Development and Validation of a Novel 11-Gene Prognostic Model for Serous Ovarian Carcinomas Based on Lipid Metabolism Expression Profile

 , , ,
, , ,
Abstract
:1. Introduction
2. Results
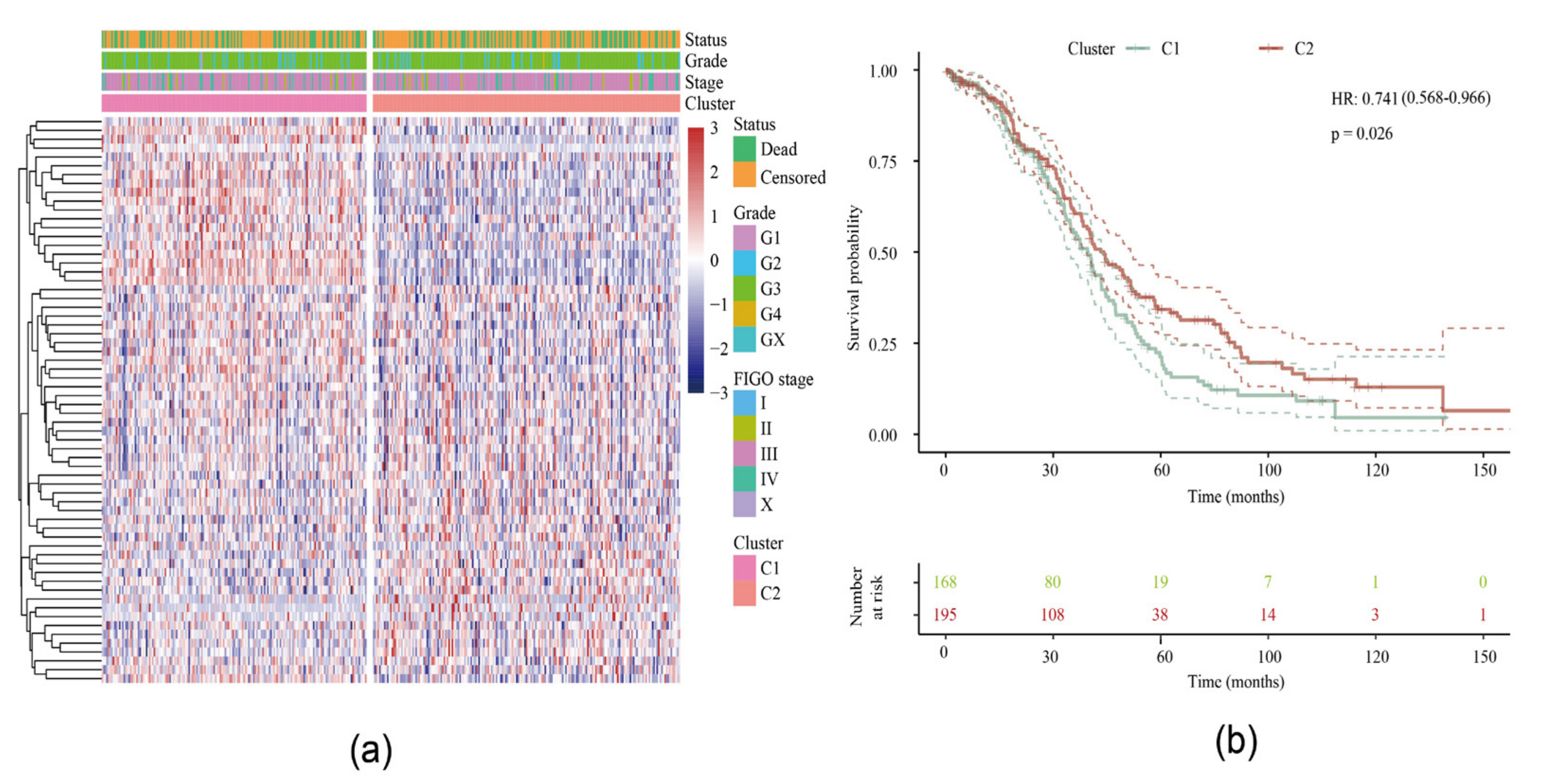
2.1. Molecular Subtypes of Lipid Metabolism-Related Genes
2.1.1. Identification of Two Molecular Subtypes
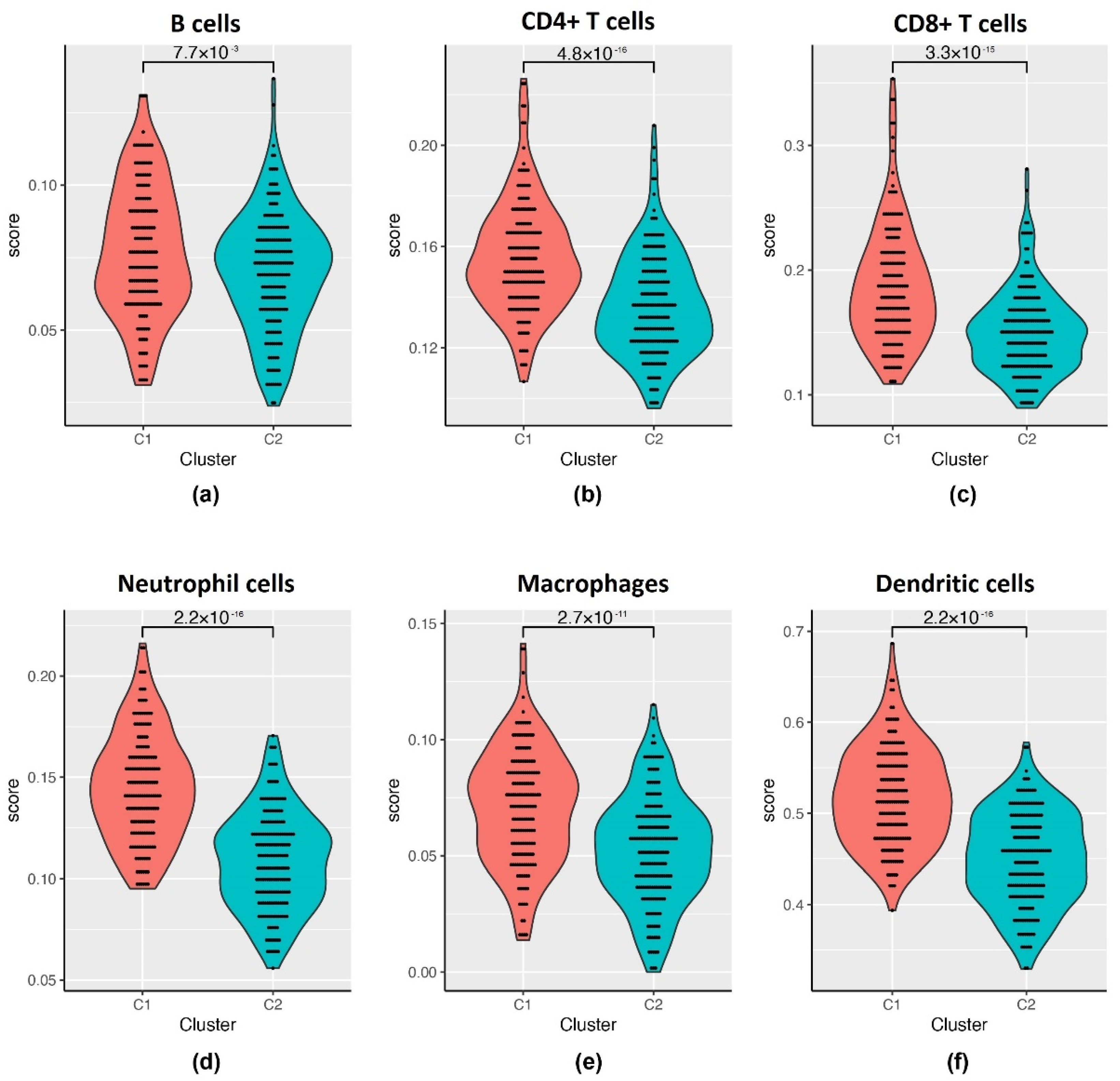
2.1.2. Relationship between Two Subtypes and Immunity
