Physiological and Differential Proteomic Analyses of Imitation Drought Stress Response in Sorghum bicolor Root at the Seedling Stage



Abstract
:1. Introduction
2. Results
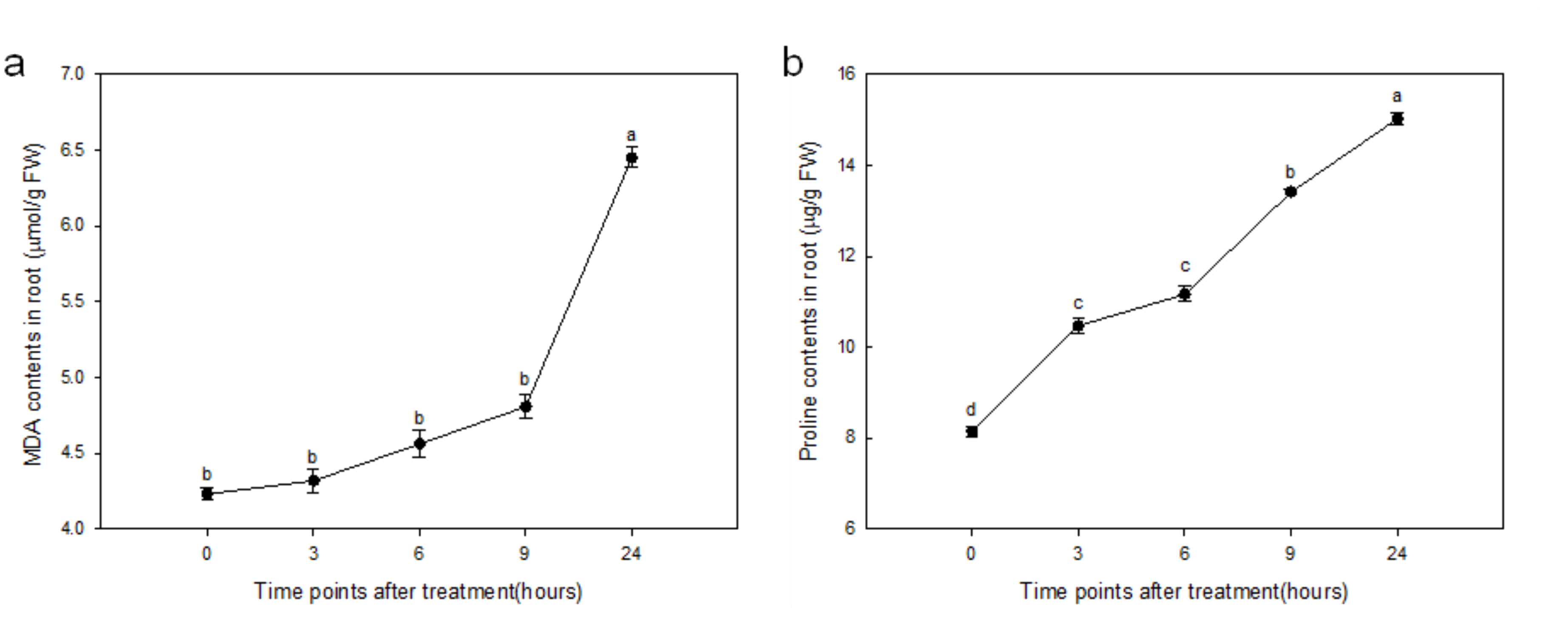
2.1. Effect of Drought Stress on MDA and Proline Contents in Roots
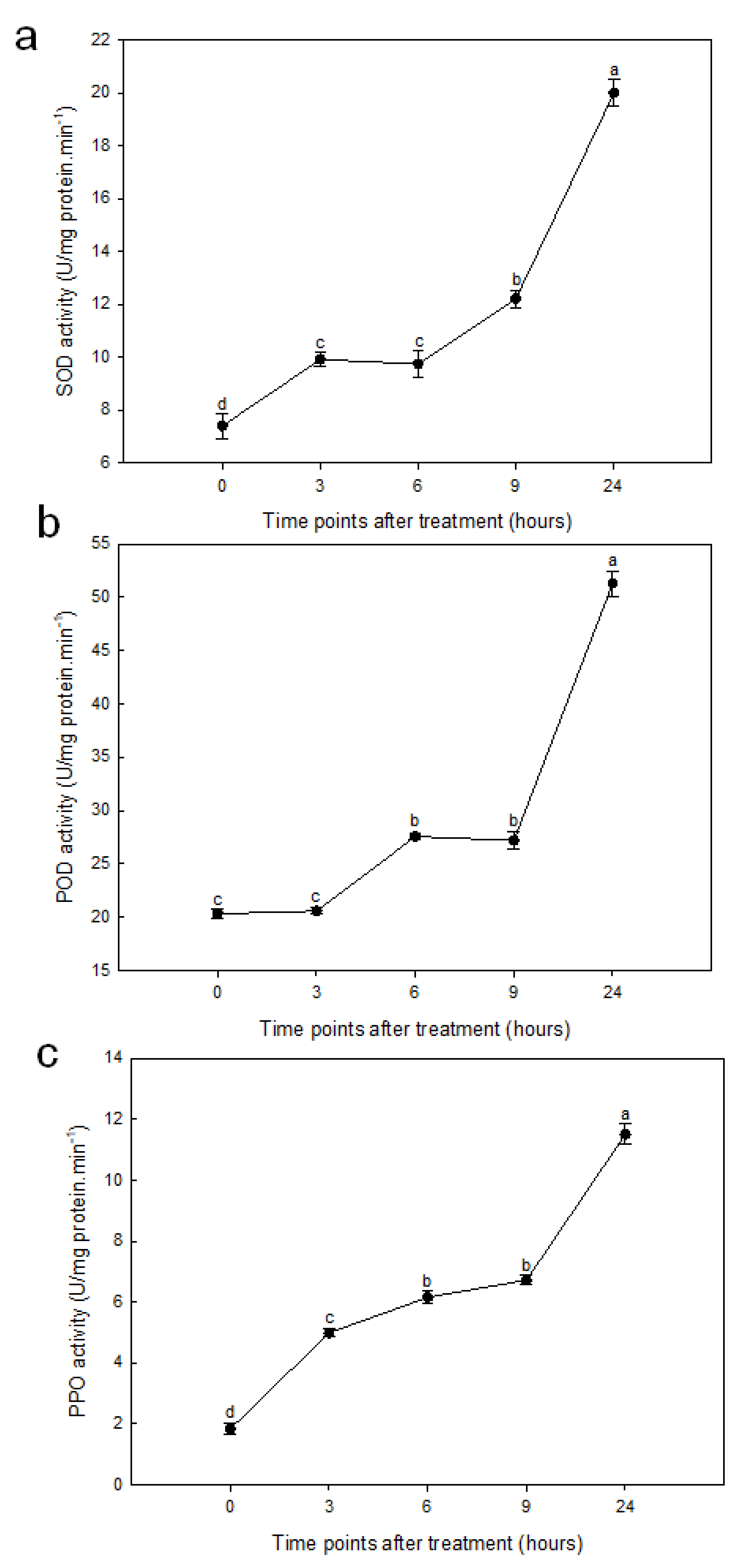
2.2. Drought Stress Increased Antioxidant Enzyme Activity in Roots
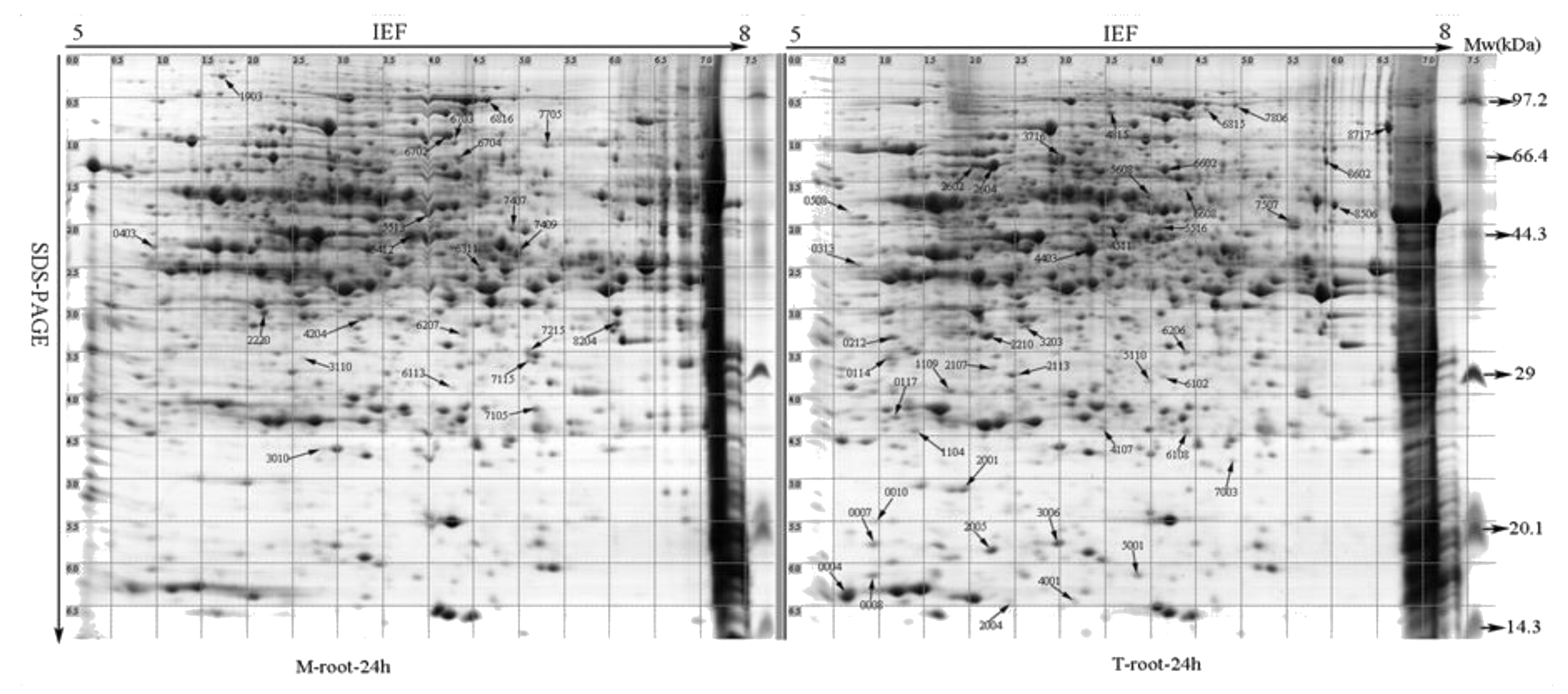
2.3. Changes in Proteomic Expression Patterns of Roots in Response to Imitation Drought Stress
2.4. Identification of Drought-Responsive Proteins by MALDI-TOF-TOF MS Analysis
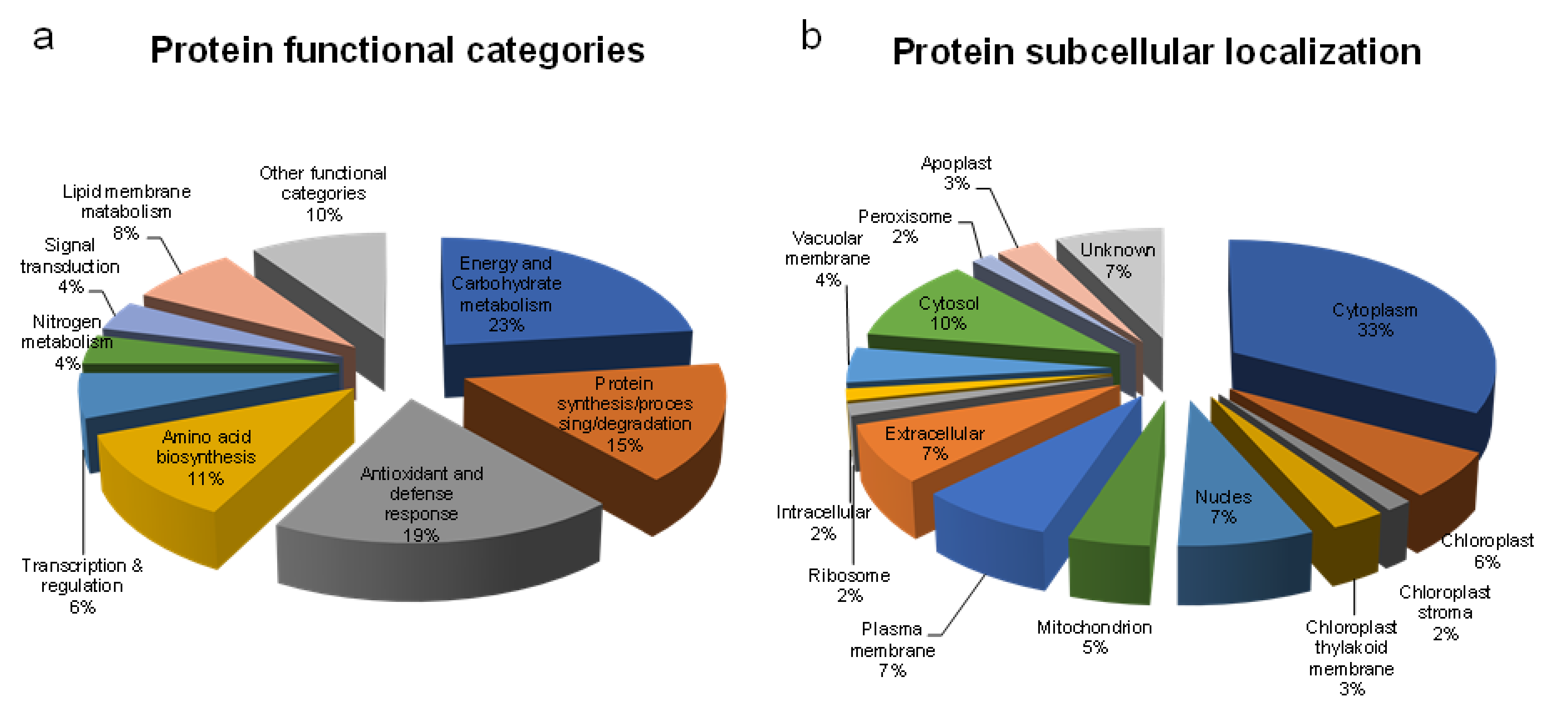
2.5. Functional Classification and Subcellular Localization of Identified Proteins
3. Discussion
3.1. Physiological Alteration in Roots of Sorghum in Response to Drought Stress
3.2. Proteins Involved in Energy and Carbohydrate Metabolism
3.3. Proteins Involved in Antioxidant and Defense Response
3.4. Proteins Involved in Protein Synthesis/Processing/Degradation
3.5. Proteins Involved in Transcriptional and Regulation
3.6. Proteins Involved in Amino Acid Biosynthesis and Nitrogen Metabolism
3.7. Proteins Involved in Lipid Membrane Metabolic
3.8. Proteins Involved in Signal Transduction
3.9. Other Functional Categories and Uncharacterized Protein
4. Materials and Methods
4.1. Seedling Cultivation, Polyethylene Glycol (PEG) Treatment
4.2. MDA Contents Measurement
4.3. Antioxidative Enzyme Activity Assays
4.4. Measurement of Free Proline Content
4.5. Protein Extraction and Quantification
4.6. Isoelectric Focusing (IEF) and SDS–PAGE Conditions
4.7. Protein Visualization and Gel-Image Analysis
4.8. Protein In-Gel Digestion
4.9. MALDI-TOF-TOF MS Analysis and Database Query
4.10. DEP Identification, Annotation, and Subcellular Localization
4.11. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Bian, Y.W.; Deng, X.; Yan, X.; Zhou, J.X.; Yuan, L.L.; Yan, Y.M. Integrated proteomic analysis of Brachypodium distachyon roots and leaves reveals a synergistic network in the response to drought stress and recovery. Sci. Rep. 2017, 7, 46183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boyer, J.S. Plant productivity and environment. Science 1982, 218, 443–448. [Google Scholar] [CrossRef] [PubMed]
- Pennisi, E. Plant genetics: The blue revolution, drop by drop, gene by gene. Science 2008, 320, 171–173. [Google Scholar] [CrossRef] [PubMed]
- Shao, H.B.; Chu, L.Y.; Jaleel, C.A.; Manivannan, P.; Panneerselvam, R.; Shao, M.A. Understanding water deficit stress-induced changes in the basic metabolism of higher plants—Biotechnologically and sustainably improving agriculture and the ecoenvironment in arid regions of the globe. Crit. Rev. Biotechnol. 2009, 29, 131–151. [Google Scholar] [CrossRef]
- Breshears, D.D.; Cobb, N.S.; Rich, P.M.; Price, K.P.; Allen, C.D.; Balice, R.G.; Romme, W.H.; Kastens, J.H.; Floyd, M.L.; Belnap, J.; et al. Regional vegetation die-off in response to global-change-type drought. Proc. Natl. Acad. Sci. USA 2005, 102, 15144–15148. [Google Scholar] [CrossRef] [Green Version]
- Bartels, D.; Furini, A.; Ingram, J.; Salamini, F. Responses of plants to dehydration stress: A molecular analysis. Plant Growth Regul. 1996, 20, 111–118. [Google Scholar] [CrossRef]
- Gowda, V.R.P.; Henry, A.; Yamauchi, A.; Shashidhar, H.E.; Serraj, R. Root biology and genetic improvement for drought avoidance in rice. Field Crop. Res. 2011, 122, 1–13. [Google Scholar] [CrossRef]
- Atkinson, N.J.; Urwin, P.E. The interaction of plant biotic and abiotic stresses: From genes to the field. J. Exp. Bot. 2012, 63, 3523–3543. [Google Scholar] [CrossRef] [Green Version]
- Fujita, M.; Fujita, Y.; Noutoshi, Y.; Takahashi, F.; Narusaka, Y.; Yamaguchi-Shinozaki, K.; Shinozaki, K. Crosstalk between abiotic and biotic stress responses: A current view from the points of convergence in the stress signaling networks. Curr. Opin. Plant Biol. 2006, 9, 436–442. [Google Scholar] [CrossRef]
- Dietz, K.J.; Mittler, R.; Noctor, G. Recent Progress in Understanding the Role of Reactive Oxygen Species in Plant Cell Signaling. Plant Physiol. 2016, 171, 1535–1539. [Google Scholar] [CrossRef] [Green Version]
- Petricka, J.J.; Schauer, M.A.; Megraw, M.; Breakfield, N.W.; Thompson, J.W.; Georgiev, S.; Soderblom, E.J.; Ohler, U.; Moseley, M.A.; Grossniklaus, U.; et al. The protein expression landscape of the Arabidopsis root. Proc. Natl. Acad. Sci. USA 2012, 109, 6811–6818. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeng, W.J.; Peng, Y.L.; Zhao, X.Q.; Wu, B.Y.; Chen, F.Q.; Ren, B.; Zhuang, Z.L.; Gao, Q.H.; Ding, Y.F. Comparative Proteomics Analysis of the Seedling Root Response of Drought-sensitive and Drought-tolerant Maize Varieties to Drought Stress. Int. J. Mol. Sci. 2019, 20, 2793. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Labuschagne, M.T. A review of cereal grain proteomics and its potential for sorghum improvement. J. Cereal Sci. 2018, 84, 151–158. [Google Scholar] [CrossRef]
