Binding of Kingella kingae RtxA Toxin Depends on Cell Surface Oligosaccharides, but Not on β2 Integrins

 ,
,  and
and 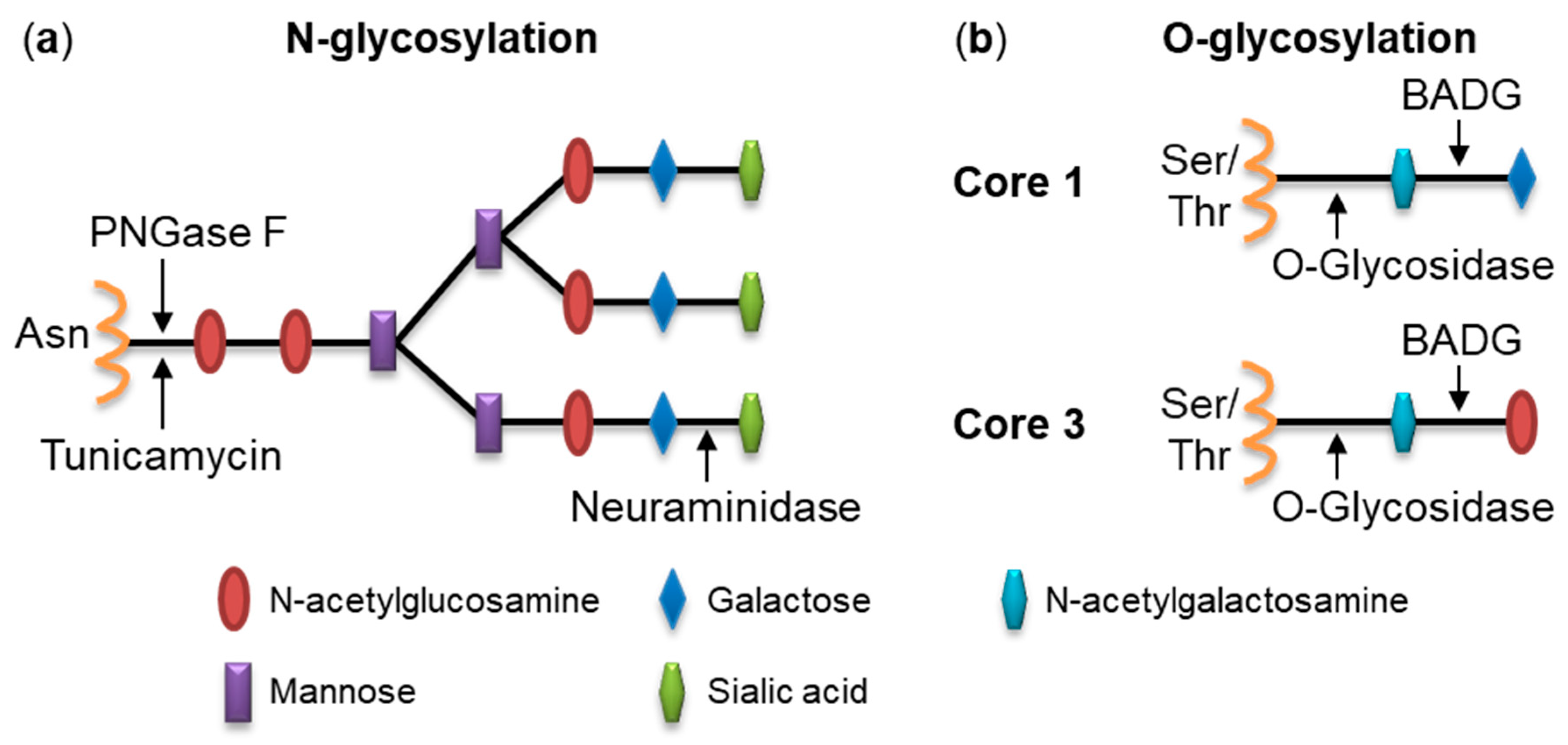{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
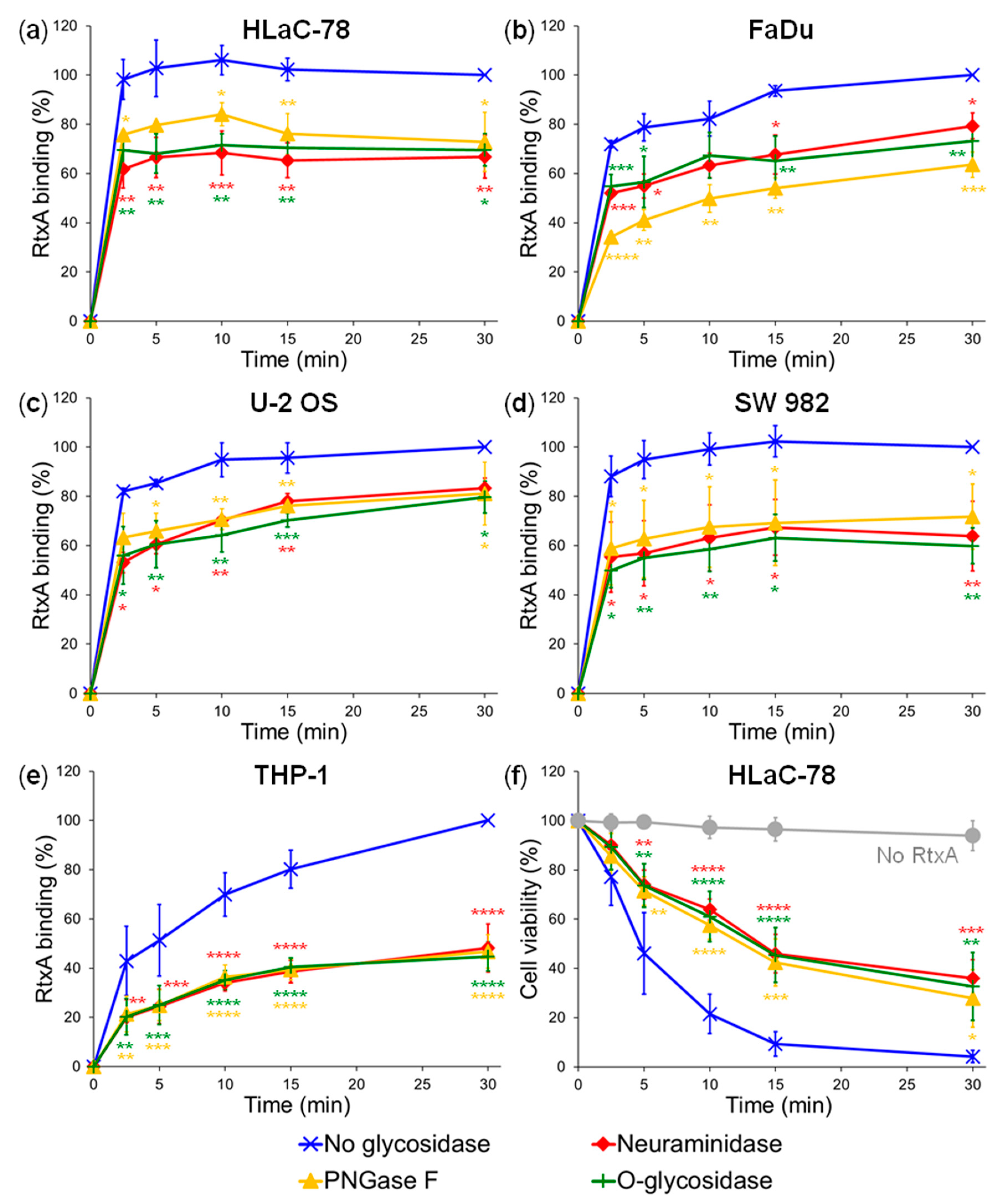
2.1. Pretreatment of Cells with Glycosidases Reduces Binding of RtxA
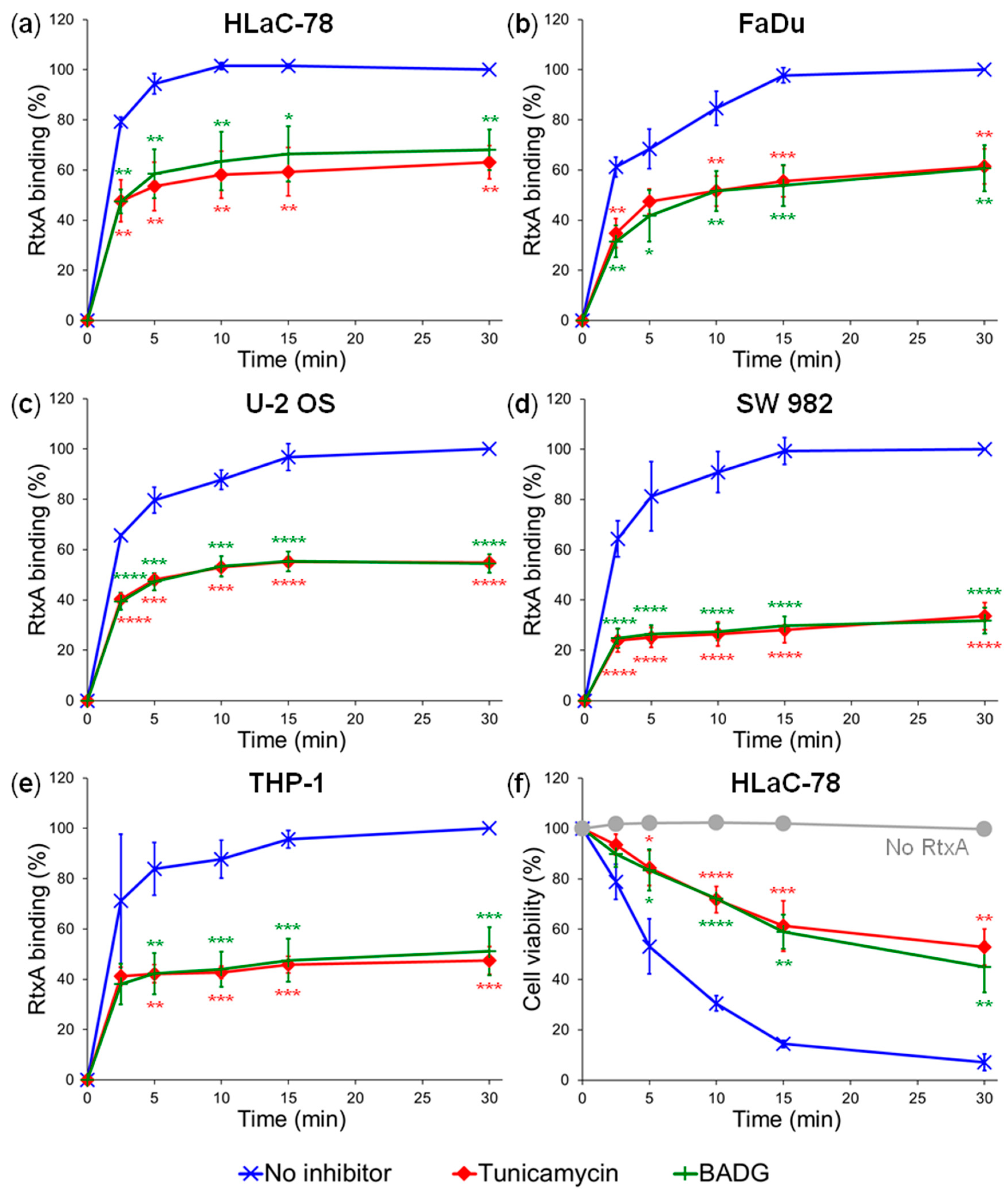
2.2. Inhibition of Protein Glycosylation Reduces Binding of RtxA to Cells
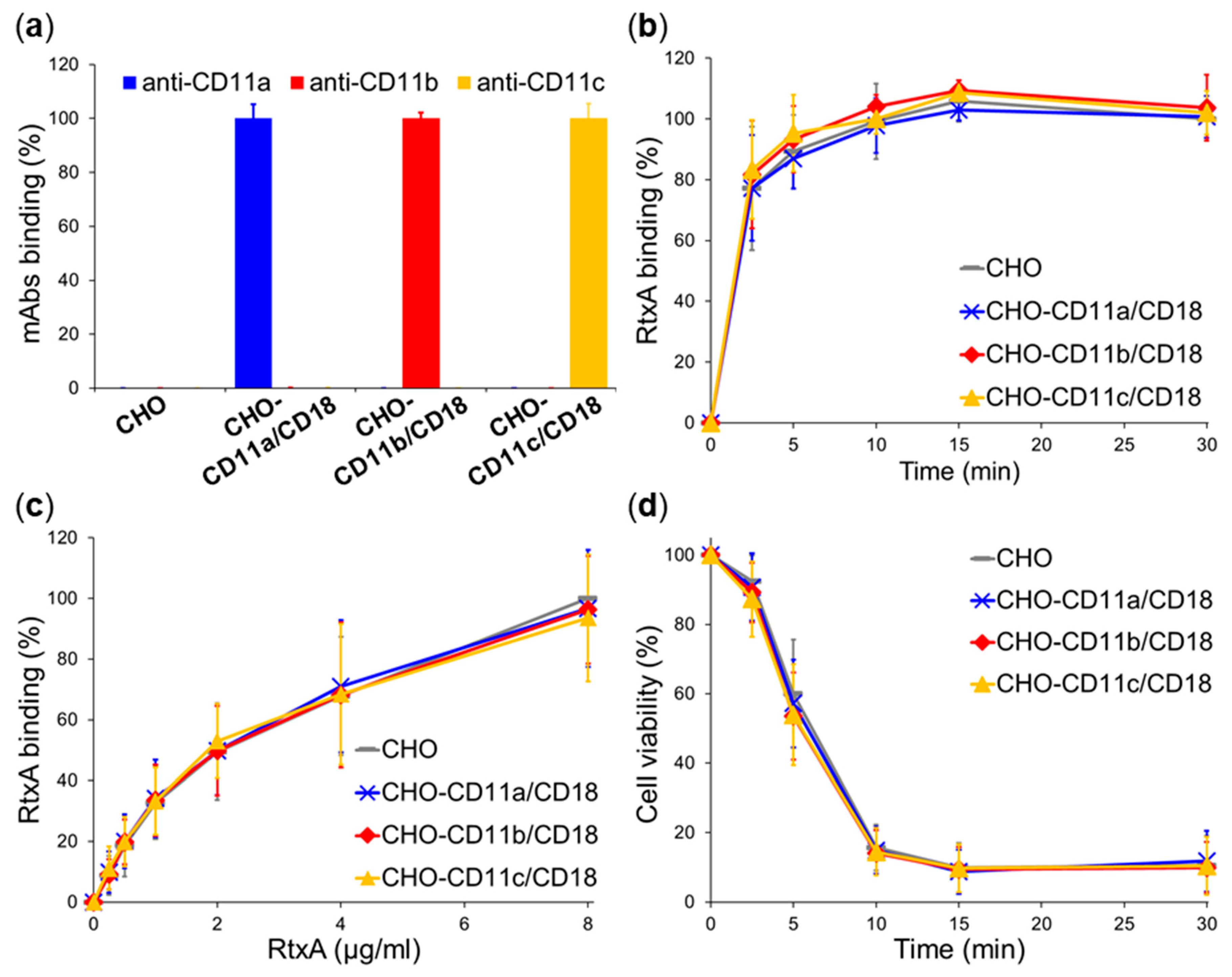
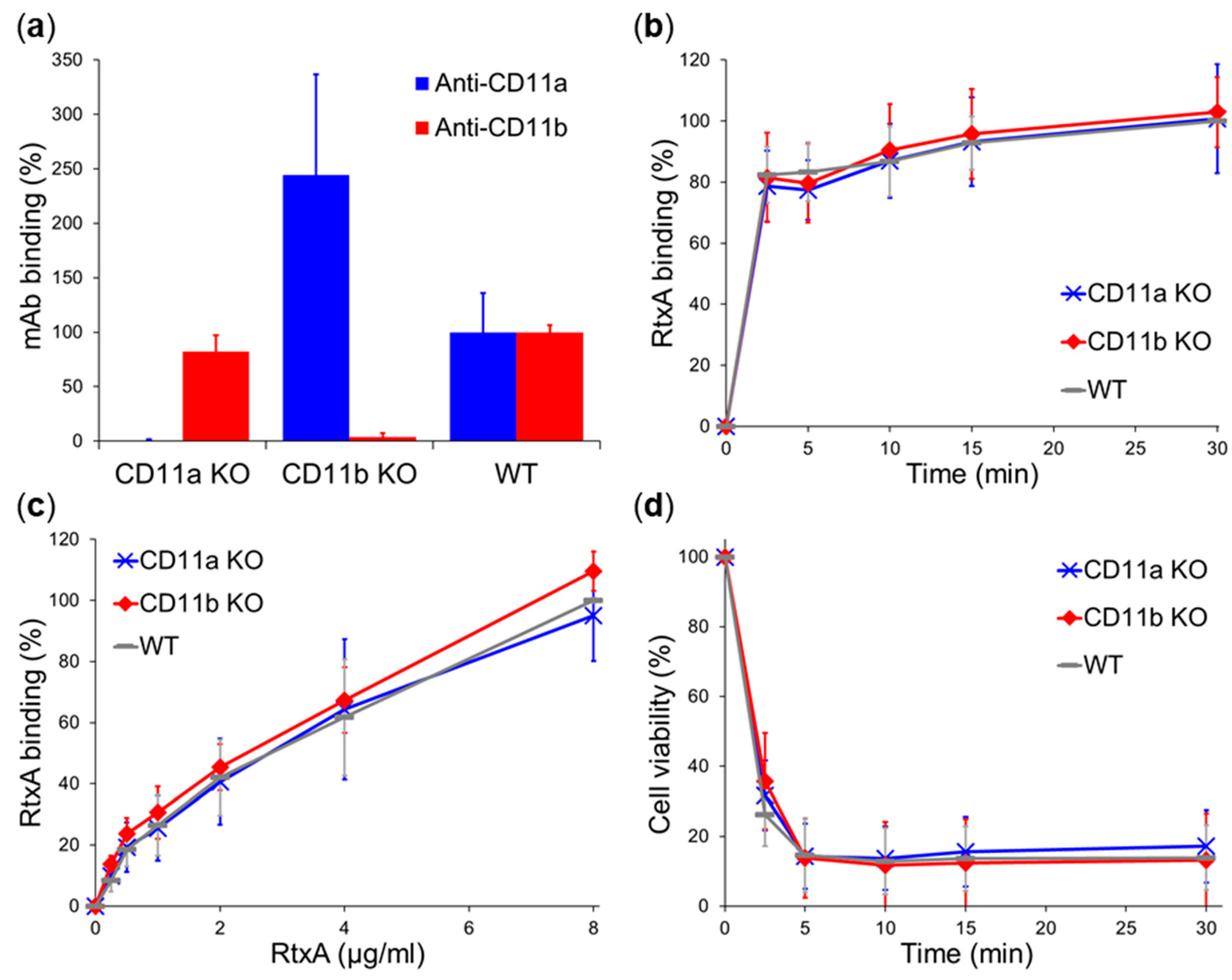
2.3. β2 Integrins Are Not Cellular Receptors of RtxA
3. Discussion
4. Materials and Methods
4.1. Antibodies
4.2. Cell Lines and Growth Conditions
4.3. Animal Studies
4.4. Isolation of Bone Marrow Cells and Their Differentiation to Macrophages
4.5. Protein Production, Purification, and Labeling
4.6. Deglycosylation of Cell Surface Structures with Glycosidases
4.7. Inhibition of Glycosylation Using Chemical Inhibitors
4.8. RtxA Binding to Cells
4.9. Cytotoxicity Assay
4.10. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| BADG | Benzyl-2-acetamido-2-deoxy-α-D-galactopyranoside |
| BMC | Bone marrow cell |
| CHO | Chinese hamster ovary |
| mAb | Monoclonal antibody |
| M-CSF | Macrophage colony-stimulating factor |
| MFI | Mean fluorescence intensity |
| O-glycosidase | Endo-α-N-acetylgalactosaminidase |
| PNGase | Peptide-N-glycosidase F |
