The Role of β-Arrestins in Regulating Stem Cell Phenotypes in Normal and Tumorigenic Cells


Abstract
:1. Introduction
2. The Role of β-Arrestins (ARRBs) in Normal Stem Cell Maintenance
2.1. Embryonic Stem Cells
2.2. Hematopoietic Stem/Progenitor Cells
2.3. Neural Precursors
2.4. Mesenchymal Stem Cells
2.5. Cardiac Progenitors
2.6. Myoblast Cells
3. The Role of β-Arrestins (ARRBs) in the Maintenance of Cancer Stem Cells
3.1. Bladder Cancer
3.2. Non-Small Cell Lung Cancer
3.3. Leukemia
3.4. Medulloblastoma
4. Targeting β-Arrestins (ARRBs) as a Therapeutic Strategy
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| ARRB1 | β-Arrestin1 |
| ARRB2 | β-Arrestin2 |
| CSC | Cancer stem cells |
| ESC | Embryonic stem cells |
| GPCR | G-protein-coupled receptor |
References
- Ma, L.; Pei, G. β-arrestin signaling and regulation of transcription. J. Cell Sci. 2007, 120, 213–218. [Google Scholar] [CrossRef] [Green Version]
- Buchanan, F.G.; Dubois, R.N. Emerging Roles of β-Arrestins. Cell Cycle 2006, 5, 2060–2063. [Google Scholar] [CrossRef] [Green Version]
- DeWire, S.M.; Ahn, S.; Lefkowitz, R.J.; Shenoy, S.K. β-Arrestins and Cell Signaling. Annu. Rev. Physiol. 2007, 69, 483–510. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sobolesky, P.M.; Moussa, O. The Role of β-Arrestins in Cancer. Prog. Mol. Biol. Transl. Sci. 2013, 118, 395–411. [Google Scholar] [PubMed]
- Vazin, T.; Freed, W.J. Human embryonic stem cells: Derivation, culture, and differentiation: A review. Restor. Neurol. Neurosci. 2010, 28, 589–603. [Google Scholar] [CrossRef] [PubMed]
- Ohtsuka, S.; Dalton, S. Molecular and biological properties of pluripotent embryonic stem cells. Gene Ther. 2007, 15, 74–81. [Google Scholar] [CrossRef]
- Donati, C.; Cencetti, F.; Nincheri, P.; Bernacchioni, C.; Brunelli, S.; Clementi, E.; Cossu, G.; Bruni, P. Sphingosine 1-Phosphate Mediates Proliferation and Survival of Mesoangioblasts. Stem Cells 2007, 25, 1713–1719. [Google Scholar] [CrossRef]
- Kleger, A.; Busch, T.; Liebau, S.; Prelle, K.; Paschke, S.; Beil, M.; Rolletschek, A.; Wobus, A.; Wolf, E.; Adler, G.; et al. The bioactive lipid sphingosylphosphorylcholine induces differentiation of mouse embryonic stem cells and human promyelocytic leukaemia cells. Cell. Signal. 2007, 19, 367–377. [Google Scholar] [CrossRef]
- Wong, R.C.; Tellis, I.; Jamshidi, P.; Pera, M.F.; Pébay, A. Anti-Apoptotic Effect of Sphingosine-1-Phosphate and Platelet-Derived Growth Factor in Human Embryonic Stem Cells. Stem Cells Dev. 2007, 16, 989–1002. [Google Scholar] [CrossRef]
- Ryu, J.M.; Bin Baek, Y.; Shin, M.S.; Park, J.H.; Park, S.H.; Lee, J.H.; Han, H.J. Sphingosine-1-phosphate-induced Flk-1 transactivation stimulates mouse embryonic stem cell proliferation through S1P1/S1P3-dependent β-arrestin/c-Src pathways. Stem Cell Res. 2014, 12, 69–85. [Google Scholar] [CrossRef] [Green Version]
- Freedman, N.J.; Shenoy, S.K. Regulation of inflammation by β-arrestins: Not just receptor tales. Cell. Signal. 2018, 41, 41–45. [Google Scholar] [CrossRef] [PubMed]