- Duodu, K.G.; Taylor, J.R.N.; Belton, P.S.; Hamaker, B.R. Factors affecting sorghum protein digestibility. J. Cereal Sci. 2003, 38, 117–131. [Google Scholar] [CrossRef] [Green Version]
- Borrell, A.K.; Mullet, J.E.; George-Jaeggli, B.; van Oosterom, E.J.; Hammer, G.L.; Klein, P.E.; Jordan, D.R. Drought adaptation of stay-green sorghum is associated with canopy development, leaf anatomy, root growth, and water uptake. J. Exp. Bot. 2014, 65, 6251–6263. [Google Scholar] [CrossRef]
- Ngara, R.; Ndimba, B.K. Understanding the complex nature of salinity and drought-stress response in cereals using proteomics technologies. Proteomics 2014, 14, 611–621. [Google Scholar] [CrossRef]
- Kailasa, S.K.; Wu, H.F. Recent developments in nanoparticle-based MALDI mass spectrometric analysis of phosphoproteomes. Microchim. Acta 2014, 181, 853–864. [Google Scholar] [CrossRef]
- Ghosh, D.; Xu, J. Abiotic stress responses in plant roots: A proteomics perspective. Front. Plant Sci. 2014, 5, 6. [Google Scholar] [CrossRef] [Green Version]
- Vincent, D.; Lapierre, C.; Pollet, B.; Cornic, G.; Negroni, L.; Zivy, M. Water deficits affect caffeate O-methyltransferase, lignification, and related enzymes in maize leaves. A proteomic investigation. Plant Physiol. 2005, 137, 949–960. [Google Scholar] [CrossRef] [Green Version]
- Jorge, I.; Navarro, R.M.; Lenz, C.; Ariza, D.; Jorrin, J. Variation in the holm oak leaf proteome at different plant developmental stages, between provenances and in response to drought stress. Proteomics 2006, 6, S207–S214. [Google Scholar] [CrossRef]
- Alam, I.; Sharmin, S.A.; Kim, K.H.; Yang, J.K.; Choi, M.S.; Lee, B.H. Proteome analysis of soybean roots subjected to short-term drought stress. Plant Soil 2010, 333, 491–505. [Google Scholar] [CrossRef]
- Sengupta, D.; Kannan, M.; Reddy, A.R. A root proteomics-based insight reveals dynamic regulation of root proteins under progressive drought stress and recovery in Vigna radiata (L.) Wilczek. Planta 2011, 233, 1111–1127. [Google Scholar] [CrossRef] [PubMed]
- Salekdeh, G.H.; Siopongco, J.; Wade, L.J.; Ghareyazie, B.; Bennett, J. Proteomic analysis of rice leaves during drought stress and recovery. Proteomics 2002, 2, 1131–1145. [Google Scholar] [CrossRef]
- Hu, Y.; Li, W.C.; Xu, Y.Q.; Li, G.J.; Liao, Y.; Fu, F.L. Differential expression of candidate genes for lignin biosynthesis under drought stress in maize leaves. J. Appl. Genet. 2009, 50, 213–223. [Google Scholar] [CrossRef] [PubMed]
- Muthurajan, R.; Shobbar, Z.S.; Jagadish, S.V.K.; Bruskiewich, R.; Ismail, A.; Leung, H.; Bennett, J. Physiological and Proteomic Responses of Rice Peduncles to Drought Stress. Mol. Biotechnol. 2011, 48, 173–182. [Google Scholar] [CrossRef]
- Ashoub, A.; Beckhaus, T.; Berberich, T.; Karas, M.; Bruggemann, W. Comparative analysis of barley leaf proteome as affected by drought stress. Planta 2013, 237, 771–781. [Google Scholar] [CrossRef]
- Yang, L.M.; Jiang, T.B.; Fountain, J.C.; Scully, B.T.; Lee, R.D.; Kemerait, R.C.; Chen, S.X.; Guo, B.Z. Protein Profiles Reveal Diverse Responsive Signaling Pathways in Kernels of Two Maize Inbred Lines with Contrasting Drought Sensitivity. Int. J. Mol. Sci. 2014, 15, 18892–18918. [Google Scholar] [CrossRef]
- Qin, N.; Xu, W.G.; Hu, L.; Li, Y.; Wang, H.W.; Qi, X.L.; Fang, Y.H.; Hua, X. Drought tolerance and proteomics studies of transgenic wheat containing the maize C4 phosphoenolpyruvate carboxylase (PEPC) gene. Protoplasma 2016, 253, 1513. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.; Li, Z.; Muhammad, W.; Lin, M.H.; Azeem, S.; Zhao, H.; Lin, S.; Chen, T.; Fang, C.X.; Letuma, P.; et al. Proteomic analysis of positive influence of alternate wetting and moderate soil drying on the process of rice grain filling. Plant Growth Regul. 2018, 84, 533–548. [Google Scholar] [CrossRef]
- Thangella, P.A.V.; Pasumarti, S.N.B.S.; Pullakhandam, R.; Geereddy, B.R.; Daggu, M.R. Differential expression of leaf proteins in four cultivars of peanut (Arachis hypogaea L.) under water stress. 3 Biotech 2018, 8, 157. [Google Scholar] [CrossRef]
- Paterson, A.H.; Bowers, J.E.; Bruggmann, R.; Dubchak, I.; Grimwood, J.; Gundlach, H.; Haberer, G.; Hellsten, U.; Mitros, T.; Poliakov, A.; et al. The Sorghum bicolor genome and the diversification of grasses. Nature 2009, 457, 551–556. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abdel-Ghany, S.E.; Ullah, F.; Ben-Hur, A.; Reddy, A.S.N. Transcriptome Analysis of Drought-Resistant and Drought-Sensitive Sorghum (Sorghum bicolor) Genotypes in Response to PEG-Induced Drought Stress. Int. J. Mol. Sci. 2020, 21, 772. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ngara, R.; Ndimba, B.K. Mapping and characterisation of the sorghum cell suspension culture secretome. Afr. J. Biotechnol. 2011, 10, 253–266. [Google Scholar]
- Ngara, R.; Ramulifho, E.; Movahedi, M.; Shargie, N.G.; Brown, A.P.; Chivasa, S. Identifying differentially expressed proteins in sorghum cell cultures exposed to osmotic stress. Sci. Rep. 2018, 8, 8671. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fadoul, H.E.; El Siddig, M.A.; Abdalla, A.W.H.; El Hussein, A.A. Physiological and proteomic analysis of two contrasting Sorghum bicolor genotypes in response to drought stress. Aust. J. Crop Sci. 2018, 12, 1543–1551. [Google Scholar] [CrossRef]