| RTX | Repeats in toxin |
References
- Yagupsky, P. Kingella kingae: Carriage, transmission, and disease. Clin. Microbiol. Rev. 2015, 28, 54–79. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ceroni, D.; Dubois-Ferriere, V.; Cherkaoui, A.; Lamah, L.; Renzi, G.; Lascombes, P.; Wilson, B.; Schrenzel, J. 30 years of study of Kingella kingae: Post tenebras, lux. Future Microbiol. 2013, 8, 233–245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Principi, N.; Esposito, S. Kingella kingae infections in children. BMC Infect. Dis. 2015, 15, 260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gene, A.; Garcia-Garcia, J.J.; Sala, P.; Sierra, M.; Huguet, R. Enhanced culture detection of Kingella kingae, a pathogen of increasing clinical importance in pediatrics. Pediatr. Infect. Dis. J. 2004, 23, 886–888. [Google Scholar] [CrossRef]
- Moumile, K.; Merckx, J.; Glorion, C.; Berche, P.; Ferroni, A. Osteoarticular infections caused by Kingella kingae in children: Contribution of polymerase chain reaction to the microbiologic diagnosis. Pediatr. Infect. Dis. J. 2003, 22, 837–839. [Google Scholar] [CrossRef]
- Verdier, I.; Gayet-Ageron, A.; Ploton, C.; Taylor, P.; Benito, Y.; Freydiere, A.M.; Chotel, F.; Berard, J.; Vanhems, P.; Vandenesch, F. Contribution of a broad range polymerase chain reaction to the diagnosis of osteoarticular infections caused by Kingella kingae: Description of twenty-four recent pediatric diagnoses. Pediatr. Infect. Dis. J. 2005, 24, 692–696. [Google Scholar] [CrossRef]