- McDonald, P.H. β-Arrestin 2: A Receptor-Regulated MAPK Scaffold for the Activation of JNK3. Science 2000, 290, 1574–1577. [Google Scholar] [CrossRef] [PubMed]
- Yue, R.; Kang, J.; Zhao, C.; Hu, W.; Tang, Y.; Liu, X.; Pei, G. β-Arrestin1 Regulates Zebrafish Hematopoiesis through Binding to YY1 and Relieving Polycomb Group Repression. Cell 2009, 139, 535–546. [Google Scholar] [CrossRef] [Green Version]
- Torossian, F.; Anginot, A.; Chabanon, A.; Clay, D.; Guerton, B.; Desterke, C.; Boutin, L.; Marullo, S.; Scott, M.G.; Lataillade, J.-J.; et al. CXCR7 participates in CXCL12-induced CD34+ cell cycling through β-arrestin–dependent Akt activation. Blood 2014, 123, 191–202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chabanon, A.; Desterke, C.; Rodenburger, E.; Clay, D.; Guerton, B.; Boutin, L.; Bennaceur-Griscelli, A.; Pierre-Louis, O.; Uzan, G.; Abecassis, L.; et al. A Cross-Talk Between Stromal Cell-Derived Factor-1 and Transforming Growth Factor-β Controls the Quiescence/Cycling Switch of CD34+Progenitors Through FoxO3 and Mammalian Target of Rapamycin. Stem Cells 2008, 26, 3150–3161. [Google Scholar] [CrossRef] [PubMed]
- Fereshteh, M.; Ito, T.; Kovacs, J.J.; Zhao, C.; Kwon, H.Y.; Tornini, V.; Konuma, T.; Chen, M.; Lefkowitz, R.J.; Reya, T. β-Arrestin2 mediates the initiation and progression of myeloid leukemia. Proc. Natl. Acad. Sci. USA 2012, 109, 12532–12537. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, J.; Shi, Y.; Xiang, B.; Qu, B.; Su, W.; Zhu, M.; Zhang, M.; Bao, G.; Wang, F.; Zhang, X.; et al. A Nuclear Function of β-Arrestin1 in GPCR Signaling: Regulation of Histone Acetylation and Gene Transcription. Cell 2005, 123, 833–847. [Google Scholar] [CrossRef] [Green Version]
- Miyazawa, K.; Himi, T.; Garcia, V.; Yamagishi, H.; Sato, S.; Ishizaki, Y. A Role for p27/Kip1 in the Control of Cerebellar Granule Cell Precursor Proliferation. J. Neurosci. 2000, 20, 5756–5763. [Google Scholar] [CrossRef]
- Parathath, S.R.; Mainwaring, L.A.; Fernandez-L, A.; Guldal, C.G.; Nahlé, Z.; Kenney, A.M. β-Arrestin-1 links mitogenic sonic hedgehog signaling to the cell cycle exit machinery in neural precursors. Cell Cycle 2010, 9, 4013–4024. [Google Scholar] [CrossRef] [Green Version]
- Tao, Y.; Ma, L.; Liao, Z.; Le, Q.; Yu, J.; Liu, X.; Li, H.; Chen, Y.; Zheng, P.; Yang, Z.; et al. Astroglial β-Arrestin1-mediated Nuclear Signaling Regulates the Expansion of Neural Precursor Cells in Adult Hippocampus. Sci. Rep. 2015, 5, 15506. [Google Scholar] [CrossRef] [Green Version]
- Mira, H.; Andreu, Z.; Suh, H.; Lie, D.C.; Jessberger, S.; Consiglio, A.; Emeterio, J.S.; Hortigüela, R.; Marqués-Torrejón, M.Á.; Nakashima, K.; et al. Signaling through BMPR-IA Regulates Quiescence and Long-Term Activity of Neural Stem Cells in the Adult Hippocampus. Cell Stem Cell 2010, 7, 78–89. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pittenger, M.F.; Mackay, A.M.; Beck, S.C.; Jaiswal, R.K.; Douglas, R.; Mosca, J.D.; Moorman, M.A.; Simonetti, D.W.; Craig, S.; Marshak, D.R. Multilineage Potential of Adult Human Mesenchymal Stem Cells. Science 1999, 284, 143–147. [Google Scholar] [CrossRef] [Green Version]
