Altered PPARγ Expression Promotes Myelin-Induced Foam Cell Formation in Macrophages in Multiple Sclerosis

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
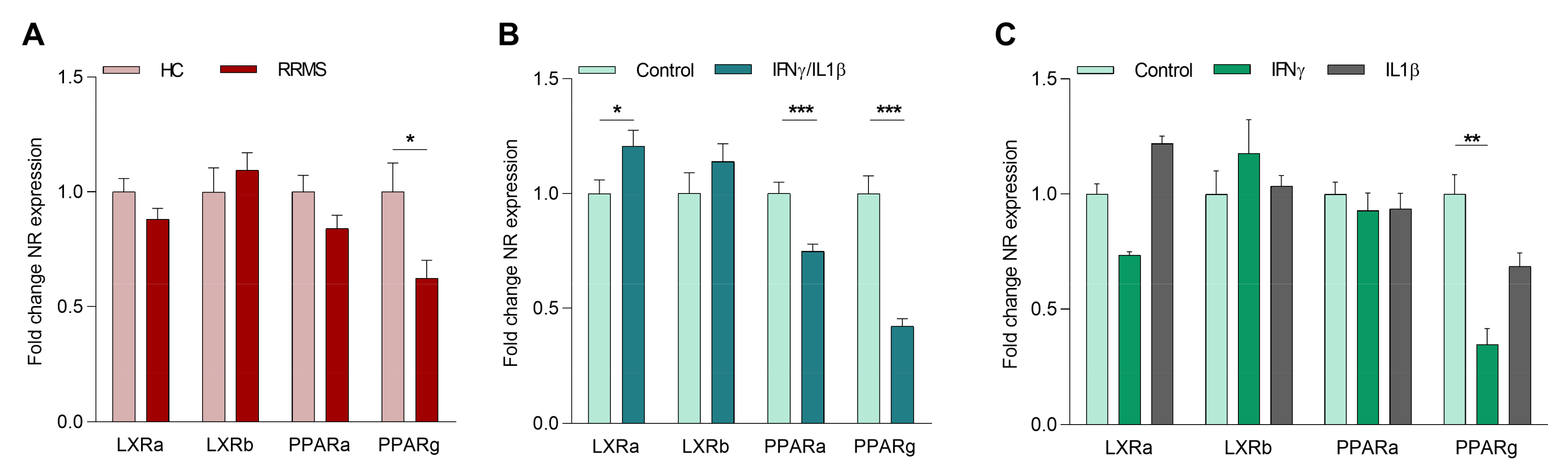
2.1. The Inflammatory Environment in MS Reduces Macrophage PPARγ Expression
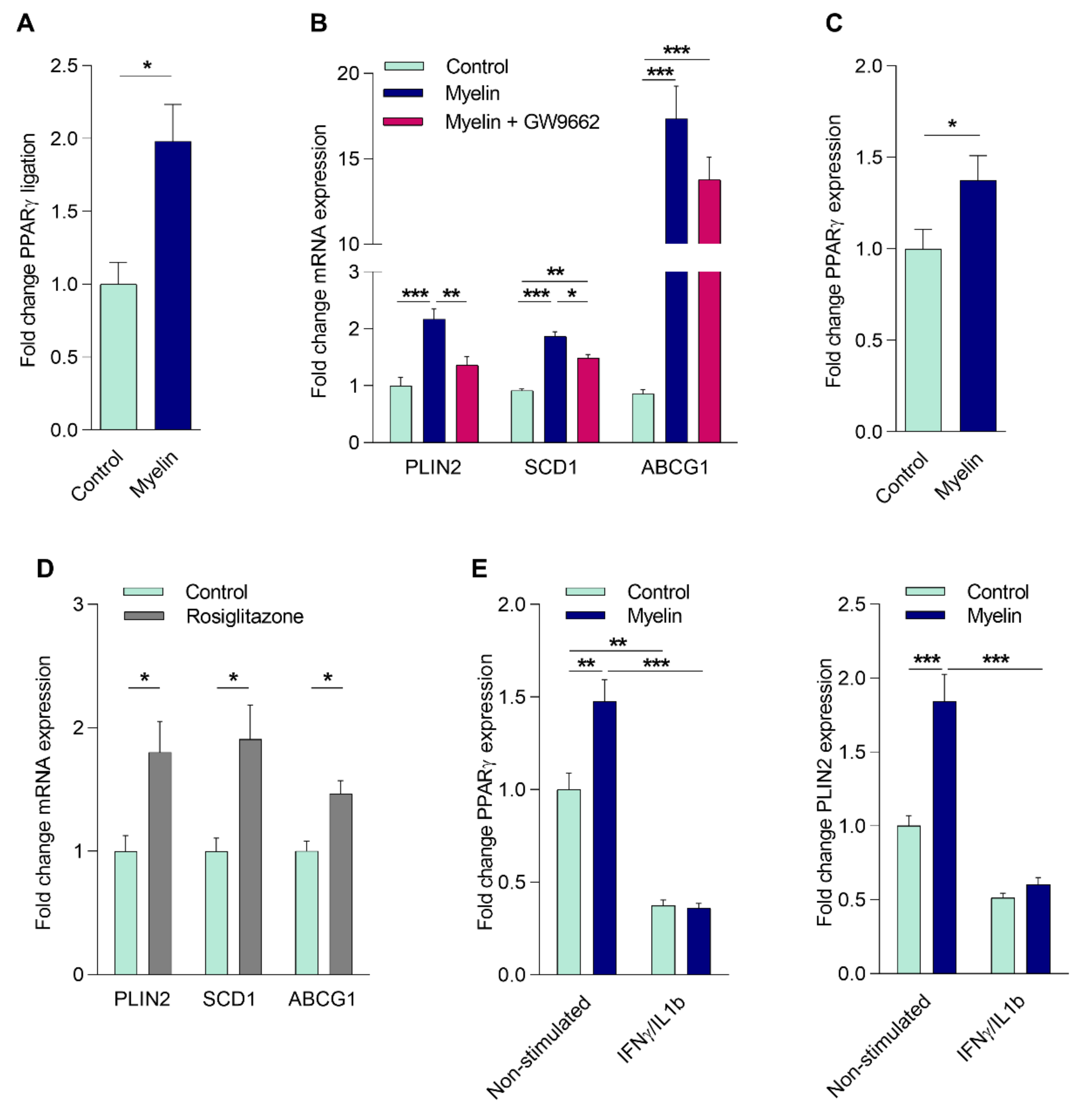
2.2. Myelin Uptake Activates PPARγ in Phagocytes
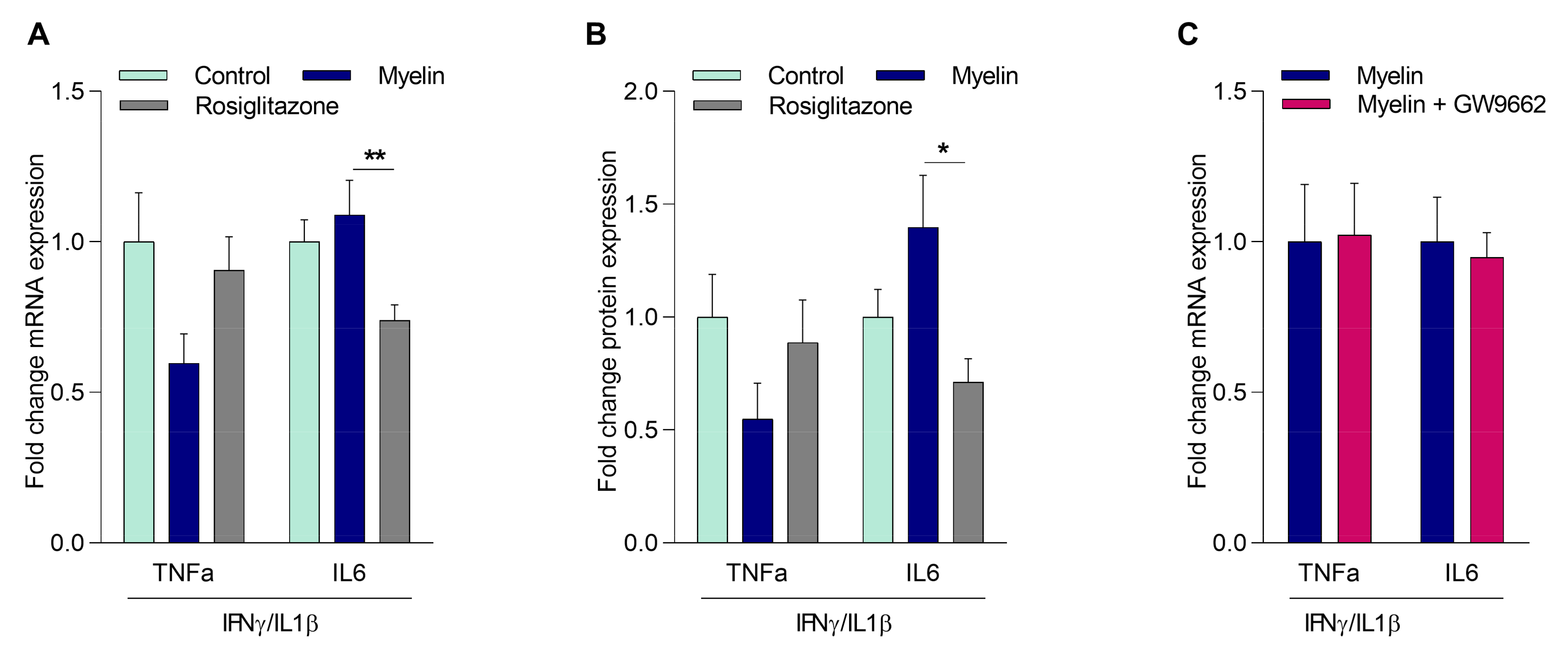
2.3. Myelin Controls the Inflammatory Phenotype of Macrophages Independently of PPARγ
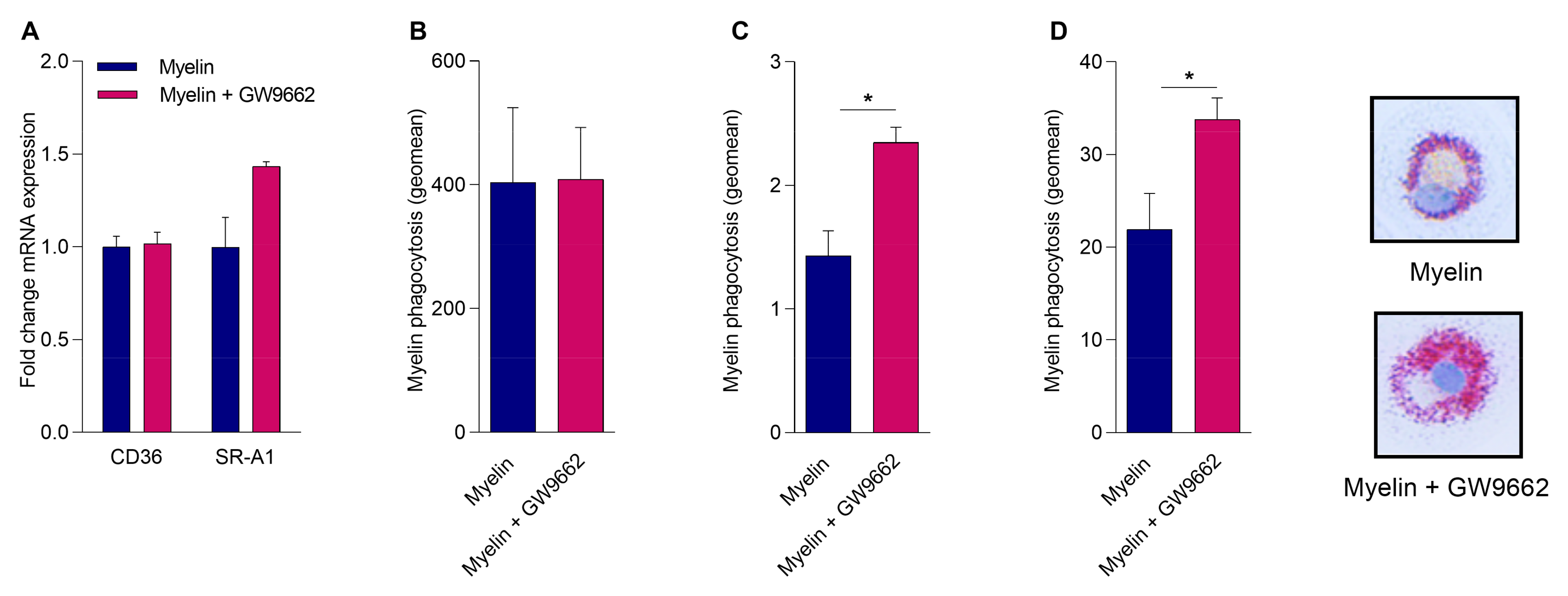
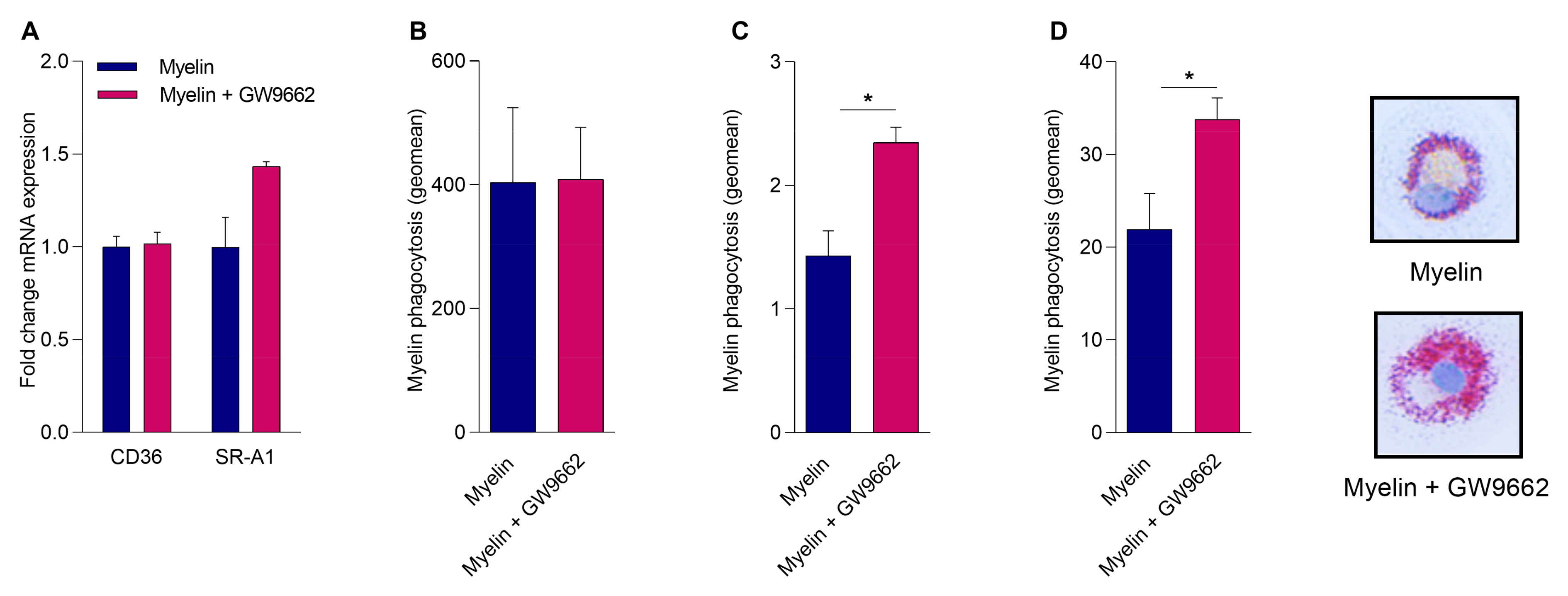
2.4. Reduced PPARγ Activity Does Not Impact Myelin Phagocytosis but Hampers Intracellular Lipid Processing
3. Discussion
4. Materials and Methods
4.1. Human Subjects
4.2. Cell Culture
4.3. Human Myelin Isolation
4.4. Macrophage Stimulation and Pharmacological Treatments
4.5. RNA Extraction and Real-Time Quantitative PCR (RT-qPCR)
4.6. Enzyme-Linked Immunosorbent Assay (ELISA)
4.7. Luciferase Assay
4.8. Flow Cytometry
4.9. Cholesteryl Ester (CE) Determination
4.10. Oil Red O (ORO) Staining and Quantification
4.11. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| ABCA1 | ATP-binding cassette transporter A1 |
| ABCG1 | ATP-binding cassette transporter G1 |
| ANOVA | Analysis of variance |
| CD36 | Cluster of differentiation 36 |
| CE | Cholesteryl ester |
| CNS | Central nervous system |
| CPT1 | Carnitine palmitoyl transferase 1 |
| CSF | Cerebrospinal fluid |
| DiI | 1,1″-diotadecyl-3,3,3′,3′,-tetramethylindocarbocyanide perchlorate |
| DMSO | Dimethylsulfoxide |
| EAE | Experimental autoimmune encephalomyelitis |
| ELISA | Enzyme-linked immunosorbent assay |
| FA | Fatty acids |
| HC | Healthy control |
| LD | Lipid droplet |
| LXR | Liver X receptor |
| MDM | Monocyte-derived macrophage |
| MS | Multiple sclerosis |
| NCoR | Nuclear receptor co-repressor |
| NR | Nuclear receptor |
