Identification of Potent CD19 scFv for CAR T Cells through scFv Screening with NK/T-Cell Line

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
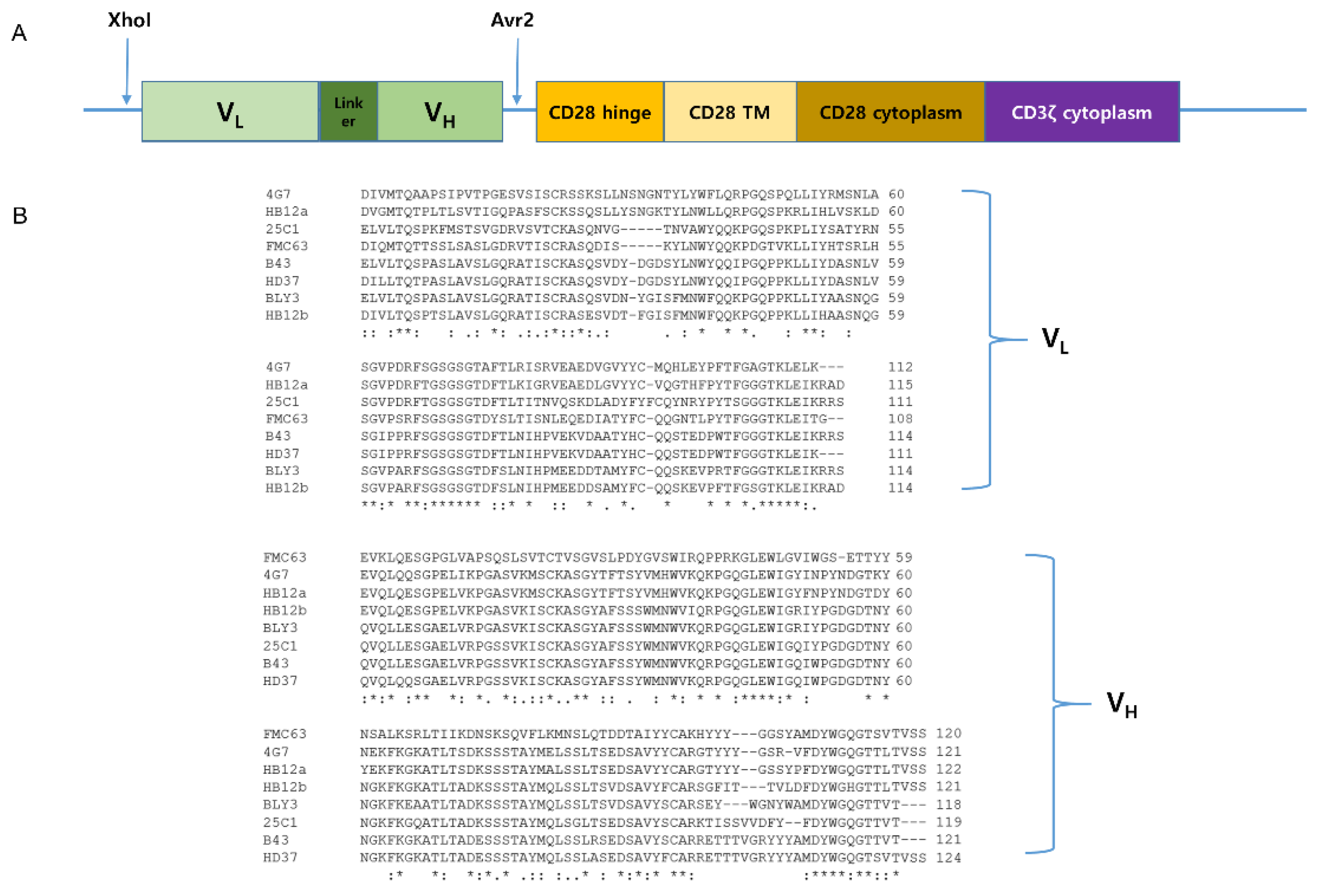
2.1. Usage of the KHYG-1 Cell Line to Screen CD19 scFvs for CD19 CAR T Cells
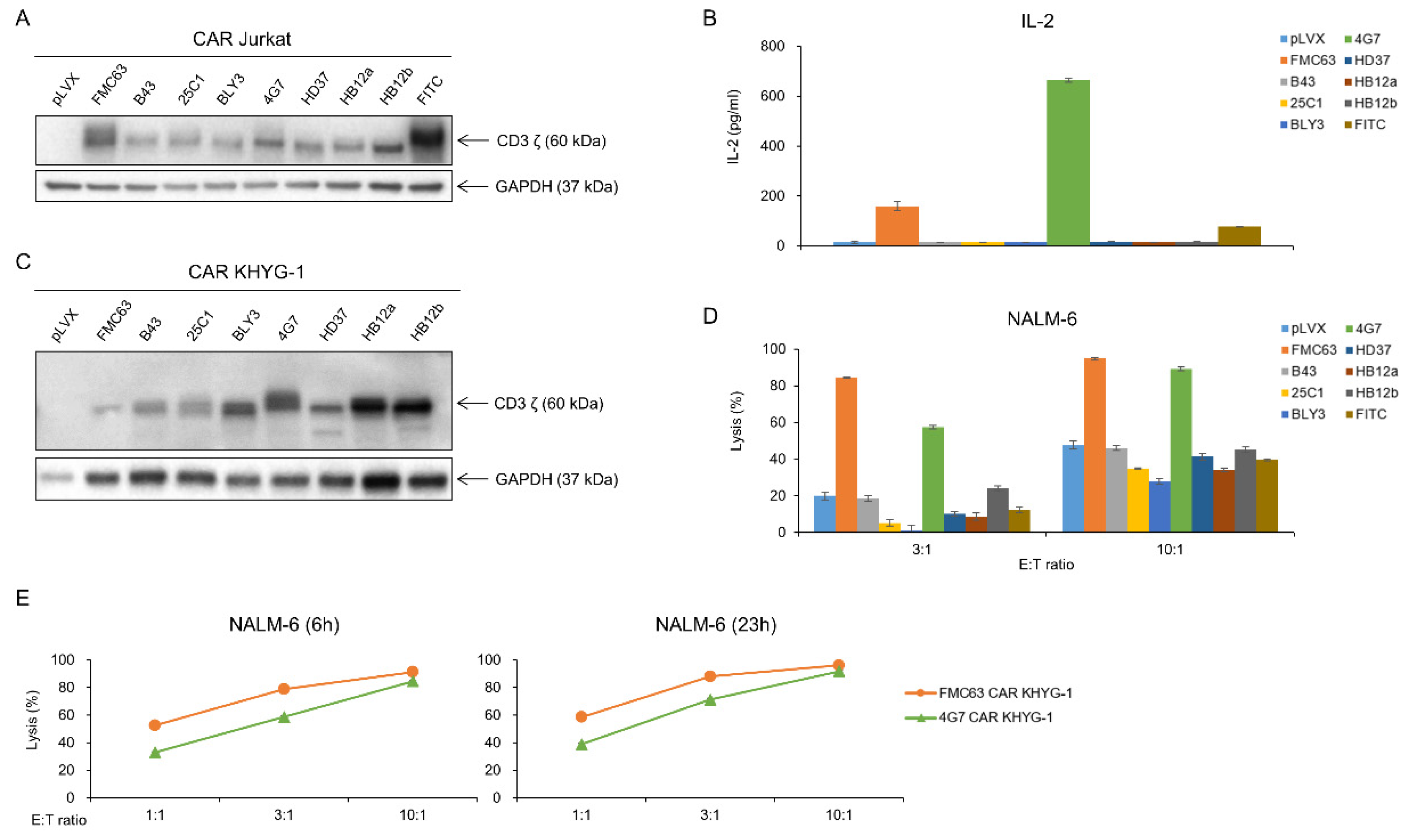
2.2. FMC63 and 4G7 scFvs Are Proved to Be Applicable for Anti-CD19 CAR Construct
2.3. 4G7 CAR T Cells Are as Effective as FMC63 CAR T Cells in Eradicating Leukemic Tumor Cells
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Construct of CARs
4.3. Lentivirus Production and Generation of CAR KHYG-1
4.4. Human T Cell Isolation and Generation of CAR T Cells
4.5. Proliferation Assay
4.6. Cytotoxicity Assay
4.7. Cytokine Release Assay
4.8. Western Blotting
4.9. Mouse Xenograft Model
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ALL | Acute lymphoblastic leukemia |
| CAR | Chimeric antigen receptor |
| CLL | Chronic lymphocytic leukemia |
| CMV | Cytomegalovirus |
| DLBCL | Diffuse large B-cell lymphoma |
| EF1α | Elongation factor-1 alpha |
| FL | Follicular lymphoma |
| HRP | Horseradish peroxidase |
| IFN-γ | Interferon-gamma |
| IL-2 | Interleukin-2 |
| mAb | Monoclonal antibody |
| MNC | Mononuclear cells |
| NCI | National Cancer Institute |
| NK | Natural killer |
| NSG | NOD.Cg-PrkdcscidIl2rgtm1Wjl/SzJ |
| PBMC | Peripheral blood mononuclear cell |
| PBS | Phosphate buffered saline |
| PES | Polyethersulfone |
| PGK | Phosphoglycerate kinase |
| scFv | Single-chain variable fragment |
| TCR | T-cell receptor |
| UPenn | University of Pennsylvania |
References
- Hartmann, J.; Schussler-Lenz, M.; Bondanza, A.; Buchholz, C.J. Clinical development of CAR T cells-challenges and opportunities in translating innovative treatment concepts. EMBO Mol. Med. 2017, 9, 1183–1197. [Google Scholar] [CrossRef] [PubMed]
- Van der Stegen, S.J.; Hamieh, M.; Sadelain, M. The pharmacology of second-generation chimeric antigen receptors. Nat. Rev. Drug Discov. 2015, 14, 499–509. [Google Scholar] [CrossRef] [PubMed]
