Autophagy—A Hidden but Important Actor on Oral Cancer Scene

 ,
,
Abstract
:1. Introduction
2. Oral Cancer
3. Autophagy
- (1)
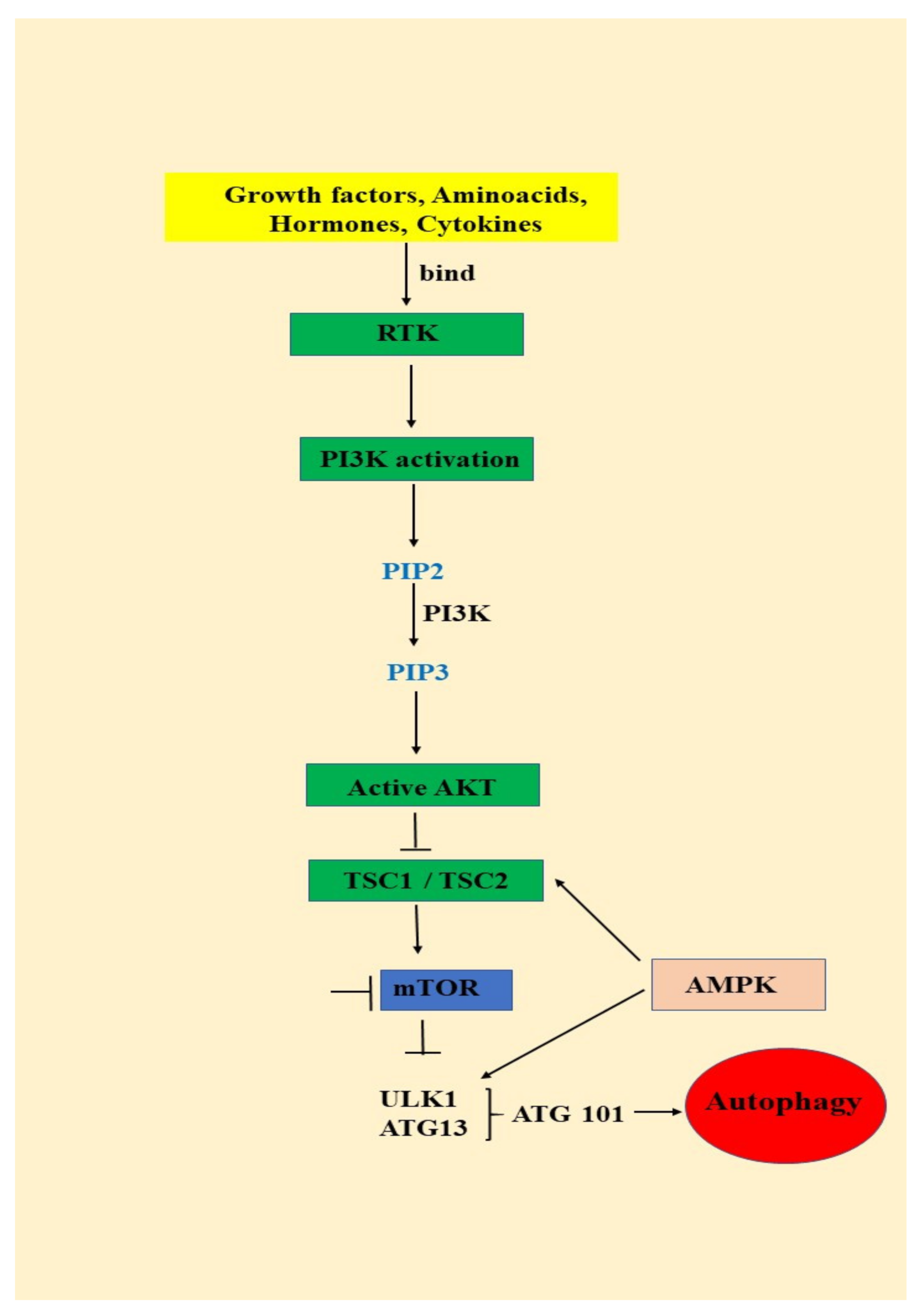
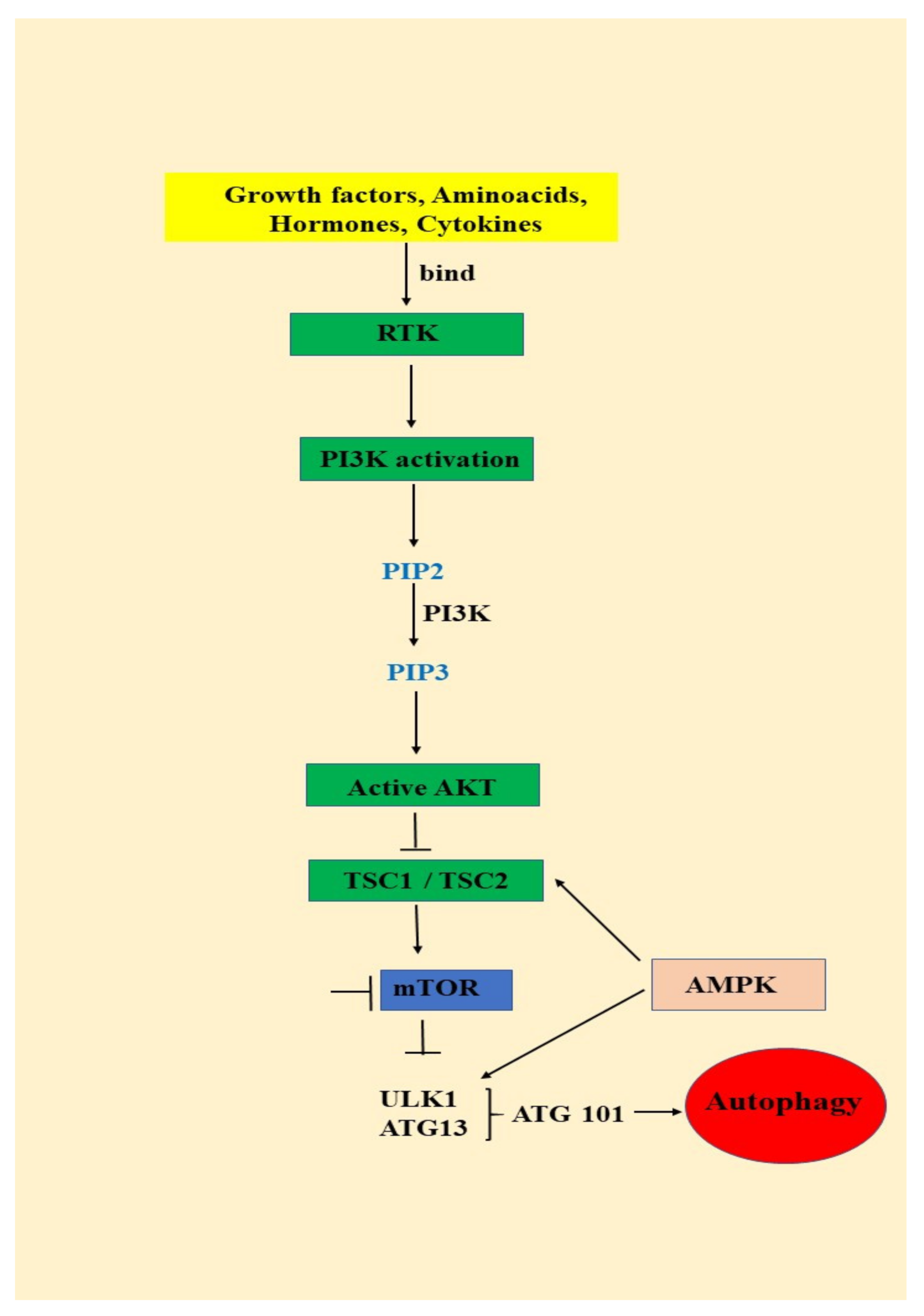
- signals such as starvation activate the ULK complex, which will bind to the PtdIns3K complex following AMPK activation or mTOR suppression [35];
- (2)
- following induction, the ULK complex, PtdIns3K complex and the ATG9 complex orchestrated action will trigger the phagophore assembly at the phagophore assembly site [35];
- (3)
- ATG12 and LC3 conjugation systems are key players in regulating the phagophore elongation to the autophagosome. mTOR, the major autophagy inhibitory factor, suppresses autophagy as response to abundant nutrients conditions. This suppressive action is mediated by class I PI3K and AKT signaling [35];
- (4)
- SQSTM1/p62 (sequestosome 1) receptor protein will consequently interact with both LC3 and ubiquitin chains [35];
- (5)
- Further, the autophagosome will fuse with a lysosome, resulting the autolysosome formation. Inside autolysosome, the autophagosome constituents will be hydrolytically degraded. The trapped SQSTM1 will be degraded in the autolysosome, which highlight SQSTM1′s role as an autophagy flux marker [35].
