neoplasms. Oncotarget 2017, 8, 33416–33421. [Google Scholar] [CrossRef] [Green Version]
- Kralovics, R. Genetic complexity of myeloproliferative neoplasms. Leukemia 2008, 22, 1841–1848. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nangalia, J.; Nice, F.L.; Wedge, D.C.; Godfrey, A.L.; Grinfeld, J.; Thakker, C.; Massie, C.E.; Baxter, J.; Sewell, D.; Silber, Y.; et al. DNMT3A mutations occur early or late in patients with myeloproliferative neoplasms and mutation order influences phenotype. Haematologica 2015, 100, e438–e442. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ortmann, C.A.; Kent, D.G.; Nangalia, J.; Silber, Y.; Wedge, D.C.; Grinfeld, J.; Baxter, E.J.; Massie, C.E.; Papaemmanuil, E.; Menon, S.; et al. Effect of mutation order on myeloproliferative neoplasms. N. Engl. J. Med. 2015, 372, 601–612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hinds, D.A.; Barnholt, K.E.; Mesa, R.A.; Kiefer, A.K.; Do, C.B.; Eriksson, N.; Mountain, J.L.; Francke, U.; Tung, J.Y.; Nguyen, H.M.; et al. Germ line variants predispose to both JAK2 V617F clonal hematopoiesis and myeloproliferative neoplasms. Blood 2016, 128, 1121–1128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hernández-Boluda, J.C.; Dupriez, B.; Pereira, A.; Passamonti, F.; Reilly, J.T.; Morra, E.; Vannucchi, A.M.; Mesa, R.A.; Demory, J.-L.; Barosi, G.; et al. New prognostic scoring system for primary myelofibrosis based on a study of the International Working Group for Myelofibrosis Research and Treatment. Blood 2009, 113, 2895–2901. [Google Scholar]
- Passamonti, F.; Cervantes, F.; Vannucchi, A.M.; Morra, E.; Rumi, E.; Pereira, A.; Guglielmelli, P.; Pungolino, E.; Caramella, M.; Maffioli, M.; et al. A dynamic prognostic model to predict survival in primary myelofibrosis: A study by the IWG-MRT (International Working Group for Myeloproliferative Neoplasms Research and Treatment). Blood 2010, 115, 1703–1708. [Google Scholar] [CrossRef] [PubMed]
- Gangat, N.; Caramazza, D.; Vaidya, R.; George, G.; Begna, K.; Schwager, S.; Van Dyke, D.; Hanson, C.; Wu, W.; Pardanani, A.; et al. DIPSS Plus: A Refined Dynamic International Prognostic Scoring System for Primary Myelofibrosis That Incorporates Prognostic Information From Karyotype, Platelet Count, and Transfusion Status. J. Clin. Oncol. 2011, 29, 392–397. [Google Scholar] [CrossRef]
- Tefferi, A.; Lasho, T.L.; Finke, C.M.; Knudson, R.; Ketterling, R.P.; Hanson, C.H.; Maffioli, M.; Caramazza, D.; Passamonti, F.; Pardanani, A. CALR vs. JAK2 vs. MPL-mutated or triple-negative myelofibrosis: Clinical, cytogenetic and molecular comparisons. Leukemia 2014, 28, 1472–1477. [Google Scholar] [CrossRef]
- Tefferi, A.; Guglielmelli, P.; Lasho, T.L.; Rotunno, G.; Finke, C.; Mannarelli, C.; Belachew, A.; Pancrazzi, A.; Wassie, E.; Ketterling, R.P.; et al. CALR and ASXL1 mutations-based molecular prognostication in primary myelofibrosis: An international study of 570 patients. Leukemia 2014, 28, 1494–1500. [Google Scholar] [CrossRef]