| ORO | Oil red O |
| PBMC | Peripheral blood mononuclear cell |
| PHA | Phytohemagglutinin |
| PLIN2 | Perilipin 2 |
| PPAR | Peroxisome proliferator-activated receptor |
| RR-MS | Relapsing-remitting MS |
| RT-qPCR | Real-time quantitative PCR |
| RXR | Retinoid X receptor |
| SCD1 | Stearoyl-CoA desaturase-1 |
| S.E.M. | Standard error of the mean |
| SP-MS | Secondary progressive MS |
| SR-A1 | Scavenger receptor-A1 |
| SR-B1 | Scavenger receptor-B1 |
References
- Bogie, J.F.; Stinissen, P.; Hendriks, J.J. Macrophage subsets and microglia in multiple sclerosis. Acta Neuropathol. 2014, 128, 191–213. [Google Scholar] [CrossRef]
- Jiang, Z.; Jiang, J.X.; Zhang, G.X. Macrophages: A double-edged sword in experimental autoimmune encephalomyelitis. Immunol. Lett. 2014, 160, 17–22. [Google Scholar] [CrossRef] [PubMed]
- Kotter, M.R.; Zhao, C.; van Rooijen, N.; Franklin, J.M.R. Macrophage-depletion induced impairment of experimental CNS remyelination is associated with a reduced oligodendrocyte progenitor cell response and altered growth factor expression. Neurobiol. Dis. 2005, 18, 166–175. [Google Scholar] [CrossRef] [PubMed]
- Nally, F.K.; De Santi, C.; McCoy, C.E. Nanomodulation of macrophages in multiple sclerosis. Cells 2019, 8, 543. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grajchen, E.; Hendriks, J.J.A.; Bogie, J.F.J. The physiology of foamy phagocytes in multiple sclerosis. Acta Neuropathol. Commun. 2018, 6, 124. [Google Scholar] [CrossRef]
- Bogie, J.F.; Timmermans, S.; Huynh-Thu, V.A.; Irrthum, A.; Smeets, J.M.H.; Gustafsson, J.-A.; Steffensen, K.R.; Mulder, M.; Stinissen, P.; Hellings, N. Myelin-derived lipids modulate macrophage activity by liver X receptor activation. PLoS ONE 2012, 7, e44998. [Google Scholar] [CrossRef] [Green Version]
- Kuhlmann, T.; Ludwin, S.; Prat, A.; Antel, J.; Bruck, W.; Lassmann, H. An updated histological classification system for multiple sclerosis lesions. Acta Neuropathol. 2017, 133, 13–24. [Google Scholar] [CrossRef]