3.1. Autophagy—An Important AKT/mTOR Pathway Target
3.2. Autophagy—Important Actor on Oral Cancers Scene
3.2.1. Oncogenes and Tumor Suppressors that Control the Autophagy Pathway
3.2.2. Autophagy Regarded as a Tumor Suppressor
Autophagic Cell Death
Autophagic Senescence
Inflammation
Oxidative Stress and Genomic Instability
3.2.3. Autophagy Regarded as a Tumor Growth Promoter
- -
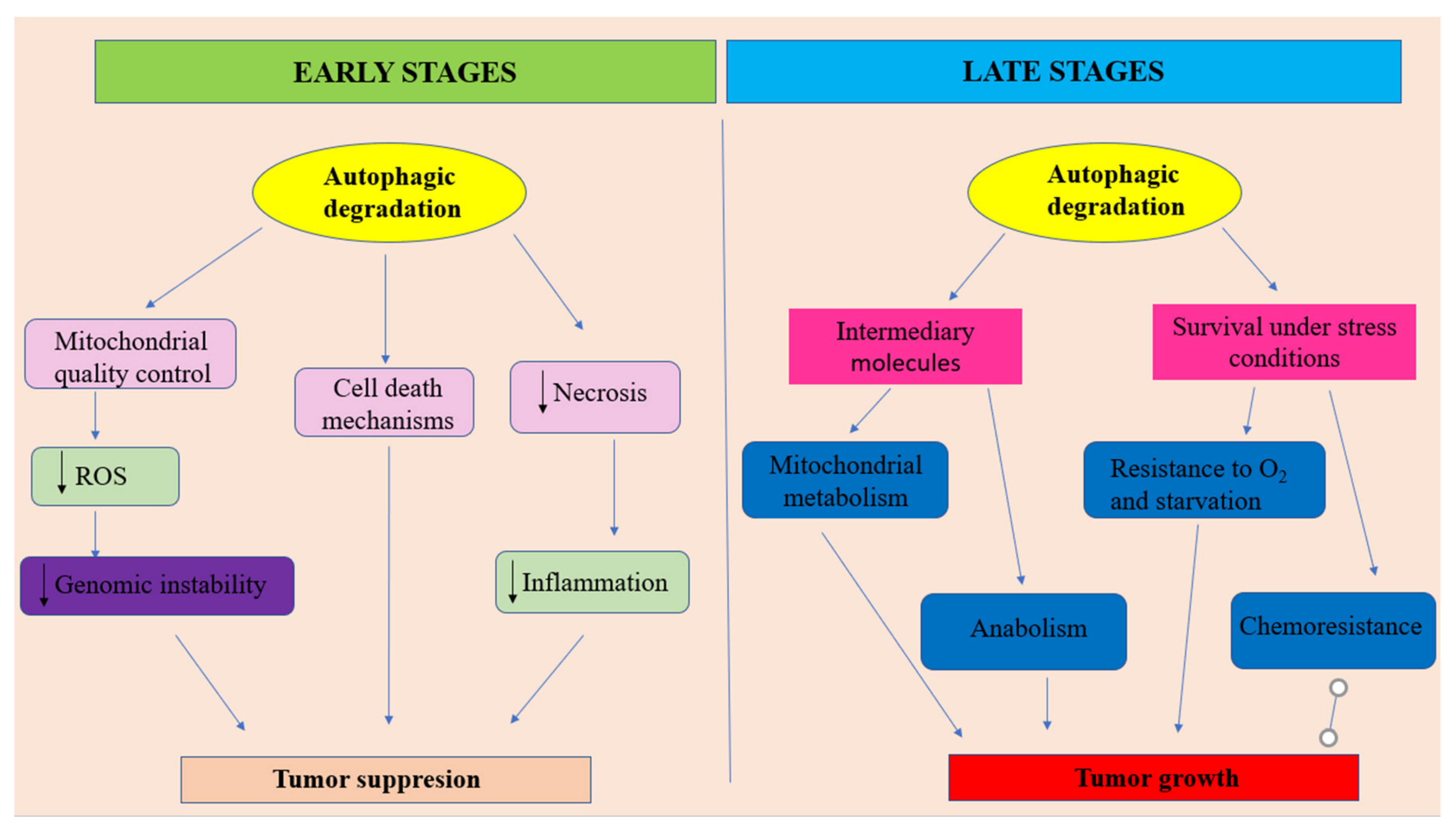
- at early stages of tumor development, autophagy plays the role of a tumor suppressor by ensuring damaged proteins and organelles degradation. In this context, autophagy should be regarded as controlling system, able to decreases ROS production and, consequently, maintaining genomic stability. Autophagy also can prevent necrotic cell death in apoptosis-defective cells, ensuring in this way the decrease of local inflammation and tumor growth. In some situations autophagy can lead to apoptotic cell death.
- -
- at later stages of tumor evolution, activated autophagy plays the role of cancer cell survival and tumor growth promoter, by suppling metabolic stressed tumor cells with nutrients, in order to sustain energy generation in mitochondria and biosynthetic pathways. Unfortunately, autophagy represents one of the main actors in developing the resistance to cancer therapy. Adapted from [100].