- Bullinger, L.; Döhner, K.; Döhner, H. Genomics of Acute Myeloid Leukemia Diagnosis and Pathways. J. Clin. Oncol. 2017, 35, 934–946. [Google Scholar] [CrossRef]
- Guglielmelli, P.; Lasho, T.L.; Biamonte, F.; Score, J.; Mannarelli, C.; Rotunno, G.; Pancrazzi, A.; Pardanani, A.; Zoi, K.; Reiter, A.; et al. Effect of the Number of Prognostically Relevant Mutated Genes on Survival and Leukemia Progression in Primary Myelofibrosis. Blood 2013, 122, 104. [Google Scholar] [CrossRef]
- Guglielmelli, P.; Pacilli, A.; Rotunno, G.; Rumi, E.; Rosti, V.; Delaini, F.; Maffioli, M.; Fanelli, T.; Pancrazzi, A.; Pietra, D.; et al. Presentation and outcome of patients with 2016 WHO diagnosis of prefibrotic and overt primary myelofibrosis. Blood 2017, 129, 3227–3236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spiegel, J.Y.; McNamara, C.; Kennedy, J.A.; Panzarella, T.; Arruda, A.; Stockley, T.; Sukhai, M.; Thomas, M.; Bartoszko, J.; Ho, M.J.; et al. Impact of genomic alterations on outcomes in myelofibrosis patients undergoing JAK1/2 inhibitor therapy. Blood Adv. 2017, 1, 1729–1738. [Google Scholar] [CrossRef] [PubMed]
- Coltro, G.; Rotunno, G.; Mannelli, L.; Mannarelli, C.; Fiaccabrino, S.; Romagnoli, S.; Bartalucci, N.; Ravenda, E.; Gelli, E.; Sant’Antonio, E.; et al. RAS/CBL mutations predict resistance to JAK inhibitors in myelofibrosis and are associated with poor prognostic features. Blood Adv. 2020, 4, 3677–3687. [Google Scholar] [CrossRef]
- Guglielmelli, P.; Lasho, T.; Rotunno, G.; Mudireddy, M.; Mannarelli, C.; Nicolosi, M.; Pacilli, A.; Pardanani, A.D.; Rumi, E.; Rosti, V.; et al. MIPSS70: Mutation-Enhanced International Prognostic Score System for Transplantation-Age Patients With Primary Myelofibrosis. J. Clin. Oncol. 2018, 36, 310–318. [Google Scholar] [CrossRef]
- Tefferi, A.; Nicolosi, M.; Mudireddy, M.; Lasho, T.L.; Gangat, N.; Begna, K.H.; Hanson, C.A.; Ketterling, R.P.; Pardanani, A. Revised cytogenetic risk stratification in primary myelofibrosis: Analysis based on 1002 informative patients. Leukemia 2018, 32, 1189–1199. [Google Scholar] [CrossRef]
- Tefferi, A.; Guglielmelli, P.; Lasho, T.; Gangat, N.; Ketterling, R.P.; Pardanani, A.D.; Vannucchi, A.M. MIPSS70+ Version 2.0: Mutation and Karyotype-Enhanced International Prognostic Scoring System for Primary Myelofibrosis. J. Clin. Oncol. 2018, 36, 1769–1770. [Google Scholar] [CrossRef]
- Tefferi, A.; Finke, C.M.; Lasho, T.L.; Hanson, C.A.; Ketterling, R.P.; Gangat, N.; Pardanani, A. U2AF1 mutation types in primary myelofibrosis: Phenotypic and prognostic distinctions. Leukemia 2018, 32, 2274–2278. [Google Scholar] [CrossRef]