- Sauter, C.S.; Senechal, B.; Riviere, I.; Ni, A.; Bernal, Y.; Wang, X.; Purdon, T.; Hall, M.; Singh, A.N.; Szenes, V.Z.; et al. CD19 CAR T cells following autologous transplantation in poor-risk relapsed and refractory B-cell non-Hodgkin lymphoma. Blood 2019, 134, 626–635. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Munoz, J.; Goy, A.; Locke, F.L.; Jacobson, C.A.; Hill, B.T.; Timmerman, J.M.; Holmes, H.; Jaglowski, S.; Flinn, I.W.; et al. KTE-X19 CAR T-cell therapy in relapsed or refractory mantle-cell lymphoma. N. Engl. J. Med. 2020, 382, 1331–1342. [Google Scholar] [CrossRef]
- Hirayama, A.V.; Gauthier, J.; Hay, K.A.; Voutsinas, J.M.; Wu, Q.; Pender, B.S.; Hawkins, R.M.; Vakil, A.; Steinmetz, R.N.; Riddell, S.R.; et al. High rate of durable complete remission in follicular lymphoma after CD19 CAR-T cell immunotherapy. Blood 2019, 134, 636–640. [Google Scholar] [CrossRef]
- Locke, F.L.; Neelapu, S.S.; Bartlett, N.L.; Siddiqi, T.; Chavez, J.C.; Hosing, C.M.; Ghobadi, A.; Budde, L.E.; Bot, A.; Rossi, J.M.; et al. Phase 1 results of ZUMA-1: A multicenter study of KTE-C19 Anti-CD19 CAR T cell therapy in refractory aggressive lymphoma. Mol. Ther. J. Am. Soc. Gene Ther. 2017, 25, 285–295. [Google Scholar] [CrossRef] [Green Version]
- Frey, N.V.; Gill, S.; Hexner, E.O.; Schuster, S.; Nasta, S.; Loren, A.; Svoboda, J.; Stadtmauer, E.; Landsburg, D.J.; Mato, A.; et al. Long-term outcomes from a randomized dose optimization study of chimeric antigen receptor modified T cells in relapsed chronic lymphocytic leukemia. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2020. [Google Scholar] [CrossRef]
- Park, J.H.; Riviere, I.; Gonen, M.; Wang, X.; Senechal, B.; Curran, K.J.; Sauter, C.; Wang, Y.; Santomasso, B.; Mead, E.; et al. Long-Term Follow-up of CD19 CAR therapy in acute lymphoblastic leukemia. N. Engl. J. Med. 2018, 378, 449–459. [Google Scholar] [CrossRef]
- Hu, Y.; Wu, Z.; Luo, Y.; Shi, J.; Yu, J.; Pu, C.; Liang, Z.; Wei, G.; Cui, Q.; Sun, J.; et al. Potent anti-leukemia activities of chimeric antigen receptor-modified T cells against CD19 in Chinese patients with relapsed/refractory acute lymphocytic leukemia. Clin. Cancer Res. 2017, 23, 3297–3306. [Google Scholar] [CrossRef] [Green Version]
