Changes of Coenzyme A and Acetyl-Coenzyme A Concentrations in Rats after a Single-Dose Intraperitoneal Injection of Hepatotoxic Thioacetamide Are Not Consistent with Rapid Recovery


Abstract
:
1. Introduction
1.1. CoA and Acetyl CoA
1.2. Development of Pathophysiology in Rats and Recovery after TAA Intoxication
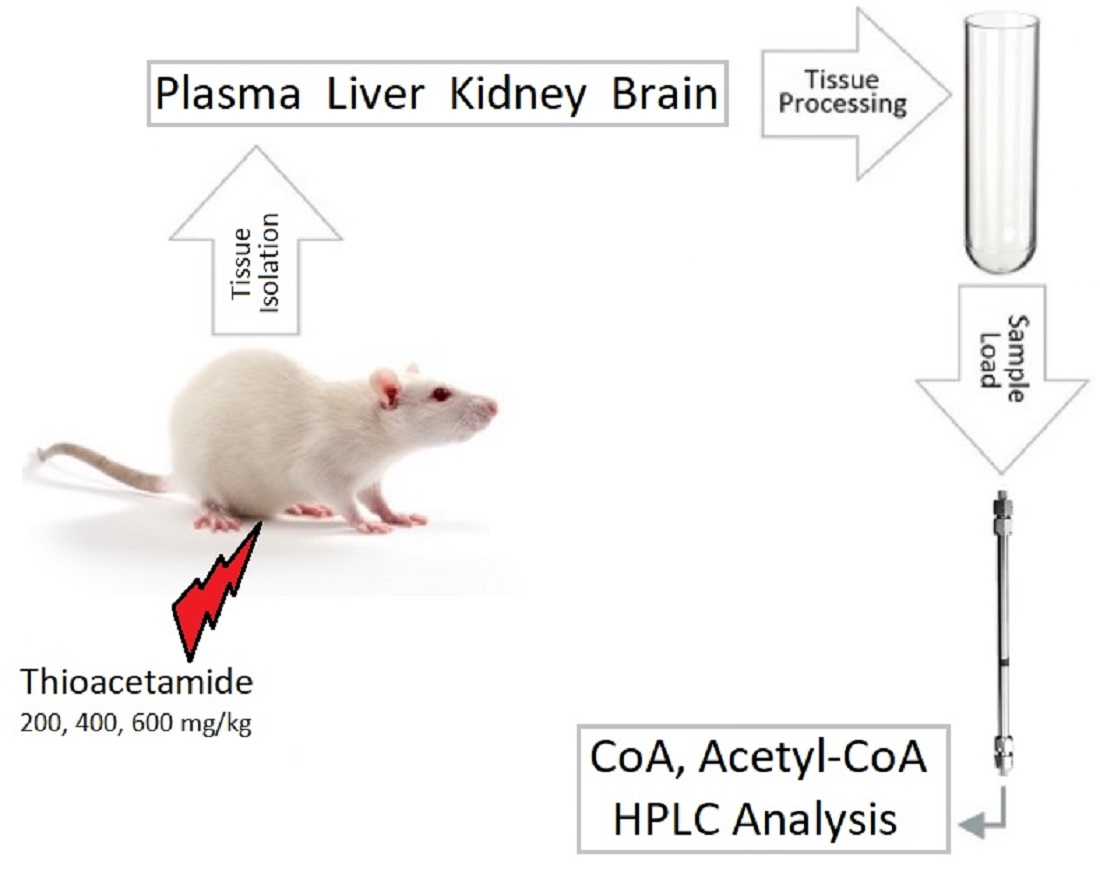
2. Materials and Methods
2.1. Chemicals
2.2. Standards
2.3. HPLC System
2.4. Mobile Phase
2.5. Preparation of Biological Samples for HPLC Analysis
2.5.1. Rat Tissues
