Genomic and Transcriptomic Survey Provides New Insight into the Organization and Transposition Activity of Highly Expressed LTR Retrotransposons of Sunflower (Helianthus annuus L.)

 ,
, 
 , , , and
, , , and
Abstract
:1. Introduction
2. Results
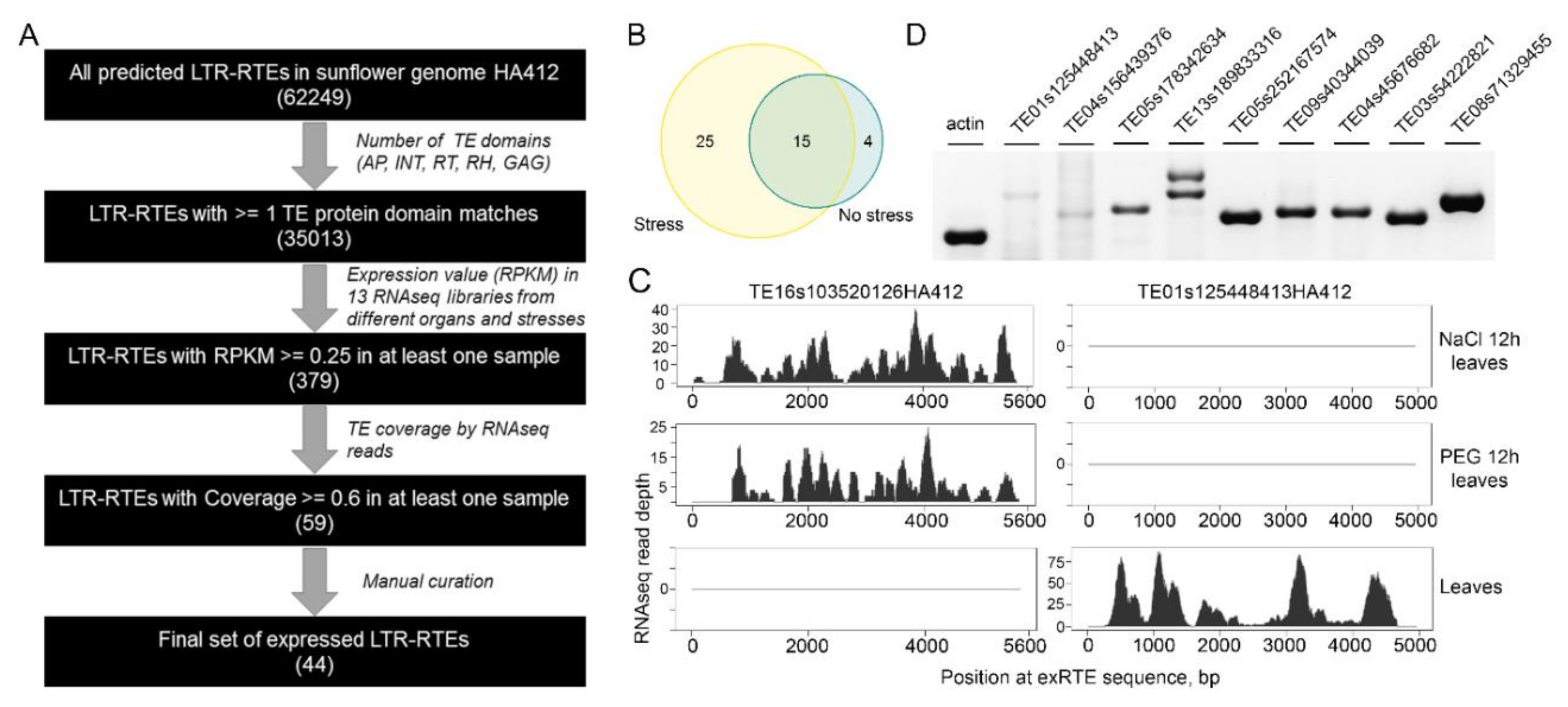
2.1. Tens of Sunflower LTR Retrotransposons Have High Expression in Stress and Normal Conditions
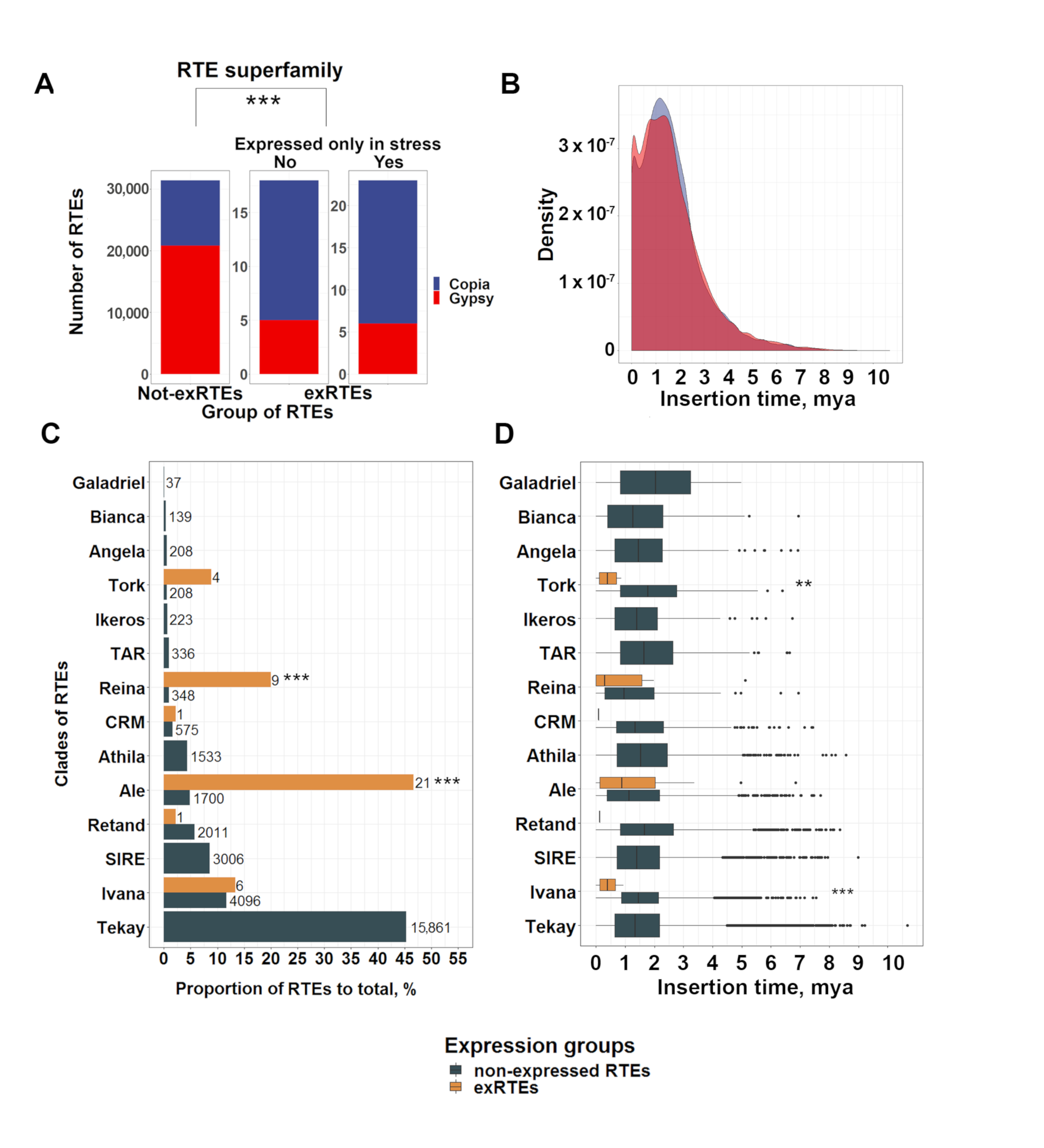
2.2. Expressed RTE Set Is Enriched by “Young” Copies and Low-Copy Clades