- Muraglia, A.; Cancedda, R.; Quarto, R. Clonal mesenchymal progenitors from human bone marrow differentiate in vitro according to a hierarchical model. J. Cell Sci. 2000, 113, 1161–1166. [Google Scholar] [PubMed]
- North, T.E.; Goessling, W.; Walkley, C.R.; Lengerke, C.; Kopani, K.R.; Lord, A.M.; Weber, G.J.; Bowman, T.V.; Jang, I.-H.; Grosser, T.; et al. Prostaglandin E2 regulates vertebrate haematopoietic stem cell homeostasis. Nature 2007, 447, 1007–1011. [Google Scholar] [CrossRef] [PubMed]
- Sugimoto, Y.; Narumiya, S. Prostaglandin E Receptors. J. Biol. Chem. 2007, 282, 11613–11617. [Google Scholar] [CrossRef] [Green Version]
- Jimenez, P.; Piazuelo, E.; Cebrian, C.; Ortego, J.; Strunk, M.; Garciaa-Gonzalez, M.A.; Santander, S.; Alcedo, J.; Lanas, A. Prostaglandin EP2 receptor expression is increased in Barrett’s oesophagus and oesophageal adenocarcinoma. Aliment. Pharmacol. Ther. 2010, 31, 440–451. [Google Scholar] [CrossRef] [PubMed]
- Yun, S.P.; Ryu, J.M.; Jang, M.W.; Han, H.J. Interaction of profilin-1 and F-actin via a β-arrestin-1/JNK signaling pathway involved in prostaglandin E2-induced human mesenchymal stem cells migration and proliferation. J. Cell. Physiol. 2011, 226, 559–571. [Google Scholar] [CrossRef]
- Liang, S.X.; Phillips, W.D. Migration of Resident Cardiac Stem Cells in Myocardial Infarction. Anat. Rec. Adv. Integr. Anat. Evol. Biol. 2012, 296, 184–191. [Google Scholar] [CrossRef]
- Le, T.; Chong, J.J. Cardiac progenitor cells for heart repair. Cell Death Discov. 2016, 2, 16052. [Google Scholar] [CrossRef] [Green Version]
- Bergmann, O.; Bhardwaj, R.D.; Bernard, S.; Zdunek, S.; Barnabé-Heider, F.; Walsh, S.; Zupicich, J.; Alkass, K.; Buchholz, B.A.; Druid, H.; et al. Evidence for Cardiomyocyte Renewal in Humans. Science 2009, 324, 98–102. [Google Scholar] [CrossRef] [Green Version]
- Attramadal, H.; Arriza, J.L.; Aoki, C.; Dawson, T.M.; Codina, J.; Kwatra, M.M.; Snyder, S.H.; Caron, M.G.; Lefkowitz, R.J. Beta-arrestin2, a novel member of the arrestin/β-arrestin gene family. J. Biol. Chem. 1992, 267, 17882–17890. [Google Scholar]
- Seo, S.-K.; Kim, N.; Lee, J.-H.; Kim, S.M.; Lee, S.Y.; Bae, J.-W.; Hwang, K.-K.; Kim, D.-W.; Koch, W.J.; Cho, M.-C. β-arrestin2 Affects Cardiac Progenitor Cell Survival through Cell Mobility and Tube Formation in Severe Hypoxia. Korean Circ. J. 2018, 48, 296–309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, J.; Feng, Y.; Yan, H.; Chen, Y.; Wang, J.; Chua, B.; Stuart, C.; Yin, D. β-arrestin2/miR-155/GSK3β regulates transition of 5′-azacytizine-induced Sca-1-positive cells to cardiomyocytes. J. Cell. Mol. Med. 2014, 18, 1562–1570. [Google Scholar] [CrossRef]
- Chal, J.; Pourquie, O. Making muscle: Skeletal myogenesis in vivo and in vitro. Development 2017, 144, 2104–2122. [Google Scholar] [CrossRef] [Green Version]