- Gardner, R.A.; Finney, O.; Annesley, C.; Brakke, H.; Summers, C.; Leger, K.; Bleakley, M.; Brown, C.; Mgebroff, S.; Kelly-Spratt, K.S.; et al. Intent-to-treat leukemia remission by CD19 CAR T cells of defined formulation and dose in children and young adults. Blood 2017, 129, 3322–3331. [Google Scholar] [CrossRef]
- Abbasi, A.; Peeke, S.; Shah, N.; Mustafa, J.; Khatun, F.; Lombardo, A.; Abreu, M.; Elkind, R.; Fehn, K.; de Castro, A.; et al. Axicabtagene ciloleucel CD19 CAR-T cell therapy results in high rates of systemic and neurologic remissions in ten patients with refractory large B cell lymphoma including two with HIV and viral hepatitis. J. Hematol. Oncol. 2020, 13, 1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chavez, J.C.; Bachmeier, C.; Kharfan-Dabaja, M.A. CAR T-cell therapy for B-cell lymphomas: Clinical trial results of available products. Ther. Adv. Hematol. 2019, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Makita, S.; Yoshimura, K.; Tobinai, K. Clinical development of anti-CD19 chimeric antigen receptor T-cell therapy for B-cell non-Hodgkin lymphoma. Cancer Sci. 2017, 108, 1109–1118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vormittag, P.; Gunn, R.; Ghorashian, S.; Veraitch, F.S. A guide to manufacturing CAR T cell therapies. Curr. Opin. Biotechnol. 2018, 53, 164–181. [Google Scholar] [CrossRef]
- Zheng, P.P.; Kros, J.M.; Li, J. Approved CAR T cell therapies: Ice bucket challenges on glaring safety risks and long-term impacts. Drug Discov. Today 2018, 23, 1175–1182. [Google Scholar] [CrossRef]
- Lichtman, E.I.; Dotti, G. Chimeric antigen receptor T-cells for B-cell malignancies. Transl. Res. J. Lab. Clin. Med. 2017, 187, 59–82. [Google Scholar] [CrossRef]
- Geyer, M.B.; Brentjens, R.J. Review: Current clinical applications of chimeric antigen receptor (CAR) modified T cells. Cytotherapy 2016, 18, 1393–1409. [Google Scholar] [CrossRef] [Green Version]
- Bejcek, B.E.; Wang, D.; Berven, E.; Pennell, C.A.; Peiper, S.C.; Poppema, S.; Uckun, F.M.; Kersey, J.H. Development and characterization of three recombinant single chain antibody fragments (scFvs) directed against the CD19 antigen. Cancer Res. 1995, 55, 2346–2351. [Google Scholar]
- Zola, H.; MacArdle, P.J.; Bradford, T.; Weedon, H.; Yasui, H.; Kurosawa, Y. Preparation and characterization of a chimeric CD19 monoclonal antibody. Immunol. Cell Biol. 1991, 69 Pt 6, 411–422. [Google Scholar] [CrossRef]