3.2.4. Autophagy Related Chemoresistance in Oral Cancer
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Jainkittivong, A.; Aneksuk, V.; Langlais, R.P. Oral Mucosal Conditions in Elderly Dental Patients. Oral Dis. 2002, 8, 218–223. [Google Scholar] [CrossRef] [PubMed]
- Gendreau, L.; Loewy, Z.G. Epidemiology and etiology of den-ture stomatitis. J. Prosthodont. 2011, 20, 251–260. [Google Scholar] [CrossRef] [PubMed]
- Bozdemir, E.; Yilmaz, H.H.; Orhan, H. Oral mucosal lesions and risk factors in elderly dental patients. J. Dent. Res. Dent. Clin. Dent. Prospect. 2019, 13, 24–30. [Google Scholar] [CrossRef] [PubMed]
- Ercalik-Yalcinkaya, S.; Özcan, M. Association between Oral Mucosal Lesions and Hygiene Habits in a Population of Re-movable Prosthesis Wearers. J. Prosthodont. 2015, 24, 271–278. [Google Scholar] [CrossRef]
- Dundar, N.; Ilhan Kal, B. Oral Mucosal Conditions and Risk Factors among Elderly in a Turkish School of Dentistry. Gerontology 2007, 53, 165–172. [Google Scholar] [CrossRef]
- Del Corso, G.; Villa, A.; Tarsitano, A.; Gohel, A. Current trends in oral cancer: A review. Cell Microenviron. 2016, 3, e1332. [Google Scholar]
- Mizushima, N.; Levine, B. Autophagy in mammalian development and differentiation. Nat. Cell Biol. 2010, 12, 823–830. [Google Scholar] [CrossRef]
- Yang, Z.; Klionsky, D.J. Eatenalive: A history of macroautophagy. Nat. Cell Biol. 2010, 12, 814–822. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.; Xia, P.; Rehm, M.; Fan, Z. Autophagy and cell reprogram-ming. Cell. Mol. Life Sci. 2015, 72, 1699–1713. [Google Scholar] [CrossRef]
- Rivera, C. Essentials of oral cancer. Int. J. Clin. Exp. Pathol. 2015, 8, 11884–11894. [Google Scholar]
- Montero, P.H.; Patel, S.G. Cancer of the oral cavity. Surg. Oncol. Clin. N. Am. 2015, 24, 491–508. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: Globocan estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, M.; Nanavati, R.; Modi, T.G.; Dobariya, C. Oral cancer: Etiology and risk factors: A review. J. Cancer Res. 2016, 12, 458–463. [Google Scholar] [CrossRef] [PubMed]
- Ong, T.K.; Murphy, C.; Smith, A.B.; Kanatas, A.N.; Mitchell, D.A. Survival after surgery for oral cancer: A 30-year experience. Br. J. Oral Maxillofac. Surg. 2017, 55, 911–916. [Google Scholar] [CrossRef] [PubMed]
- Bais, M.V. Impact of Epigenetic Regulation on Head and Neck Squamous Cell Carcinoma. J. Dent. Res. 2019, 98, 268–276. [Google Scholar] [CrossRef] [PubMed]
- Kaur, J.; Jacobs, R.; Huang, Y.; Salvo, N.; Politis, C. Salivary biomarkers for oral cancer and pre-cancer screening: A review. Clin. Oral Investig. 2018, 22, 633–640. [Google Scholar] [CrossRef] [PubMed]
- Valdez, J.A.; MAS, M.T.B. Impact of oral cancer on quality of life. Oral Cancer Issue Dent. Clin. N. Am. 2017, 62, 143–154. [Google Scholar] [CrossRef]
- D’souza, S.; Addepalli, V. Preventive measures in oral cancer: An overview. Biomed. Pharm. 2018, 107, 72–80. [Google Scholar] [CrossRef]
- Neville, B.W. Oral cancer and precancerous lesions. Fogorv. Szle. 2010, 52, 195–215. [Google Scholar] [CrossRef]
- Lee, Y.C.; Hashibe, M. Tobacco, alcohol, and cancer in low and high income countries. Ann. Glob. Health 2014, 80, 378–383. [Google Scholar]
- Jethwa, A.R.; Khariwala, S.S. Tobacco-related carcinogenesis in head and neck cancer. Cancer Metastasis Rev. 2017, 36, 411–423. [Google Scholar] [CrossRef]
- Gandini, S.E.; Botteri, S.; Iodice, M.; Boniol, A.B.; Lowenfels, P.; Maisonneuve, P.; Boyle, P. Tobacco smoking and cancer: A meta-analysis. Int. J. Cancer 2008, 122, 155–164. [Google Scholar] [CrossRef] [PubMed]
- Choudhari, S.K.; Chaudhary, M.; Gadbail, A.R.; Sharma, A.; Tekade, S. Oxidative and antioxidative mechanisms in oral cancer and precancer: A review. Oral Oncol. 2014, 50, 10–18. [Google Scholar] [CrossRef] [PubMed]
- Boffetta, P.; Hashibe, M. Alcohol and cancer. Lancet Oncol. 2006, 7, 149–156. [Google Scholar] [CrossRef]
- Gaonkar, P.P.; Patankar, S.R.; Tripathi, N.; Sridharan, G. Oral bacterial flora and oral cancer: The possible link? J. Oral Maxillofac. Pathol. 2018, 22, 234–238. [Google Scholar] [CrossRef] [PubMed]
- Candotto, V.D.; Lauritano, M.; Nardone, L.; Baggi, C.; Arcuri, R.; Gatto, R.M.; Gaudio, F.; Spadari, F.; Carinci, F. HPV infection in the oral cavity: Epidemiology, clinical manifestations and relationship with oral cancer. Oral Implant. 2017, 10, 209–220. [Google Scholar] [CrossRef] [PubMed]
- Sritippho, T.P.; Chotjumlong, A.; Iamaroon, A. Roles of Human Papillomaviruses and p16 in Oral Cancer. Asian Pac. J. Cancer Prev. 2015, 16, 6193–6200. [Google Scholar] [CrossRef] [Green Version]
- Lazos, J.P.; Piemonte, E.D.; Lanfranchi, H.E.; Brunotto, M.N. Characterization of Chronic Mechanical Irritation in Oral Cancer. Int. J. Dent. 2017, 2017, 6784526. [Google Scholar] [CrossRef]
- Piemonte, E.J.; Lazos, P.; Belardinelli, D.; Secchi, M.; Brunotto, H.; Lanfranchi-Tizeira, H. Oral cancer associated with chronic mechanical irritation of the oral mucosa. Med. Oral Patol. Oral Cir. Bucal. 2018, 23, e151–e160. [Google Scholar] [CrossRef]
- Yardimci, G.Z.; Kutlubay, B.; Engin, Y.; Tuzun, Y. Precancerous lesions of oral mucosa. World J. Clin. Cases 2014, 2, 866–872. [Google Scholar] [CrossRef]
- Al-Jaber, A.; Al-Nasser, L.; El-Metwally, A. Epidemiology of oral cancer in Arab countries. Saudi Med. J. 2016, 37, 249–255. [Google Scholar] [CrossRef] [PubMed]