- Gurriarán-Rodríguez, U.; Santos-Zas, I.; Al Massadi, O.; Mosteiro, C.S.; Beiroa, D.; Nogueiras, R.; Crujeiras, A.B.; Seoane, L.M.; Señaris, J.; Garcia-Caballero, T.; et al. The Obestatin/GPR39 System Is Up-regulated by Muscle Injury and Functions as an Autocrine Regenerative System. J. Biol. Chem. 2012, 287, 38379–38389. [Google Scholar] [CrossRef] [Green Version]
- Gurriarán-Rodríguez, U.; Santos-Zas, I.; González-Sánchez, J.; Beiroa, D.; Moresi, V.; Mosteiro, C.S.; Lin, W.; E Viñuela, J.; Señarís, J.; García-Caballero, T.; et al. Action of Obestatin in Skeletal Muscle Repair: Stem Cell Expansion, Muscle Growth, and Microenvironment Remodeling. Mol. Ther. 2015, 23, 1003–1021. [Google Scholar] [CrossRef] [Green Version]
- Santos-Zas, I.; Gurriarán-Rodríguez, U.; Cid-Díaz, T.; Figueroa, G.; González-Sánchez, J.; Bouzo-Lorenzo, M.; Mosteiro, C.S.; Señaris, J.; Casanueva, F.F.; Casabiell, J.; et al. β-Arrestin scaffolds and signaling elements essential for the obestatin/GPR39 system that determine the myogenic program in human myoblast cells. Cell. Mol. Life Sci. 2015, 73, 617–635. [Google Scholar] [CrossRef]
- Goodwin, J.G.; Willis, D.; Kamat, A. Bladder Cancer Stem Cells: Biological and Therapeutic Perspectives. Curr. Stem Cell Res. Ther. 2014, 9, 89–101. [Google Scholar]
- Acar, O.; Özkurt, E.; Demir, G.; Sarac, H.; Alkan, C.; Esen, T.; Somel, M.; Lack, N.A. Determining the origin of synchronous multifocal bladder cancer by exome sequencing. BMC Cancer 2015, 15, 871. [Google Scholar] [CrossRef] [Green Version]
- Ferreira-Teixeira, M.; Parada, B.; Rodrigues-Santos, P.; Alves, V.; Ramalho, J.S.; Caramelo, F.; Sousa, V.; Reis, F.; Gomes, C. Functional and molecular characterization of cancer stem-like cells in bladder cancer: A potential signature for muscle-invasive tumors. Oncotarget 2015, 6, 36185–36201. [Google Scholar] [CrossRef] [Green Version]
- Kallifatidis, G.; Smith, D.K.; Morera, D.S.; Gao, J.; Hennig, M.J.; Hoy, J.J.; Pearce, R.F.; Dabke, I.R.; Li, J.; Merseburger, A.S.; et al. β-Arrestins Regulate Stem Cell-Like Phenotype and Response to Chemotherapy in Bladder Cancer. Mol. Cancer Ther. 2019, 18, 801–811. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Su, Y.; Qiu, Q.; Zhang, X.; Jiang, Z.; Leng, Q.; Liu, Z.; Stass, S.A.; Jiang, F. Aldehyde Dehydrogenase 1 A1-Positive Cell Population Is Enriched in Tumor-Initiating Cells and Associated with Progression of Bladder Cancer. Cancer Epidemiol. Biomarkers Prev. 2010, 19, 327–337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chan, K.S.; Espinosa, I.; Chao, M.; Wong, D.; Ailles, L.; Diehn, M.; Gill, H.; Presti, J.; Chang, H.Y.; Van De Rijn, M.; et al. Identification, molecular characterization, clinical prognosis, and therapeutic targeting of human bladder tumor-initiating cells. Proc. Natl. Acad. Sci. USA 2009, 106, 14016–14021. [Google Scholar] [CrossRef] [Green Version]