- Bogie, J.F.; Jorissen, W.; Mailleux, J.; Nijland, P.G.; Zelcer, N.; Vanmierlo, T.; Van Horssen, J.; Stinissen, P.; Hellings, N.; Hendriks, J.J.A. Myelin alters the inflammatory phenotype of macrophages by activating PPARs. Acta Neuropathol. Commun. 2013, 1, 43. [Google Scholar] [CrossRef] [Green Version]
- Bensinger, S.J.; Tontonoz, P. Integration of metabolism and inflammation by lipid-activated nuclear receptors. Nature 2008, 454, 470–477. [Google Scholar] [CrossRef]
- Rigamonti, E.; Chinetti-Gbaguidi, G.; Staels, B. Regulation of macrophage functions by PPAR-alpha, PPAR-gamma, and LXRs in mice and men. Arterioscler. Thromb. Vasc. Biol. 2008, 28, 1050–1059. [Google Scholar] [CrossRef] [Green Version]
- Bougarne, N.; Weyers, B.; Desemet, S.J.; Deckers, J.; Ray, D.W.; Staels, B.; De Bosscher, K. Molecular actions of PPARalpha in lipid metabolism and inflammation. Endocr. Rev. 2018, 39, 760–802. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klotz, L.; Schmidt, M.; Giese, T.; Sastre, M.; Knolle, P.; Klockgether, T.; Heneka, M.T. Proinflammatory stimulation and pioglitazone treatment regulate peroxisome proliferator-activated receptor gamma levels in peripheral blood mononuclear cells from healthy controls and multiple sclerosis patients. J. Immunol. 2005, 175, 4948–4955. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bogie, J.F.J.; Grajchen, E.; Wouters, E.; Corales Garcia, A.; Dierckx, T.; Vanherle, S.; Mailleux, J.; Gervois, P.; Wolfs, E.; Dehairs, J.; et al. Stearoyl-CoA desaturase-1 impairs the reparative properties of macrophages and microglia in the brain. J. Exp. Med. 2020, 217, e20191660. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Healy, L.M.; Ho Jang, J.; Won, S.J.; Lin, H.; Touil, H.; Aljarallah, S.; Bar-Or, A.; Antel, J.P. MerTK-mediated regulation of myelin phagocytosis by macrophages generated from patients with MS. Neurol. Neuroimmunol. Neuroinflamm. 