- Mohan, M.; Jagannathan, N. Oral field cancerization: An update on current concepts. Oncol. Rev. 2014, 8, 244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paderno, A.R.; Morello, C.; Piazza, C. Tongue carcinoma in young adults: A review of the literature. Acta Otorhinolaryngol. Ital. 2018, 38, 175–180. [Google Scholar] [PubMed]
- Kerawala, C.; Roques, T.; Jeannon, J.P.; Bisase, B. Oral cavity and lip cancer: United Kingdom National Multidisciplinary Guidelines. J. Laryngol. Otol. 2016, 130, S83–S89. [Google Scholar] [CrossRef]
- Tan, Y.Q.; Zhang, J.; Zhou, G. Autophagy and its implication in human oral diseases. Autophagy 2017, 13, 225–236. [Google Scholar] [CrossRef] [Green Version]
- Mauthe, M.; Reggiori, F. ATG proteins: Are we always looking at autophagy? Autophagy 2016, 12, 2502–2503. [Google Scholar] [CrossRef] [Green Version]
- Staal, S.P. Molecular cloning of the akt oncogene and its human homologues AKT1 and AKT2: Amplification of AKT1 in a primary human gastric adenocarcinoma. Proc. Natl. Acad. Sci. USA 1987, 84, 5034–5037. [Google Scholar] [CrossRef] [Green Version]
- Kohn, A.D.; Kovacina, K.S.; Roth, R.A. Insulin stimulates the kinase activity of RAC-PK, a pleckstrin homology domain containing ser/thr kinase. Embo J. 1995, 14, 4288–4295. [Google Scholar] [CrossRef]
- Yang, Z.Z.; Tschopp, O.; Hemmings-Mieszczak, M.; Feng, J.; Brodbeck, D.; Perentes, E.; Hemmings, B.A. Protein kinase B alpha/Akt1 regulates placental development and fetal growth. J. Biol. Chem. 2003, 278, 32124–321231. [Google Scholar] [CrossRef] [Green Version]
- Qin, G.; Li, P.; Xue, Z. Triptolide induces protective autophagy and apoptosis in human cervical cancer cells by downregulating Akt/mTOR activation. Oncol. Lett. 2018, 16, 3929–3934. [Google Scholar] [CrossRef] [Green Version]
- Hers, I.; Vicent, E.E.; Tavare, J.M. AKT signaling in health and disease. Cell. Signal. 2011, 23, 1515–1527. [Google Scholar] [CrossRef] [PubMed]
- Manning, B.D.; Toker, A. AKT/PKB Signaling: Navigating the Network. Cell 2017, 169, 381–405. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Totan, A.; Miricescu, D.; Stanescu, I.I.; Didilescu, A.; Melescanu-Imre, M.; Tancu, A.M.C.; Totan, C.; Spinu, T.C.; Greabu, M. (PI3K)/AKT signalling pathway–a Pandora’s box in oral squamous cell carcinoma. Rom. J. Med. Pract. 2019, 14, 389–392. [Google Scholar] [CrossRef]
- Khalid, A.; Hussain, T.; Manzoor, S.; Saalim, M.; Khaliq, S. PTEN: A potential prognostic marker in virus-induced hepatocellular carcinoma. Tumour Biol. 2017, 39, 1010428317705754. [Google Scholar] [CrossRef] [Green Version]
- Zhao, J.; Chi, J.; Gao, M.; Zhi, J.; Li, Y.; Zheng, X. Loss of PTEN Expression is Associated with High MicroRNA 24 Level and Poor Prognosis in Patients with Tongue Squamous Cell Carcinoma. J. Oral Maxillofac. Surg. 2017, 75, 1449.e1–1449.e8. [Google Scholar] [CrossRef]
- Kurasawa, Y.; Shiiba, M.; Nakamura, M.; Fushimi, K.; Ishigami, T.; Bukawa, H.; Yokoe, H.; Uzawa, K.; Tanzawa, H. PTEN expression and methylation status in oral squamous cell carcinoma. Oncol. Rep. 2008, 19, 1429–1434. [Google Scholar]
- Roy, N.K.; Monisha, J.; Padmavathi, G.; Lalhruaitluanga, H.; Kumar, N.S.; Kumar, A.; Singh, S.D.; Baruah, M.N.; Ahmed, G.N.; Longkumar, I.; et al. Isoform-Specific Role of Akt in Oral Squamous Cell Carcinoma. Biomolecules 2019, 9, 253. [Google Scholar] [CrossRef] [Green Version]
- Xu, D.W.; Zhang, G.Q.; Wang, Z.W.; Xu, X.Y.; Liu, T.X. Autophagy in Tumorigenesis and Cancer Treatment. Asian Pac. J. Cancer Prev. 2015, 16, 2165–2175. [Google Scholar] [CrossRef]
- Wani, W.Y.; Boyer-Guittaut, M.; Dodson, M.; Chatham, J.; Darley-Usmar, V.; Zhang, J. Regulation of autophagy by protein post-translational modification. Lab. Investig. 2015, 95, 14–25. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Wang, C.; Tang, H.; Wang, M.; Weng, J.; Liu, X.; Zhang, R.; Huang, H.; Hou, J. Decrease of autophagy activity promotes malig-nant progression of tongue squamous cell carcinoma. J. Oral Pathol. Med. 2013, 42, 557–564. [Google Scholar] [CrossRef]
- Tang, J.Y.; Hsi, E.; Huang, Y.C.; Hsu, N.C.; Chu, P.Y.; Chai, C.Y. High LC3 expression correlates with poor survival in patients with oral squamous cell carcinoma. Hum. Pathol. 2013, 44, 2558–2562. [Google Scholar] [CrossRef] [PubMed]
- Yu, T.; Zuber, J.; Li, J. Targeting autophagy in skin diseases. J. Mol. Med. 2015, 93, 31–38. [Google Scholar] [CrossRef] [PubMed]
- Sakakura, K.; Takahashi, H.; Kaira, K.; Toyoda, M.; Oyama, T.; Chikamatsu, K. Immunological significance of the accumulation of autophagy components in oral squamous cell carcinoma. Cancer Sci. 2015, 106, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Tang, J.Y.; Fang, Y.Y.; Hsi, E.; Huang, Y.C.; Hsu, N.C.; Yang, W.C.; Chang, H.W.; Chai, C.Y.; Chu, P.Y. Immunopositivity of Beclin-1 and ATG5 as indicators of survival and disease recurrence in oral squamous cell carcinoma. Anticancer Res. 2013, 33, 5611–5616. [Google Scholar] [PubMed]
- Tang, J.Y.; Hsi, E.; Huang, Y.C.; Hsu, N.C.; Yang, W.C.; Chang, H.W.; Chai, C.Y.; Chu, P.Y. Overexpression of autophagy-related 16-like 1 in patients with oral squamous cell carcinoma. Pathol. Oncol. Res. 2015, 21, 301–305. [Google Scholar] [CrossRef]
- Kapoor, V.; Paliwal, D.; Baskar Singh, S.; Mohanti, B.K.; Das, S.N. Deregulation of Beclin 1 in patients with tobacco-related oral squamous cellcarcinoma. Biochem. Biophys. Res. Commun. 2012, 422, 764–769. [Google Scholar] [CrossRef]