- Tefferi, A.; Lasho, T.L.; Mudireddy, M.; Finke, C.M.; Hanson, C.A.; Ketterling, R.P.; Gangat, N.; Pardanani, A. The germline JAK2 GGCC (46/1) haplotype and survival among 414 molecularly‐annotated patients with primary myelofibrosis. Am. J. Hematol. 2019, 94, 299–305. [Google Scholar] [CrossRef]
- Guglielmelli, P.; Biamonte, F.; Rotunno, G.; Artusi, V.; Artuso, L.; Bernardis, I.; Tenedini, E.; Pieri, L.; Paoli, C.; Mannarelli, C.; et al. Impact of mutational status on outcomes in myelofibrosis patients treated with ruxolitinib in the COMFORT-II study. Blood 2014, 123, 2157–2160. [Google Scholar] [CrossRef]
- Guglielmelli, P.; Rotunno, G.; Bogani, C.; Mannarelli, C.; Giunti, L.; Provenzano, A.; Giglio, S.; Squires, M.; Stalbovskaya, V.; Gopalakrishna, P.; et al. Ruxolitinib is an effective treatment forCALR-positive patients with myelofibrosis. Br. J. Haematol. 2016, 173, 938–940. [Google Scholar] [CrossRef] [PubMed]
- Patel, K.P.; Newberry, K.J.; Luthra, R.; Jabbour, E.; Pierce, S.; Cortes, J.; Singh, R.; Mehrotra, M.; Routbort, M.J.; Luthra, M.; et al. Correlation of mutation profile and response in patients with myelofibrosis treated with ruxolitinib. Blood 2015, 126, 790–797. [Google Scholar] [CrossRef] [Green Version]
- Newberry, K.J.; Patel, K.; Masarova, L.; Luthra, R.; Manshouri, T.; Jabbour, E.; Bose, P.; Daver, N.; Cortes, J.; Kantarjian, H.; et al. Clonal evolution and outcomes in myelofibrosis after ruxolitinib discontinuation. Blood 2017, 130, 1125–1131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pacilli, A.; Rotunno, G.; Mannarelli, C.; Fanelli, T.; Pancrazzi, A.; Contini, E.; Mannelli, F.; Gesullo, F.; Bartalucci, N.; Fattori, G.C.; et al. Mutation landscape in patients with myelofibrosis receiving ruxolitinib or hydroxyurea. Blood Cancer J. 2018, 8, 122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tefferi, A.; Lasho, T.; Begna, K.H.; Patnaik, M.M.; Zblewski, D.L.; Finke, C.M.; Laborde, R.R.; Wassie, E.; Schimek, L.; Hanson, C.A.; et al. A Pilot Study of the Telomerase Inhibitor Imetelstat for Myelofibrosis. N. Engl. J. Med. 2015, 373, 908–919. [Google Scholar] [CrossRef] [Green Version]
- Tefferi, A.; Barraco, D.; Lasho, T.L.; Shah, S.; Begna, K.H.; Al-Kali, A.; Hogan, W.J.; Litzow, M.R.; Hanson, C.A.; Ketterling, R.P.; et al. Momelotinib therapy for myelofibrosis: A 7-year follow-up. Blood Cancer J. 2018, 8, 29. [Google Scholar] [CrossRef]
- Silver, R.T.; Barel, A.C.; Lascu, E.; Ritchie, E.; Roboz, G.J.; Christos, P.J.; Orazi, A.; Hassane, D.C.; Tam, W.; Cross, N.C.P. The effect of initial molecular profile on response to recombinant interferon-α (rIFNα) treatment in early myelofibrosis. Cancer 2017, 123, 2680–2687. [Google Scholar] [CrossRef]