- Zhu, D.; Wan, X.; Huang, H.; Chen, X.; Liang, W.; Zhao, F.; Lin, T.; Han, J.; Xie, W. Knockdown of Bmi1 inhibits the stemness properties and tumorigenicity of human bladder cancer stem cell-like side population cells. Oncol. Rep. 2013, 31, 727–736. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lokeshwar, B.L.; Lokeshwar, V.B.; Block, N.L. Expression of CD44 in prostate cancer cells: Association with cell proliferation and invasive potential. Anticancer. Res. 1995, 15, 1191–1998. [Google Scholar] [PubMed]
- Yang, Y.M.; Chang, J.W. Bladder Cancer Initiating Cells (BCICs) Are Among EMA−CD44v6+Subset: Novel Methods for Isolating Undetermined Cancer Stem (Initiating) Cells. Cancer Investig. 2008, 26, 725–733. [Google Scholar] [CrossRef]
- Ho, P.L.; Lay, E.J.; Jian, W.; Parra, D.; Chan, K.S. Stat3 Activation in Urothelial Stem Cells Leads to Direct Progression to Invasive Bladder Cancer. Cancer Res. 2012, 72, 3135–3142. [Google Scholar] [CrossRef] [Green Version]
- Yang, Z.; He, L.; Lin, K.; Zhang, Y.; Deng, A.; Liang, Y.; Li, C.; Wen, T. The KMT1A-GATA3-STAT3 Circuit Is a Novel Self-Renewal Signaling of Human Bladder Cancer Stem Cells. Clin. Cancer Res. 2017, 23, 6673–6685. [Google Scholar] [CrossRef] [Green Version]
- Pan, Q.; Yang, G.-L.; Yang, J.-H.; Lin, S.-L.; Liu, N.; Liu, S.-S.; Liu, M.-Y.; Zhang, L.-H.; Huang, Y.; Shen, R.-L.; et al. Metformin can block precancerous progression to invasive tumors of bladder through inhibiting STAT3-mediated signaling pathways. J. Exp. Clin. Cancer Res. 2015, 34, 77. [Google Scholar] [CrossRef] [Green Version]
- Wicha, M.S. Targeting self-renewal, an Achilles’ heel of cancer stem cells. Nat. Med. 2014, 20, 14–15. [Google Scholar] [CrossRef]
- Wang, M.-C.; Li, C.-L.; Cui, J.; Jiao, M.; Wu, T.; Jing, L.; Nan, K.-J. BMI-1, a promising therapeutic target for human cancer. Oncol. Lett. 2015, 10, 583–588. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gotti, C.; Clementi, F. Neuronal nicotinic receptors: From structure to pathology. Prog. Neurobiol. 2004, 74, 363–396. [Google Scholar] [CrossRef] [PubMed]
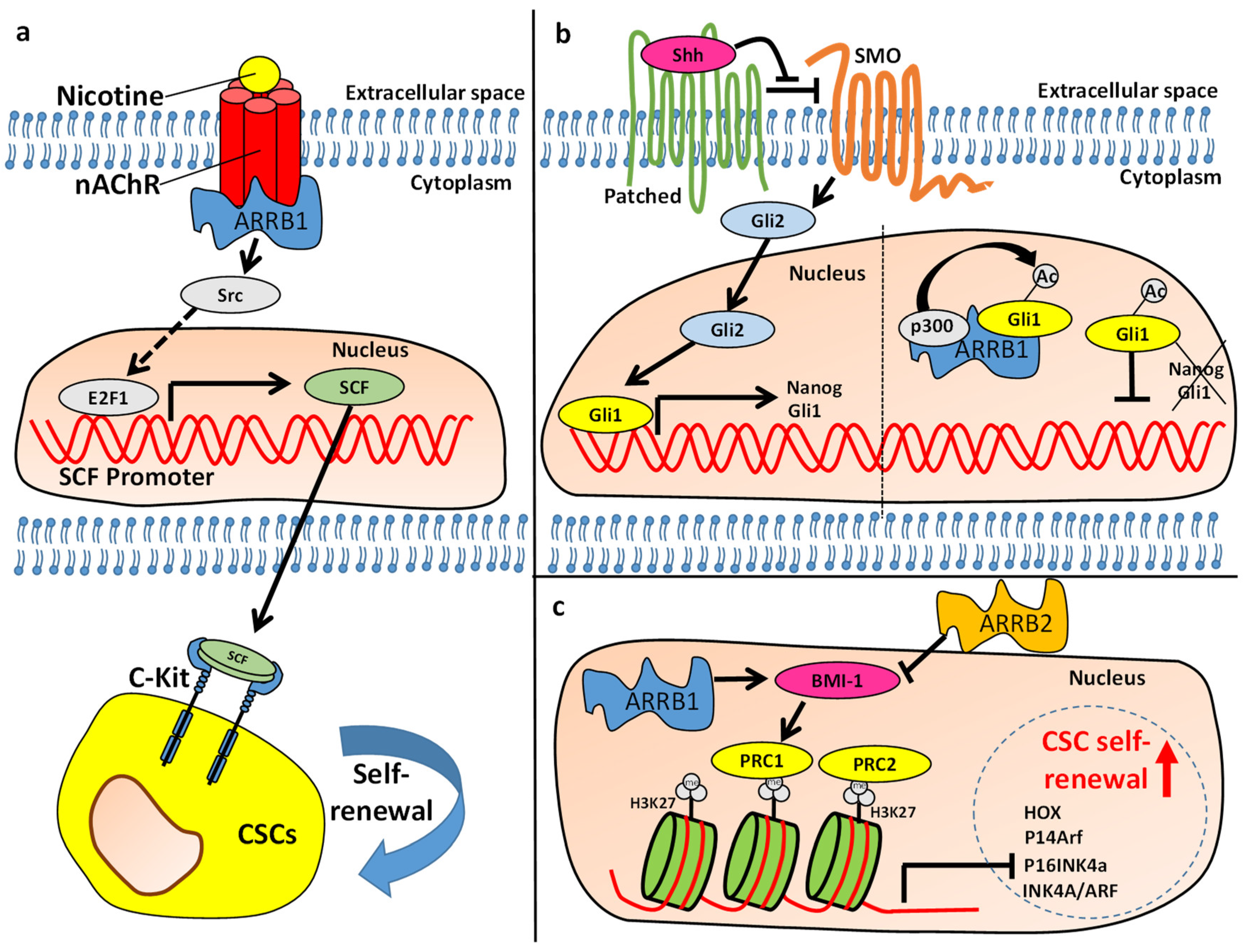
- Dasgupta, P.; Rastogi, S.; Pillai, S.; Ordonez-Ercan, D.; Morris, M.; Haura, E.; Chellappan, S. Nicotine induces cell proliferation by -arrestin-mediated activation of Src and Rb-Raf-1 pathways. J. Clin. Investig. 2006, 116, 2208–2217. [Google Scholar] [CrossRef] [PubMed]
- Schaal, C.; Chellappan, S. Nicotine-Mediated Cell Proliferation and Tumor Progression in Smoking-Related Cancers. Mol. Cancer Res. 2014, 12, 14–23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perumal, D.; Pillai, S.; Nguyen, J.; Schaal, C.; Coppola, D.; Chellappan, S. Nicotinic acetylcholine receptors induce c-Kit ligand/Stem Cell Factor and promote stemness in an ARRB1/ β-arrestin-1 dependent manner in NSCLC. Oncotarget 2014, 5, 10486–10502. [Google Scholar] [CrossRef] [Green Version]
- Singh, S.; Bora-Singhal, N.; Kroeger, J.; Laklai, H.; Chellappan, S. βArrestin-1 and Mcl-1 Modulate Self-Renewal Growth of Cancer Stem-Like Side-Population Cells in Non-Small Cell Lung Cancer. PLoS ONE 2013, 8, e55982. [Google Scholar] [CrossRef] [PubMed]