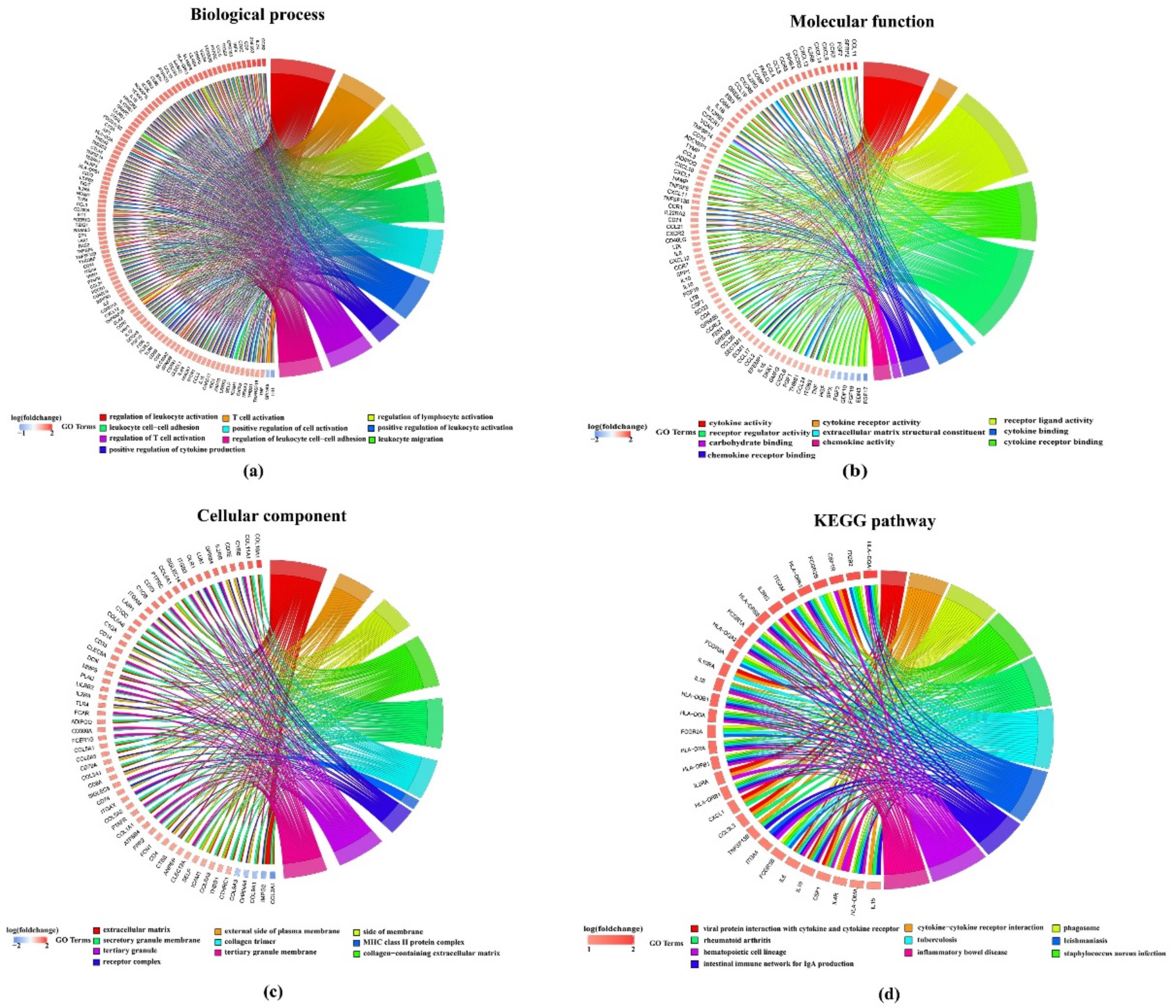
2.2. Analysis of DEGs between Subtypes
2.2.1. The DEGs in C2 Subtype Were Mainly Downregulated
2.2.2. DEGs Are Enriched in Tumor-Related Pathways
2.3. Construction of a Prognostic Risk Model
2.3.1. Randomly Grouping of Training and Testing Cohort
2.3.2. Univariable Analysis of Training Cohort
2.3.3. Construction of the 11-Gene Signature Using Lasso Regression