- Ianotto, J.-C.; Chauveau, A.; Boyer-Perrard, F.; Gyan, E.; Laribi, K.; Cony-Makhoul, P.; Demory, J.-L.; De Renzis, B.; Dosquet, C.; Rey, J.; et al. Benefits and pitfalls of pegylated interferon-α2a therapy in patients with myeloproliferative neoplasm-associated myelofibrosis: A French Intergroup of Myeloproliferative neoplasms (FIM) study. Haematologica 2017, 103, 438–446. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alchalby, H.; Badbaran, A.; Zabelina, T.; Kobbe, G.; Hahn, J.; Wolff, D.; Bornhäuser, M.; Thiede, C.; Baurmann, H.; Bethge, W.; et al. Impact of JAK2V617F mutation status, allele burden, and clearance after allogeneic stem cell transplantation for myelofibrosis. Blood 2010, 116, 3572–3581. [Google Scholar] [CrossRef] [Green Version]
- Panagiota, V.; Thol, F.; Márkus, B.; Fehse, B.; Alchalby, H.; Badbaran, A.; Lehmann, U.; Koenecke, C.; Shahswar, R.; Chaturvedi, A.; et al. Prognostic effect of calreticulin mutations in patients with myelofibrosis after allogeneic hematopoietic stem cell transplantation. Leukemia 2014, 28, 1552–1555. [Google Scholar] [CrossRef]
- Kröger, N.; Panagiota, V.; Badbaran, A.; Zabelina, T.; Triviai, I.; Cruz, M.M.A.; Shahswar, R.; Ayuk, F.; Gehlhaar, M.; Wolschke, C.; et al. Impact of Molecular Genetics on Outcome in Myelofibrosis Patients after Allogeneic Stem Cell Transplantation. Biol. Blood Marrow Transplant. 2017, 23, 1095–1101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gagelmann, N.; Ditschkowski, M.; Bogdanov, R.; Bredin, S.; Robin, M.; Cassinat, B.; Shahswar, R.; Thol, F.; Heuser, M.; Socié, G.; et al. Comprehensive clinical-molecular transplant scoring system for myelofibrosis undergoing stem cell transplantation. Blood 2019, 133, 2233–2242. [Google Scholar] [CrossRef] [PubMed]
- Kröger, N.; Badbaran, A.; Holler, E.; Hahn, J.; Kobbe, G.; Bornhäuser, M.; Reiter, A.; Zabelina, T.; Zander, A.R.; Fehse, B. Monitoring of the JAK2-V617F mutation by highly sensitive quantitative real-time PCR after allogeneic stem cell transplantation in patients with myelofibrosis. Blood 2006, 109, 1316–1321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alchalby, H.; Badbaran, A.; Bock, O.; Fehse, B.; Bacher, U.; Zander, A.R.; Kröger, N. Screening and monitoring of MPL W515L mutation with real-time PCR in patients with myelofibrosis undergoing allogeneic-SCT. Bone Marrow Transplant. 2010, 45, 1404–1407. [Google Scholar] [CrossRef] [PubMed]
- Rumi, E.; Passamonti, F.; Arcaini, L.; Bernasconi, P.; Elena, C.; Pietra, D.; Brisci, A.; Arbustini, E.; Cazzola, M.; Lazzarino, M. Molecular remission after allo-SCT in a patient with post-essential thrombocythemia myelofibrosis carrying the MPL (W515A) mutation. Bone Marrow Transplant. 2009, 45, 798–800. [Google Scholar] [CrossRef] [Green Version]
- Wolschke, C.; Badbaran, A.; Zabelina, T.; Christopeit, M.; Ayuk, F.; Triviai, I.; Zander, A.; Alchalby, H.; Bacher, U.; Fehse, B.; et al. Impact of molecular residual disease post allografting in myelofibrosis patients. Bone Marrow Transplant. 2017, 52, 1526–1529. [Google Scholar] [CrossRef]