- Kim, M.; Pyo, S.; Kang, C.H.; Lee, C.O.; Lee, H.K.; Choi, S.U.; Park, C.H. Folate receptor 1 (FOLR1) targeted chimeric antigen receptor (CAR) T cells for the treatment of gastric cancer. PLoS ONE 2018, 13, e0198347. [Google Scholar] [CrossRef]
- Kobayashi, E.; Kishi, H.; Ozawa, T.; Hamana, H.; Nakagawa, H.; Jin, A.; Lin, Z.; Muraguchi, A. A chimeric antigen receptor for TRAIL-receptor 1 induces apoptosis in various types of tumor cells. Biochem. Biophys. Res. Commun. 2014, 453, 798–803. [Google Scholar] [CrossRef] [PubMed]
- Murakami, T.; Nakazawa, T.; Natsume, A.; Nishimura, F.; Nakamura, M.; Matsuda, R.; Omoto, K.; Tanaka, Y.; Shida, Y.; Park, Y.S.; et al. Novel human NK cell line carrying CAR targeting EGFRvIII induces antitumor effects in glioblastoma cells. Anticancer Res. 2018, 38, 5049–5056. [Google Scholar] [CrossRef] [PubMed]
- Hirayama, A.V.; Gauthier, J.; Hay, K.A.; Voutsinas, J.M.; Wu, Q.; Gooley, T.; Li, D.; Cherian, S.; Chen, X.; Pender, B.S.; et al. The response to lymphodepletion impacts PFS in patients with aggressive non-Hodgkin lymphoma treated with CD19 CAR T cells. Blood 2019, 133, 1876–1887. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garfall, A.L.; Stadtmauer, E.A.; Hwang, W.T.; Lacey, S.F.; Melenhorst, J.J.; Krevvata, M.; Carroll, M.P.; Matsui, W.H.; Wang, Q.; Dhodapkar, M.V.; et al. Anti-CD19 CAR T cells with high-dose melphalan and autologous stem cell transplantation for refractory multiple myeloma. JCI Insight 2018, 3. [Google Scholar] [CrossRef] [PubMed]
- Geyer, M.B.; Riviere, I.; Senechal, B.; Wang, X.; Wang, Y.; Purdon, T.J.; Hsu, M.; Devlin, S.M.; Halton, E.; Lamanna, N.; et al. Autologous CD19-targeted CAR T cells in patients with residual CLL following initial purine analog-based therapy. Mol. Ther. J. Am. Soc. Gene Ther. 2018, 26, 1896–1905. [Google Scholar] [CrossRef] [Green Version]
- Hay, K.A.; Gauthier, J.; Hirayama, A.V.; Voutsinas, J.M.; Wu, Q.; Li, D.; Gooley, T.A.; Cherian, S.; Chen, X.; Pender, B.S.; et al. Factors associated with durable EFS in adult B-cell ALL patients achieving MRD-negative CR after CD19 CAR T-cell therapy. Blood 2019, 133, 1652–1663. [Google Scholar] [CrossRef] [Green Version]