- Adhauliya, N.; Kalappanavar, A.N.; Ali, I.M.; Annigeri, R.G. Autophagy: A boon or bane in oral cancer. Oral. Oncol. 2016, 61, 120–126. [Google Scholar] [CrossRef]
- Tang, J.Y.; Hsi, E.; Huang, Y.C.; Hsu, N.C.; Chen, Y.K.; Chu, P.Y.; Chai, C.Y. ATG9A overexpression is associated with disease recurrence and poor survival in patients with oral squamous cell carcinoma. Virchows Arch. 2013, 463, 737–742. [Google Scholar] [CrossRef]
- Liu, J.L.; Chen, F.F.; Lung, J.; Lo, C.H.; Lee, F.H.; Lu, Y.C.; Hung, C.H. Prog- nostic significance of p62/SQSTM1 subcellular localization and LC3Binoralsquamouscellcarcinoma. Br. J. Cancer 2014, 111, 944–954. [Google Scholar] [CrossRef] [Green Version]
- Nomura, H.; Uzawa, K.; Yamano, Y.; Fushimi, K.; Ishigami, T.; Kouzu, Y.; Koike, H.; Siiba, M.; Bukawa, H.; Yokoe, H.; et al. Overexpression and altered subcellular localization of autophagy-related 16-like 1 in human oral squamous-cell carcinoma: Correlation with lymphovas-cular invasion and lymph-node metastasis. Hum. Pathol. 2009, 40, 83–91. [Google Scholar] [CrossRef]
- Ha, J.; Kim, J. Novel pharmacological modulators of autophagy: An updated patent review (2012–2015). Expert Opin. Pat. 2016, 26, 1273–1289. [Google Scholar] [CrossRef] [PubMed]
- Mizushima, N.; Yoshimori, T.; Ohsumi, Y. The role of Atg proteinsin autophagosome formation. Annu. Rev. Cell Dev. Biol. 2011, 27, 107–132. [Google Scholar] [CrossRef] [PubMed]
- Wilson, M.; Dooley, H.; Tooze, S. Wipi2b and atg16l1: Setting the stage for autophagosome formation. Biochem. Soc. Trans. 2014, 42, 1327–1334. [Google Scholar] [CrossRef] [PubMed]
- Ishimura, R.; Tanaka, K.; Komatsu, M. Dissection of the role of p62/ Sqstm1 in activation of Nrf2 during xenophagy. Febs Lett. 2014, 588, 822–828. [Google Scholar] [CrossRef] [Green Version]
- Lamark, T.; Svenning, S.; Johansen, T. Regulation of selective autophagy: The p62/SQSTM1 paradigm. Essays Biochem. 2017, 61, 609–624. [Google Scholar] [CrossRef]
- Inui, T.C.T.; Takikita-Suzuki, M.; Nishikawa, M.; Yamamoto, G.; Okabe, H. Association of p62/SQSTM1 excess and oral carcinogenesis. PLoS ONE 2013, 8, e74398. [Google Scholar] [CrossRef] [Green Version]
- Wei, Y.; Zou, Z.; Becker, N.; Anderson, M.; Sumpter, R.; Xiao, G.; Kinch, L.; Koduru, P.; Christudass, C.S.; Veltri, R.W.; et al. EGFR-mediatedBeclin 1 phosphorylation in autophagy suppression, tumor progression, and tumor chemoresistance. Cell 2013, 154, 1269–1284. [Google Scholar] [CrossRef] [Green Version]
- Lozy, F.; Karantza, V. Autophagy and cancer cell metabolism. Semin. Cell Dev. Biol. 2012, 23, 395–401. [Google Scholar] [CrossRef] [Green Version]
- Levy, J.M.M.; Towers, C.G.; Thorburn, A. Targeting autophagy in cancer. Nat. Rev. Cancer 2017, 17, 528–542. [Google Scholar] [CrossRef]
- Deng, S.; Shanmugam, M.K.; Kumar, A.P.; Yap, C.T.; Sethi, G.; Bishayee, A. Targeting autophagy using natural compounds for cancer prevention and therapy. Cancer 2019, 125, 1228–1246. [Google Scholar] [CrossRef]
- Weng, J.; Wang, C.; Wang, Y.; Tang, H.; Liang, J.; Liu, X.; Huang, H.; Hou, J. Beclin1 inhibits proliferation, migration and invasion in tongue squamous cell carcinoma cell lines. Oral Oncol. 2014, 50, 983–990. [Google Scholar] [CrossRef] [PubMed]
- Jiang, L.; Huang, S.; Li, W.; Zhang, D.; Zhang, S.; Zhang, W.; Zheng, P.; Chen, Z. Expression of autophagy and ER stress-related proteins in primary salivary adenoid cystic carcinoma. Pathol. Res. Pract. 2012, 208, 635–641. [Google Scholar] [CrossRef] [PubMed]
- Liang, L.Z.; Ma, B.; Liang, Y.J.; Liu, H.C.; Zheng, G.S.; Zhang, T.H.; Chu, M.; Xu, P.P.; Su, Y.X.; Liao, G.Q. High expression of the autophagy gene Beclin-1 is associated with favorable prognosis for salivary gland adenoid cystic carcinoma. J. Oral Pathol. Med. 2012, 41, 621–629. [Google Scholar] [CrossRef] [PubMed]
- Avalos, Y.; Canales, J.; Bravo-Sagua, R.; Criollo, A.; Lavandero, S.; Quest, A.F. Tumor suppression and promotion by autophagy. Biomed. Res. Int. 2014, 2014, 603980. [Google Scholar] [CrossRef]
- Martins, F.; De Sousa, S.C.; Dos Santos, E.; Woo, S.B.; Gallottini, M. PI3K-AKT-mTOR pathway proteins are differently expressed in oral carcinogenesis. J. Oral Pathol. Med. 2016, 45, 746–752. [Google Scholar] [CrossRef]
- Choudhary, H.; Banik, K.; Ang, H.L.; Girisa, S.; Vikkurthi, R.; Parama, D.; Rana, V.; Shabnam, B.; Khatoon, E.; Kumar, A.P.; et al. Targeting AKT/mTOR in Oral Cancer: Mechanisms and Advances in Clinical Trials. Int. J. Mol. Sci. 2020, 21, 3285. [Google Scholar] [CrossRef]
- Matsuo, F.S.; Andrade, M.F.; Loyola, A.M.; Da Silva, S.J.; Silva, M.J.B.; Cardoso, S.V.; De Faria, P.R. Pathologic significance of AKT, mTOR, and GSK3beta proteins in oral squamous cell carcinoma-a_ected patients. Virchows Arch. Int. J. Pathol. 2018, 472, 983–997. [Google Scholar] [CrossRef]
- Ferreira, D.M.; Neves, T.J.; Lima, L.G.C.A.; Alves, F.A.; Begnami, M.D. Prognostic implications of the phosphatidylinositol 3-kinase/Akt signaling pathway in oral squamous cell carcinoma: Overexpression of p-mTOR indicates an adverse prognosis. Appl. Cancer Res. 2017, 37, 41. [Google Scholar] [CrossRef] [Green Version]
- Lakshminarayana, S.; Augustine, D.; Rao, R.S.; Patil, S.; Awan, K.H.; Venkatesiah, S.S.; Haragannavar, V.C.; Nambiar, S.; Prasad, K. Molecular pathways of oral cancer that predict prognosis and survival: A systematic review. J. Carcinog. 2018, 17, 7. [Google Scholar] [CrossRef]