2017, 4, e402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moore, K.J.; Rosen, E.D.; Fitzgerald, M.L.; Randow, F.; Andersson, L.P.; Altshuler, D.; Milstone, D.S.; Mortensen, R.M.; Spiegelman, B.M.; Freeman, M.W. The role of PPAR-gamma in macrophage differentiation and cholesterol uptake. Nat. Med. 2001, 7, 41–47. [Google Scholar] [CrossRef] [PubMed]
- Reichert, F.; Rotshenker, S. Complement-receptor-3 and scavenger-receptor-AI/II mediated myelin phagocytosis in microglia and macrophages. Neurobiol. Dis. 2003, 12, 65–72. [Google Scholar] [CrossRef]
- Argmann, C.A.; Sawyez, C.G.; McNeil, C.J.; Hegele, R.A.; Huff, M.W. Activation of peroxisome proliferator-activated receptor gamma and retinoid X receptor results in net depletion of cellular cholesteryl esters in macrophages exposed to oxidized lipoproteins. Arterioscler. Thromb. Vasc. Biol. 2003, 23, 475–482. [Google Scholar] [CrossRef] [Green Version]
- Chinetti, G.; Lestavel, S.; Bocher, V.; Remaley, A.T.; Neve, B.; Torra, I.P.; Teissier, E.; Minnich, A.; Jaye, M.; Duverger, N.; et al. PPAR-alpha and PPAR-gamma activators induce cholesterol removal from human macrophage foam cells through stimulation of the ABCA1 pathway. Nat. Med. 2001, 7, 53–58. [Google Scholar] [CrossRef]
- Van Boxel-Dezaire, A.H.; Hoff, S.C.; van Oosten, B.W.; Verweij, C.L.; Dräger, A.M.; Adèr, H.J.; van Houwelingen, J.C.; Barkhof, F.; Polman, C.H.; Nagelkerken, L. Decreased interleukin-10 and increased interleukin-12p40 mRNA are associated with disease activity and characterize different disease stages in multiple sclerosis. Ann. Neurol. 1999, 45, 695–703. [Google Scholar] [CrossRef]