- Bryja, V.; Gradl, D.; Schambony, A.; Arenas, E.; Schulte, G. β-Arrestin is a necessary component of Wnt/β-catenin signaling in vitro and in vivo. Proc. Natl. Acad. Sci. USA 2007, 104, 6690–6695. [Google Scholar] [CrossRef] [Green Version]
- Nusse, R. Wnt signaling in disease and in development. Cell Res. 2005, 15, 28–32. [Google Scholar] [CrossRef] [Green Version]
- Qin, R.; Li, K.; Qi, X.; Zhou, X.; Wang, L.; Zhang, P.; Zou, L. β-Arrestin1 promotes the progression of chronic myeloid leukaemia by regulating BCR/ABL H4 acetylation. Br. J. Cancer 2014, 111, 568–576. [Google Scholar] [CrossRef] [Green Version]
- Kleer, C.G.; Cao, Q.; Varambally, S.; Shen, R.; Ota, I.; Tomlins, S.A.; Ghosh, D.; Sewalt, R.G.A.B.; Otte, A.P.; Hayes, D.F.; et al. EZH2 is a marker of aggressive breast cancer and promotes neoplastic transformation of breast epithelial cells. Proc. Natl. Acad. Sci. USA 2003, 100, 11606–11611. [Google Scholar] [CrossRef] [Green Version]
- Strahl, B.D.; Allis, C.D. The language of covalent histone modifications. Nature 2000, 403, 41–45. [Google Scholar] [CrossRef] [PubMed]
- Shu, Y.; Zhou, X.; Qi, X.; Liu, S.; Li, K.; Tan, J.; Liu, Z.; Yu, J.; Zhang, P.; Zou, L. β-Arrestin1 promotes the self-renewal of the leukemia-initiating cell-enriched subpopulation in B-lineage acute lymphoblastic leukemia related to DNMT1 activity. Cancer Lett. 2015, 357, 170–178. [Google Scholar] [CrossRef] [PubMed]
- Roman-Gomez, J.; Jimenez-Velasco, A.; Castillejo, J.A.; Agirre, X.; Barrios, M.; Navarro, G.; Molina, F.J.; Calasanz, M.J.; Prósper, F.; Heiniger, A.; et al. Promoter hypermethylation of cancer-related genes: A strong independent prognostic factor in acute lymphoblastic leukemia. Blood 2004, 104, 2492–2498. [Google Scholar] [CrossRef] [PubMed]
- Román-Gómez, J.; Jiménez-Velasco, A.; Barrios, M.; Prósper, F.; Heiniger, A.; Torres, A.; Agirre, X. Poor prognosis in acute lymphoblastic leukemia may relate to promoter hypermethylation of cancer-related genes. Leuk. Lymphoma 2007, 48, 1269–1282. [Google Scholar] [CrossRef] [PubMed]
- Okano, M.; Bell, D.W.; A Haber, D.; Li, E. DNA Methyltransferases Dnmt3a and Dnmt3b Are Essential for De Novo Methylation and Mammalian Development. Cell 1999, 99, 247–257. [Google Scholar] [CrossRef] [Green Version]
- Rhee, I.; Bachman, K.E.; Park, B.H.; Jair, K.-W.; Yen, R.-W.C.; Schuebel, K.E.; Cui, H.; Feinberg, A.P.; Lengauer, C.; Kinzler, K.W.; et al. DNMT1 and DNMT3b cooperate to silence genes in human cancer cells. Nature 2002, 416, 552–556. [Google Scholar] [CrossRef]
- Viré, E.; Brenner, C.; Deplus, R.; Blanchon, L.; Fraga, M.; Didelot, C.; Morey, L.; Van Eynde, A.; Bernard, D.; Vanderwinden, J.-M.; et al. The Polycomb group protein EZH2 directly controls DNA methylation. Nature 2006, 439, 871–874. [Google Scholar] [CrossRef]