- Monisha, J.; Roy, N.K.; Padmavathi, G.; Banik, K.; Bordoloi, D.; Khwairakpam, A.D.; Arfuso, F.; Chinnathambi, A.; Alahmadi, T.A.; Alharbi, S.A.; et al. NGAL is Downregulated in Oral Squamous Cell Carcinoma and Leads to Increased Survival, Proliferation, Migration and Chemoresistance. Cancers 2018, 10, 228. [Google Scholar] [CrossRef] [Green Version]
- De Amicis, A.; Sanctis, S.D.; Cristofaro, S.D.; Franchini, V.; Lista, F.; Regalbuto, E.; Giovenale, E.; Gallerano, G.P.; Nenzi, P.; Bei, R.; et al. Biological effects of in vitro THz radiation exposure in human foetal fibroblasts. Mutat. Res. Genet. Toxicol. Environ. Mutagen. 2015, 793, 150–160. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Chen, L.; Guo, L.; Hupp, T.R.; Lin, Y. Evaluating DAPK as a therapeutic target. Apoptosis 2014, 19, 371–386. [Google Scholar] [CrossRef] [PubMed]
- He, M.X.; He, Y.W. A role for c-FLIP(L) in the regulation of apoptosis, autophagy, and necroptosis in T lymphocytes. Cell Death Differ. 2013, 20, 188–197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.S.; Li, Q.; Lee, J.Y.; Lee, S.H.; Jeong, J.H.; Lee, H.R.; Chang, H.; Zhou, F.C.; Gao, S.J.; Liang, C.; et al. FLIP-mediated autophagy regulation in cell death control. Nat. Cell Biol. 2009, 11, 1355–1362. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sasahira, T.; Kirita, T. Hallmarks of Cancer-Related Newly Prognostic Factors of Oral Squamous Cell Carcinoma. Int. J. Mol. Sci. 2018, 19, 2413. [Google Scholar] [CrossRef] [Green Version]
- Oikawa, Y.; Morita, K.I.; Kayamori, K.; Tanimoto, K.; Sakamoto, K.; Katoh, H.; Ishikawa, S.; Inazawa, J.; Harada, H. Receptor tyros-ine kinase amplification is predictive of distant metas-tasis in patients with oral squamous cell carcinoma. Cancer Sci. 2017, 108, 256–266. [Google Scholar] [CrossRef]
- Mrakovcic, M.; Fröhlich, L.F. P53-Mediated Molecular Control of Autophagy in Tumor Cells. Biomolecules 2018, 8, 14. [Google Scholar] [CrossRef] [Green Version]
- Guo, W.; Yan, L.; Yang, L.; Liu, X.; Qiukai, E.; Gao, P.; Ye, X.; Liu, W.; Zuo, J. Targeting GRP75 improves HSP90 inhibitor efficacy by enhancing P53-mediated apoptosis in hepatocellular carcinoma. PLoS ONE 2014, 9, e85766. [Google Scholar] [CrossRef]
- Liang, X.H.; Jackson, S.; Seaman, M.; Brown, K.; Kempkes, B.; Hibshoosh, H.; Levine, B. Induction of autophagy and inhibition of tumorigenesis by beclin 1. Nature 1999, 402, 672–676. [Google Scholar] [CrossRef]
- Menon, M.B.; Dhamija, S. Beclin 1 phosphorylation–at the center of autophagy regulation. Front. Cell Dev. Biol. 2018, 6, 137. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Zhang, W.; Wang, Y.F.; Liu, B.; Zhang, W.F.; Zhao, Y.F.; Kulkarni, A.B.; Sun, Z.J. Dual induction of apoptotic and autophagic cell death by targeting survivin in head neck squamous cell carcinoma. Cell Death Dis. 2015, 6, e1771. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hsieh, M.T.; Chen, H.P.; Lu, C.C.; Chiang, J.H.; Wu, T.S.; Kuo, D.H.; Huang, L.J.; Kuo, S.C.; Yang, J.S. The novel pterostilbene derivative ANK-199 induces autophagic cell death through regulating PI3 kinase class III/beclin 1/Atgrelated proteins in cisplatinresistant CAR human oral cancer cells. Int. J. Oncol. 2014, 45, 782–794. [Google Scholar] [CrossRef] [PubMed]
- Han, H.Y.; Kim, H.; Jeong, S.H.; Lim, D.S.; Ryu, M.H. Sulfasalazine induces autophagic cell death in oral cancer cells via Akt and ERK pathways. Asian Pac. J. Cancer Prev. 2014, 15, 6939–6944. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kroemer, G.; Levine, B. Autophagic cell death: The story of a misnomer. Nat. Rev. Mol. Cell Biol. 2008, 9, 1004–1010. [Google Scholar] [CrossRef]
- Pattingre, S.; Tassa, A.; Qu, X.; Garuti, R.; Liang, X.H.; Mizushima, N.; Packer, M.; Schneider, M.D.; Levine, B. Bcl-2 antiapoptotic proteins inhibit Beclin 1-dependent autophagy. Cell 2005, 122, 927–939. [Google Scholar] [CrossRef] [Green Version]
- Young, A.R.; Narita, M.; Ferreira, M.; Kirschner, K.; Sadaie, M.; Darot, J.F.; Tavaré, S.; Arakawa, S.; Shimizu, S.; Watt, F.M.; et al. Autophagy mediates the mitotic senescence transition. Genes Dev. 2009, 23, 798–803. [Google Scholar] [CrossRef] [Green Version]
- Kimmelman, A.C. The dynamic nature of autophagy in cancer. Genes Dev. 2011, 25, 1999–2010. [Google Scholar] [CrossRef] [Green Version]
- Candido, J.; Hagemann, T. Cancer-related inflammation. J. Clin. Immunol. 2013, 33 (Suppl. S1), S79–S84. [Google Scholar] [CrossRef]
- Degenhardt, K.; Mathew, R.; Beaudoin, B.; Bray, K.; Anderson, D.; Chen, G.; Mukherjee, C.; Shi, Y.; Gélinas, C.; Fan, Y.; et al. Autophagy promotes tumor cell survival and restricts necrosis, inflammation, and tumorigenesis. Cancer Cell 2006, 10, 51–64. [Google Scholar] [CrossRef] [Green Version]
- Yun, C.; Lee, S. The roles of autophagy in cancer. Int. J. Mol. Sci. 2018, 19, 3466. [Google Scholar] [CrossRef] [Green Version]
- Levine, B.; Mizushima, N.; Virgin, H.W. Autophagy in immunity and inflammation. Nature 2011, 469, 323–335. [Google Scholar] [CrossRef] [PubMed]
- Jounai, N.; Takeshita, F.; Kobiyama, K.; Sawano, A.; Miyawaki, A.; Xin, K.; Ishii, K.J.; Kawai, T.; Akira, S.; Suzuki, K.; et al. The Atg5–Atg12 conjugate associates with innate antiviral immune responses. Proc. Natl. Acad. Sci. USA 2007, 104, 14050–14055. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saitoh, T.; Fujita, N.; Jang, M.H.; Uematsu, S.; Yang, B.; Satoh, T.; Omori, H.; Noda, T.; Yamamoto, N.; Komatsu, M.; et al. Loss of the autophagy protein Atg16L1 enhances endotoxin-induced IL-1β production. Nature 2008, 456, 264–268. [Google Scholar] [CrossRef] [PubMed]