- Rieckmann, P.; Albertch, M.; Kitze, B.; Weber, T.; Tumani, H.; Broocks, A.; Luer, W.; Helvig, A.; Poser, S. Tumor necrosis factor-alpha messenger RNA expression in patients with relapsing-remitting multiple sclerosis is associated with disease activity. Ann. Neurol. 1995, 37, 82–88. [Google Scholar] [CrossRef]
- Khaibullin, T.; Ivanova, V. Elevated levels of proinflammatory cytokines in cerebrospinal fluid of multiple sclerosis patients. Front. Immunol. 2017, 8, 531. [Google Scholar] [CrossRef] [PubMed]
- Lees, J.R.; Cross, A.H. A little stress is good: IFN-gamma, demyelination, and multiple sclerosis. J. Clin. Investig. 2007, 117, 297–299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chu, C.Q.; Wittmer, S.; Dalton, D.K. Failure to suppress the expansion of the activated CD4 T cell population in interferon gamma-deficient mice leads to exacerbation of experimental autoimmune encephalomyelitis. J. Exp. Med. 2000, 192, 123–128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Panitch, H.S.; Haley, A.S. Exacerbations of multiple sclerosis in patients treated with gamma interferon. Lancet 1987, 1, 893–895. [Google Scholar] [CrossRef]
- Panitch, H.S.; Hirsch, R.L.; Schindler, J.; Johnson, K.P. Treatment of multiple sclerosis with gamma interferon: Exacerbations associated with activation of the immune system. Neurology 1987, 37, 1097–1102. [Google Scholar] [CrossRef] [Green Version]
- Niino, M.; Iwabuchi, K.; Kikuchi, S.; Ato, M.; Morohashi, T.; Ogata, A.; Tashiro, K.; Onoe, K. Amelioration of experimental autoimmune encephalomyelitis in C57BL/6 mice by an agonist of peroxisome proliferator-activated receptor-gamma. J. Neuroimmunol. 2001, 116, 40–48. [Google Scholar] [CrossRef]
- Storer, P.D.; Xu, J.; Chavis, J.; Drew, P.D. Peroxisome proliferator-activated receptor-gamma agonists inhibit the activation of microglia and astrocytes: Implications for multiple sclerosis. J. Neuroimmunol. 2005, 161, 113–122. [Google Scholar] [CrossRef]