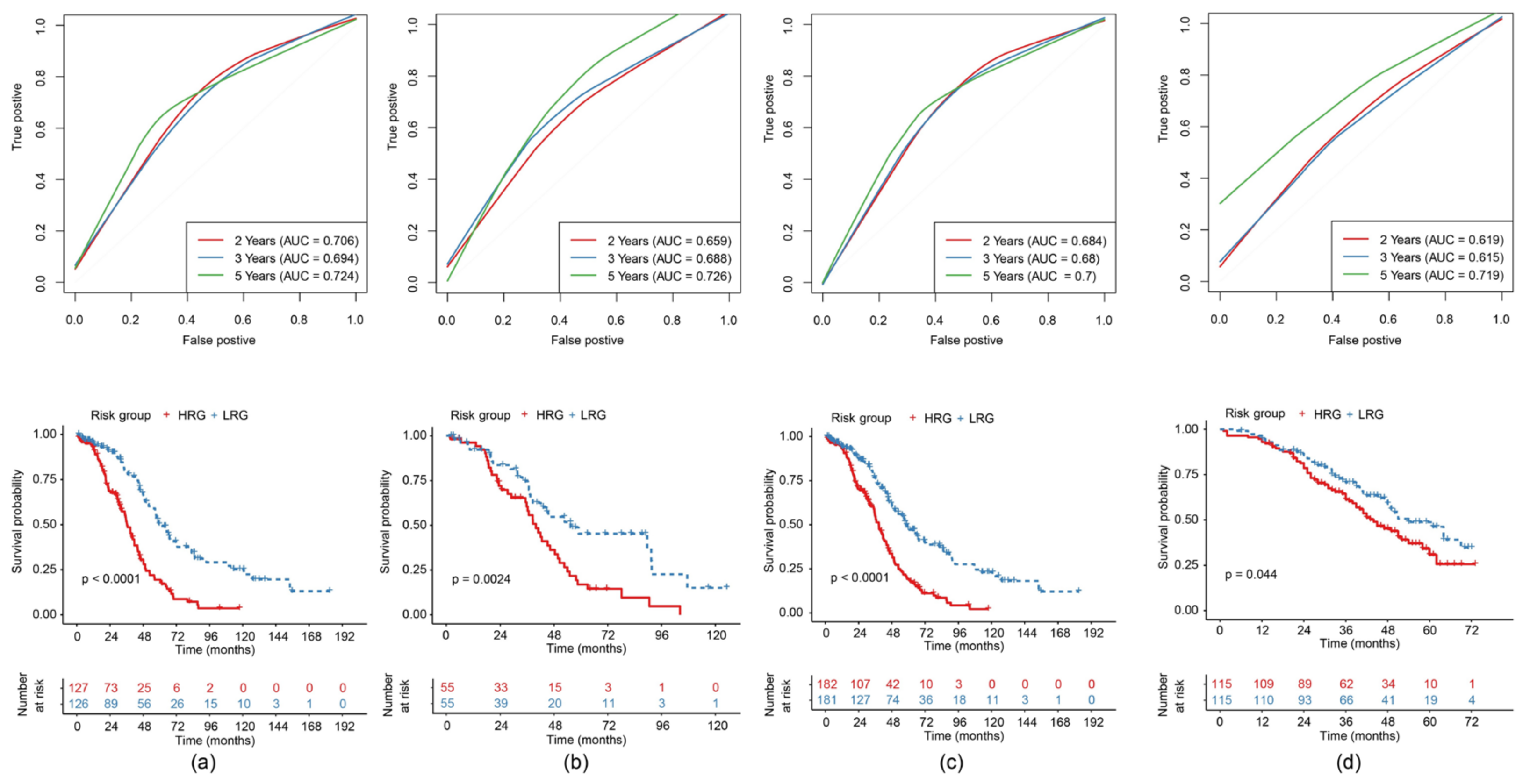
2.3.4. Construction and Evaluation of a Risk Model
2.4. Univariable and Multivariable Analysis of Gene Signature
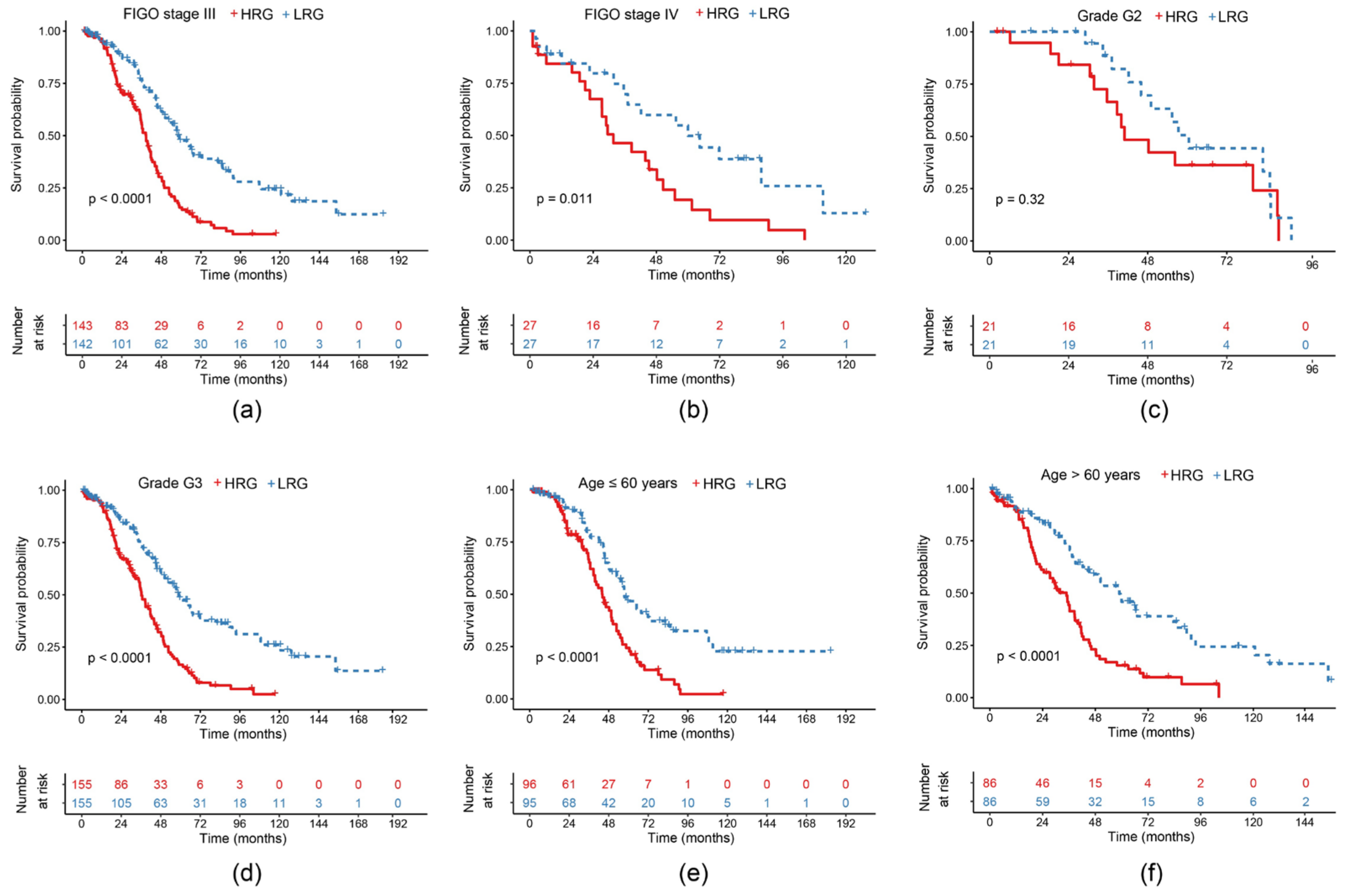
2.5. Survival Curves of Risk Models in Different Clinical Subgroups
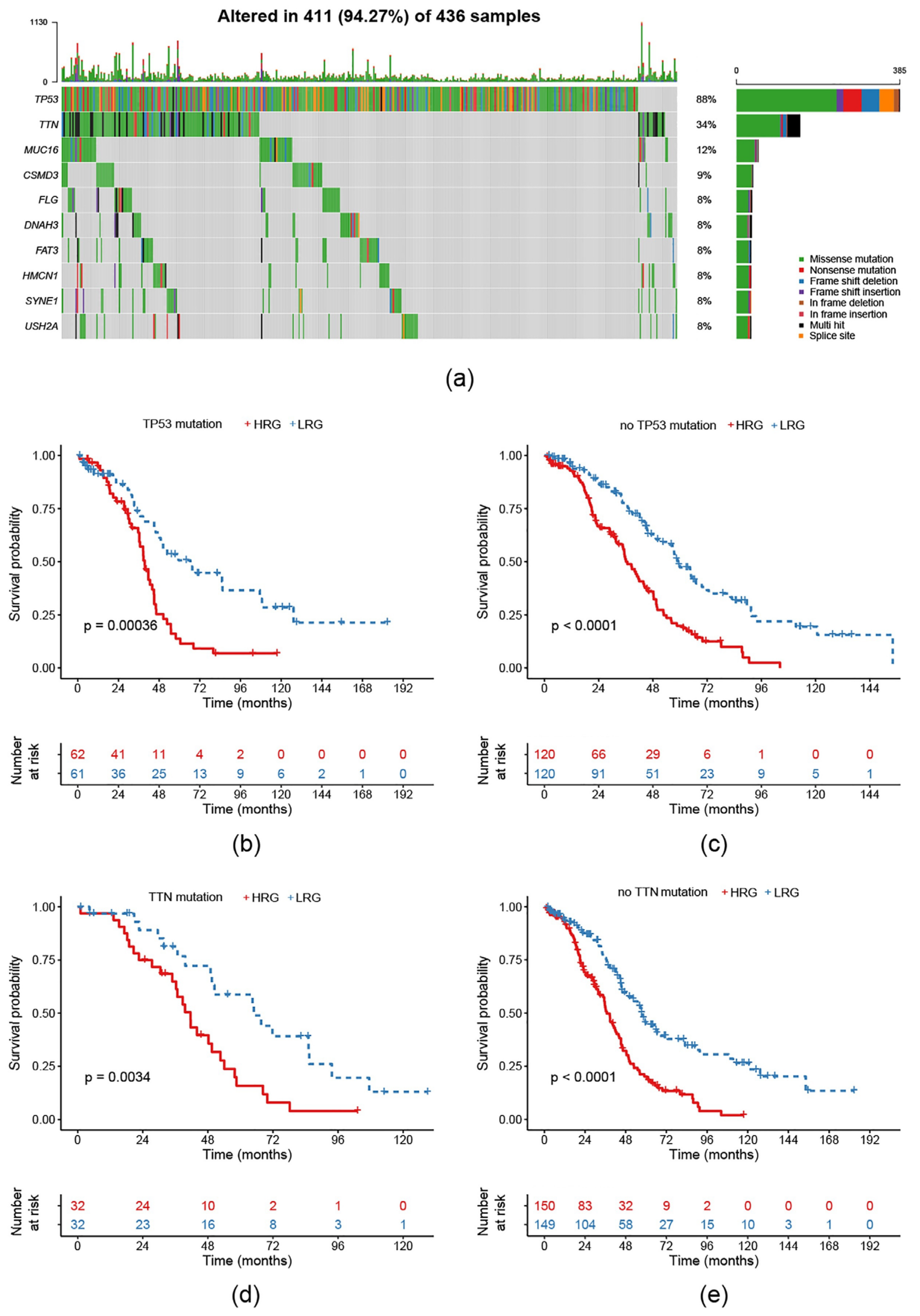
2.6. Survival Prognosis on Different Mutation Subtypes in the Risk Model
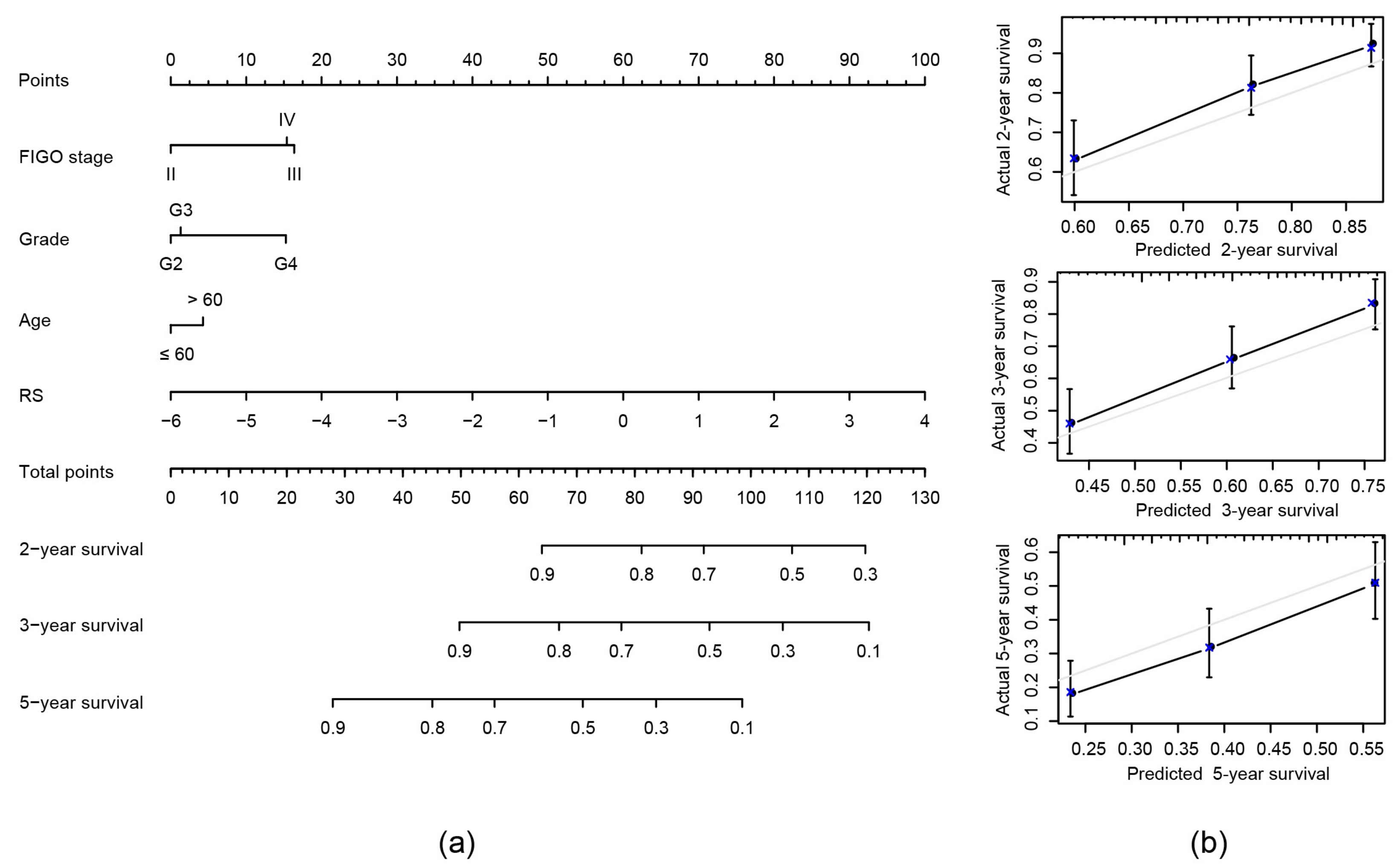