2.5.2. Rat Plasma
2.6. Protein Analysis
2.7. Data and Statistical Analysis
3. Results
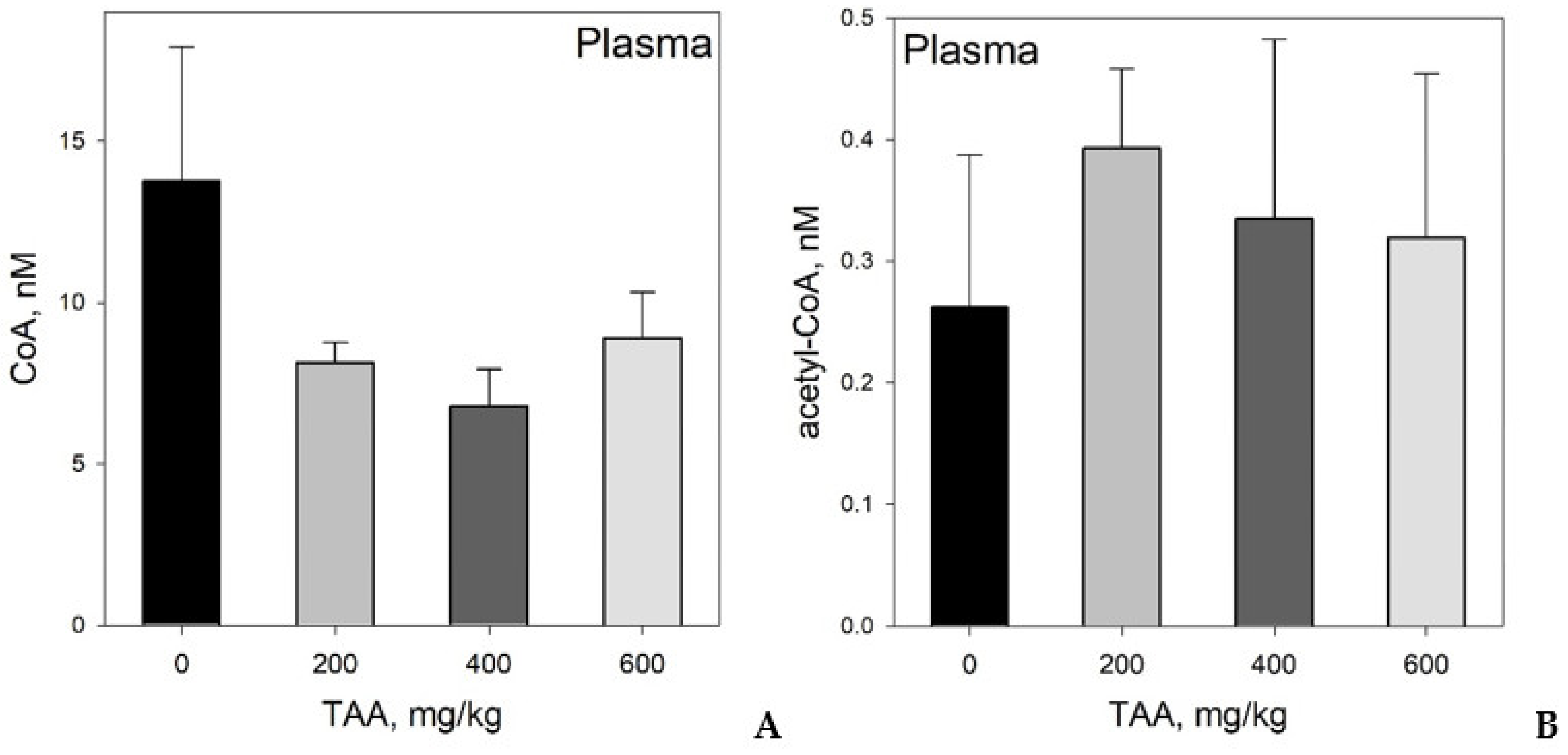
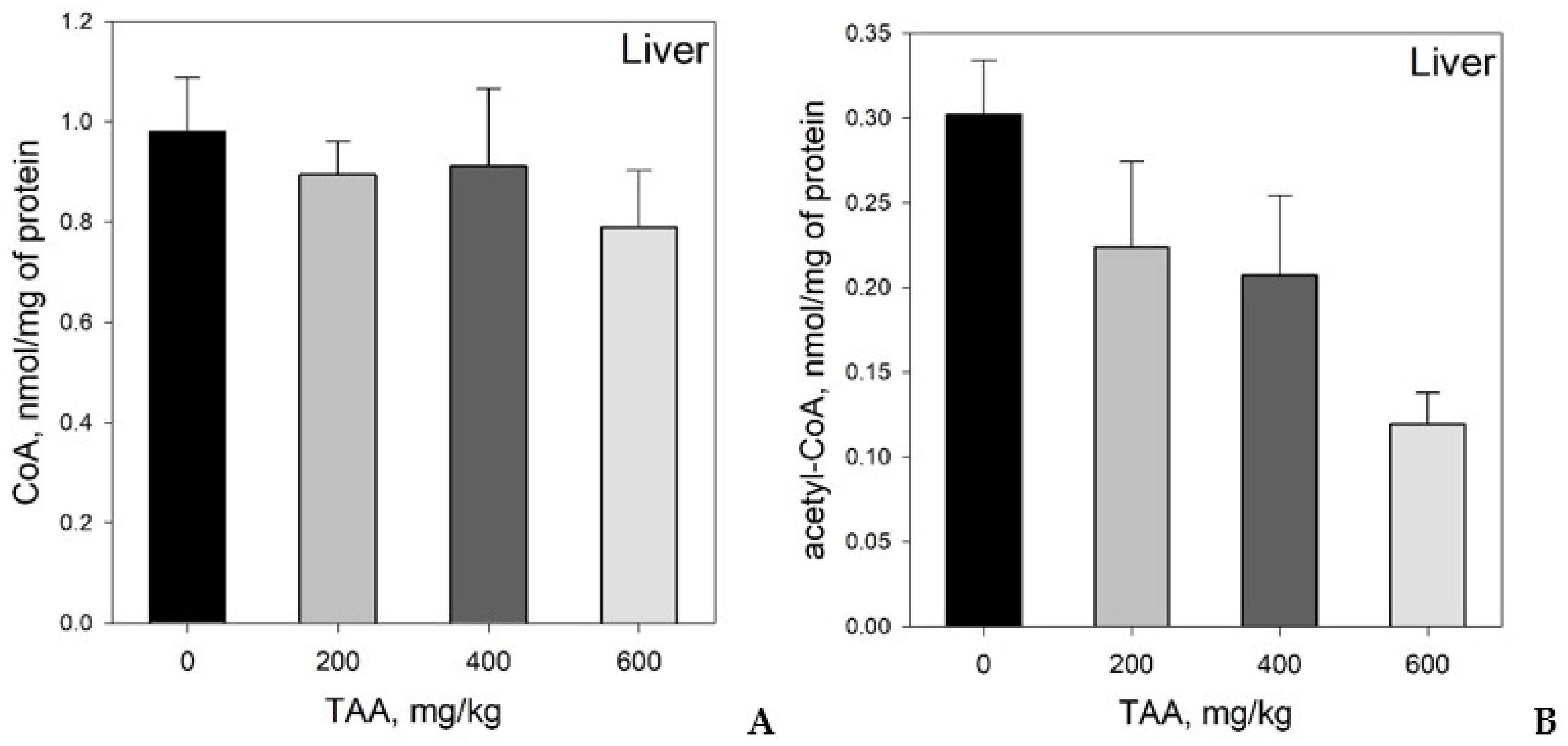
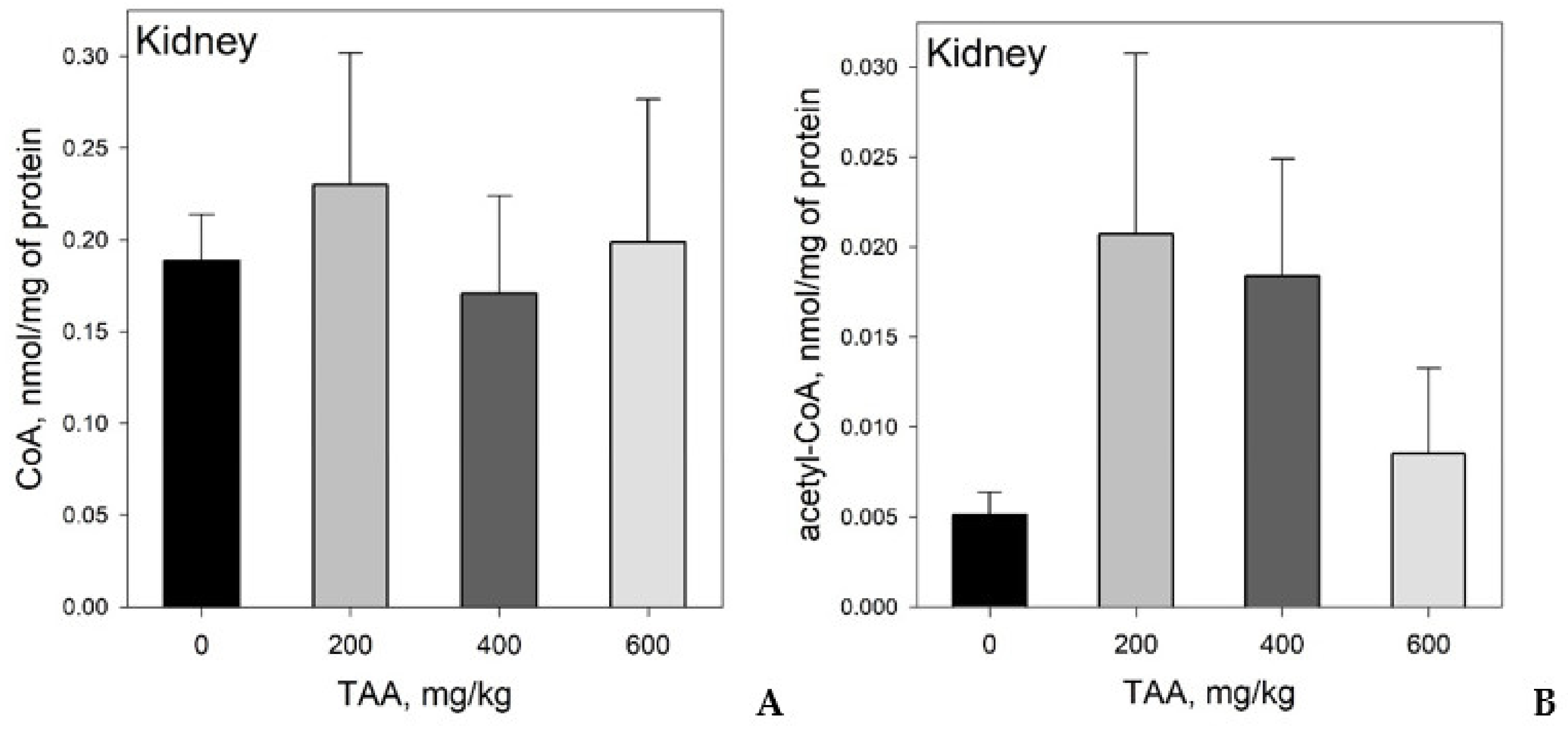
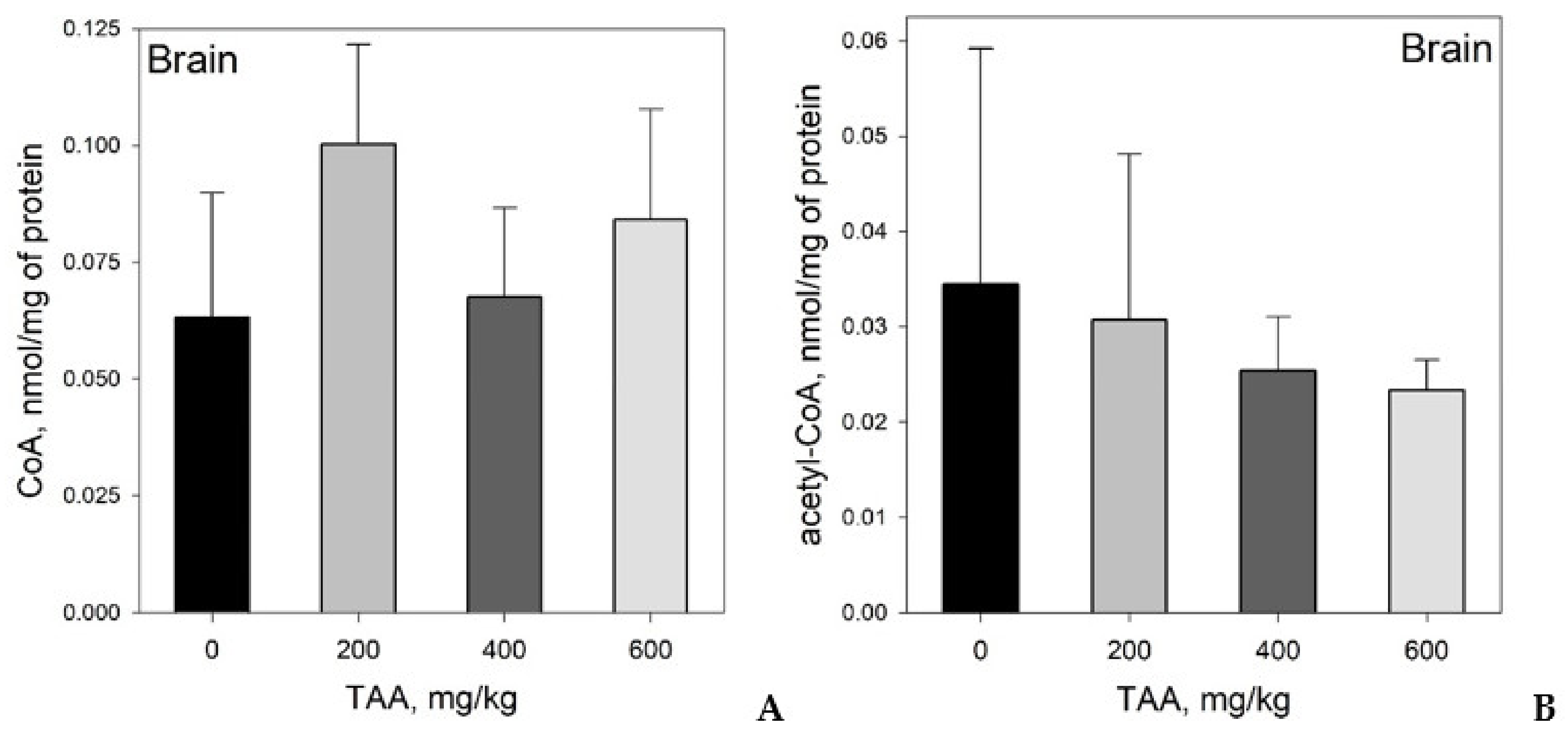