- Goche, T.; Shargie, N.G.; Cummins, I.; Brown, A.P.; Chivasa, S.; Ngara, R. Comparative physiological and root proteome analyses of two sorghum varieties responding to water limitation. Sci. Rep. 2020, 10, 11835. [Google Scholar] [CrossRef]
- Swami, A.K.; Alam, S.I.; Sengupta, N.; Sarin, R. Differential proteomic analysis of salt stress response in Sorghum bicolor leaves. Environ. Exp. Bot. 2011, 71, 321–328. [Google Scholar] [CrossRef]
- Ngara, R.; Ndimba, R.; Borch-Jensen, J.; Jensen, O.N.; Ndimba, B. Identification and profiling of salinity stress-responsive proteins in Sorghum bicolor seedlings. J. Proteom. 2012, 75, 4139–4150. [Google Scholar] [CrossRef] [Green Version]
- Jedmowski, C.; Ashoub, A.; Beckhaus, T.; Berberich, T.; Karas, M.; Bruggemann, W. Comparative Analysis of Sorghum bicolor Proteome in Response to Drought Stress and following Recovery. Int. J. Proteom. 2014, 2014, 395905. [Google Scholar] [CrossRef] [Green Version]
- Roy, S.K.; Cho, S.W.; Kwon, S.J.; Kamal, A.M.; Kim, S.W.; Oh, M.W.; Lee, M.S.; Chung, K.Y.; Xin, Z.G.; Woo, S.H. Morpho-Physiological and Proteome Level Responses to Cadmium Stress in Sorghum. PLoS ONE 2016, 11, e0150431. [Google Scholar] [CrossRef]
- Roy, S.K.; Kwon, S.J.; Cho, S.W.; Kamal, A.H.M.; Kim, S.W.; Sarker, K.; Oh, M.W.; Lee, M.S.; Chung, K.Y.; Xin, Z.G.; et al. Leaf proteome characterization in the context of physiological and morphological changes in response to copper stress in sorghum. Biometals 2016, 29, 495–513. [Google Scholar] [CrossRef] [PubMed]
- Koca, H.; Bor, M.; Ozdemir, F.; Turkan, I. The effect of salt stress on lipid peroxidation, antioxidative enzymes and proline content of sesame cultivars. Environ. Exp. Bot. 2007, 60, 344–351. [Google Scholar] [CrossRef]
- Veeranagamallaiah, G.; Chandraobulreddy, P.; Jyothsnakumari, G.; Sudhakar, C. Glutamine synthetase expression and pyrroline-5-carboxylate reductase activity influence proline accumulation in two cultivars of foxtail millet (Setaria italica L.) with differential salt sensitivity. Environ. Exp. Bot. 2007, 60, 239–244. [Google Scholar] [CrossRef]
- Maggio, A.; Miyazaki, S.; Veronese, P.; Fujita, T.; Ibeas, J.I.; Damsz, B.; Narasimhan, M.L.; Hasegawa, P.M.; Joly, R.J.; Bressan, R.A. Does proline accumulation play an active role in stress-induced growth reduction? Plant J. 2002, 31, 699–712. [Google Scholar] [CrossRef] [Green Version]
- Claussen, W. Proline as a measure of stress in tomato plants. Plant Sci. 2005, 168, 241–248. [Google Scholar] [CrossRef]
- Kishor, P.B.K.; Sangam, S.; Amrutha, R.N.; Laxmi, P.S.; Naidu, K.R.; Rao, K.R.S.S.; Rao, S.; Reddy, K.J.; Theriappan, P.; Sreenivasulu, N. Regulation of proline biosynthesis, degradation, uptake and transport in higher plants: Its implications in plant growth and abiotic stress tolerance. Curr. Sci. India 2005, 88, 424–438. [Google Scholar]
- Ben Rejeb, K.; Abdelly, C.; Savoure, A. How reactive oxygen species and proline face stress together. Plant Physiol. Biochem. 2014, 80, 278–284. [Google Scholar] [CrossRef]
- Deeba, F.; Pandey, A.K.; Ranjan, S.; Mishra, A.; Singh, R.; Sharma, Y.K.; Shirke, P.A.; Pandey, V. Physiological and proteomic responses of cotton (Gossypium herbaceum L.) to drought stress. Plant Physiol. Biochem. 2012, 53, 6–18. [Google Scholar] [CrossRef]
- Horvath, E.; Szalai, G.; Janda, T. Induction of abiotic stress tolerance by salicylic acid signaling. J. Plant Growth Regul. 2007, 26, 290–300. [Google Scholar] [CrossRef]
- Moller, I.M.; Jensen, P.E.; Hansson, A. Oxidative modifications to cellular components in plants. Annu. Rev. Plant Biol. 2007, 58, 459–481. [Google Scholar] [CrossRef] [Green Version]
- Sharma, P.; Dubey, R.S. Drought induces oxidative stress and enhances the activities of antioxidant enzymes in growing rice seedlings. Plant Growth Regul. 2005, 46, 209–221. [Google Scholar] [CrossRef]
- Parida, A.K.; Das, A.B. Salt tolerance and salinity effects on plants: A review. Ecotoxicol. Environ. Saf. 2005, 60, 324–349. [Google Scholar] [CrossRef] [PubMed]
- Zorb, C.; Schmitt, S.; Muhling, K.H. Proteomic changes in maize roots after short-term adjustment to saline growth conditions. Proteomics 2010, 10, 4441–4449. [Google Scholar] [CrossRef] [PubMed]
- Mirzaei, M.; Soltani, N.; Sarhadi, E.; George, I.S.; Neison, K.A.; Pascovici, D.; Shahbazian, S.; Haynes, P.A.; Atwell, B.J.; Salekdeh, G.H. Manipulating Root Water Supply Elicits Major Shifts in the Shoot Proteome. J. Proteome Res. 2014, 13, 517–526. [Google Scholar] [CrossRef]
- Huang, S.B.; Greenway, H.; Colmer, T.D.; Millar, A.H. Protein synthesis by rice coleoptiles during prolonged anoxia: Implications for glycolysis, growth and energy utilization. Ann. Bot.-Lond. 2005, 96, 703–715. [Google Scholar] [CrossRef] [Green Version]