- Liu, S.; Liu, H.; Qin, R.; Shu, Y.; Liu, Z.; Zhang, P.; Duan, C.; Hong, D.; Yu, J.; Zou, L. The cellular senescence of leukemia-initiating cells from acute lymphoblastic leukemia is postponed by β-Arrestin1 binding with P300-Sp1 to regulate hTERT transcription. Cell Death Dis. 2017, 8, e2756. [Google Scholar] [CrossRef] [Green Version]
- Clarke, M.F.; Dick, J.E.; Dirks, P.B.; Eaves, C.J.; Jamieson, C.H.; Jones, D.L.; Visvader, J.; Weissman, I.L.; Wahl, G.M. Cancer Stem Cells—Perspectives on Current Status and Future Directions: AACR Workshop on Cancer Stem Cells. Cancer Res. 2006, 66, 9339–9344. [Google Scholar] [CrossRef] [Green Version]
- Sangkhae, V.; Etheridge, S.L.; Kaushansky, K.; Hitchcock, I.S. The thrombopoietin receptor, MPL, is critical for development of a JAK2V617F-induced myeloproliferative neoplasm. Blood 2014, 124, 3956–3963. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rein, L.A.; Wisler, J.W.; Kim, J.; Theriot, B.; Huang, L.; Price, T.; Yang, H.; Chen, M.; Chen, W.; Sipkins, D.A.; et al. β-Arrestin2 mediates progression of murine primary myelofibrosis. JCI Insight 2017, 2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kotula, J.W.; Sun, J.; Li, M.; Pratico, E.D.; Fereshteh, M.P.; Ahrens, D.P.; Sullenger, B.A.; Kovacs, J.J. Targeted Disruption of β-Arrestin 2-Mediated Signaling Pathways by Aptamer Chimeras Leads to Inhibition of Leukemic Cell Growth. PLoS ONE 2014, 9, e93441. [Google Scholar] [CrossRef] [PubMed] [Green Version]
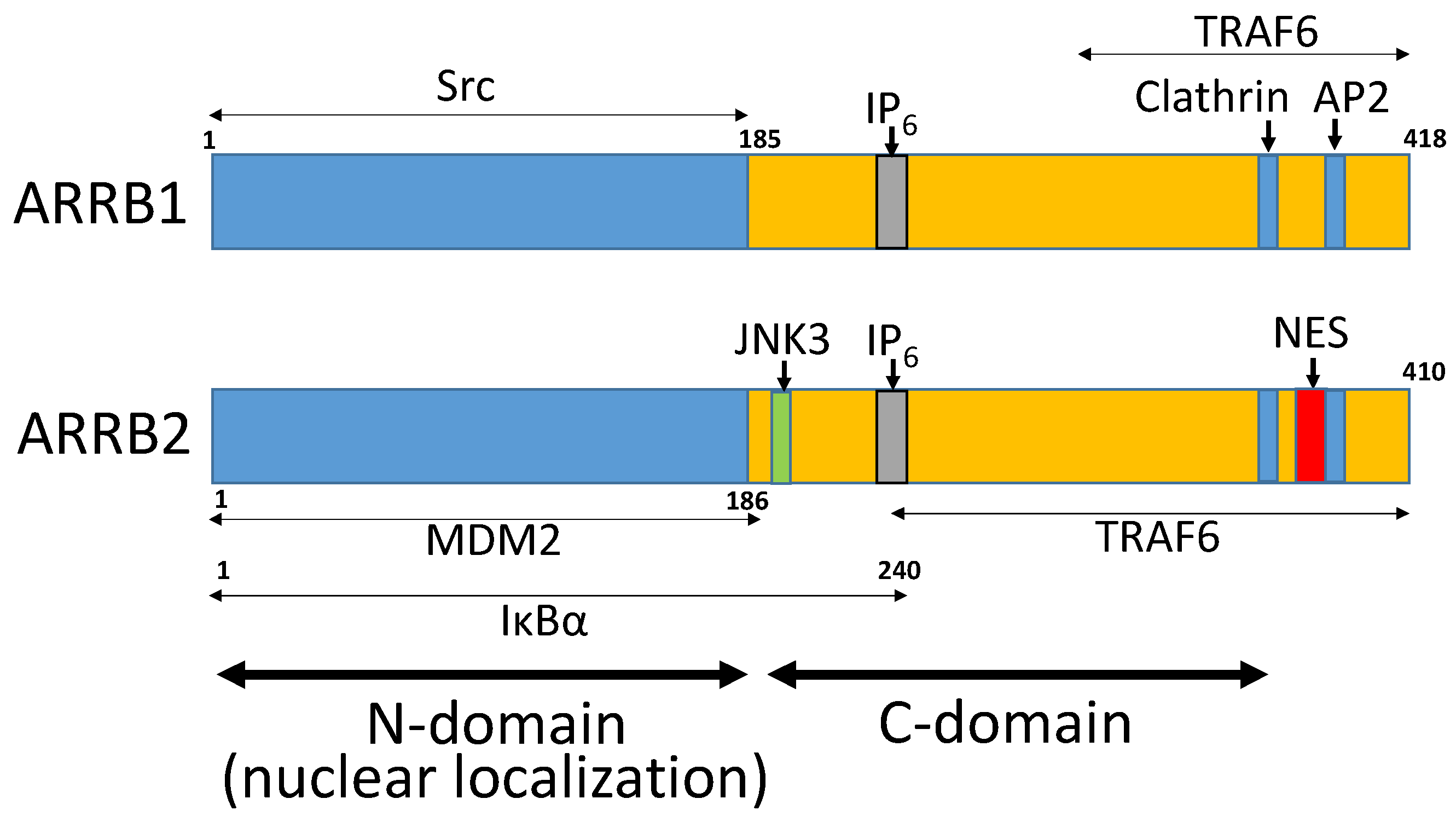
- Scott, M.G.; Le Rouzic, E.; Périanin, A.; Pierotti, V.; Enslen, H.; Benichou, S.; Marullo, S.; Benmerah, A. Differential Nucleocytoplasmic Shuttling of β-Arrestins. J. Biol. Chem. 2002, 277, 37693–37701. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, P.; Wu, Y.; Ge, X.; Ma, L.; Pei, G. Subcellular Localization of β-Arrestins Is Determined by Their Intact N Domain and the Nuclear Export Signal at the C Terminus. J. Biol. Chem. 2003, 278, 11648–11653. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bahmad, H.F.; Poppiti, R. Medulloblastoma cancer stem cells: Molecular signatures and therapeutic targets. J. Clin. Pathol. 2020, 73, 243–249. [Google Scholar] [CrossRef]
- Po, A.; Abballe, L.; Sabato, C.; Gianno, F.; Chiacchiarini, M.; Catanzaro, G.; De Smaele, E.; Giangaspero, F.; Ferretti, E.; Miele, E.; et al. Sonic Hedgehog Medulloblastoma Cancer Stem Cells Mirnome and Transcriptome Highlight Novel Functional Networks. Int. J. Mol. Sci. 2018, 19, 2326. [Google Scholar] [CrossRef] [Green Version]
- Louis, D.N.; Perry, A.; Reifenberger, G.; Von Deimling, A.; Figarella-Branger, D.; Cavenee, W.K.; Ohgaki, H.; Wiestler, O.D.; Kleihues, P.; Ellison, D.W. The 2016 World Health Organization Classification of Tumors of the Central Nervous System: A summary. Acta Neuropathol. 2016, 131, 803–820. [Google Scholar] [CrossRef] [Green Version]
- Miele, E.; Po, A.; Begalli, F.; Antonucci, L.; Mastronuzzi, A.; Marras, C.E.; Carai, A.; Cucchi, D.; Abballe, L.; Besharat, Z.M.; et al. β-arrestin1-mediated acetylation of Gli1 regulates Hedgehog/Gli signaling and modulates self-renewal of SHH medulloblastoma cancer stem cells. BMC Cancer 2017, 17, 488. [Google Scholar] [CrossRef] [Green Version]