- Ma, F.; Ho, J.Y.; Du, H.; Xuan, F.; Wu, X.; Wang, Q.; Wang, L.; Liu, Y.; Ba, M.; Wang, Y.; et al. Evidence of long-lasting anti-CD19 activity of engrafted CD19 chimeric antigen receptor-modified T cells in a phase I study targeting pediatrics with acute lymphoblastic leukemia. Hematol. Oncol. 2019, 37, 601–608. [Google Scholar] [CrossRef]
- Biondi, A.; Magnani, C.F.; Tettamanti, S.; Gaipa, G.; Biagi, E. Redirecting T cells with Chimeric Antigen Receptor (CAR) for the treatment of childhood acute lymphoblastic leukemia. J. Autoimmun. 2017, 85, 141–152. [Google Scholar] [CrossRef]
- Wang, J.; Mou, N.; Yang, Z.; Li, Q.; Jiang, Y.; Meng, J.; Liu, X.; Deng, Q. Efficacy and safety of humanized anti-CD19-CAR-T therapy following intensive lymphodepleting chemotherapy for refractory/relapsed B acute lymphoblastic leukaemia. Br. J. Haematol. 2020. [Google Scholar] [CrossRef] [Green Version]
- Heng, G.; Jia, J.; Li, S.; Fu, G.; Wang, M.; Qin, D.; Li, Y.; Pei, L.; Tian, X.; Zhang, J.; et al. Sustained therapeutic efficacy of humanized Anti-CD19 chimeric antigen receptor T cells in relapsed/refractory acute lymphoblastic leukemia. Clin. Cancer Res. An Off. J. Am. Assoc. Cancer Res. 2020, 26, 1606–1615. [Google Scholar] [CrossRef] [Green Version]
- Ghorashian, S.; Kramer, A.M.; Onuoha, S.; Wright, G.; Bartram, J.; Richardson, R.; Albon, S.J.; Casanovas-Company, J.; Castro, F.; Popova, B.; et al. Enhanced CAR T cell expansion and prolonged persistence in pediatric patients with ALL treated with a low-affinity CD19 CAR. Nat. Med. 2019, 25, 1408–1414. [Google Scholar] [CrossRef] [PubMed]
- Pezzutto, A.; Dorken, B.; Rabinovitch, P.S.; Ledbetter, J.A.; Moldenhauer, G.; Clark, E.A. CD19 monoclonal antibody HD37 inhibits anti-immunoglobulin-induced B cell activation and proliferation. J. Immunol. 1987, 138, 2793–2799. [Google Scholar] [PubMed]