2.7. Construction of Nomogram Model Based on RS and Clinical Features
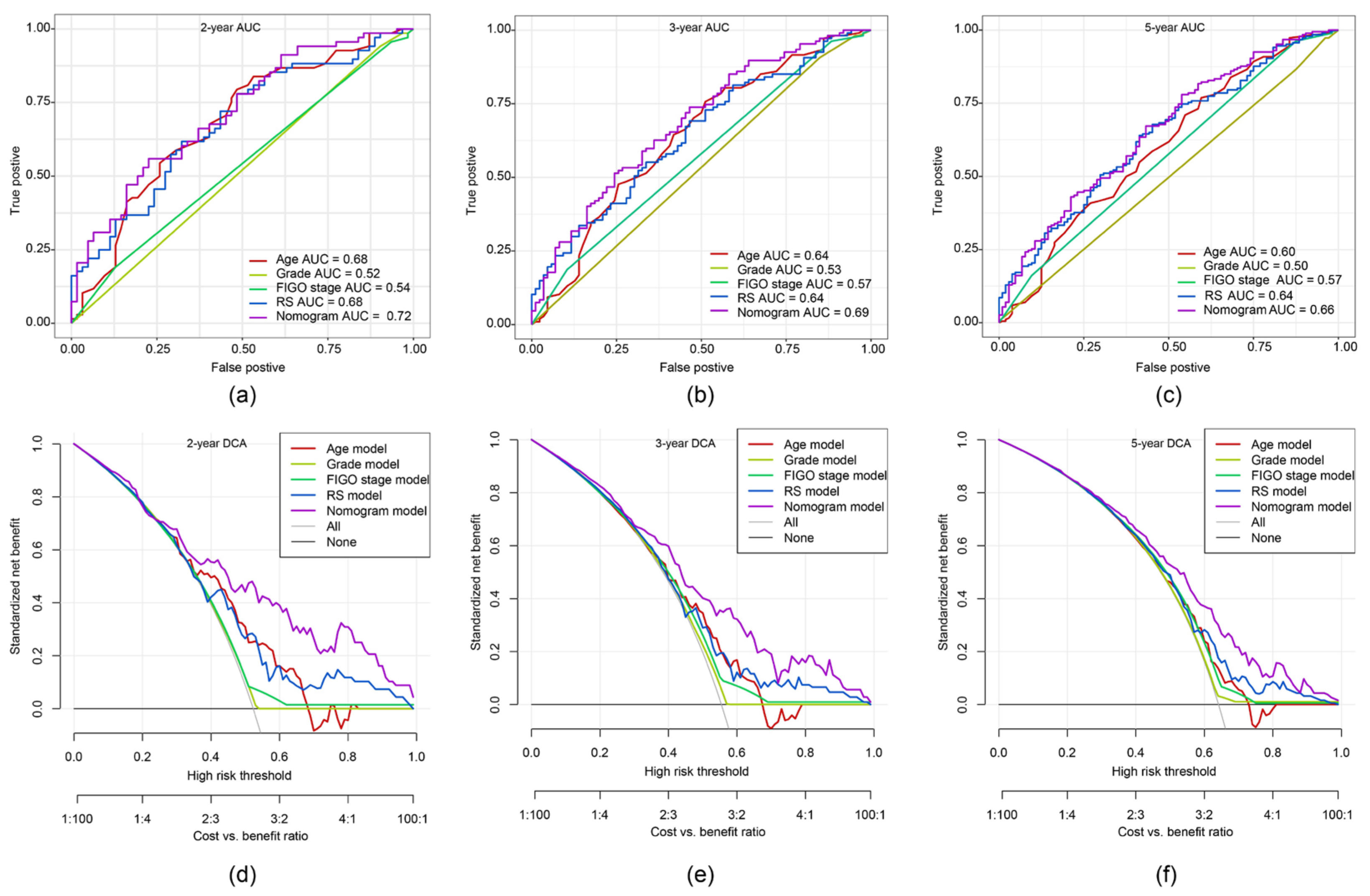
2.8. ROC Curve and DCA of Nomogram Model
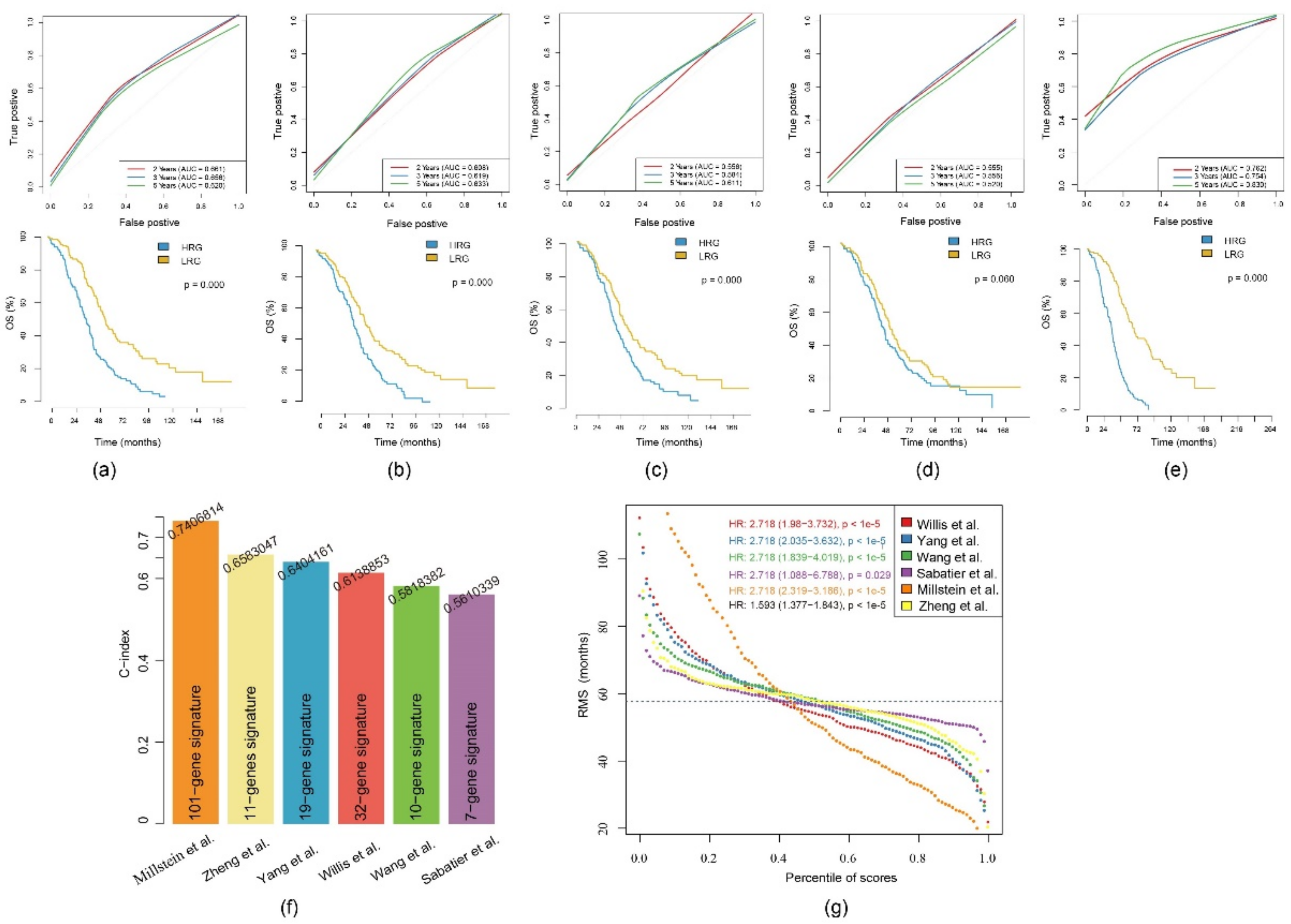
2.9. Comparison of the 11-Gene Risk Model with Other Models
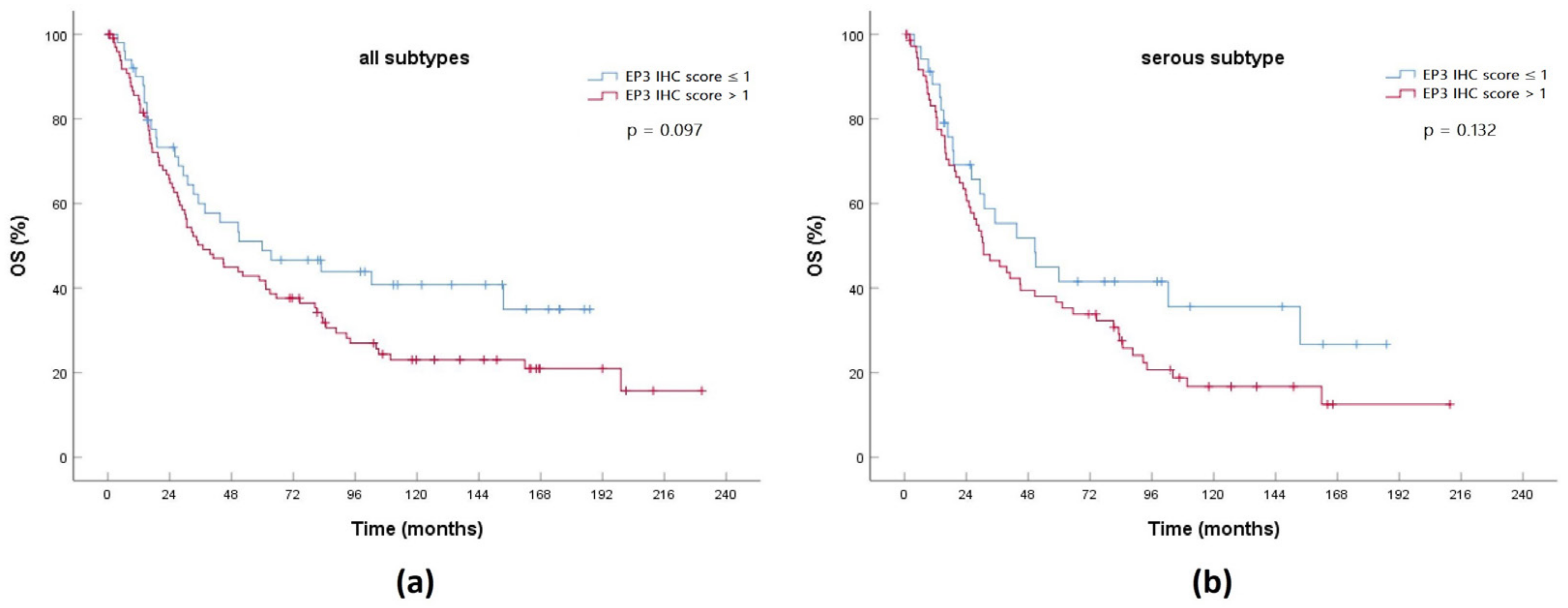
2.10. Expression of a Gene Product from the 11-Gene Signature in an EOC Cohort
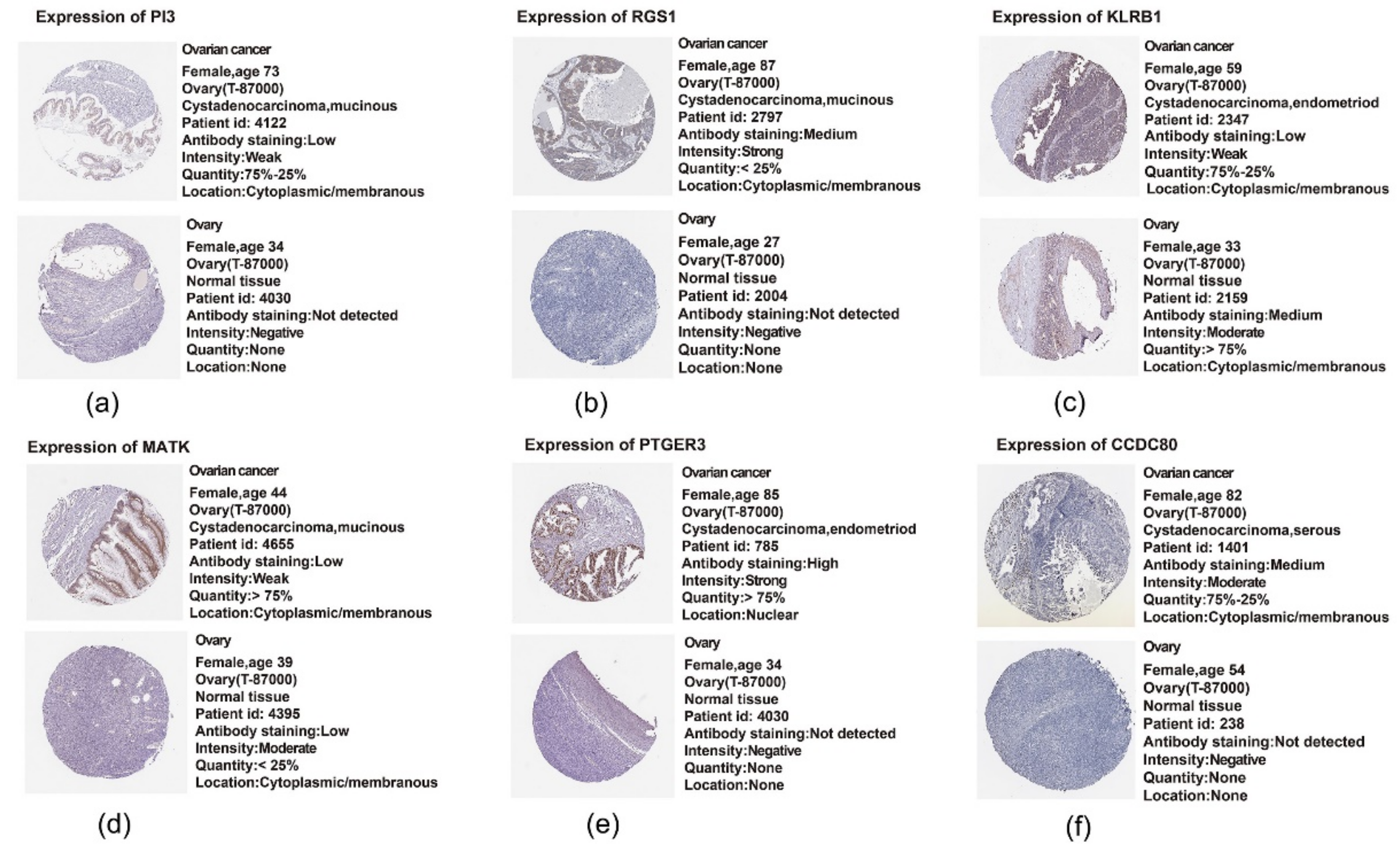
2.11. Translational Level Validation Related to Signature Genes
3. Discussion
4. Materials and Methods
4.1. Ovarian Cancer Cohort Source and Preprocessing
4.2. Identification of Molecular Subtypes Using NMF Algorithm
4.3. Comparison of Molecular Subtype Immune Scores with TIMER
4.4. Functional Analysis of DEGs
4.5. Sample Preparation
- The two cohorts are similar in age distribution, FIGO stage, follow-up time and death rate of patients.