- Schett, G. Physiological effects of modulating the interleukin-6 axis. Rheumatology 2018, 57 (Suppl. S2), ii43–ii50. [Google Scholar] [CrossRef] [Green Version]
- Mailleux, J.; Vanmierlo, T.; Bogie, J.F.J.; Wouters, E.; Lutjohann, D.; Hendriks, J.J.A.; van Horssen, J. Active liver X receptor signaling in phagocytes in multiple sclerosis lesions. Mult. Scler. 2018, 24, 279–289. [Google Scholar] [CrossRef]
- Kotter, M.R.; Li, W.W.; Zhao, C.; Franklin, R.J.M. Myelin impairs CNS remyelination by inhibiting oligodendrocyte precursor cell differentiation. J. Neurosci. 2006, 26, 328–332. [Google Scholar] [CrossRef]
- Zhu, Y.; Lyapichev, D.H.L.; Motti, D.; Ferraro, N.M.; Zhang, Y.; Yahn, S.; Soderblom, C.; Zha, J.; Bethea, Z.J.R.; Spiller, K.L. Macrophage transcriptional profile identifies lipid catabolic pathways that can be therapeutically targeted after spinal cord injury. J. Neurosci. 2017, 37, 2362–2376. [Google Scholar] [CrossRef] [PubMed]
- Natrajan, M.S.; Komori, M.; Kosa, P.; Johnson, K.R.; Wu, T.; Franklin, R.J.M.; Bielekova, B. Pioglitazone regulates myelin phagocytosis and multiple sclerosis monocytes. Ann. Clin. Transl. Neurol. 2015, 2, 1071–1084. [Google Scholar] [CrossRef] [PubMed]
- Wang, N.; Yin, R.; Liu, Y.; Guangmei, Y.L.; Mao, G.; Xi, F. Role of peroxisome proliferator-activated receptor-gamma in atherosclerosis: An update. Circ. J. 2011, 75, 528–535. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chinetti, G.; Lestavel, S.; Fruchart, J.-C.; Clavey, V.; Staels, B. Peroxisome proliferator-activated receptor alpha reduces cholesterol esterification in macrophages. Circ. Res. 2003, 92, 212–217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boren, J.; Brindle, K.M. Apoptosis-induced mitochondrial dysfunction causes cytoplasmic lipid droplet formation. Cell Death Differ. 2012, 19, 1561–1570. [Google Scholar] [CrossRef] [Green Version]