- Chitteti, B.R.; Peng, Z.H. Proteome and phosphoproteome differential expression under salinity stress in rice (Oryza sativa) roots. J. Proteome Res. 2007, 6, 1718–1727. [Google Scholar] [CrossRef]
- Wang, M.C.; Peng, Z.Y.; Li, C.L.; Li, F.; Liu, C.; Xia, G.M. Proteomic analysis on a high salt tolerance introgression strain of Triticum aestivum/Thinopyrum ponticum. Proteomics 2008, 8, 1470–1489. [Google Scholar] [CrossRef]
- Manaa, A.; Ben Ahmed, H.; Valot, B.; Bouchet, J.P.; Aschi-Smiti, S.; Causse, M.; Faurobert, M. Salt and genotype impact on plant physiology and root proteome variations in tomato. J. Exp. Bot. 2011, 62, 2797–2813. [Google Scholar] [CrossRef] [Green Version]
- Xu, C.P.; Sibicky, T.; Huang, B.R. Protein profile analysis of salt-responsive proteins in leaves and roots in two cultivars of creeping bentgrass differing in salinity tolerance. Plant Cell Rep. 2010, 29, 595–615. [Google Scholar] [CrossRef]
- Jiang, Y.; Yang, B.; Harris, N.S.; Deyholos, M.K. Comparative proteomic analysis of NaCl stress-responsive proteins in Arabidopsis roots. J. Exp. Bot. 2007, 58, 3591–3607. [Google Scholar] [CrossRef] [Green Version]
- Witzel, K.; Weidner, A.; Surabhi, G.K.; Borner, A.; Mock, H.P. Salt stress-induced alterations in the root proteome of barley genotypes with contrasting response towards salinity. J. Exp. Bot. 2009, 60, 3545–3557. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peng, Z.Y.; Wang, M.C.; Li, F.; Lv, H.J.; Li, C.L.; Xia, G.M. A Proteomic Study of the Response to Salinity and Drought Stress in an Introgression Strain of Bread Wheat. Mol. Cell Proteom. 2009, 8, 2676–2686. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, S.P.; Sauve, R.J.; Liu, Z.; Reddy, S.; Bhatti, S.; Hucko, S.D.; Fish, T.; Thannhauser, T.W. Identification of Salt-induced Changes in Leaf and Root Proteomes of the Wild Tomato, Solanum chilense. J. Am. Soc. Hortic. Sci. 2011, 136, 288–302. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Q.; Zhang, H.; Wang, T.; Chen, S.X.; Dai, S.J. Proteomics-based investigation of salt-responsive mechanisms in plant roots. J. Proteom. 2013, 82, 230–253. [Google Scholar] [CrossRef] [PubMed]
- Koh, J.; Chen, G.; Yoo, M.J.; Zhu, N.; Dufresne, D.; Erickson, J.E.; Shao, H.B.; Chen, S.X. Comparative Proteomic Analysis of Brassica napus in Response to Drought Stress. J. Proteome Res. 2015, 14, 3068–3081. [Google Scholar] [CrossRef]
- Xu, J.; Xing, X.J.; Tian, Y.S.; Peng, R.H.; Xue, Y.; Zhao, W.; Yao, Q.H. Transgenic Arabidopsis Plants Expressing Tomato Glutathione S-Transferase Showed Enhanced Resistance to Salt and Drought Stress. PLoS ONE 2015, 10, e0136960. [Google Scholar] [CrossRef]
- Rodrigues, S.M.; Andrade, M.O.; Gomes, A.P.S.; DaMatta, F.M.; Baracat-Pereira, M.C.; Fontes, E.P.B. Arabidopsis and tobacco plants ectopically expressing the soybean antiquitin-like ALDH7 gene display enhanced tolerance to drought, salinity, and oxidative stress. J. Exp. Bot. 2006, 57, 1909–1918. [Google Scholar] [CrossRef] [Green Version]
- Sunkar, R.; Bartels, D.; Kirch, H.H. Overexpression of a stress-inducible aldehyde dehydrogenase gene from Arabidopsis thaliana in transgenic plants improves stress tolerance. Plant J. 2003, 35, 452–464. [Google Scholar] [CrossRef]
- Zhang, H.; Han, B.; Wang, T.; Chen, S.X.; Li, H.Y.; Zhang, Y.H.; Dai, S.J. Mechanisms of Plant Salt Response: Insights from Proteomics. J. Proteome Res. 2012, 11, 49–67. [Google Scholar] [CrossRef]
- Singh, R.; Jwa, N.S. Understanding the Responses of Rice to Environmental Stress Using Proteomics. J. Proteome Res. 2013, 12, 4652–4669. [Google Scholar] [CrossRef]
- Kosova, K.; Vitamvas, P.; Urban, M.O.; Prasil, I.T.; Renaut, J. Plant Abiotic Stress Proteomics: The Major Factors Determining Alterations in Cellular Proteome. Front. Plant Sci. 2018, 9, 122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hand, S.C.; Menze, M.A.; Toner, M.; Boswell, L.; Moore, D. LEA Proteins During Water Stress: Not Just for Plants Anymore. Annu. Rev. Physiol. 2011, 73, 115–134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buchanan, C.D.; Lim, S.; Salzman, R.A.; Kagiampakis, I.; Morishige, D.T.; Weers, B.D.; Klein, R.R.; Pratt, L.H.; Cordonnier-Pratt, M.M.; Klein, P.E.; et al. Sorghum bicolor’s transcriptome response to dehydration, high salinity and ABA. Plant Mol. Biol. 2005, 58, 699–720. [Google Scholar] [CrossRef] [PubMed]
- Dugas, D.V.; Monaco, M.K.; Olsen, A.; Klein, R.R.; Kumari, S.; Ware, D.; Klein, P.E. Functional annotation of the transcriptome of Sorghum bicolor in response to osmotic stress and abscisic acid. BMC Genom. 2011, 12, 514. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnson, S.M.; Lim, F.L.; Finkler, A.; Fromm, H.; Slabas, A.R.; Knight, M.R. Transcriptomic analysis of Sorghum bicolor responding to combined heat and drought stress. BMC Genom. 2014, 15, 456. [Google Scholar] [CrossRef] [Green Version]
- Zhang, W.; Zhang, H.L.; Ning, L.Y.; Li, B.; Bao, M.Z. Quantitative Proteomic Analysis Provides Novel Insights into Cold Stress Responses in Petunia Seedlings. Front. Plant Sci. 2016, 7, 136. [Google Scholar] [CrossRef] [Green Version]