- Dubnov-Raz, G.; Ephros, M.; Garty, B.Z.; Schlesinger, Y.; Maayan-Metzger, A.; Hasson, J.; Kassis, I.; Schwartz-Harari, O.; Yagupsky, P. Invasive pediatric Kingella kingae Infections: A nationwide collaborative study. Pediatr. Infect. Dis. J. 2010, 29, 639–643. [Google Scholar] [CrossRef]
- Yagupsky, P.; Porsch, E.; St Geme, J.W., 3rd. Kingella kingae: An emerging pathogen in young children. Pediatrics 2011, 127, 557–565. [Google Scholar] [CrossRef]
- Osickova, A.; Balashova, N.; Masin, J.; Sulc, M.; Roderova, J.; Wald, T.; Brown, A.C.; Koufos, E.; Chang, E.H.; Giannakakis, A.; et al. Cytotoxic activity of Kingella kingae RtxA toxin depends on post-translational acylation of lysine residues and cholesterol binding. Emerg. Microbes Infect. 2018, 7, 178. [Google Scholar] [CrossRef] [Green Version]
- Kehl-Fie, T.E.; St Geme, J.W., 3rd. Identification and characterization of an RTX toxin in the emerging pathogen Kingella kingae. J. Bacteriol. 2007, 189, 430–436. [Google Scholar] [CrossRef] [Green Version]