- Kröger, N.; Alchalby, H.; Klyuchnikov, E.; Badbaran, A.; Hildebrandt, Y.; Ayuk, F.; Bacher, U.; Bock, O.; Kvasnicka, M.; Fehse, B.; et al. JAK2-V617F–triggered preemptive and salvage adoptive immunotherapy with donor-lymphocyte infusion in patients with myelofibrosis after allogeneic stem cell transplantation. Blood 2009, 113, 1866–1868. [Google Scholar] [CrossRef]
- Larsen, T.S.; Christensen, J.H.; Hasselbalch, H.C.; Pallisgaard, N. The JAK2 V617F mutation involves B- and T-lymphocyte lineages in a subgroup of patients with Philadelphia-chromosome negative chronic myeloproliferative disorders. Br. J. Haematol. 2007, 136, 745–751. [Google Scholar] [CrossRef]
- Jovanovic, J.V.; Ivey, A.; Vannucchi, A.M.; Lippert, E.; Leibundgut, E.O.; Cassinat, B.; Pallisgaard, N.; Maroc, N.; Hermouet, S.; Nickless, G.; et al. Establishing optimal quantitative-polymerase chain reaction assays for routine diagnosis and tracking of minimal residual disease in JAK2-V617F-associated myeloproliferative neoplasms: A joint European LeukemiaNet/MPN&MPNr-EuroNet (COST action BM0902) study. Leukemia 2013, 27, 2032–2039. [Google Scholar]
- Asp, J.; Skov, V.; Bellosillo, B.; Kristensen, T.; Lippert, E.; Dicker, F.; Schwarz, J.; Wojtaszewska, M.; Palmqvist, L.; Akiki, S.; et al. International external quality assurance of JAK2 V617F quantification. Ann. Hematol. 2018, 98, 1111–1118. [Google Scholar] [CrossRef] [Green Version]
- Kjær, L.; Cordua, S.; Holmström, M.O.; Thomassen, M.; Kruse, T.; Pallisgaard, N.; Larsen, T.S.; De Stricker, K.; Skov, V.; Hasselbalch, H.C. Differential Dynamics of CALR Mutant Allele Burden in Myeloproliferative Neoplasms during Interferon Alfa Treatment. PLoS ONE 2016, 11, e0165336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mansier, O.; Migeon, M.; Saint-Lézer, A.; James, C.; Verger, E.; Robin, M.; Socié, G.; Bidet, A.; Mahon, F.-X.; Cassinat, B.; et al. Quantification of the Mutant CALR Allelic Burden by Digital PCR. J. Mol. Diagn. 2016, 18, 68–74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cordua, S.; Kjær, L.; Skov, V.; Pallisgaard, N.; Hasselbalch, H.C.; Ellervik, C. Prevalence and phenotypes of JAK2 V617F and calreticulin mutations in a Danish general population. Blood 2019, 134, 469–479. [Google Scholar] [CrossRef] [PubMed]
- Guglielmelli, P.; Pietra, D.; Pane, F.; Pancrazzi, A.; Cazzola, M.; Vannucchi, A.M.; Tura, S.; Barosi, G. Recommendations for molecular testing in classical Ph1-neg myeloproliferative disorders–A consensus project of the Italian Society of Hematology. Leuk. Res. 2017, 58, 63–72. [Google Scholar] [CrossRef]


{kind=link}
| Overt Primary Myelofibrosis (All 3 Major and at Least 1 Minor are Required) | Prefibrotic Primary Myelofibrosis (All 3 Major and at Least 1 Minor are Required) |
|---|---|
Major criteria
| Major criteria
|
Minor criteria
| Minor criteria
|
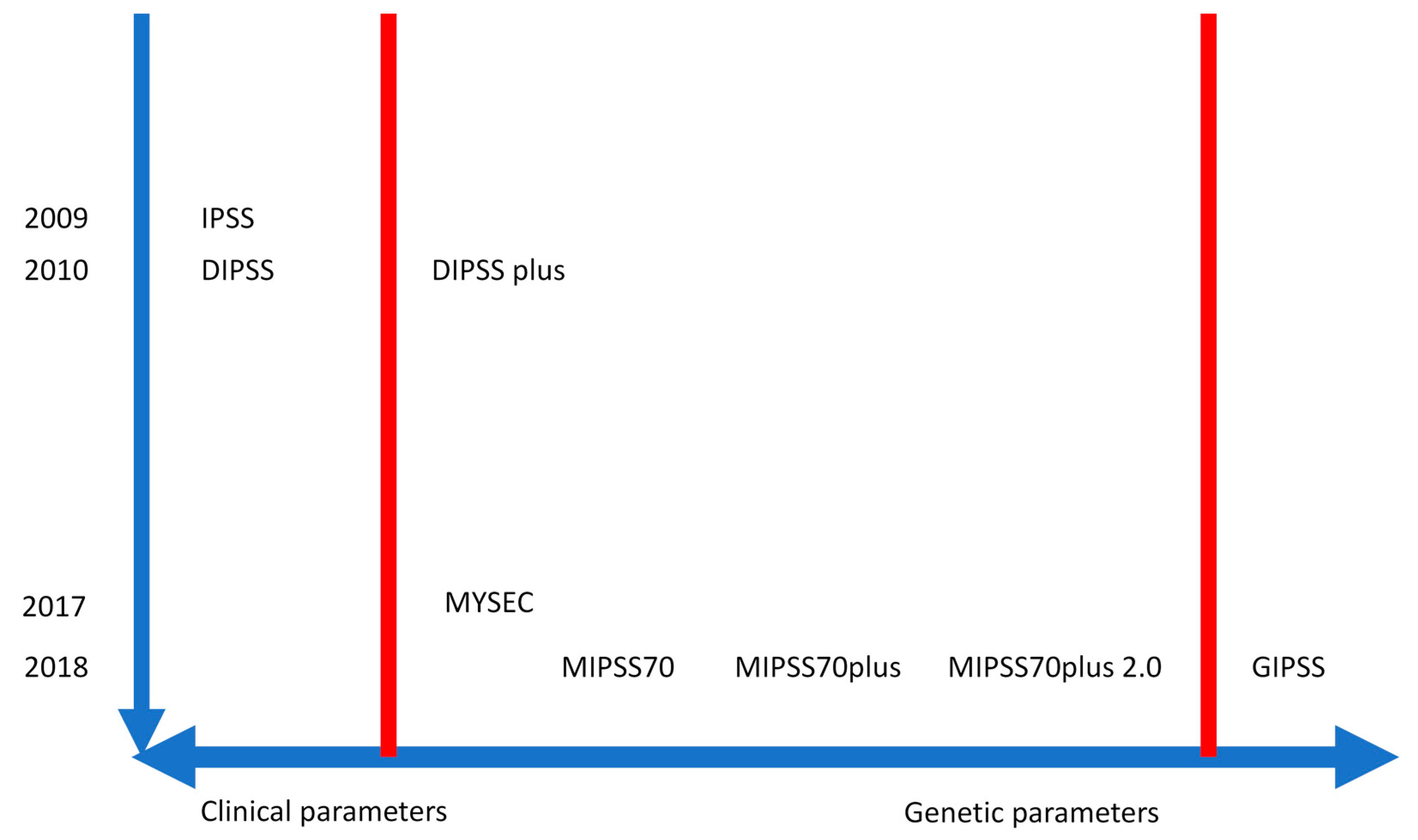