- Shukla, A.K.; Dwivedi-Agnihotri, H. Structure and function of β-arrestins, their emerging role in breast cancer, and potential opportunities for therapeutic manipulation. Adv. Cancer Res. 2020, 145, 139–156. [Google Scholar]
- Beautrait, A.; Paradis, J.S.; Zimmerman, B.; Giubilaro, J.; Nikolajev, L.; Armando, S.; Kobayashi, H.; Yamani, L.; Namkung, Y.; Heydenreich, F.M.; et al. A new inhibitor of the β-arrestin/AP2 endocytic complex reveals interplay between GPCR internalization and signalling. Nat. Commun. 2017, 8, 15054. [Google Scholar] [CrossRef]
- Ghosh, E.; Srivastava, A.; Baidya, M.; Kumari, P.; Dwivedi, H.; Nidhi, K.; Ranjan, R.; Dogra, S.; Koide, A.; Yadav, P.N.; et al. A synthetic intrabody-based selective and generic inhibitor of GPCR endocytosis. Nat. Nanotechnol. 2017, 12, 1190–1198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McMahon, C.; Baier, A.S.; Pascolutti, R.; Wegrecki, M.; Zheng, S.; Ong, J.X.; Erlandson, S.C.; Hilger, D.; Rasmussen, S.G.F.; Ring, A.M.; et al. Yeast surface display platform for rapid discovery of conformationally selective nanobodies. Nat. Struct. Mol. Biol. 2018, 25, 289–296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Michel, M.; Charlton, S.J. Biased Agonism in Drug Discovery—Is It Too Soon to Choose a Path? Mol. Pharmacol. 2018, 93, 259–265. [Google Scholar] [CrossRef] [PubMed] [Green Version]


{kind=link}
{kind=link}
{kind=link}
| Cancer | Role of ARRB1 | Role of ARRB2 | Reference No. |
|---|---|---|---|
| Bladder | Stimulates growth and stem-cell phenotype/CSC-self-renewal | Inhibits growth and CSC phenotype, inhibits motility and invasion | [41] |
| Non-small cell lung cancer | Promotes nicotine-mediated self-renewal of CSCs | - | [55] |
| Myeloproliferative neoplasms (e.g., leukemia) | Inhibits senescence, promotes self-renewal of CSCs, promotes progression | Anti-apoptotic effects, promotes CSC maintenance, mediates initiation and progression of disease | [16,59,62,68,71] |
| Medulloblastoma | Inhibits self-renewal of CSCs | - | [78] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kallifatidis, G.; Mamouni, K.; Lokeshwar, B.L. The Role of β-Arrestins in Regulating Stem Cell Phenotypes in Normal and Tumorigenic Cells. Int. J. Mol. Sci. 2020, 21, 9310. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21239310
Kallifatidis G, Mamouni K, Lokeshwar BL. The Role of β-Arrestins in Regulating Stem Cell Phenotypes in Normal and Tumorigenic Cells. International Journal of Molecular Sciences. 2020; 21(23):9310. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21239310
Chicago/Turabian StyleKallifatidis, Georgios, Kenza Mamouni, and Bal L. Lokeshwar. 2020. "The Role of β-Arrestins in Regulating Stem Cell Phenotypes in Normal and Tumorigenic Cells" International Journal of Molecular Sciences 21, no. 23: 9310. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21239310