- Chang, D.W.; Nudell, Y.A.; Lau, J.; Zakharian, E.; Balashova, N.V. RTX toxin plays a key role in Kingella kingae virulence in an infant rat model. Infect. Immun. 2014, 82, 2318–2328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Linhartova, I.; Bumba, L.; Masin, J.; Basler, M.; Osicka, R.; Kamanova, J.; Prochazkova, K.; Adkins, I.; Hejnova-Holubova, J.; Sadilkova, L.; et al. RTX proteins: A highly diverse family secreted by a common mechanism. FEMS Microbiol. Rev. 2010, 34, 1076–1112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Osickova, A.; Khaliq, H.; Masin, J.; Jurnecka, D.; Sukova, A.; Fiser, R.; Holubova, J.; Stanek, O.; Sebo, P.; Osicka, R. Acyltransferase-mediated selection of the length of the fatty acyl chain and of the acylation site governs activation of bacterial RTX toxins. J. Biol. Chem. 2020, 295, 9268–9280. [Google Scholar] [CrossRef] [PubMed]
- Barcena-Uribarri, I.; Benz, R.; Winterhalter, M.; Zakharian, E.; Balashova, N. Pore forming activity of the potent RTX-toxin produced by pediatric pathogen Kingella kingae: Characterization and comparison to other RTX-family members. Biochim. Biophys. Acta 2015, 1848, 1536–1544. [Google Scholar] [CrossRef] [Green Version]
- Mazzone, A.; Ricevuti, G. Leukocyte CD11/CD18 integrins: Biological and clinical relevance. Haematologica 1995, 80, 161–175. [Google Scholar]
- Lally, E.T.; Kieba, I.R.; Sato, A.; Green, C.L.; Rosenbloom, J.; Korostoff, J.; Wang, J.F.; Shenker, B.J.; Ortlepp, S.; Robinson, M.K.; et al. RTX toxins recognize a beta2 integrin on the surface of human target cells. J. Biol. Chem. 1997, 272, 30463–30469. [Google Scholar] [CrossRef] [Green Version]