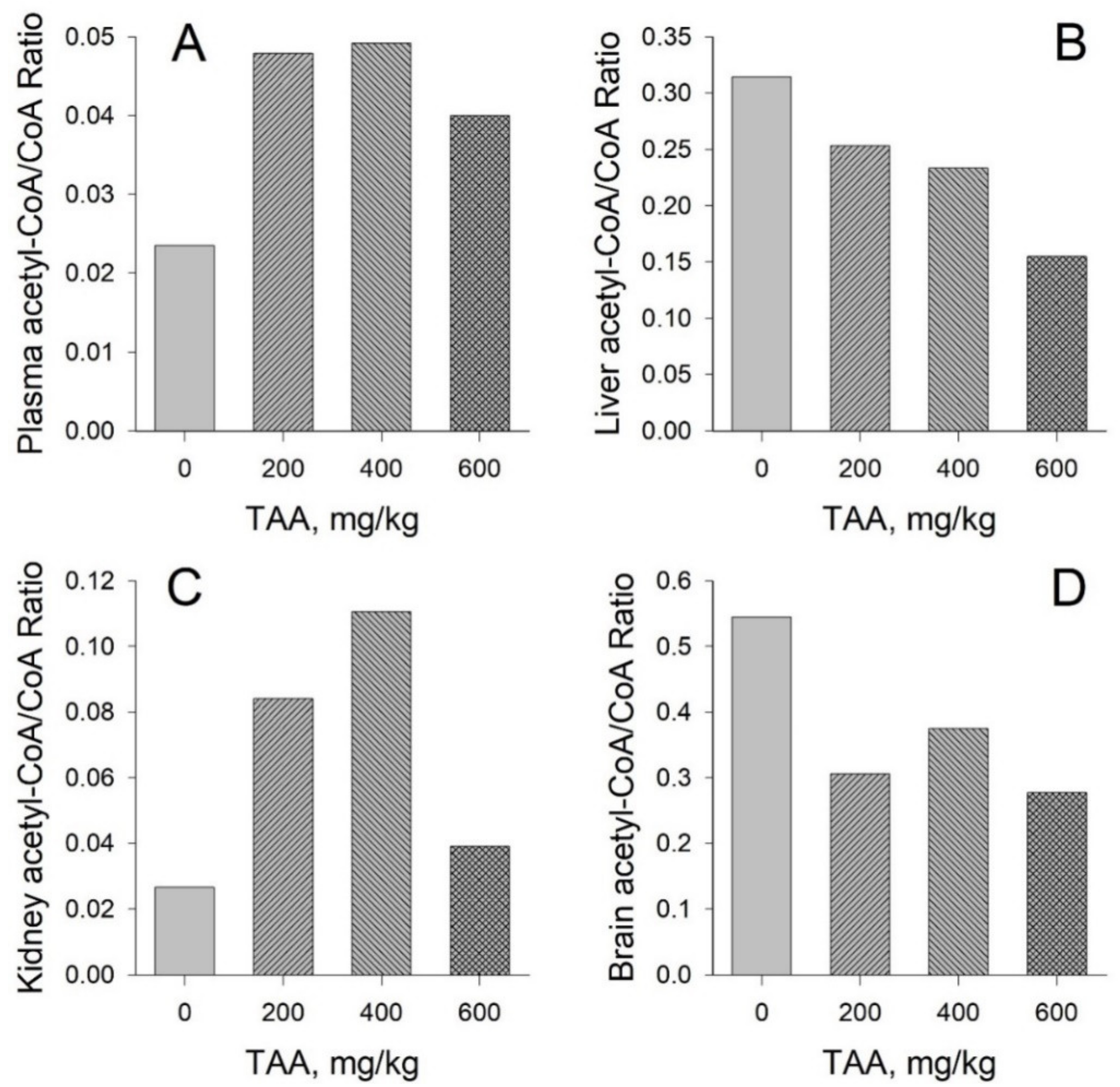
Concentration of CoA and Acetyl-CoA in Rat Tissues and Plasma
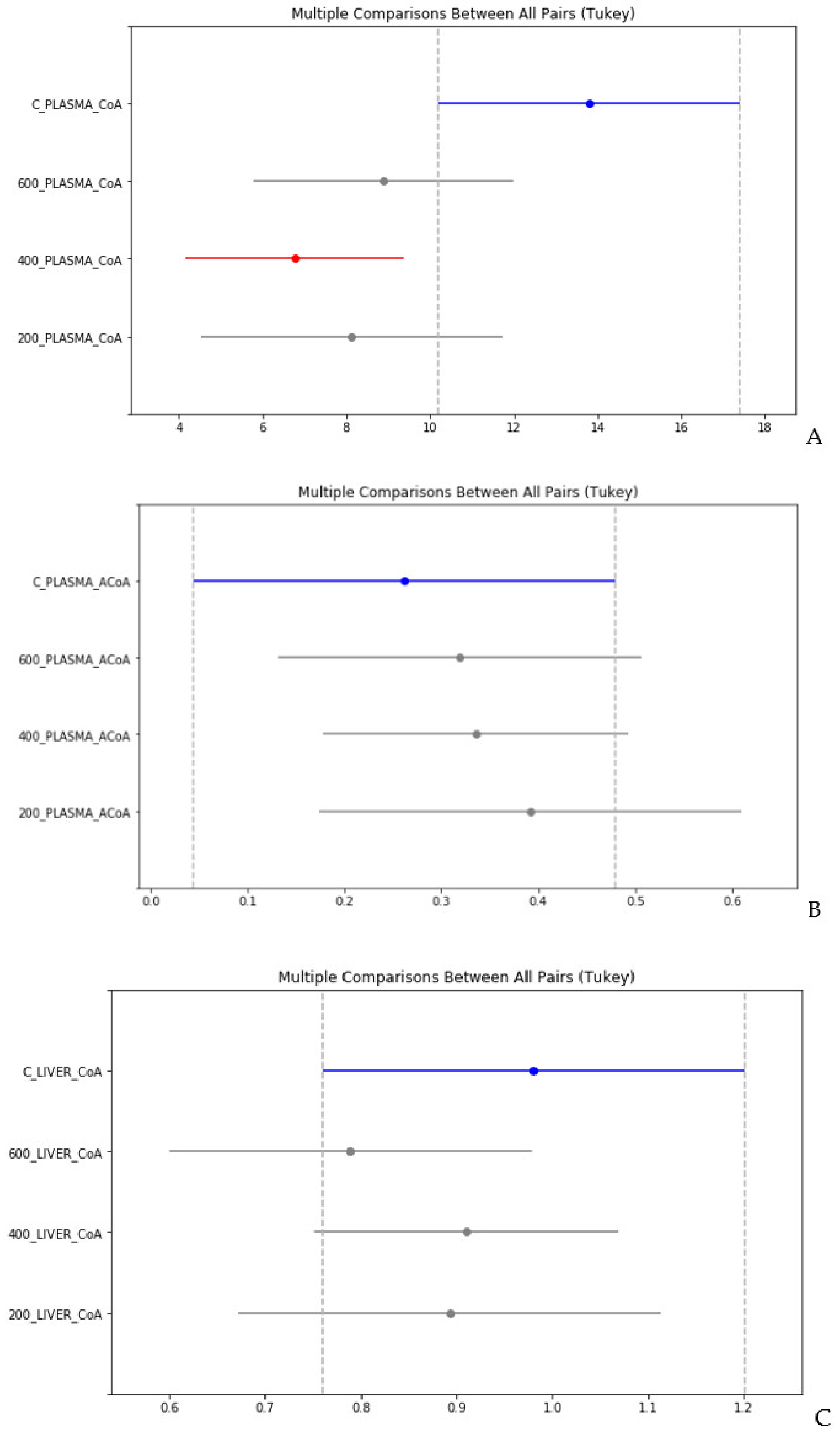
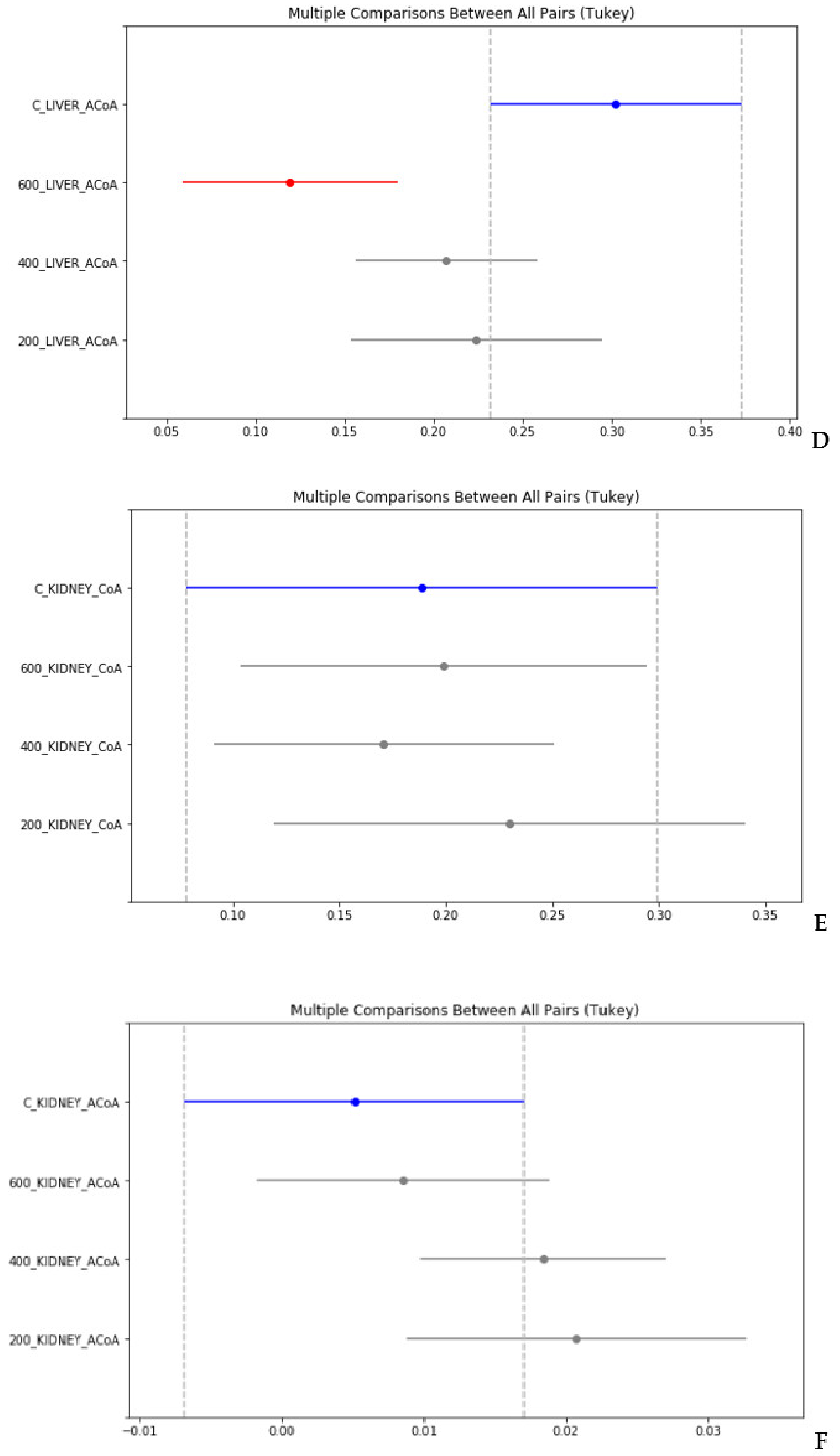
4. Statistical Evaluation of the Data
5. Discussion
CoA and Acetyl CoA in Control and TAA-Treated Rats
6. Conclusions
- CoA and acetyl-CoA concentrations in various rat organs differ markedly. Thus, a decrease in CoA levels was observed in the order: Liver → kidney → brain, and acetyl-CoA levels in the order: Liver → brain → kidney.