- The gene expression profiles of the two data sets that were randomly grouped were close in the number of classified samples.
4.6. Lasso Regression Analysis
4.7. Stability Assessment of Risk Model
4.8. Construction of Nomogram Combined with RS and Clinical Features
4.9. Analysis of DCA
4.10. IHC of EP3 in an EOC Patient Cohort
4.11. Translational Level Validation of Signature Genes
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| AUC | area under the curve |
| C1 | cluster 1 |
| C2 | cluster 2 |
| C-index | concordance index |
| CI | confidence interval |
| DCA | decision curve analysis |
| DEG | differentially expressed gene |
| EOC | epithelial ovarian cancer |
| FDR | false discovery rate |
| FIGO | Fédération Internationale de Gynécologie et d’Obstétrique |
| GEO | Gene Expression Omnibus |
| GO | Gene Ontology |
| GSE32026 | GEO Series 32026 |
| HPA | Human Protein Atlas |
| HR | hazard ratio |
| HRG | high-risk group |
| IHC | immunohistochemistry |
| KEGG | Kyoto Encyclopedia of Genes and Genomes |
| KM | Kaplan-Meier |
| LRG | low-risk group |
| NMF | non-negative matrix factorization |
| OS | overall survival |
| EP3 | prostaglandin E2 receptor 3 |
| RMS | restricted mean survival |
| ROC | receiver operating characteristic |
| RS | risk score |
| TCGA | The Cancer Genome Atlas |
| TIMER | Tumor Immune Estimation Resource |
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics 2019. CA Cancer J. Clin. 2019, 69, 7–34. [Google Scholar] [PubMed] [Green Version]
- Waldmann, A.; Eisemann, N.; Katalinic, A. Epidemiology of malignant cervical, corpus uteri and ovarian tumours-current data and epidemiological trends. Geburtshilfe Frauenheilkd 2013, 73, 123–129. [Google Scholar] [PubMed] [Green Version]
- Jayson, G.C.; Kohn, E.C.; Kitchener, H.C.; Ledermann, J.A. Ovarian cancer. Lancet 2014, 384, 1376–1388. [Google Scholar] [PubMed]
- Howlader, N.; Noone, A.M.; Krapcho, M.; Miller, D.; Brest, A.; Yu, M.; Ruhl, J.; Tatalovich, Z.; Mariotto, A.; Lewis, D.R.; et al. (Eds.) SEER Cancer Statistics Review, 1975–2016; based on November 2018 SEER data submission; National Cancer Institute: Bethesda, MD, USA, 2019.
- Lech, A.; Daneva, T.; Pashova, S.; Gagov, H.; Crayton, R.; Kukwa, W.; Czarnecka, A.M.; Szczylik, C. Ovarian cancer as a genetic disease. Front. Biosci. 2013, 18, 543–563. [Google Scholar]
- Eng, K.H.; Weir, I.; Tsuji, T.; Odunsi, K. Immuno-stimultory/regulatory gene expression patterns in advanced ovarian cancer. Genes Cancer 2015, 6, 399–407. [Google Scholar]
- Mittempergher, L. Genomic characterization of high-grade serous ovarian cancer: Dissecting its molecular heterogeneity as a road towards effective therapeutic strategies. Curr. Oncol. Rep. 2016, 18, 44. [Google Scholar]
- Cai, S.Y.; Yang, T.; Chen, Y.; Wang, J.W.; Li, L.; Xu, M.J. Gene expression profiling of ovarian carcinomas and prognostic analysis of outcome. J. Ovarian Res. 2015, 8, 50. [Google Scholar]
- Tassi, R.A.; Todeschini, P.; Siegel, E.R.; Calza, S.; Cappella, P.; Ardighieri, L.; Cadei, M.; Bugatti, M.; Romani, C.; Bandiera, E.; et al. FOXM1 expression is significantly associated with chemotherapy resistance and adverse prognosis in non-serous epithelial ovarian cancer patients. J. Exp. Clin. Cancer Res. 2017, 36, 63. [Google Scholar]
- Niemi, R.J.; Braicu, E.I.; Kulbe, H.; Koistinen, K.M.; Sehouli, J.; Puistola, U.; Mäenpää, U.J.; Hilvo, M. Ovarian tumours of different histologic type and clinical stage induce similar changes in lipid metabolism. Br. J. Cancer 2018, 119, 847–854. [Google Scholar]
- Nickels, J.T. New links between lipid accumulation and cancer progression. J. Biol. Chem. 2018, 293, 6635–6636. [Google Scholar]
- Xu, Y. Lysophospholipid signaling in the epithelial ovarian cancer tumor microenvironment. Cancers 2018, 10, 227. [Google Scholar]
- Zhao, G.; Cardenas, H.; Matei, D. Ovarian cancer-why lipids matter. Cancers 2019, 10, 1870. [Google Scholar]
- Braicu, E.I.; Darb-Esfahani, S.; Schmitt, W.D.; Koistinen, K.M.; Heiskanen, L.; Pöhö, P.; Budczies, J.; Kuhberg, M.; Dietel, M.; Frezza, C.; et al. High-grade ovarian serous carcinoma patients exhibit profound alterations in lipid metabolism. Oncotarget 2017, 8, 102912–102922. [Google Scholar] [PubMed] [Green Version]
- Chen, R.R.; Yung, M.M.; Xuan, Y.; Zhan, S.; Leung, L.L.; Liang, R.R.; Leung, T.H.Y.; Yang, H.; Xu, D.; Sharma, R.; et al. Targeting of lipid metabolism with a metabolic inhibitor cocktail eradicates peritoneal metastases in ovarian cancer cells. Commun. Biol. 2019, 2, 281. [Google Scholar]
- Cui, T.; Zhang, L.; Huang, Y.; Yi, Y.; Tan, P.; Zhao, Y.; Hu, Y.; Xu, L.; Li, E.; Wang, D. MNDR v2.0: An updated resource of ncRNA–disease associations in mammals. Nucleic Acids Res. 2017, 46, D371–D374. [Google Scholar]
- Liang, Z.Y.; Lai, H.Y.; Yang, H.; Zhang, C.J.; Yang, H.; Wei, H.H.; Chen, X.X.; Zhao, Y.W.; Su, Z.D.; Li, W.C.; et al. Pro54DB: A database for experimentally verified sigma-54 promoters. Bioinformatics 2017, 33, 467–469. [Google Scholar]
- Hu, B.; Zheng, L.; Long, C.; Song, M.; Li, T.; Yang, L.; Zuo, Y. EmExplorer: A database for exploring time activation of gene expression in mammalian embryos. Open Biol. 2019, 9, 190054. [Google Scholar]
- Wang, Z.; Gerstein, M.; Snyder, M. RNA-Seq: A revolutionary tool for transcriptomics. Nat. Rev. Genet. 2009, 10, 57–63. [Google Scholar]
- Yang, H.; Lv, H.; Ding, H.; Chen, W.; Lin, H. iRNA-2OM: A sequence-based predictor for identifying 2′-O-methylation sites in homo sapiens. J. Comput. Biol. 2018, 25, 1266–1277. [Google Scholar]
- Safonov, A.; Jiang, T.; Bianchini, G.; Győrffy, B.; Karn, T.; Hatzis, C.; Pusztai, L. Immune gene expression is associated with genomic aberrations in breast cancer. Cancer Res. 2017, 77, 3317–3324. [Google Scholar]
- Yang, L.; Wang, S.; Zhang, Q.; Pan, Y.; Lv, Y.; Chen, X.; Zuo, Y.; Hao, D. Clinical significance of the immune microenvironment in ovarian cancer patients. Mol. Omics 2018, 14, 341–351. [Google Scholar] [CrossRef] [PubMed]
- Yang, R.; Xiong, J.; Deng, D.; Wang, Y.; Liu, H.; Jiang, G.; Peng, Y.; Peng, X.; Zeng, X. An integrated model of clinical information and gene expression for prediction of survival in ovarian cancer patients. Transl. Res. 2016, 172, 84–95. [Google Scholar] [CrossRef] [PubMed]
- Willis, S.; Villalobos, V.M.; Gevaert, O.; Abramovitz, M.; Williams, C.; Sikic, B.I.; Leyland-Jones, B. Single gene prognostic biomarkers in ovarian cancer: A meta-analysis. PLoS ONE 2016, 11, e0149183. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Wang, L.; Ma, L.; Liu, J.; Ma, S. Identifying gene signature for the detection of ovarian cancer based on the achieved related genes. Gynecol. Obstet. Investig. 2017, 82, 361–370. [Google Scholar] [CrossRef] [PubMed]
- Sabatier, R.; Finetti, P.; Bonensea, J.; Jacquemier, J.; Adelaide, J.; Lambaudie, E.; Viens, P.; Birnbaum, D.; Bertucci, F. A seven-gene prognostic model for platinum-treated ovarian carcinomas. Br. J. Cancer 2011, 105, 304–311. [Google Scholar] [CrossRef] [PubMed]