- Ambagala, T.C.; Ambagala, A.P.; Srikumaran, S. The leukotoxin of Pasteurella haemolytica binds to beta(2) integrins on bovine leukocytes. FEMS Microbiol. Lett. 1999, 179, 161–167. [Google Scholar]
- Li, J.; Clinkenbeard, K.D.; Ritchey, J.W. Bovine CD18 identified as a species specific receptor for Pasteurella haemolytica leukotoxin. Vet. Microbiol. 1999, 67, 91–97. [Google Scholar] [CrossRef]
- Jeyaseelan, S.; Hsuan, S.L.; Kannan, M.S.; Walcheck, B.; Wang, J.F.; Kehrli, M.E.; Lally, E.T.; Sieck, G.C.; Maheswaran, S.K. Lymphocyte function-associated antigen 1 is a receptor for Pasteurella haemolytica leukotoxin in bovine leukocytes. Infect. Immun. 2000, 68, 72–79. [Google Scholar] [CrossRef] [Green Version]
- Thumbikat, P.; Dileepan, T.; Kannan, M.S.; Maheswaran, S.K. Characterization of Mannheimia (Pasteurella) haemolytica leukotoxin interaction with bovine alveolar macrophage beta2 integrins. Vet. Res. 2005, 36, 771–786. [Google Scholar] [CrossRef] [Green Version]
- Valeva, A.; Walev, I.; Kemmer, H.; Weis, S.; Siegel, I.; Boukhallouk, F.; Wassenaar, T.M.; Chavakis, T.; Bhakdi, S. Binding of Escherichia coli hemolysin and activation of the target cells is not receptor-dependent. J. Biol. Chem. 2005, 280, 36657–36663. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guermonprez, P.; Khelef, N.; Blouin, E.; Rieu, P.; Ricciardi-Castagnoli, P.; Guiso, N.; Ladant, D.; Leclerc, C. The adenylate cyclase toxin of Bordetella pertussis binds to target cells via the alpha(M)beta(2) integrin (CD11b/CD18). J. Exp. Med. 2001, 193, 1035–1044. [Google Scholar] [CrossRef] [PubMed]
- Osicka, R.; Osickova, A.; Hasan, S.; Bumba, L.; Cerny, J.; Sebo, P. Bordetella adenylate cyclase toxin is a unique ligand of the integrin complement receptor 3. Elife 2015, 4, e10766. [Google Scholar] [CrossRef] [PubMed]
- Wald, T.; Osickova, A.; Masin, J.; Liskova, P.M.; Petry-Podgorska, I.; Matousek, T.; Sebo, P.; Osicka, R. Transmembrane segments of complement receptor 3 do not participate in cytotoxic activities but determine receptor structure required for action of Bordetella adenylate cyclase toxin. Pathog. Dis. 2016, 74, ftw008. [Google Scholar] [CrossRef] [Green Version]
- Balashova, N.V.; Crosby, J.A.; Al Ghofaily, L.; Kachlany, S.C. Leukotoxin confers beta-hemolytic activity to Actinobacillus actinomycetemcomitans. Infect. Immun. 2006, 74, 2015–2021. [Google Scholar] [CrossRef] [Green Version]
- Morova, J.; Osicka, R.; Masin, J.; Sebo, P. RTX cytotoxins recognize beta2 integrin receptors through N-linked oligosaccharides. Proc. Natl. Acad. Sci. USA 2008, 105, 5355–5360. [Google Scholar] [CrossRef] [Green Version]