- Chawla, A.; Boisvert, W.A.; Lee, C.-H.; Laffitte, B.A.; Barak, Y.; Joseph, S.B.; Liao, D.; Nagy, L.; Edwards, P.A. A PPAR gamma-LXR-ABCA1 pathway in macrophages is involved in cholesterol efflux and atherogenesis. Mol. Cell 2001, 7, 161–171. [Google Scholar] [CrossRef]
- Chinetti, G.; Gbaguidi, F.C.; Griglio, S.; Mallat, Z.; Antonucci, M.; Poulain, P.; Chapman, J.; Frucgart, J.C.; Tedgui, A.; Fruchart, J.N. CLA-1/SR-BI is expressed in atherosclerotic lesion macrophages and regulated by activators of peroxisome proliferator-activated receptors. Circulation 2000, 101, 2411–2417. [Google Scholar] [CrossRef] [Green Version]
- Diab, A.; Deng, C.; Smith, J.D.; Hussain, R.Z.; Phanavanh, B.; Lovett-Racke, A.E.; Drew, P.D.; Racke, K.M. Peroxisome proliferator-activated receptor-gamma agonist 15-deoxy-Delta(12,14)-prostaglandin J(2) ameliorates experimental autoimmune encephalomyelitis. J. Immunol. 2002, 168, 2508–2515. [Google Scholar] [CrossRef] [Green Version]
- Szalardy, L.; Zadori, D.; Tanzos, E.; Simu, M.; Bencsik, K.; Vecsei, L.; Klivenyi, P. Elevated levels of PPAR-gamma in the cerebrospinal fluid of patients with multiple sclerosis. Neurosci. Lett. 2013, 554, 131–134. [Google Scholar] [CrossRef] [Green Version]
- Jorissen, W.; Wouters, E.; Bogie, J.F.; Vanmierlo, T.; Noben, J.-P.; Sviridov, D.; Hellings, N.; Somers, V.; Valcke, R.; Vanwijmeersch, B. Relapsing-remitting multiple sclerosis patients display an altered lipoprotein profile with dysfunctional HDL. Sci Rep. 2017, 7, 43410. [Google Scholar] [CrossRef]
- Bogie, J.F.; Mailleux, J.; Wouters, E.; Jorissen, W.; Grajchen, E.; Vanmol, J.; Wouters, K.; Hellings, N.; van Horssen, J.; Vanmierlo, T. Scavenger receptor collectin placenta 1 is a novel receptor involved in the uptake of myelin by phagocytes. Sci. Rep. 2017, 7, 44794. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bogie, J.F.; Stinissen, P.; Hellings, N.; Hendriks, J.J.A. Myelin-phagocytosing macrophages modulate autoreactive T cell proliferation. J. Neuroinflam. 