- Liver, kidney, brain, and blood plasma, in response to TAA exposure, are characterized by individual and unique patterns of distribution of CoA and acetyl-CoA compared to the physiological state. The patterns of these distributions may reflect the different metabolic roles of these organs.
- The ratios of acetyl-CoA to CoA in the organs and blood plasma of healthy and TAA-treated rats reflect the balance between anabolic and catabolic processes in organs during the period of their active recovery from the toxic effect of TAA.
- Despite an adequate recovery period after TAA intoxication, the concentrations of CoA and acetyl CoA in the rat liver and blood plasma, especially at higher doses of TAA, remained significantly reduced relative to the controls.
Author Contributions
Funding
Conflicts of Interest
References
- Leonardi, R.; Zhang, Y.M.; Rock, C.O.; Jackowski, S. Coenzyme A: Back in action. Prog. Lipid Res. 2005, 44, 125–153. [Google Scholar] [CrossRef] [PubMed]
- Randle, P.J.; Garland, P.B.; Hales, C.N.; Newsholme, E.A. The glucose fatty-acid cycle: Its role in insulin sensitivity and the metabolic disturbances of diabetes mellitus. Lancet 1963, 281, 785–789. [Google Scholar] [CrossRef]
- Neely, J.R.; Morgan, H.E. Relationship between carbohydrate and lipid metabolism and the energy balance of heart muscle. Annu. Rev. Physiol. 1974, 36, 413–459. [Google Scholar] [CrossRef] [PubMed]
- Lopaschuk, G.D.; Ussher, J.R.; Folmes, C.D.; Jaswal, J.S.; Stanley, W.C. Myocardial fatty acid metabolism in health and disease. Physiol. Rev. 2010, 90, 207–258. [Google Scholar]
- Mufson, E.J.; Counts, S.E.; Perez, S.E.; Ginsberg, S.D. Cholinergic system during the progression of Alzheimer’s disease: Therapeutic implications. Expert Rev. Neurother. 2008, 8, 1703–1718. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szutowicz, A.; Bielarczyk, H.; Jankowska-Kulawy, A.; Pawełczyk, T.; Ronowska, A. Acetyl-CoA the key factor for survival or death of cholinergic neurons in course of neurodegenerative diseases. Neurochem. Res. 2013, 38, 1523–1542. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szutowicz, A.; Bielarczyk, H.; Zyśk, M.; Dyś, A.; Ronowska, A.; Gul-Hinc, S. Early and late pathomechanisms in Alzheimer’s disease. From zinc to amyloid-β neurotoxicity. Neurochem. Res. 2017, 42, 891–904. [Google Scholar] [CrossRef] [Green Version]
- Pietrocola, F.; Galluzzi, L.; Bravo-San Pedro, J.M.; Madeo, F.; Kroeme, G. Acetyl coenzyme A: A central metabolite and second messenger. Cell Metab. 2015, 21, 805–821. [Google Scholar] [CrossRef] [Green Version]
- Ronowska, A.; Szutowicz, A.; Bielarczyk, H.; Gul-Hinc, S.; Klimaszewska-Łata, J.; Dyś, A.; Zyśk, M.; Jankowska-Kulawy, A. The regulatory effects of acetyl-CoA distribution in the healthy and diseased brain. Front. Cell. Neurosci. 2018, 12, 169, 1–20. [Google Scholar] [CrossRef] [Green Version]
- Gout, I. Coenzyme A, protein CoAlation and redox regulation in mammalian cells. Biochem. Soc. Trans. 2018, 46, 721–728. [Google Scholar] [CrossRef] [Green Version]
- Pettit, F.H.; Pelley, J.W.; Reed, L.J. Regulation of pyruvate dehydrogenase kinase and phosphatase by acetyl-CoA/CoA and NADH/NAD ratios. Biochem. Biophys. Res. Commun. 1975, 65, 575–582. [Google Scholar] [CrossRef]
- Yeh, L.A.; Song, C.S.; Kim, K.H. Coenzyme A activation of acetyl-CoA carboxylase. J. Biol. Chem. 1981, 256, 2289–2296. [Google Scholar] [PubMed]
- Tahiliani, A.G.; Beinlich, C.J. Pantothenic acid in health and disease. Vitam. Horm. 1991, 46, 165–228. [Google Scholar]
- Martinez, D.L.; Tsuchiya, Y.; Gout, I. Coenzyme A biosynthetic machinery in mammalian cells. Biochem. Soc. Trans. 2014, 42, 1112–1117. [Google Scholar] [CrossRef] [PubMed]
- Di Meo, I.; Carecchio, M.; Tiranti, V. Inborn errors of coenzyme A metabolism and neurodegeneration. J. Inherit. Metab. Dis. 2019, 42, 49–56. [Google Scholar] [CrossRef] [Green Version]
- Venco, P.; Dusi, S.; Valletta, L.; Tiranti, V. Alteration of the coenzyme A biosynthetic pathway in neurodegeneration with brain iron accumulation syndromes. Biochem. Soc. Trans. 2014, 42, 1069–1074. [Google Scholar] [CrossRef] [PubMed]
- Currais, A.; Huang, L.; Goldberg, J.; Petrascheck, M.; Ates, G.; Pinto-Duarte, A.; Shokhirev, M.N.; Schubert, D.; Maher, P. Elevating acetyl-CoA levels reduces aspects of brain aging. eLife 2019, 19, 1–21. [Google Scholar] [CrossRef]
- Hajovsky, H.; Hu, G.; Koen, Y.; Sarma, D.; Cui, W.; Moore, D.S.; Staudinger, J.L.; Hanzlik, R.P. Metabolism and toxicity of thioacetamide and thioacetamide S-oxide in rat hepatocytes. Chem. Res. Toxicol. 2012, 25, 1955–1963. [Google Scholar] [CrossRef] [Green Version]
- Zargar, S.; Wani, T.A.; Alamro, A.A.; Ganaie, M.A. Amelioration of thioacetamide-induced liver toxicity in Wistar rats by rutin. Int. J. Immunopathol. Pharmacol. 2017, 30, 207–214. [Google Scholar] [CrossRef]