- Hasan, S.; Osickova, A.; Bumba, L.; Novak, P.; Sebo, P.; Osicka, R. Interaction of Bordetella adenylate cyclase toxin with complement receptor 3 involves multivalent glycan binding. FEBS Lett. 2015, 589, 374–379. [Google Scholar] [CrossRef] [Green Version]
- El-Azami-El-Idrissi, M.; Bauche, C.; Loucka, J.; Osicka, R.; Sebo, P.; Ladant, D.; Leclerc, C. Interaction of Bordetella pertussis adenylate cyclase with CD11b/CD18: Role of toxin acylation and identification of the main integrin interaction domain. J. Biol. Chem. 2003, 278, 38514–38521. [Google Scholar] [CrossRef] [Green Version]
- Zenner, H.P.; Lehner, W.; Herrmann, I.F. Establishment of carcinoma cell lines from larynx and submandibular gland. Arch. Otorhinolaryngol. 1979, 225, 269–277. [Google Scholar] [CrossRef]
- Ding, Z.M.; Babensee, J.E.; Simon, S.I.; Lu, H.; Perrard, J.L.; Bullard, D.C.; Dai, X.Y.; Bromley, S.K.; Dustin, M.L.; Entman, M.L.; et al. Relative contribution of LFA-1 and Mac-1 to neutrophil adhesion and migration. J. Immunol. 1999, 163, 5029–5038. [Google Scholar]
- Coxon, A.; Rieu, P.; Barkalow, F.J.; Askari, S.; Sharpe, A.H.; von Andrian, U.H.; Arnaout, M.A.; Mayadas, T.N. A novel role for the beta 2 integrin CD11b/CD18 in neutrophil apoptosis: A homeostatic mechanism in inflammation. Immunity 1996, 5, 653–666. [Google Scholar] [CrossRef] [Green Version]