- Zhou, G.; Yang, L.T.; Li, Y.R.; Zou, C.L.; Huang, L.P.; Qiu, L.H.; Huang, X.; Srivastava, M.K. Proteomic Analysis of Osmotic Stress-Responsive Proteins in Sugarcane Leaves. Plant Mol. Biol. Rep. 2012, 30, 349–359. [Google Scholar] [CrossRef]
- Liu, C.W.; Hsu, Y.K.; Cheng, Y.H.; Yen, H.C.; Wu, Y.P.; Wang, C.S.; Lai, C.C. Proteomic analysis of salt-responsive ubiquitin-related proteins in rice roots. Rapid Commun. Mass Spectrom. 2012, 26, 1649–1660. [Google Scholar] [CrossRef]
- Yang, L.; Ma, C.Q.; Wang, L.L.; Chen, S.X.; Li, H.Y. Salt stress induced proteome and transcriptome changes in sugar beet monosomic addition line M14. J. Plant Physiol. 2012, 169, 839–850. [Google Scholar] [CrossRef]
- Pang, Q.Y.; Chen, S.X.; Dai, S.J.; Chen, Y.Z.; Wang, Y.; Yan, X.F. Comparative Proteomics of Salt Tolerance in Arabidopsis thaliana and Thellungiella halophila. J. Proteome Res. 2010, 9, 2584–2599. [Google Scholar] [CrossRef]
- Britto, D.T.; Kronzucker, H.J. NH4+ toxicity in higher plants: A critical review. J. Plant Physiol. 2002, 159, 567–584. [Google Scholar] [CrossRef] [Green Version]
- Jacoby, R.P.; Millar, A.H.; Taylor, N.L. Wheat Mitochondrial Proteomes Provide New Links between Antioxidant Defense and Plant Salinity Tolerance. J. Proteome Res. 2010, 9, 6595–6604. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, J.J.; Chen, S.X.; Zhao, Q.; Wang, T.; Yang, C.P.; Diaz, C.; Sun, G.R.; Dai, S.J. Physiological and Proteomic Analysis of Salinity Tolerance in Puccinellia tenuiflora. J. Proteome Res. 2011, 10, 3852–3870. [Google Scholar] [CrossRef] [PubMed]
- Ahn, C.S.; Lee, J.H.; Hwang, A.R.; Kim, W.T.; Pai, H.S. Prohibitin is involved in mitochondrial biogenesis in plants. Plant J. 2006, 46, 658–667. [Google Scholar] [CrossRef]
- Long, R.C.; Li, M.N.; Zhang, T.J.; Kang, J.M.; Sun, Y.; Cong, L.L.; Gao, Y.L.; Liu, F.Q.; Yang, Q.C. Comparative Proteomic Analysis Reveals Differential Root Proteins in Medicago sativa and Medicago truncatula in Response to Salt Stress. Model Legume Med. Truncatula 2016, 7, 1102–1111. [Google Scholar] [CrossRef] [Green Version]
- Horie, T.; Motoda, J.; Kubo, M.; Yang, H.; Yoda, K.; Horie, R.; Chan, W.Y.; Leung, H.Y.; Hattori, K.; Konomi, M.; et al. Enhanced salt tolerance mediated by AtHKT1 transporter-induced Na+ unloading from xylem vessels to xylem parenchyma cells. Plant. J. 2005, 44, 928–938. [Google Scholar]
- Bradford, M.M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976, 72, 248–254. [Google Scholar] [CrossRef]
- Beyer, W.F.; Fridovich, I. Assaying for superoxide dismutase activity: Some large consequences of minor changes in conditions. Anal. Biochem. 1987, 161, 559–566. [Google Scholar] [CrossRef]
- Kwak, S.S.; Kim, S.K.; Lee, M.S.; Jung, K.H.; Park, I.H.; Liu, J.R. Acidic Peroxidases from Suspension-Cultures of Sweet-Potato. Phytochemistry 1995, 39, 981–984. [Google Scholar] [CrossRef]
- Bates, L.; Waldren, R.; Teare, I. Rapid determination of free proline for water stress studies. Plant Soil 1973, 39, 205–207. [Google Scholar] [CrossRef]
- Wang, W.; Scali, M.; Vignani, R.; Spadafora, A.; Sensi, E.; Mazzuca, S.; Cresti, M. Protein extraction for two-dimensional electrophoresis from olive leaf, a plant tissue containing high levels of interfering compounds. Electrophoresis 2003, 24, 2369–2375. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Mock | PEG Treated | |
|---|---|---|
| Total number of spots | 906 | 653 |
| Replicates | 494 | |
| Up-regulated (Quality score > 80 and fold > 2.0) | 43 | |
| Up-regulated (Quality score < 80 and fold > 2.0) | 17 | |
| Down-regulated (Quality score > 80 and fold > 2.0) | 22 | |
| Down-regulated (Quality score < 80 and fold > 2.0) | 23 | |
| Other spots (fold < 2.0) | 389 |
| Spot No. (ssp) a | Protein Function b | Subcellular Location c | Mock Average Qty | PEG-6000 Treated Average Qty | Fold Change d |
|---|---|---|---|---|---|
| 0004 | abscisic acid-activated signaling pathway, defense response | cytoplasm, nucleus | 2417.1 ± 389.0 | 5547.8 ± 755.3 | 2.30 |
| 0007 | failed to identify spot | unknown | 384.5 ± 38.5 | 811.6 ± 47.9 | 2.11 |
| 0008 | superoxide metabolic process, toxin catabolic process, oxidation-reduction process | apoplast, chloroplast stroma, cytoplasm, extracellular space, thylakoid | 367.2 ± 15.4 | 766.1 ± 89.6 | 2.09 |
| 0010 | failed to identify | unknown | N | Y | ≥10 |
| 0114 | response to oxidative stress, hydrogen peroxide catabolic process | extracellular region or secreted | 408.3 ± 45.5 | 823.5 ± 65.1 | 2.02 |
| 0117 | failed to identify | unknown | 203.1 ± 11.2 | 527.9 ± 20.6 | 2.62 |
| 0212 | intracellular protein transport, membrane fusion | snare complex, vacuolar membrane | 375.6 ± 25.6 | 779.5 ± 46.0 | 2.08 |
| 0313 | racemase and epimerase activity, acting on amino acids and derivatives | unknown | 283.8 ± 23.7 | 570.5 ± 36.9 | 2.01 |
| 0508 | failed to identify | unknown | N | Y | ≥10 |
| 1104 | phosphorelay signal transduction system | intracellular | 145.2 ± 8.2 | 457.7 ± 37.0 | 3.15 |