- Millstein, J.; Budden, T.; Goode, E.L.; Anglesio, M.S.; Talhouk, A.; Intermaggio, M.P.; Leong, H.S.; Chen, S.; Elatre, W.; Gilks, B.; et al. Prognostic gene expression signature for high-grade serous ovarian cancer. Ann. Oncol. 2020, 31, 1240–1250. [Google Scholar] [CrossRef]
- Royston, P.; Parmar, M.K.B. Restricted mean survival time: An alternative to the hazard ratio for the design and analysis of randomized trials with a time-to-event outcome. BMC Med. Res. Methodol. 2013, 13, 152. [Google Scholar] [CrossRef] [Green Version]
- Herzog, T.J. Recurrent ovarian cancer: How important is it to treat to disease progression? Clin. Cancer Res. 2004, 10, 7439–7449. [Google Scholar] [CrossRef] [Green Version]
- Nagasawa, S.; Ikeda, K.; Horie-Inoue, K.; Sato, S.; Itakura, A.; Takeda, S.; Hasegawa, K.; Inoue, S. Systematic identification of characteristic genes of ovarian clear cell carcinoma compared with high-grade serous carcinoma based on RNA-sequencing. Int. J. Mol. Sci. 2019, 20, 4330. [Google Scholar] [CrossRef] [Green Version]
- Seborova, K.; Vaclavikova, R.; Soucek, P.; Elsnerova, K.; Bartakova, A.; Cernaj, P.; Bouda, J.; Rob, L.; Hruda, M.; Dvorak, P. Association of ABC gene profiles with time to progression and resistance in ovarian cancer revealed by bioinformatics analyses. Cancer Med. 2019, 8, 606–616. [Google Scholar] [CrossRef]
- Zhang, Z.; Huang, K.; Gu, C.; Zhao, L.; Wang, N.; Wang, X.; Zhao, D.; Zhang, C.; Lu, Y.; Meng, Y. Molecular subtyping of serous ovarian cancer based on multi-omics data. Sci. Rep. 2016, 6, 26001. [Google Scholar] [CrossRef] [PubMed]
- Tothill, R.W.; Tinker, A.V.; George, J.; Brown, R.; Fox, S.B.; Lade, S.; Johnson, D.S.; Trivett, M.K.; Etemadmoghadam, D.; Locandro, B.; et al. Novel molecular subtypes of serous and endometrioid ovarian cancer linked to clinical outcome. Clin. Cancer Res. 2008, 14, 5198–5208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, D.; Chen, P.; Zheng, C.H.; Xia, J. Identification of ovarian cancer subtype-specific network modules and candidate drivers through an integrative genomics approach. Oncotarget 2016, 7, 4298–4309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clemente, G.C.; Mihm, C.M.; Zurrida, S.; Collini, P.; Cascineelli, N. Prognostic value of tumor infiltrating lymphocytes in the vertical growth phase of primary cutaneous melanoma. Cancer 1996, 77, 1303–1310. [Google Scholar] [CrossRef]
- Rozek, L.S.; Schmit, S.L.; Greenson, J.K.; Tomsho, L.P.; Rennert, H.S.; Rennert, G.; Gruber, S.B. Tumor-infiltrating lymphocytes, crohn’s-like lymphoid reaction, and survival from colorectal cancer. J. Natl. Cancer Inst. 2016, 108, djw027. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Naito, Y.; Saito, K.; Shiiba, K.; Ohuchi, A.; Saigenji, K.; Nagura, H.; Ohtani, H. CD8+ T cells infiltrated within cancer cell nests as a prognostic factor in human colorectal cancer. Cancer Res. 1998, 58, 3491–3494. [Google Scholar]
- Nakano, O.; Sato, M.; Naito, Y.; Suzuki, K.; Orikasa, S.; Aizawa, M.; Suzuki, Y.; Shintaku, I.; Nagura, H.; Ohtani, H. Proliferative activity of intratumoral CD8(+) T-lymphocytes as a prognostic factor in human renal cell carcinoma: Clinicopathologic demonstration of antitumor immunity. Cancer Res 2001, 61, 5132–5136. [Google Scholar]
- Sato, E.; Olson, S.H.; Ahn, J.; Bundy, B.; Nishikawa, H.; Qian, F.; Junbluth, A.A.; Frosina, D.; Gnjatic, S.; Ambrosone, C.; et al. Intraepithelial CD8+ tumor-infiltrating lymphocytes and a high CD8+/regulatory T cell ratio are associated with favorable prognosis in ovarian cancer. Proc. Natl. Acad. Sci. USA 2005, 102, 18538–18543. [Google Scholar] [CrossRef] [Green Version]
- Diederichsen, A.C.; Hjelmborg, J.V.B.; Christensen, P.B.; Zeuthen, J.; Fenger, C. Prognostic value of the CD4+/CD8+ ratio of tumour infiltrating lymphocytes in colorectal cancer and HLA-DR expression on tumour cells. Cancer Immunol. Immunother. 2003, 52, 423–428. [Google Scholar] [CrossRef]
- Wefers, C.; Duiveman-de Boer, T.; Zusterzeel, P.L.M.; Massuger, L.F.A.G.; Fuchs, D.; Torensma, R.; Wheelock, C.E.; de Vries, I.J.M. Different lipid regulation in ovarian cancer: Inhibition of the immune system. Int. J. Mol. Sci. 2018, 19, 273. [Google Scholar] [CrossRef] [Green Version]
- Schalkwijk, J.; Wiedow, O.; Hirose, S. The trappin gene family: Proteins defined by an N-terminal transglutaminase substrate domain and a C-terminal four-disulphide core. Biochem. J. 1999, 340, 569–577. [Google Scholar] [CrossRef] [PubMed]
- Clauss, A.; Lilja, H.; Lundwall, Å. A locus on human chromosome 20 contains several genes expressing protease inhibitor domains with homology to whey acidic protein. Biochem. J. 2002, 368, 233–242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Labidi-Galy, S.I.; Clauss, A.; Ng, V.; Duraisamy, S.; Elias, K.M.; Piao, H.Y.; Bilal, E.; Davidowitz, R.A.; Lu, Y.; Badalian-Very, G.; et al. Elafin drives poor outcome in high-grade serous ovarian cancers and basal-like breast tumors. Oncogene 2015, 34, 373–383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clauss, A.; Ng, V.; Liu, J.; Piao, H.; Russo, M.; Vena, N.; Sheng, Q.; Hirsch, M.S.; Bonome, T.; Matulonis, U.; et al. Overexpression of elafin in ovarian carcinoma is driven by genomic gains and activation of the nuclear factor κB pathway and is associated with poor overall survival. Neoplasia 2010, 12, 161–172. [Google Scholar] [CrossRef] [Green Version]
- Wei, H.; Hellström, K.E.; Hellström, I. Elafin selectively regulates the sensitivity of ovarian cancer cells to genotoxic drug-induced apoptosis. Gynecol. Oncol. 2012, 125, 727–733. [Google Scholar] [CrossRef] [Green Version]
- Bou, D.J.; Lee, J.K. Regulator of G-Protein signaling 1 (RGS1). In Encyclopedia of Signaling Molecules, 2nd ed.; Choi, S., Ed.; Springer International Publishing: Basel, Switzerland, 2018. [Google Scholar]
- Sethakorn, N.; Dulin, N.O. RGS expression in cancer: Oncomining the cancer microarray data. J. Recept. Signal. Transduct. Res. 2013, 33, 166–171. [Google Scholar] [CrossRef]
- Wong, Y.F.; Cheung, T.H.; Tsao, G.S.; Lo, K.W.; Yim, S.F.; Wang, V.W.; Heung, M.M.; Chan, S.C.; Chan, L.K.; Ho, T.W.; et al. Genome-wide gene expression profiling of cervical cancer in Hong Kong women by oligonucleotide microarray. Int. J. Cancer 2006, 118, 2461–2469. [Google Scholar] [CrossRef]
- Fredholm, B.B.; IJzerman, A.P.I.; Jacobson, K.A.; Klotz, K.N.; Linden, J. International Union of Pharmacology–XXV. Nomenclature and classification of adenosine receptors. Pharmacol. Rev. 2001, 53, 527–552. [Google Scholar]
- Riccardo, F.; Arigoni, M.; Buson, G.; Zago, E.; Iezzi, M.; Longo, D.; Carrara, M.; Fiore, A.; Nuzzo, S.; Bicciato, S.; et al. Characterization of a genetic mouse model of lung cancer: A promise to identify non-small cell lung cancer therapeutic targets and biomarkers. BMC Genom. 2014, 15, S1. [Google Scholar] [CrossRef] [Green Version]