- Osicka, R.; Osickova, A.; Basar, T.; Guermonprez, P.; Rojas, M.; Leclerc, C.; Sebo, P. Delivery of CD8(+) T-cell epitopes into major histocompatibility complex class I antigen presentation pathway by Bordetella pertussis adenylate cyclase: delineation of cell invasive structures and permissive insertion sites. Infect Immun. 2000, 68, 247–256. [Google Scholar] [CrossRef] [PubMed] [Green Version]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rahman, W.U.; Osickova, A.; Klimova, N.; Lora, J.; Balashova, N.; Osicka, R. Binding of Kingella kingae RtxA Toxin Depends on Cell Surface Oligosaccharides, but Not on β2 Integrins. Int. J. Mol. Sci. 2020, 21, 9092. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21239092
Rahman WU, Osickova A, Klimova N, Lora J, Balashova N, Osicka R. Binding of Kingella kingae RtxA Toxin Depends on Cell Surface Oligosaccharides, but Not on β2 Integrins. International Journal of Molecular Sciences. 2020; 21(23):9092. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21239092
Chicago/Turabian StyleRahman, Waheed Ur, Adriana Osickova, Nela Klimova, Jinery Lora, Nataliya Balashova, and Radim Osicka. 2020. "Binding of Kingella kingae RtxA Toxin Depends on Cell Surface Oligosaccharides, but Not on β2 Integrins" International Journal of Molecular Sciences 21, no. 23: 9092. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21239092