- Díez-Fernández, C.; Boscá, L.; Fernández-Simón, L.; Alvarez, A.; Cascales, M. Relationship between genomic DNA ploidy and parameters of liver damage during necrosis and regeneration induced by thioacetamide. Hepatology 1993, 18, 912–918. [Google Scholar] [CrossRef]
- Zaky, A.; Bassiouny, A.R. Thioacetamide-induced liver cirrhosis alters oxidative stress balance and induces mitochondrial respiratory chain inhibition in the brain of cirrhotic rats. J. Biomol. Res. Ther. 2015, 4, 1–5. [Google Scholar]
- Grant, S.; McMillin, M.; Frampton, G.; Petrescu, A.D.; Williams, E.; Jaeger, V.; Kain, J.; DeMorrow, S. Direct comparison of the thioacetamide and azoxymethane models of type A hepatic encephalopathy in mice. Gene Expr. 2018, 18, 171–185. [Google Scholar] [CrossRef] [PubMed]
- Butterworth, R.F. Pathophysiology of hepatic encephalopathy: A new look at ammonia. Metab. Brain Dis. 2002, 17, 221–227. [Google Scholar] [CrossRef] [PubMed]
- Hindfelt, B.; Plum, F.; Duffy, T.E. Effect of acute ammonia intoxication on cerebral metabolism in rats with portacaval shunts. J. Clin. Investig. 1977, 59, 386–396. [Google Scholar] [CrossRef]
- Cooper, A.J.L.; Mora, S.N.; Cruz, N.F.; Gelbard, A.S. Cerebral ammonia metabolism in hyperammonemic rats. J. Neurochem. 1985, 44, 1716–1723. [Google Scholar] [CrossRef]
- Strauss, G.I.; Moller, K.; Larsen, F.S.; Kondrup, J.; Knudsen, G.M. Cerebral glucose and oxygen metabolism in patients with fulminant hepatic failure. Liver Transpl. 2003, 9, 1244–1252. [Google Scholar] [CrossRef]
- Iversen, P.; Sorensen, M.; Bak, L.K.; Waagepetersen, H.S.; Vafaee, M.S.; Borghammer, P.; Mouridsen, K.; Jensen, S.B.; Vilstrup, H.; Schousboe, A.; et al. Low cerebral oxygen consumption and blood flow in patients with cirrhosis and an acute episode of hepatic encephalopathy. Gastroenterology 2009, 136, 863–871. [Google Scholar] [CrossRef]
- Dam, G.; Keiding, S.; Munk, O.L.; Ott, P.; Vilstrup, H.; Bak, L.K.; Waagepetersen, H.S.; Schousboe, A.; Sorensen, M. Hepatic encephalopathy is associated with decreased cerebral oxygen metabolism and blood flow, not increased ammonia uptake. Hepatology 2013, 57, 258–265. [Google Scholar] [CrossRef]
- Rao, K.V.; Norenberg, M.D. Cerebral energy metabolism in hepatic encephalopathy and hyperammonemia. Metab. Brain Dis. 2001, 16, 67–78. [Google Scholar] [CrossRef]
- Ott, P.; Clemmesen, O.; Larsen, F.S. Cerebral metabolic disturbances in the brain during acute liver failure: From hyperammonemia to energy failure and proteolysis. Neurochem Int. 2005, 47, 13–18. [Google Scholar] [CrossRef]
- Bessman, S.P.; Bessman, A.N. The cerebral and peripheral uptake of ammonia in liver disease with a hypothesis for the mechanism of hepatic coma. J. Clin. Investig. 1955, 34, 622–628. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cooper, A.J.L.; Plum, F. Biochemistry and physiology of brain ammonia. Physiol. Rev. 1987, 67, 440–519. [Google Scholar] [CrossRef] [PubMed]
- Lai, J.C.; Cooper, A.J.L. Brain αalpha-ketoglutarate dehydrogenase complex: Kinetic properties, regional distribution, and effects of inhibitors. J. Neurochem. 1986, 47, 1376–1386. [Google Scholar] [CrossRef] [PubMed]
- Zwingmann, C.; Chatauret, N.; Leibfritz, D.; Butterworth, R.F. Selective increase of brain lactate synthesis in experimental acute liver failure: Results of a [H-C] nuclear magnetic resonance study. Hepatology 2003, 37, 420–428. [Google Scholar] [CrossRef] [PubMed]
- Siesjo, B.K.; Plum, F. Cerebral energy metabolism in normoxia and in hypoxia. Acta Anaesthesiol. Scand. Suppl. 1971, 45, 81–101. [Google Scholar] [CrossRef] [PubMed]
- Kosenko, E.; Kaminsky, Y.G.; Felipo, V.; Miñana, M.D.; Grisolía, S. Chronic hyperammonemia prevents changes in brain energy and ammonia metabolites induced by acute ammonium intoxication. Biochim. Biophys. Acta 1993, 1180, 321–326. [Google Scholar] [CrossRef]
- Katanuma, N.; Okada, M.; Nishii, Y. Regulation of urea cycle and TCA cycle by ammonia. Adv. Enzym. Regul. 1966, 4, 317–335. [Google Scholar] [CrossRef]
- Schousboe, A.; Waagepetersen, H.S.; Leke, R.; Bak, L.K. Effects of hyperammonemia on brain energy metabolism: Controversial findings in vivo and in vitro. Metab. Brain Dis. 2014, 29, 913–917. [Google Scholar] [CrossRef]
- Shurubor, Y.I.; D’Aurelio, M.; Clark-Matott, J.; Isakova, E.P.; Deryabina, Y.I.; Beal, M.F.; Cooper, A.J.L.; Krasnikov, B.F. Determination of coenzyme A and acetyl-coenzyme A in biological samples using HPLC with UV detection. Molecules 2017, 22, 2–13. [Google Scholar]
- Alberts, B.; Johnson, A.; Lewis, J.; Raff, M.; Roberts, K.; Walter, P. How cells obtain energy from food. In Molecular Biology of the Cell, 4th ed.; Garland Science: New York, NY, USA, 2002. [Google Scholar]