- Doms, A.; Sanabria, T.; Hansen, J.N.; Altan-Bonnet, N.; Holm, G.H. 25-Hydroxycholesterol production by the cholesterol-25-Hydroxylase interferon-stimulated gene restricts mammalian Reovirus infection. J. Virol. 2018, 92, e01047-18. [Google Scholar] [CrossRef] [Green Version]
- Bauman, D.R.; Bitmansour, A.D.; McDonald, J.G.; Thompson, B.M.; Liang, G.; Russell, D.W. 25-Hydroxycholesterol secreted by macrophages in response to toll-like receptor activation suppresses immunoglobulin A production. Proc. Natl. Acad. Sci. USA 2009, 106, 16764–16769. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mittempergher, L.; Saghatchian, M.; Wolf, D.M.; Michiels, S.; Canisius, S.; Dessen, P.; Delaloge, S.; Lazar, V.; Benz, S.C.; Tursz, T.; et al. A gene signature for late distant metastasis in breast cancer identifies a potential mechanism of late recurrences. Mol. Oncol. 2013, 7, 987–999. [Google Scholar] [CrossRef] [PubMed]
- Sugimoto, Y.; Narumiya, S. Prostaglandin E receptors. J. Biol. Chem. 2007, 282, 11613–11617. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Czogalla, B.; Kuhn, C.; Heublein, S.; Schmöckel, E.; Mayr, D.; Kolben, T.; Trillsch, F.; Burges, A.; Mahner, S.; Jeschke, U.; et al. EP3 receptor is a prognostic factor in TA-MUC1-negative ovarian cancer. J. Cancer Res. Clin. Oncol. 2019, 145, 2519–2527. [Google Scholar] [CrossRef] [PubMed]
- Heidegger, H.; Dietlmeier, S.; Ye, Y.; Kuhn, C.; Vattai, A.; Aberl, C.; Jeschke, U.; Mahner, S.; Kost, B. The prostaglandin EP3 receptor is an independent negative prognostic factor for cervical cancer patients. Int. J. Mol. Sci. 2017, 18, 1571. [Google Scholar] [CrossRef] [Green Version]
- Zhu, J.; Trillsch, F.; Mayr, D.; Kuhn, C.; Rahmeh, M.; Hofmann, S.; Vogel, M.; Mahner, S.; Jeschke, U.; Schönfeldt, V. Prostaglandin receptor EP3 regulates cell proliferation and migration with impact on survival of endometrial cancer patients. Oncotarget 2018, 9, 982–994. [Google Scholar] [CrossRef] [Green Version]
- Semmlinger, A.; von Schoenfeldt, V.; Wolf, V.; Meuter, A.; Kolben, T.M.; Kolben, T.; Zeder-Goess, C.; Weis, F.; Gallwas, J.; Wuerstlein, R.; et al. EP3 (prostaglandin E2 receptor 3) expression is a prognostic factor for progression-free and overall survival in sporadic breast cancer. BMC Cancer 2018, 18, 431. [Google Scholar] [CrossRef] [Green Version]
- Gentles, A.J.; Newman, A.M.; Liu, C.L.; Bratman, S.V.; Feng, W.; Kim, D.; Nair, V.S.; Xu, Y.; Khuong, A.; Hoang, C.D.; et al. The prognostic landscape of genes and infiltrating immune cells across human cancers. Nat. Med. 2015, 21, 938–945. [Google Scholar] [CrossRef]
- Li, T.; Fan, J.; Wang, B.; Traugh, N.; Chen, Q.; Liu, J.S.; Li, B.; Liu, X.S. TIMER: A web server for comprehensive analysis of tumor-infiltrating immune cells. Cancer Res. 2017, 77, e108–e110. [Google Scholar] [CrossRef] [Green Version]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. Limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef]
- Yu, G.; Wang, L.G.; Han, Y.; He, Q.Y. ClusterProfiler: An R package for comparing biological themes among gene clusters. Omics 2012, 16, 284–287. [Google Scholar] [CrossRef] [PubMed]
- Tibshirani, R. Regression shrinkage and selection via the lasso. J. R. Stat. Soc. B 1996, 58, 267–288. [Google Scholar] [CrossRef]
- Friedman, J.; Hastie, T.; Tibshirani, R. Regularization paths for generalized linear models via coordinate descent. J. Stat. Softw. 2010, 33, 1–22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Remmele, W.; Stegner, H.E. Recommendation for uniform definition of an immunoreactive score (IRS) for immunohistochemical estrogen receptor detection (ER-ICA) in breast cancer tissue. Pathologe 1987, 8, 138–140. [Google Scholar] [PubMed]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Clinical feature | Training Cohort | Testing Cohort | X-Squared | p-Value | |
|---|---|---|---|---|---|
| Event | Censored Dead | 99 154 | 42 68 | 0.0028 | 0.9575 |
| FIGO stage | I II III IV None | 0 15 198 37 3 | 1 5 87 17 0 | 3.9129 | 0.4179 |
| Age | ≤60 >60 | 132 121 | 59 51 | 0.0202 | 0.8870 |
| Grade | G1 G2 G3 G4 None | 1 29 217 0 6 | 0 13 93 1 3 | 2.7958 | 0.5926 |
| Clinical Feature | Univariable Analysis | Multivariable Analysis | ||||
|---|---|---|---|---|---|---|
| HR | 95% CI | p-Value | HR | 95% CI | p-Value | |
| FIGO stage | 1.929 | 0.856–4.349 | 0.1130 | 1.705 | 0.742–3.917 | 0.2080 |
| Age | 1.186 | 0.791–1.778 | 0.4090 | 1.087 | 0.718–1.646 | 0.6940 |
| Grade | 1.291 | 0.992–1.680 | 0.0576 | 1.193 | 0.910–1.566 | 0.2020 |
| RS | 1.593 | 1.377–1.843 | <0.001 | 1.534 | 1.322–1.780 | <0.001 |
| Variables | C-Index | 95% CI of C-Index | p-Value |
|---|---|---|---|
| FIGO stage | 0.609 | 0.523–0.696 | 0.013 |
| Age | 0.619 | 0.547–0.690 | 0.001 |
| Grade | 0.593 | 0.508–0.678 | 0.032 |
| RS | 0.658 | 0.602–0.684 | <0.001 |
| Nomogram | 0.663 | 0.625–0.701 | <0.001 |
| Pathway | Database | Gene Count |
|---|---|---|
| Peroxisome proliferator activated receptor alpha | Reactome | 119 |
| Metabolism of lipids | Reactome | 738 |
| Sphingolipid metabolism | Reactome | 89 |
| Transcriptional regulation of white adipocyte differentiation | Reactome | 84 |
| Glycerophospholipid metabolism | KEGG | 77 |
| Fatty acid metabolism | Reactome | 77 |
| Total: 1184 Unique: 776 |
| Clinical Feature | TCGA | GSE32026 | |
|---|---|---|---|
| Event | Censored Dead | 141 222 | 116 114 |
| FIGO stage | I II III IV None | 1 20 285 54 3 | |
| Age | ≤60 >60 | 191 172 | |
| Grade | G1 G2 G3 G4 None | 1 42 310 1 9 | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zheng, M.; Mullikin, H.; Hester, A.; Czogalla, B.; Heidegger, H.; Vilsmaier, T.; Vattai, A.; Chelariu-Raicu, A.; Jeschke, U.; Trillsch, F.; et al. Development and Validation of a Novel 11-Gene Prognostic Model for Serous Ovarian Carcinomas Based on Lipid Metabolism Expression Profile. Int. J. Mol. Sci. 2020, 21, 9169. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21239169
Zheng M, Mullikin H, Hester A, Czogalla B, Heidegger H, Vilsmaier T, Vattai A, Chelariu-Raicu A, Jeschke U, Trillsch F, et al. Development and Validation of a Novel 11-Gene Prognostic Model for Serous Ovarian Carcinomas Based on Lipid Metabolism Expression Profile. International Journal of Molecular Sciences. 2020; 21(23):9169. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21239169
Chicago/Turabian StyleZheng, Mingjun, Heather Mullikin, Anna Hester, Bastian Czogalla, Helene Heidegger, Theresa Vilsmaier, Aurelia Vattai, Anca Chelariu-Raicu, Udo Jeschke, Fabian Trillsch, and et al. 2020. "Development and Validation of a Novel 11-Gene Prognostic Model for Serous Ovarian Carcinomas Based on Lipid Metabolism Expression Profile" International Journal of Molecular Sciences 21, no. 23: 9169. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21239169