- Saha, S.; Panigrahi, D.P.; Patil, S.; Bhutia, S.K. Autophagy in health and disease: A comprehensive review. Biomed. Pharm. 2018, 104, 485–495. [Google Scholar] [CrossRef]
- Cadwell, K.; Patel, K.K.; Maloney, N.S.; Liu, T.; Ng, A.C.Y.; Storer, C.E.; Head, R.D.; Xavier, R.; Stappenbeck, T.S.; Virgin, H.W. Virus-plus-susceptibility gene interaction determines Crohn’s disease gene Atg16L1 phenotypes in intestine. Cell 2010, 141, 1135–1145. [Google Scholar] [CrossRef] [Green Version]
- Goetz, M.E.; Luch, A. Reactive species: A cell damaging rout assisting to chemical carcinogens. Cancer Lett. 2008, 266, 73–83. [Google Scholar] [CrossRef]
- Galluzzi, L.; Morselli, E.; Kepp, O.; Vitale, I.; Rigoni, A.; Vacchelli, E.; Michaud, M.; Zischka, H.; Castedo, M.; Kroemer, G. Mitochondrial gateways to cancer. Mol. Asp. Med. 2010, 31, 1–20. [Google Scholar] [CrossRef]
- Mathew, R.; Kongara, S.; Beaudoin, B.; Karp, C.M.; Bray, K.; Degenhardt, K.; Chen, G.; Jin, S.; White, E. Autophagy suppresses tumor progression by limiting chromosomal instability. Genes Dev. 2007, 21, 1367–1381. [Google Scholar] [CrossRef] [Green Version]
- Morselli, E.; Galluzzi, L.; Kepp, O.; Mariño, G.; Michaud, M.; Vitale, I.; Maiuri, M.C.; Kroemer, G. Oncosuppressive functions of autophagy. Antioxid. Redox Signal. 2011, 14, 2251–2269. [Google Scholar] [CrossRef]
- Geisler, S.; Holmström, K.M.; Skujat, D.; Fiesel, F.C.; Rothfuss, O.C.; Kahle, P.J.; Springer, W. PINK1/Parkin-mediated mitophagy is dependent on VDAC1 and p62/SQSTM1. Nat. Cell Biol. 2010, 12, 119–131. [Google Scholar] [CrossRef]
- Lau, A.; Wang, X.J.; Zhao, F.; Villeneuve, N.F.; Wu, T.; Jiang, T.; Sun, Z.; White, E.; Zhang, D.D. A noncanonical mechanism of Nrf2 activation by autophagy deficiency: Direct interaction between Keap1 and p62. Mol. Cell. Biol. 2010, 30, 3275–3285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huo, Y.; Cai, H.; Teplova, I.; Bowman-Colin, C.; Chen, G.; Price, S.; Barnard, N.; Ganesan, S.; Karantza, V.; White, E.; et al. Autophagy opposes p53-mediated tumor barrier to facilitate tumorigenesis in a model of PALB2-associated hereditary breast cancer. Cancer Discov. 2013, 3, 894–907. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nam, H.Y.; Han, M.W.; Chang, H.W.; Lee, Y.S.; Lee, M.; Lee, H.J.; Lee, B.W.; Lee, K.E.; Jung, M.K.; Jeon, H.; et al. Radioresistant cancer cells can beconditioned to enter senescence by mTOR inhibition. Cancer Res. 2013, 73, 4267–4277. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, L.; Huang, S.; Zhang, D.; Zhang, B.; Li, K.; Li, W.; Zhang, S.; Zhang, W.; Zheng, P. Inhibition of autophagy augments chemotherapy in human salivary adenoid cystic carcinoma. J. Oral Pathol. Med. 2014, 43, 265–272. [Google Scholar] [CrossRef] [PubMed]
- Yen, C.Y.; Chiang, W.F.; Liu, S.Y.; Cheng, P.C.; Lee, S.Y.; Hong, W.Z.; Lin, P.Y.; Lin, M.H.; Liu, Y.C. Long-term stimulation of areca nut components results in increased chemoresistance through elevated autophagic activity. J. Oral Pathol. Med. 2014, 43, 91–96. [Google Scholar] [CrossRef]
- Shi, T.T.; Yu, X.X.; Yan, L.J.; Xiao, H.T. Research progress of hydroxychloroquine and autophagy inhibitors on cancer. Cancer Chemother. Pharm. 2017, 79, 287–294. [Google Scholar] [CrossRef] [PubMed]
- Quan, H.Y.; Quan, H.Y.; Zhou, L.J.; Li, A.D.; Zhang, Z.B. Mechanism of chloroquine in promoting sensitivity of chemotherapeutics inoral squamous cell carcinoma CAL-27 cell line to cisplatin. Shanghai Kou Qiang Yi Xue 2015, 24, 30–36. [Google Scholar]
- Zhao, X.G.; Sun, R.J.; Yang, X.Y.; Liu, D.; Lei, D.; Jin, T.; Pan, X. Chloroquine-enhanced efficacy of cisplatin in the treatment of hypopharyngeal carcinoma in xenograft mice. PLoS ONE 2015, 10, e0126147. [Google Scholar] [CrossRef]
- Carew, J.S.; Kelly, K.R.; Nawrocki, S.T. Autophagy as a target for cancer therapy: New developments. Cancer Manag. Res. 2012, 4, 357–365. [Google Scholar]
- Notte, A.; Leclere, L.; Michiels, C. Autophagy as a mediator of chemotherapy-induced cell death in cancer. Biochem. Pharm. 2011, 82, 427–434. [Google Scholar] [CrossRef] [Green Version]
- Tsai, C.W.; Lai, F.J.; Sheu, H.M.; Lin, Y.S.; Chang, T.H.; Jan, M.S.; Chen, S.M.; Hsu, P.C.; Huang, T.T.; Huang, T.C.; et al. WWOX suppresses autophagy for inducing apoptosis in methotrexate-treated human squamous cell carcinoma. Cell Death Dis. 2013, 4, e792. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, R.; Wang, Z.H.; Wang, B.Q.; Zhang, C.; Gao, W.; Feng, Y.; Bai, T.; Zhang, H.; Huang-Pu, H.; Wen, S. Inhibition of autophagy-potentiated chemosensitivity to cisplatin in laryngeal cancer Hep-2cells. Am. J. Otolaryngol. 2012, 33, 678–684. [Google Scholar] [CrossRef]
- Han, H.Y.; Park, B.S.; Lee, G.S.; Jeong, S.H.; Kim, H.; Ryu, M.H. Autophagic cell death by Poncirus trifoliata Rafin., a traditional oriental medicine, in human oral cancer HSC-4 cells. Evid. Based Complement. Altern. Med. 2015, 394263. [Google Scholar] [CrossRef] [Green Version]