- Uckun, F.M.; Jaszcz, W.; Ambrus, J.L.; Fauci, A.S.; Gajl-Peczalska, K.; Song, C.W.; Wick, M.R.; Myers, D.E.; Waddick, K.; Ledbetter, J.A. Detailed studies on expression and function of CD19 surface determinant by using B43 monoclonal antibody and the clinical potential of anti-CD19 immunotoxins. Blood 1988, 71, 13–29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shiozawa, M.; Chang, C.H.; Huang, Y.C.; Chen, Y.C.; Chi, M.S.; Hao, H.C.; Chang, Y.C.; Takeda, S.; Chi, K.H.; Wang, Y.S. Pharmacologically upregulated carcinoembryonic antigen-expression enhances the cytolytic activity of genetically-modified chimeric antigen receptor NK-92MI against colorectal cancer cells. BMC Immunol. 2018, 19, 27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mitwasi, N.; Feldmann, A.; Arndt, C.; Koristka, S.; Berndt, N.; Jureczek, J.; Loureiro, L.R.; Bergmann, R.; Mathe, D.; Hegedus, N.; et al. “UniCAR”-modified off-the-shelf NK-92 cells for targeting of GD2-expressing tumour cells. Sci. Rep. 2020, 10, 2141. [Google Scholar] [CrossRef] [Green Version]
- Zhang, P.; Zhao, S.; Wu, C.; Li, J.; Li, Z.; Wen, C.; Hu, S.; An, G.; Meng, H.; Zhang, X.; et al. Effects of CSF1R-targeted chimeric antigen receptor-modified NK92MI & T cells on tumor-associated macrophages. Immunotherapy 2018, 10, 935–949. [Google Scholar] [CrossRef]
- Zhang, C.; Oberoi, P.; Oelsner, S.; Waldmann, A.; Lindner, A.; Tonn, T.; Wels, W.S. Chimeric antigen receptor-engineered NK-92 cells: An off-the-shelf cellular therapeutic for targeted elimination of cancer cells and induction of protective antitumor immunity. Front. Immunol. 2017, 8, 533. [Google Scholar] [CrossRef]
- Oelsner, S.; Friede, M.E.; Zhang, C.; Wagner, J.; Badura, S.; Bader, P.; Ullrich, E.; Ottmann, O.G.; Klingemann, H.; Tonn, T.; et al. Continuously expanding CAR NK-92 cells display selective cytotoxicity against B-cell leukemia and lymphoma. Cytotherapy 2017, 19, 235–249. [Google Scholar] [CrossRef] [Green Version]
- Nowakowska, P.; Romanski, A.; Miller, N.; Odendahl, M.; Bonig, H.; Zhang, C.; Seifried, E.; Wels, W.S.; Tonn, T. Clinical grade manufacturing of genetically modified, CAR-expressing NK-92 cells for the treatment of ErbB2-positive malignancies. Cancer Immunol. Immunother. CII 2018, 67, 25–38. [Google Scholar] [CrossRef]
- Klesmith, J.R.; Wu, L.; Lobb, R.R.; Rennert, P.D.; Hackel, B.J. Fine epitope mapping of the CD19 extracellular domain promotes design. Biochemistry 2019, 58, 4869–4881. [Google Scholar] [CrossRef]