| 1109 | transcription, transcription regulation | nucleus | 395.2 ± 7.7 | 831.7 ± 52.5 | 2.10 |
| 2001 | failed to identify | unknown | 837.8 ± 24.6 | 1780.5 ± 35.8 | 2.13 |
| 2004 | failed to identify | unknown | 345.7 ± 26.1 | 715.2 ± 65.3 | 2.07 |
| 2005 | protein biosynthesis | cytoplasm | 257.9 ± 17.6 | 1441.4 ± 95.9 | 5.59 |
| 2107 | actin binding | unknown | 165.8 ± 14.0 | 721.3 ± 64.6 | 4.35 |
| 2113 | failed to identify | unknown | 338.8 ± 14.3 | 686.9 ± 53.1 | 2.03 |
| 2210 | unknown | unknown | 182.5 ± 13.8 | 1803.4 ± 126.4 | 9.88 |
| 2602 | glucose catabolic process, cellular carbohydrate metabolic process | cytoplasm | 737.7 ± 23.1 | 1511.4 ± 153.0 | 2.05 |
| 2604 | glucose catabolic process, cellular carbohydrate metabolic process | cytoplasm | 1770.5 ± 104.4 | 3642.4 ± 446.6 | 2.06 |
| 3006 | protein biosynthesis | cytoplasm | 697.0 ± 12.2 | 1415.6 ± 65.7 | 2.03 |
| 3203 | glyoxylate cycle | cytoplasm | 130.5 ± 21.2 | 336.5 ± 46.0 | 2.58 |
| 3716 | carboxylyase activity, thiamine pyrophosphate | cytosol | 337.3 ± 65.4 | 1496.2 ± 85.5 | 4.44 |
| 4001 | posttranscriptional gene silencing by RNA, gene silencing by RNA | nucleus, cytoplasm, risc complex, | 186.8 ± 8.5 | 392.0 ± 18.2 | 2.10 |
| 4107 | fail to identify | unknown | 243.4 ± 19.1 | 494.5 ± 23.0 | 2.03 |
| 4403 | formaldehyde catabolic process | cytosol | 1132.8 ± 182.3 | 3760.6 ± 206.4 | 3.32 |
| 4511 | regulation of abscisic acid biosynthetic process | unknown | 143.5 ± 11.0 | 329.3 ± 31.9 | 2.29 |
| 4815 | glycosyltransferase, sucrose metabolic process | plasma membrane, vacuole | 149.0 ± 14.4 | 313.6 ± 31.7 | 2.10 |
| 5001 | nitrite reductase [NAD(P)H] activity, nitrate assimilation | chloroplast envelope, chloroplast thylakoid membrane | 312.5 ± 19.6 | 628.9 ± 64.1 | 2.01 |
| 5110 | failed to identify | unknown | 110.5 ± 6.2 | 222.2 ± 29.0 | 2.01 |
| 5516 | lipid metabolic process, lipid degradation | cytoplasm | 249.4 ± 11.6 | 506.6 ± 18.3 | 2.03 |
| 5608 | oxidoreductase activity, acting on the aldehyde or oxo group of donors, NAD or NADP as acceptor | cytoplasm, membrane | 82.2 ± 12.4 | 311.5 ± 56.9 | 3.79 |
| 6102 | ammonia assimilation cycle, glutamate biosynthetic process | cytoplasm | 111.4 ± 11.8 | 232.3 ± 30.6 | 2.09 |
| 6108 | response to stress | unknown | 171.1 ± 23.6 | 538.3 ± 61.9 | 3.15 |
| 6206 | glycolysis, fructose 6-phosphate metabolic process | cytoplasm | 158.5 ± 15.3 | 325.9 ± 16.7 | 2.06 |
| 6602 | glycolysis, fructose 6-phosphate metabolic process | cytoplasm | 385.3 ± 10.3 | 1041.5 ± 177.1 | 2.70 |
| 6608 | UDP-N-acetylglucosamine biosynthetic process | cytoplasm | 125.6 ± 16.8 | 253.9 ± 26.1 | 2.02 |
| 6815 | fail to identify | unknown | 42.3 ± 2.8 | 206.5 ± 13.6 | 4.90 |
| 7003 | fail to identify | unknown | 140.3 ± 16.9 | 281.4 ± 25.7 | 2.01 |
| 7507 | ribonucleoprotein, translation | small ribosomal subunit, chloroplast | N | Y | ≥10 |
| 7806 | oxylipin biosynthetic process, fatty acid biosynthesis, fatty acid metabolism, lipid biosynthesis, lipid metabolism | chloroplast | 240.1 ± 22.1 | 483.5 ± 32.3 | 2.01 |
| 8506 | oxidation-reduction process, response to oxidative stress, hydrogen peroxide catabolic process | peroxisome, plasma membrane, cytoplasm | 152.1 ± 17.1 | 1006.0 ± 24 | 6.60 |
| 8602 | response to drought stress | cytoplasm | N | Y | ≥10 |
| 8717 | unknown | unknown | 153.2 ± 14.9 | 1520.0 ± 133.9 | 9.92 |
| Spot No (ssp) a | Protein Function b | Subcellular Location c | Mock Average Qty | PEG-6000 Treated Average Qty | Fold Change d |
|---|---|---|---|---|---|
| 0403 | gluconeogenesis, glycolytic process | cytosol | 298.5 ± 17.2 | 145.3 ± 11.6 | 0.49 |
| 1903 | ubiquitin-dependent protein catabolic process | nucleus, cytoplasm | 395.2 ± 30.2 | 116.8 ± 12.1 | 0.30 |
| 2220 | cysteine biosynthetic, amino-acid biosynthesis | cytoplasm | 736.5 ± 71.1 | 357.3 ± 36.3 | 0.49 |
| 3010 | photosynthesis | extrinsic component of membrane, photosystem II oxygen evolving complex | Y | N | ≤0.1 |
| 3110 | protein serine/threonine phosphatase activity, protein dephosphorylation | cytoplasm | 219.3 ± 19.5 | 108.6 ± 13.0 | 0.50 |
| 4204 | amino-acid biosynthesis, cysteine biosynthesis | cytoplasm | 825.4 ± 82.6 | 401.7 ± 25.7 | 0.49 |
| 5412 | GDP-mannose 3,5-epimerase activity, steroid biosynthetic process, | cytoplasm | 2592.4 ± 135.8 | 1237.1 ± 45.5 | 0.48 |
| 5513 | pentose-phosphate shunt, gluconate utilization, oxidation-reduction process | cytosol | 817.1 ± 20.0 | 405.4 ± 37.4 | 0.50 |
| 6113 | glutamine metabolic process | cytosol | Y | N | ≤0.1 |
| 6207 | UDP-rhamnose and dTDP-rhamnose biosynthetic process | apoplast, cytosol, plasma membrane, plasmodesma | 411.5 ± 32.1 | 202.8 ± 10.9 | 0.49 |
| 6311 | protein biosynthesis | cytoplasm | 784.0 ± 79.3 | 382.1 ± 23.3 | 0.49 |
| 6702 | Hsp90 protein binding | cytoplasm | 2373.8 ± 285.7 | 1186.0 ± 171.2 | 0.50 |
| 6703 | oxidation-reduction process, stress response | chloroplast stroma, mitochondrion | 460.0 ± 26.3 | 225.5 ± 13.3 | 0.49 |
| 6704 | aspartyl-tRNA aminoacylation, protein biosynthesis | cytoplasm | 799.6 ± 74.3 | 396.2 ± 39.5 | 0.50 |