- Paetzke-Brunner, I.; Schön, H.; Wieland, O.H. Insulin activates pyruvate dehydrogenase by lowering the mitochondrial acetyl-CoA/CoA ratio as evidenced by digitonin fractionation of isolated fat cells. FEBS Lett. 1978, 93, 307–311. [Google Scholar] [CrossRef] [Green Version]
- Jitrapakdee, S.; St. Maurice, M.; Rayment, I.; Cleland, W.W.; Wallace, J.G.; Attwood, P.V. Structure, mechanism and regulation of pyruvate carboxylase. Biochem. J. 2008, 413, 369–387. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- White, H. The role of TCA cycle anaplerosis in ketosis and fatty liver in periparturient dairy cows. Animals 2015, 5, 793–802. [Google Scholar] [CrossRef] [PubMed]
- Alrob, O.A.; Lopaschuk, G.D. Role of CoA and acetyl-CoA in regulating cardiac fatty acid and glucose oxidation. Biochem. Soc. Trans. 2014, 42, 1043–1051. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Zhang, Y.; Yang, C.; Xiong, H.; Lin, Y.; Yao, J.; Li, H.; Xie, L.; Zhao, W.; Yao, Y.; et al. Acetylation of metabolic enzymes coordinates carbon source utilization and metabolic flux. Science 2010, 327, 1004–1007. [Google Scholar] [CrossRef] [Green Version]
- Lin, R.; Zhou, X.; Huang, W.; Zhao, D.; Lv, L.; Xiong, Y.; Guan, K.L.; Lei, Q.Y. Acetylation control of cancer cell metabolism. Curr. Pharm. 2014, 20, 2627–2633. [Google Scholar] [CrossRef] [PubMed]
- Jina, E.S.; Morenoa, K.X.; Wang, J.-X.; Fidelinoa, L.; Merritta, M.E.; Sherrya, A.D.; Malloya, C.R. Metabolism of hyperpolarized [1-(13)C]pyruvate through alternate pathways in rat liver. NMR Biomed. 2016, 29, 466–474. [Google Scholar] [CrossRef]
- Krahenbuhl, S.; Mang, G.; Kupferschmidt, H.; Meier, P.J.; Krause, M. Plasma and hepatic carnitine and coenzyme A pools in a patient with fatal, valproate induced hepatotoxicity. Gut 1995, 37, 140–143. [Google Scholar] [CrossRef]
- Kala, G.; Hertz, L. Ammonia effects on pyruvate/lactate production in astrocytes interaction with glutamate. Neurochem. Int. 2005, 47, 4–12. [Google Scholar] [CrossRef]
- Simpson, I.A.; Carruthers, A.; Vanucci, S. Supply and demand in cerebral energy metabolism: The role of nutrient transporters. J. Cereb. Blood Flow Metab. 2007, 27, 1766–1791. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| TAA mg/kg | PLASMA | LIVER | KIDNEY | BRAIN | ||||
|---|---|---|---|---|---|---|---|---|
| CoA | ACoA | CoA | ACoA | CoA | ACoA | CoA | ACoA | |
| 0 | 20.0 | 0.074 | 0.917 | 0.278 | 0.151 | 0.003 | 0.054 | 0.017 |
| 0 | 10.2 | 0.344 | 0.883 | 0.350 | 0.210 | 0.006 | 0.103 | 0.015 |
| 0 | 11.2 | 0.368 | 1.14 | 0.279 | 0.206 | 0.006 | 0.032 | 0.072 |
| 200 | 9.02 | 0.466 | 0.995 | 0.204 | 0.261 | 0.019 | 0.132 | 0.014 |
| 200 | 8.19 | 0.417 | 0.834 | 0.168 | 0.122 | 0.008 | 0.094 | 0.022 |
| 200 | 7.17 | 0.294 | 0.851 | 0.300 | 0.307 | 0.036 | 0.075 | 0.057 |
| 400 | 5.39 | 0.221 | 0.781 | 0.173 | 0.278 | 0.030 | 0.077 | 0.028 |
| 400 | 6.47 | 0.368 | 1.24 | 0.195 | 0.151 | 0.017 | 0.080 | 0.021 |
| 400 | 7.96 | 0.589 | 0.715 | 0.247 | 0.124 | 0.021 | 0.018 | 0.016 |
| 400 | 8.74 | 0.172 | 0.944 | 0.270 | 0.175 | 0.023 | 0.071 | 0.026 |
| 400 | 7.09 | 0.491 | 1.02 | 0.245 | 0.219 | 0.012 | 0.060 | 0.022 |
| 400 | 4.99 | 0.172 | 0.770 | 0.113 | 0.077 | 0.006 | 0.099 | 0.039 |
| 600 | 6.06 | 0.442 | 0.726 | 0.100 | 0.179 | 0.006 | 0.091 | 0.028 |
| 600 | 9.99 | 0.172 | 0.627 | 0.130 | 0.268 | 0.008 | 0.124 | 0.019 |
| 600 | 9.18 | 0.466 | 0.922 | 0.146 | 0.285 | 0.018 | 0.048 | 0.025 |
| 600 | 10.3 | 0.196 | 0.882 | 0.102 | 0.063 | 0.002 | 0.072 | 0.021 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shurubor, Y.I.; Cooper, A.J.L.; Krasnikov, A.B.; Isakova, E.P.; Deryabina, Y.I.; Beal, M.F.; Krasnikov, B.F. Changes of Coenzyme A and Acetyl-Coenzyme A Concentrations in Rats after a Single-Dose Intraperitoneal Injection of Hepatotoxic Thioacetamide Are Not Consistent with Rapid Recovery. Int. J. Mol. Sci. 2020, 21, 8918. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21238918
Shurubor YI, Cooper AJL, Krasnikov AB, Isakova EP, Deryabina YI, Beal MF, Krasnikov BF. Changes of Coenzyme A and Acetyl-Coenzyme A Concentrations in Rats after a Single-Dose Intraperitoneal Injection of Hepatotoxic Thioacetamide Are Not Consistent with Rapid Recovery. International Journal of Molecular Sciences. 2020; 21(23):8918. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21238918
Chicago/Turabian StyleShurubor, Yevgeniya I., Arthur J. L. Cooper, Andrey B. Krasnikov, Elena P. Isakova, Yulia I. Deryabina, M. Flint Beal, and Boris F. Krasnikov. 2020. "Changes of Coenzyme A and Acetyl-Coenzyme A Concentrations in Rats after a Single-Dose Intraperitoneal Injection of Hepatotoxic Thioacetamide Are Not Consistent with Rapid Recovery" International Journal of Molecular Sciences 21, no. 23: 8918. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21238918