- Hombach, A.; Hombach, A.A.; Abken, H. Adoptive immunotherapy with genetically engineered T cells: Modification of the IgG1 Fc ‘spacer’ domain in the extracellular moiety of chimeric antigen receptors avoids ‘off-target’ activation and unintended initiation of an innate immune response. Gene Ther. 2010, 17, 1206–1213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guedan, S.; Posey, A.D., Jr.; Shaw, C.; Wing, A.; Da, T.; Patel, P.R.; McGettigan, S.E.; Casado-Medrano, V.; Kawalekar, O.U.; Uribe-Herranz, M.; et al. Enhancing CAR T cell persistence through ICOS and 4-1BB costimulation. JCI Insight 2018, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morin, S.O.; Giroux, V.; Favre, C.; Bechah, Y.; Auphan-Anezin, N.; Roncagalli, R.; Mege, J.L.; Olive, D.; Malissen, M.; Nunes, J.A. In the absence of its cytosolic domain, the CD28 molecule still contributes to T cell activation. Cell. Mol. Life Sci. 2015, 72, 2739–2748. [Google Scholar] [CrossRef] [PubMed]
- Alabanza, L.; Pegues, M.; Geldres, C.; Shi, V.; Wiltzius, J.J.W.; Sievers, S.A.; Yang, S.; Kochenderfer, J.N. Function of novel Anti-CD19 chimeric antigen receptors with human variable regions is affected by hinge and transmembrane domains. Mol. Ther. 2017, 25, 2452–2465. [Google Scholar] [CrossRef] [Green Version]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kang, C.H.; Kim, Y.; Lee, H.K.; Lee, S.M.; Jeong, H.G.; Choi, S.U.; Park, C.H. Identification of Potent CD19 scFv for CAR T Cells through scFv Screening with NK/T-Cell Line. Int. J. Mol. Sci. 2020, 21, 9163. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21239163
Kang CH, Kim Y, Lee HK, Lee SM, Jeong HG, Choi SU, Park CH. Identification of Potent CD19 scFv for CAR T Cells through scFv Screening with NK/T-Cell Line. International Journal of Molecular Sciences. 2020; 21(23):9163. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21239163
Chicago/Turabian StyleKang, Chung Hyo, Yeongrin Kim, Heung Kyoung Lee, So Myoung Lee, Hye Gwang Jeong, Sang Un Choi, and Chi Hoon Park. 2020. "Identification of Potent CD19 scFv for CAR T Cells through scFv Screening with NK/T-Cell Line" International Journal of Molecular Sciences 21, no. 23: 9163. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21239163