| 6816 | fail to identify | unknown | 719.1 ± 40.1 | 355.6 ± 24.1 | 0.49 |
| 7105 | fail to identify | unknown | 718.0 ± 46.2 | 355.8 ± 23.6 | 0.50 |
| 7115 | a lipid bilayer along with all the proteins and protein complexes embedded in it an attached to it | mitochondrion inner membrane | Y | N | ≤0.1 |
| 7215 | plant secondary metabolism | apoplast | Y | N | ≤0.1 |
| 7407 | regulation of RNA metabolic process | nucleolus | 663.8 ± 40.2 | 327.2 ± 13.9 | 0.49 |
| 7409 | oxidation-reduction process, cellular amino acid metabolic process | mitochondrion, vacuolar membrane | 1261.4 ± 181.5 | 281.9 ± 26.6 | 0.22 |
| 7705 | purine nucleobase biosynthetic process, 10-formyltetrahydrofolate biosynthetic process | cytoplasm | 335.4 ± 12.2 | 164.3 ± 2.3 | 0.49 |
| 8204 | siderophore biosynthetic process, oxidation-reduction process | cytosol | 907.6 ± 58.2 | 431.6 ± 25.7 | 0.48 |
| Spot No (ssp) a | Protein Identification b | Mascot Score c | Sequence Coverage (%) d | Estimated Mw(kDa)/PI e | Experimental Mw(kDa)/PI f | Gene/Locus g | Accession No h | Taxonomy i |
|---|---|---|---|---|---|---|---|---|
| 0004 | Bet_v_1 domain-containing protein | 137 | 76 | 17.1/5.05 | 15.6/5.3 | Sb01g037940 | XP_002468006 | Sorghum bicolor |
| 0008 | Superoxide dismutase (Cu-Zn SOD EC = 1.15.1.1) | 81 | 42 | 20.8/5.30 | 16.6/5.45 | Sb07g023950 | XP_002445671 | Sorghum bicolor |
| 0114 | Peroxidase (EC = 1.11.1.7) | 114 | 45 | 36.8/4.99 | 30.4/5.53 | Sb06g033850 | XP_002448823 | Sorghum bicolor |
| 0212 | Uncharacterized protein | 210 | 68 | 32.6/5.04 | 32.4/5.54 | Sb07g018430 | XP_002444250 | Sorghum bicolor |
| 0313 | MR_MLE domain-containing protein | 80 | 37 | 46.0/5.10 | 40.5/5.40 | Sb03g006670 | XP_002455229 | Sorghum bicolor |
| 0403 | Phosphoglycerate kinase (EC = 2.7.2.3) | 135 | 45 | 50.2/5.89 | 41.5/5.41 | Sb09g024340 | XP_002441313 | Sorghum bicolor |
| 1104 | Uncharacterized protein | 105 | 50 | 19.0/5.28 | 25.2/5.65 | N/A | ABK21830 | Picea sitchensis |
| 1109 | NAC domain-containing protein | 73 | 30 | 35.6/6.54 | 27.9/5.83 | Sb05g001590 | XP_002448920 | Sorghum bicolor |
| 1903 | UBA_e1_C domain-containing protein | 247 | 42 | 117.7/5.18 | 111.7/5.7 | Sb08g000540 | XP_002442655 | Sorghum bicolor |
| 2005 | Eukaryotic translation initiation factor 5A (eIF-5A) | 92 | 50 | 17.7/5.61 | 18.5/6.02 | TIF5A | NP_001105606 | Zea mays |
| 2107 | NAB domain-containing protein | 84 | 17 | 105.9/5.30 | 29.9/6.03 | Sb03g047500 | XP_002457032 | Sorghum bicolor |
| 2210 | Putative uncharacterized protein | 90 | 18 | 122.4/6.05 | 33.0/6.00 | Sb07g001185 | XP_002443741 | Sorghum bicolor |
| 2220 | Cysteine synthase | 186 | 59 | 42.3/8.48 | 34.7/5.9 | Sb03g009260 | XP_002457554 | Sorghum bicolor |
| 2602 | Uncharacterized protein | 134 | 43 | 60.4/5.41 | 64.8/5.92 | Sb03g038020 | KXG33615 | Sorghum bicolor |
| 2604 | Uncharacterized protein | 147 | 38 | 60.4/5.41 | 64.86/6.01 | Sb03g038020 | KXG33615 | Sorghum bicolor |
| 3006 | Eukaryotic translation initiation factor 5A(eIF-5A) | 117 | 59 | 17.7/5.61 | 19.0/6.30 | TIF5A | NP_001105606 | Zea mays |
| 3010 | PsbP domain-containing protein | 107 | 47 | 27.7/8.63 | 24.4/6.24 | Sb02g002690 | XP_002461438 | Sorghum bicolor |
| 3110 | Probable protein phosphatase 2C 76 isoform X2 | 86 | 31 | 39.4/5.55 | 30.4/6.13 | LOC105115317 | XP_011010457 | Populus euphratica |
| 3203 | Uncharacterized protein | 98 | 41 | 33.5/5.43 | 34.1/6.16 | Sb06g013750 | XP_002447699 | Sorghum bicolor |
| 3716 | Uncharacterized protein | 187 | 52 | 66.4/5.67 | 69/6.30 | Sb01g038360 | XP_002465414 | Sorghum bicolor |
| 4001 | Ribonuclease | 104 | 20 | 100/6.82 | 15.4/6.38 | Sb04g021280 | XP_002453908 | Sorghum bicolor |
| 4204 | Cysteine synthase | 96 | 36 | 28.3/5.86 | 35.6/6.4 | Sb08g022280 | KXG24064 | Sorghum bicolor |
| 4403 | Uncharacterized protein | 149 | 67 | 41.9/5.72 | 43.9/6.43 | Sb05g009350 | XP_002449392 | Sorghum bicolor |
| 4511 | Uncharacterized protein | 156 | 53 | 44.9/5.72 | 49.5/6.52 | Sb01g043260 | XP_002465659 | Sorghum bicolor |
| 4815 | Sucrose synthase EC = 2.4.1.13 | 111 | 18 | 92.1/5.82 | 94.2/6.50 | Sb10g006330 | ACM69042 | Sorghum bicolor |
| 5001 | Rieske-like [2Fe-2S] domain containing protein | 75 | 30 | 29.8/5.49 | 17.1/6.62 | RADI4G21270 | XP_003577663 | Brachypodium distachyon |
| 5412 | Epimerase domain-containing protein | 138 | 39 | 43.3/5.94 | 46.8/6.6 | Sb01g021890 | XP_002467242 | Sorghum bicolor |
| 5513 | 6-phosphogluconate dehydrogenase, decarboxylating (EC = 1.1.1.44)000 | 97 | 23 | 54.3/6.00 | 53.6/6.67 | Sb05g016740 | XP_002449496 | Sorghum bicolor |
| 5516 | Patatin (EC:3.1.1.-) | 78 | 22 | 47.0/6.27 | 49.0/6.70 | Sb07g023210 | XP_002445639 | Sorghum bicolor |
| 5608 | Mitochondrial aldehyde dehydrogenase | 97 | 23 | 59.5/6.65 | 57.9/6.67 | Sb10g009790 | BAB92019 | Sorghum bicolor |
| 6102 | Glutamine amidotransferase type-2 domain-containing protein | 74 | 9 | 235.8/5.49 | 29.8/6.73 | Sb03g031310 | XP_002458326 | Sorghum bicolor |