- Chu, S.C.; Hsieh, Y.S.; Yu, C.C.; Lai, Y.Y.; Chen, P.N. Thymoquinone induces cell death in human squamous carcinoma cells via caspase activation-dependent apoptosis and LC3-II activation-dependent autophagy. PLoS ONE 2014, 9, e101579. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, A.C.; Lien, J.C.; Lin, M.W.; Yang, J.; Wu, P.; Chang, S.; Lai, T. Tetrandrine induces cell death in SAS human oral cancer cells through caspase activation- dependent apoptosis and LC3-I and LC3-II activation-dependent autophagy. Int. J. Oncol. 2013, 43, 485–494. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vander Broek, R.; Mohan, S.; Eytan, D.F.; Chen, Z.; Van Waes, C. The PI 3 K/A kt/m TOR axis in head and neck cancer: Functions, aberrations, cross-talk, and therapies. Oral Dis. 2015, 21, 815–825. [Google Scholar] [CrossRef] [PubMed]
- Khan, T.; Relitti, N.; Brindisi, M.; Magnano, S.; Zisterer, D.; Gemma, S.; Butini, S.; Campiani, G. Autophagy modulators for the treatment of oral and esophageal squamous cell carcinomas. Med. Res. Rev. 2020, 40, 1002–1060. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
| Oncogene Product or Tumor Suppressor | Effect on Autophagy Pathway | Reference |
|---|---|---|
| (1) mTOR | Negative regulator | [74,75,76,77,78,79,80] |
| (2) PTEN | Inducer | [46,81] |
| (3) Beclin-1 | Inducer | [74] |
| (4) DAPK | Inducer | [82] |
| (5) BCL-2; BCL-XL | Negative regulator | [74] |
| (6) c-FLIP | Negative regulator | [83,84] |
| (7) P53 | Negative regulator/Inducer | [85,86,87,88] |
| Publication Title | Proposed Molecular Mechanism for Sustaining the Tumor Suppressor Role of Autophagy | Reference |
|---|---|---|
| Autophagic cell death: the story of a misnomer | Autophagic cell death | [94] |
| Bcl-2 antiapoptotic proteins inhibit Beclin 1-dependent autophagy | Autophagic cell death | [95] |
| Anti-neoplastic activity of the cytosolic FoxO1 results from autophagic cell death | Autophagic cell death | [45] |
| Autophagy mediates the mitotic senescence transition | Autophagic senescence | [96] |
| The dynamic nature of autophagy in cancer | Autophagic senescence | [97] |
| Cancer-related inflammation | Inflammation downregulation | [98] |
| Autophagy promotes tumor cell survival and restricts necrosis, inflammation, and tumorigenesis | Inflammation downregulation | [99] |
| The double-edged sword of autophagy modulation in cancer | Inflammation downregulation | [74] |
| The Roles of Autophagy in Cancer. | Inflammation downregulation | [100] |
| Autophagy in immunity and inflammation | Inflammation downregulation | [101] |
| The Atg5–Atg12 conjugate associates with innate antiviral immune responses. | Inflammation downregulation | [102] |
| Loss of the autophagy protein Atg16L1 enhances endotoxin-induced IL-1β production | Inflammation downregulation | [103] |
| Autophagy in mammalian development and differentiation | Inflammation downregulation | [7] |
| Autophagy in health and disease: A comprehensive review. | Inflammation downregulation | [104] |
| Virus-plus-susceptibility gene interaction determines Crohn’s disease gene Atg16L1 phenotypes in intestine | Inflammation downregulation | [105] |
| Reactive species: a cell damaging rout assisting to chemical carcinogens | Oxidative stress and genome instability | [106] |
| Mitochondrial gateways to cancer | Oxidative stress and genome instability | [107] |
| Autophagy suppresses tumor progression by limiting chromosomal instability | Oxidative stress and genome instability | [108] |
| Oncosuppressive functions of autophagy | Oxidative stress and genome instability | [109] |
| PINK1/Parkin-mediated mitophagy is dependent on VDAC1 and p62/SQSTM1 | Oxidative stress and genome instability | [110] |
| A noncanonical mechanism of Nrf2 activation by autophagy deficiency: direct interaction between Keap1 and p62 | Oxidative stress and genome instability | [111] |
| Publication Title | Proposed Molecular Mechanism | Reference |
|---|---|---|
| Autophagy promotes tumor cell survival and restricts necrosis, inflammation, and tumorigenesis | Beclin-1 dependent regulation | [99] |
| The double-edged sword of autophagy modulation in cancer. | Stress tolerance increase | [74] |
| The Roles of Autophagy in Cancer. | Inflammation downregulation | [100] |
| Targeting GRP75 improves HSP90 inhibitor efficacy by enhancing P53-mediated apoptosis in hepatocellular carcinoma | Ras activation | [88] |
| Autophagy suppresses tumor progression by limiting chromosomal instability | Ras activation | [108] |
| Autophagy opposes P53-mediated tumor barrier to facilitate tumorigenesis in a model of PALB2-associated hereditary breast cancer | P53 suppression | [112] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alexandra, T.; Marina, I.M.; Daniela, M.; Ioana, S.I.; Maria, B.; Radu, R.; Maria, T.A.; Tudor, S.; Maria, G. Autophagy—A Hidden but Important Actor on Oral Cancer Scene. Int. J. Mol. Sci. 2020, 21, 9325. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21239325
Alexandra T, Marina IM, Daniela M, Ioana SI, Maria B, Radu R, Maria TA, Tudor S, Maria G. Autophagy—A Hidden but Important Actor on Oral Cancer Scene. International Journal of Molecular Sciences. 2020; 21(23):9325. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21239325
Chicago/Turabian StyleAlexandra, Totan, Imre Melescanu Marina, Miricescu Daniela, Stanescu Iulia Ioana, BencZe Maria, Radulescu Radu, Tancu Ana Maria, Spinu Tudor, and Greabu Maria. 2020. "Autophagy—A Hidden but Important Actor on Oral Cancer Scene" International Journal of Molecular Sciences 21, no. 23: 9325. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21239325