| Prognostic Model | Risk Groups and Clinical Relevance |
|---|---|
| International Prognostic Scoring System (IPSS) [45] IPSS estimates survival at the time of diagnosis. | |
Risk factors (weight):
| Low risk: 0 (median survival 11.3 years) Intermediate-1 risk: 1 point (7.9 years) Intermediate-2 risk: 2 points (4.0 years) High risk: ≥3 points (2.3 years) |
| Dynamic International Prognostic Scoring System (DIPSS) [46] DIPSS can be applied anytime during clinical course. | |
Risk factors (weight):
| Low risk: 0 (median survival: not reached) Intermediate-1 risk: 1 point (14.2 years) Intermediate-2 risk: 2 points (4.0 years) High risk: ≥3 points (1.5 years) |
| DIPSS-plus [47] DIPSS-plus can be applied anytime during the clinical course. | |
Risk factors (weight):
| Low risk: 0 (median survival: 15.4 years) Intermediate-1 risk: 1 point (6.5 years) Intermediate-2 risk: 2 points (2.9 years) High risk: ≥3 points (1.3 years) |
| MIPSS70 [55] MIPSS70 is used to better select patients <70 years as candidates for allogeneic stem cell transplantation. | |
Risk factors (weight):
| Low risk: 0–1 points (median survival: 27.7 years) Intermediate risk: 2–4 points (7.1 years) High risk: ≥5 points (2.3 years) |
| MIPSS70+ version 2.0 [57] MIPSS70+ version 2.0 incorporates the revised cytogenetic risk levels, U2AF1Q157 as an additional HMR mutation, and new sex- and severity-adjusted hemoglobin thresholds. | |
Risk factors (weight):
| Very low risk: 0 (median survival: not reached) Low risk: 1–2 points (16.4 years) Intermediate risk: 3–4 points (7.7 years) High risk: 5–8 points (4.1 years) Very high risk: ≥9 points (1.8 years) |
| GIPSS [59] GIPSS may be useful in early-stage patients because it can predict outcomes in the absence of clinical signs of progressive disease. | |
Risk factors (weight):
| Low risk: 0 (median survival: 26.4 years) Intermediate-1 risk: 1 point (8 years) Intermediate-2 risk: 2 points (4.2 years) High risk: ≥3 points (2 years) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rumi, E.; Trotti, C.; Vanni, D.; Casetti, I.C.; Pietra, D.; Sant’Antonio, E. The Genetic Basis of Primary Myelofibrosis and Its Clinical Relevance. Int. J. Mol. Sci. 2020, 21, 8885. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21238885
Rumi E, Trotti C, Vanni D, Casetti IC, Pietra D, Sant’Antonio E. The Genetic Basis of Primary Myelofibrosis and Its Clinical Relevance. International Journal of Molecular Sciences. 2020; 21(23):8885. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21238885
Chicago/Turabian StyleRumi, Elisa, Chiara Trotti, Daniele Vanni, Ilaria Carola Casetti, Daniela Pietra, and Emanuela Sant’Antonio. 2020. "The Genetic Basis of Primary Myelofibrosis and Its Clinical Relevance" International Journal of Molecular Sciences 21, no. 23: 8885. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21238885