2011, 8, 85. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van der Laan, L.J.; Ruuls, S.R.; Weber, K.S.; Lodder, I.J.; Dopp, E.A.; Dijkstra, C.D. Macrophage phagocytosis of myelin in vitro determined by flow cytometry: Phagocytosis is mediated by CR3 and induces production of tumor necrosis factor-alpha and nitric oxide. J. Neuroimmunol. 1996, 70, 145–152. [Google Scholar] [CrossRef]
- Thompson, A.J.; Banwell, B.L.; Barkof, B.; Caroll, W.M.; Coetzee, T.; Comi, G.; Correale, J.; Fazekas, F.; Filippi, M.; Fujihara, K. Diagnosis of multiple sclerosis: 2017 revisions of the McDonald criteria. Lancet Neurol. 2018, 17, 162–173. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wouters, E.; Grajchen, E.; Jorissen, W.; Dierckx, T.; Wetzels, S.; Loix, M.; Tulleners, M.P.; Staels, B.; Stinissen, P.; Haidar, M.; et al. Altered PPARγ Expression Promotes Myelin-Induced Foam Cell Formation in Macrophages in Multiple Sclerosis. Int. J. Mol. Sci. 2020, 21, 9329. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21239329
Wouters E, Grajchen E, Jorissen W, Dierckx T, Wetzels S, Loix M, Tulleners MP, Staels B, Stinissen P, Haidar M, et al. Altered PPARγ Expression Promotes Myelin-Induced Foam Cell Formation in Macrophages in Multiple Sclerosis. International Journal of Molecular Sciences. 2020; 21(23):9329. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21239329
Chicago/Turabian StyleWouters, Elien, Elien Grajchen, Winde Jorissen, Tess Dierckx, Suzan Wetzels, Melanie Loix, Marie Paule Tulleners, Bart Staels, Piet Stinissen, Mansour Haidar, and et al. 2020. "Altered PPARγ Expression Promotes Myelin-Induced Foam Cell Formation in Macrophages in Multiple Sclerosis" International Journal of Molecular Sciences 21, no. 23: 9329. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21239329