| 6108 | Uncharacterized protein | 126 | 64 | 29.7/8.33 | 25.8/6.81 | Sb01g039360 | XP_002465466 | Sorghum bicolor |
| 6113 | DUF3700 domain-containing protein | 112 | 47 | 28.2/6.95 | 28.8/6.77 | Sb06g033080 | XP_002448783 | Sorghum bicolor |
| 6206 | Pyrophosphate--fructose 6-phosphate 1-phosphotransferase subunit beta | 110 | 29 | 61.7/6.07 | 32.1/6.81 | Sb10g008850 | XP_002438147 | Sorghum bicolor |
| 6207 | RmlD_sub_bind domain-containing protein | 106 | 46 | 34.2/6.15 | 33.6/6.82 | Sb04g031900 | XP_002454480 | Sorghum bicolor |
| 6311 | Eukaryotic translation initiation factor 3 subunit I(eIF3i) | 131 | 47 | 36.3/6.06 | 40.8/6.89 | Sb02g003760 | XP_002461510 | Sorghum bicolor |
| 6602 | Pyrophosphate--fructose 6-phosphate 1-phosphotransferase subunit beta | 239 | 58 | 61.7/6.07 | 66.6/6.73 | Sb10g008850 | XP_002438147 | Sorghum bicolor |
| 6608 | Uncharacterized protein | 152 | 45 | 54.8/6.08 | 59.9/6.81 | Sb06g028340 | KXG27103 | Sorghum bicolor |
| 6702 | Uncharacterized protein | 283 | 64 | 65.4/6.07 | 80/6.75 | Sb04g033510 | XP_002454564 | Sorghum bicolor |
| 6703 | Pyr_redox_2 domain-containing protein | 74 | 26 | 51.6/8.57 | 76.7/6.77 | Sb07g003280 | KXG24413 | Sorghum bicolor |
| 6704 | Aspartate--tRNA ligase 2 cytoplasmic | 163 | 31 | 61.2/5.92 | 70.9/6.79 | LOC100279253 | NP_001145746 | Zea mays |
| 7115 | Prohibitin | 101 | 44 | 30.7/6.55 | 30.8/7.06 | Sb06g019110 | XP_002447973 | Sorghum bicolor |
| 7215 | Dirigent protein | 126 | 47 | 32.6/6.75 | 31.6/7.06 | Sb09g001880 | XP_002439182 | Sorghum bicolor |
| 7407 | Heterogeneous nuclear ribonucleoprotein 1 | 86 | 30 | 44.2/6.17 | 47.0/7.01 | LOC100382683 | NP_001168878 | Zea mays |
| 7409 | Glutamate dehydrogenase | 146 | 48 | 44.8/6.24 | 72.6/7.03 | Sb06g024150 | XP_002446878 | Sorghum bicolor |
| 7507 | Small ribosomal protein 4 (rps4) | 77 | 47 | 23.0/10.9 | 48.5/7.26 | rps4 | AAL26212 | Pinus thunbergii |
| 7705 | Uncharacterized protein | 119 | 28 | 81.0/8.50 | 74.1/7.15 | Sb02g026140 | OQU89595 | Sorghum bicolor |
| 7806 | Lipoxygenase (EC = 1.13.11.58) | 109 | 24 | 100.6/6.26 | 104.6/6.85 | Sb01g011040 | XP_002466613 | Sorghum bicolor |
| 8204 | Aldo_ket_red domain-containing protein | 203 | 61 | 39.6/8.44 | 33.8/7.4 | Sb01g041640 | XP_002468202 | Sorghum bicolor |
| 8506 | Catalase EC = 1.11.1.6 | 193 | 54 | 57.3/6.62 | 52.5/7.43 | Sb10g030840 | XP_002437631 | Sorghum bicolor |
| 8602 | LEA-like protein | 94 | 42 | 24.2/4.82 | 66.9/7.4 | LP28 | BAB97392 | Lilium longiflorum |
| 8717 | Uncharacterized protein | 153 | 33 | 81.5/6.82 | 79.0/7.68 | Sb06g000930 | XP_002446045 | Sorghum bicolor |
| Categories | Classification Basis | Protein Spot Serial Number (Total 52) |
|---|---|---|
| Biological function | Energy and Carbohydrate metabolism | 2602, 2604, 3203, 3716, 4815, 6206, 6602, 6608 0403, 3010, 5513, 6207 |
| Protein synthesis/processing/degradation | 1903, 3110, 6311, 6704, 0212, 2005, 3006, 7507 | |
| Antioxidant and defense response | 0008, 0114, 6108, 8506, 8602, 4403, 5608, 6702, 6703, 8204 | |
| Amino acid biosynthesis | 0313, 2220, 4204, 6113, 7409 7705 | |
| Transcription and regulation | 1109, 4001, 7407 | |
| Nitrogen metabolism | 5001, 6102 | |
| Signal transduction | 0004, 1104 | |
| Lipid membrane metabolism | 5412, 7115, 5516, 7806 | |
| Other functional categories | 2107, 4511, 8717, 7215, 2210 | |
| Subcellular localization | Cytoplasm | 0004, 0008, 1903, 2005, 2220, 2602, 2604, 3006, 3110, 3203, 4001, 4204, 5412, 5516, 5608, 6102, 6206, 6602, 6608, 6704, 7705, 8602 |
| Cytosol | 0403, 3716, 4403, 5513, 6113, 6207, 8204 | |
| Chloroplast | 0008, 5001, 7507, 7806 | |
| Chloroplast thylakoid membrane | 0008, 5001 | |
| Nucleus | 0004, 1109, 1903, 4001, 7407 | |
| Vacuolar membrane | 0212, 4815, 7409 | |
| Plasma membrane | 3010, 4815, 5608, 8506, 6207 | |
| Ribosome | 7507 | |
| Extracellular | 0008, 0114, 2210, 4511, 6108 | |
| Intracellular | 1104 | |
| Peroxisome | 8506 | |
| Chloroplast stroma | 6703 | |
| Mitochondrion | 6703, 7115, 7409 | |
| Apoplast | 6207, 7215 | |
| Uncharacterized | 0313, 2107, 8717 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, H.; Li, Y.; Ke, Q.; Kwak, S.-S.; Zhang, S.; Deng, X. Physiological and Differential Proteomic Analyses of Imitation Drought Stress Response in Sorghum bicolor Root at the Seedling Stage. Int. J. Mol. Sci. 2020, 21, 9174. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21239174
Li H, Li Y, Ke Q, Kwak S-S, Zhang S, Deng X. Physiological and Differential Proteomic Analyses of Imitation Drought Stress Response in Sorghum bicolor Root at the Seedling Stage. International Journal of Molecular Sciences. 2020; 21(23):9174. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21239174
Chicago/Turabian StyleLi, Hongbing, Yulin Li, Qingbo Ke, Sang-Soo Kwak, Suiqi Zhang, and Xiping Deng. 2020. "Physiological and Differential Proteomic Analyses of Imitation Drought Stress Response in Sorghum bicolor Root at the Seedling Stage" International Journal of Molecular Sciences 21, no. 23: 9174. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21239174