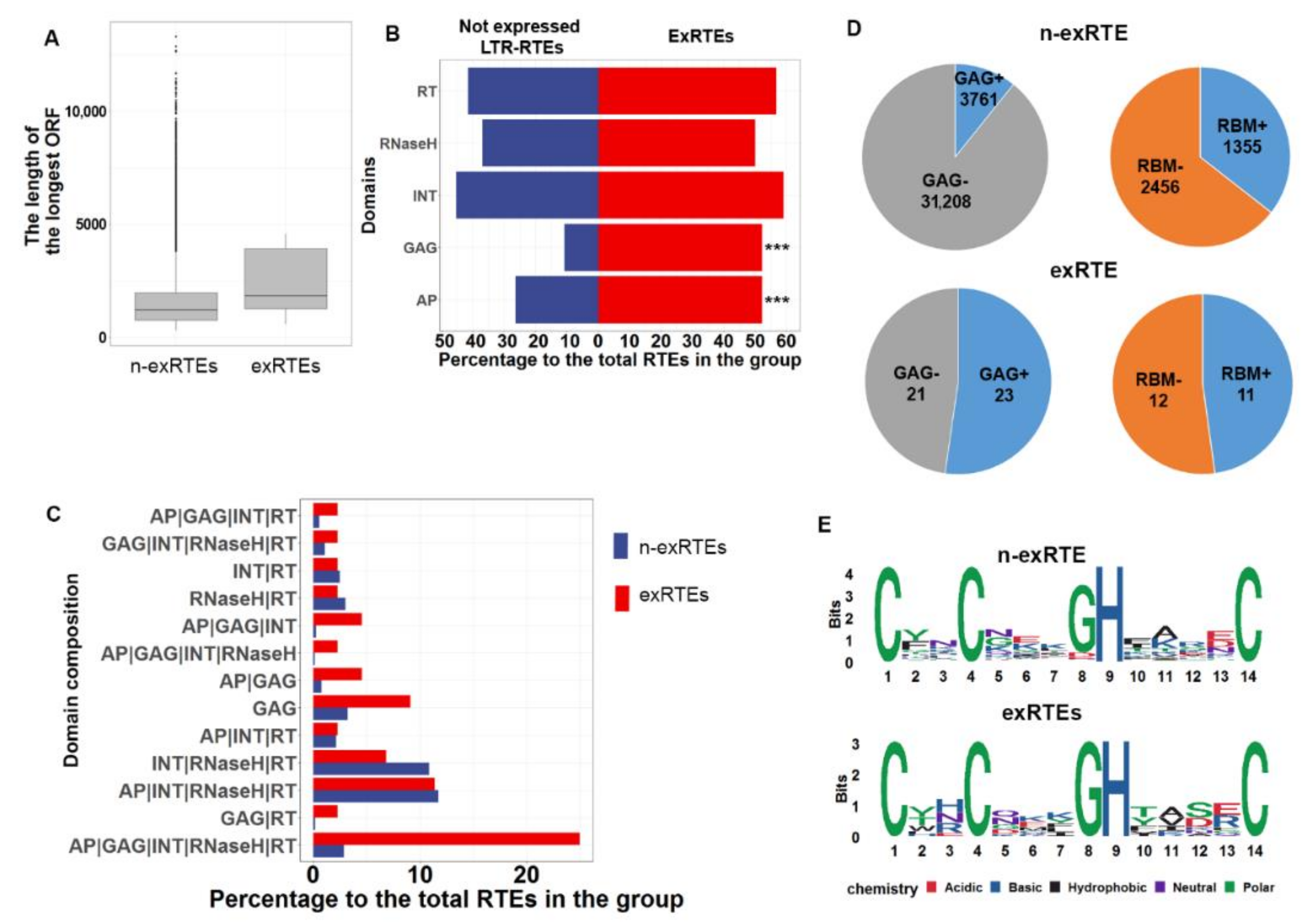
2.3. ExRTEs Domain Composition Is Biased Toward GAG Protein
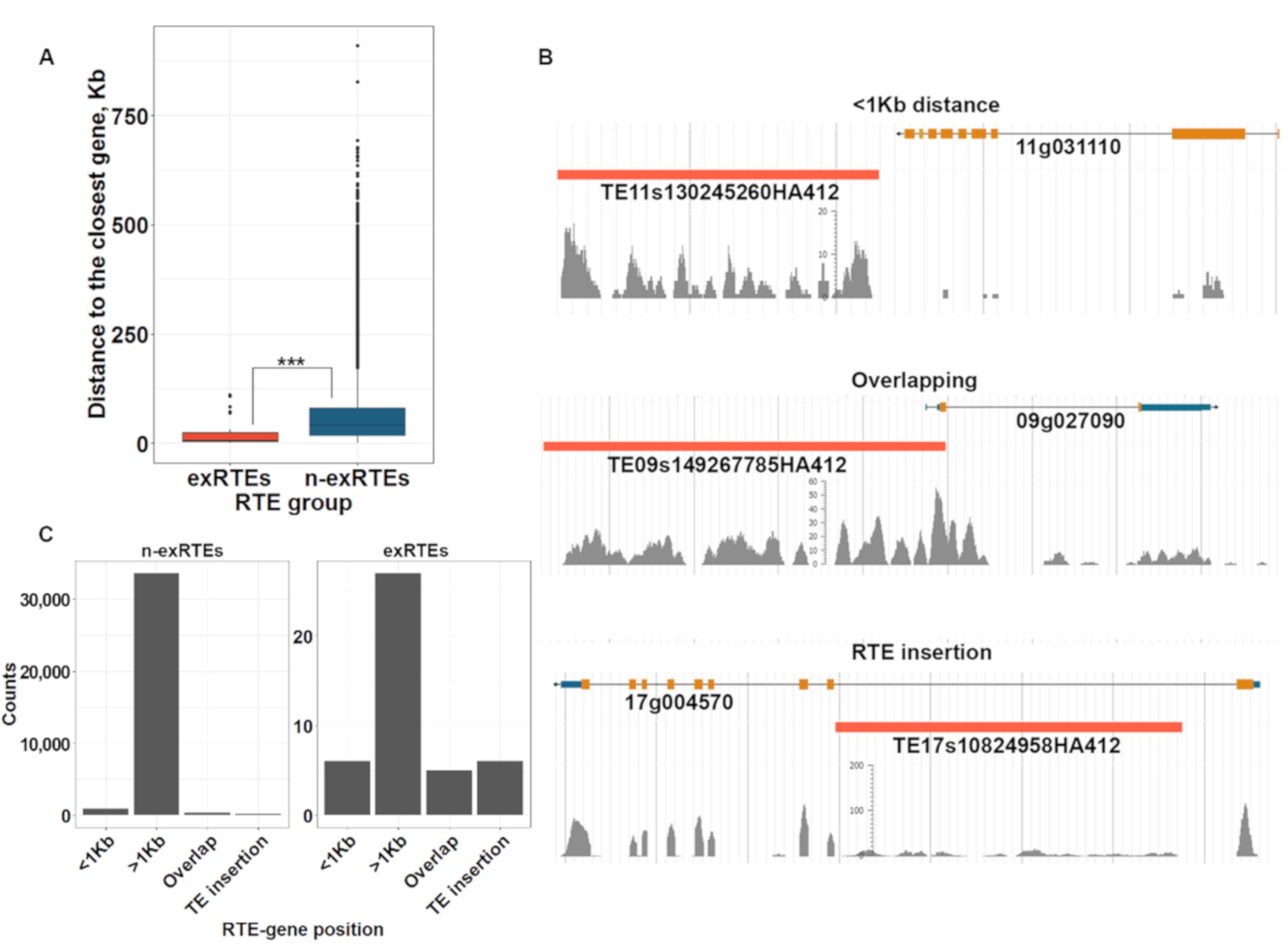
2.4. The Proximity of exRTEs to Genes
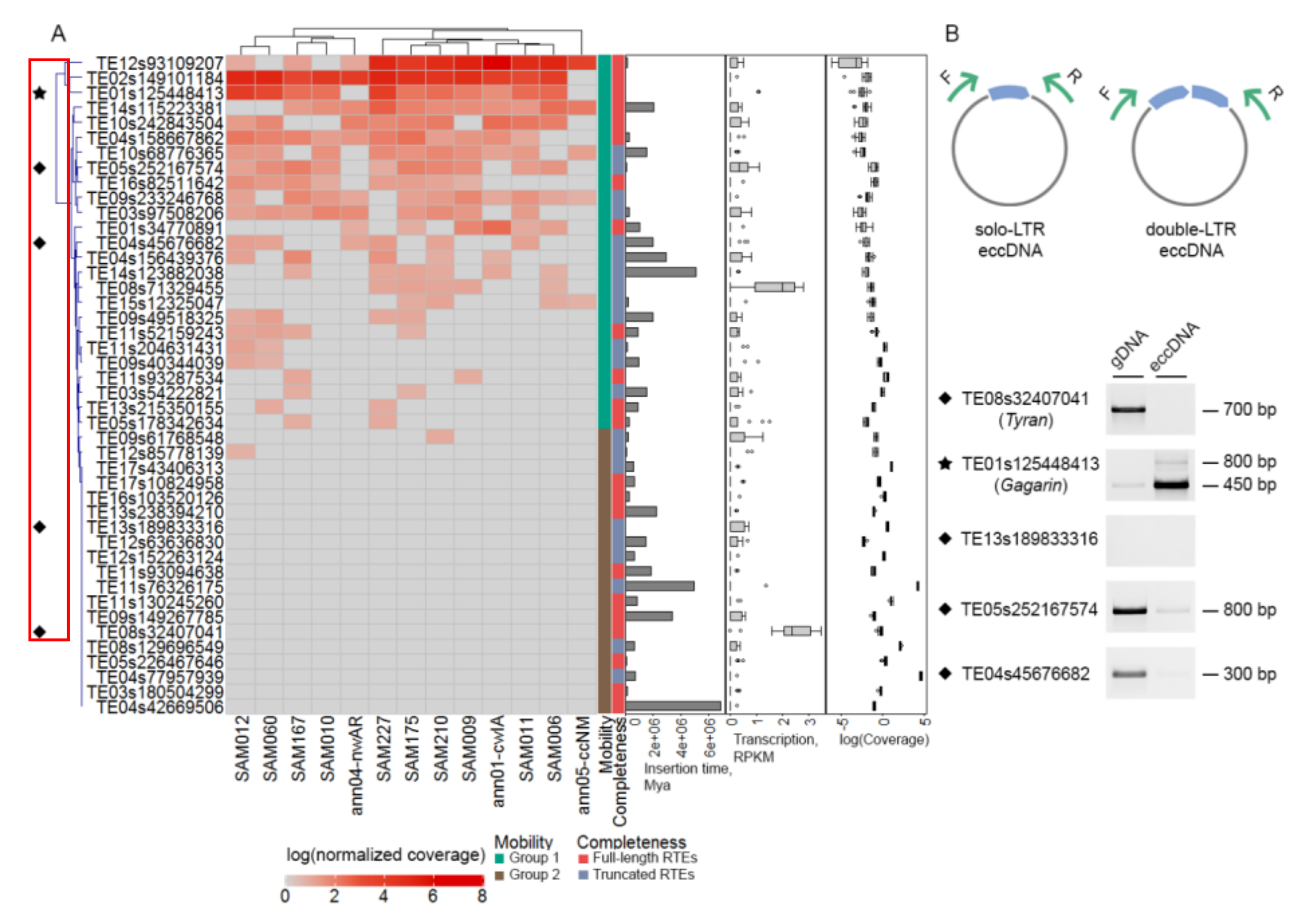
2.5. Most exRTEs Have Recent and/or Ongoing Mobilome Activity
2.6. Nanopore Direct RNA Sequencing Revealed Alternative Splicing of exRTE Transcripts
3. Discussion
3.1. Tens of RTEs with Distinct Features Are Expressed in Plant Somatic Tissues
3.2. The shGAG Isoform Originated via Splicing and Premature Transcription Termination Is Conserved Feature among Plant RTEs
3.3. Ongoing Transcription and Transposition Activity Are Weakly Connected
3.4. RNAseq and Nanopore RNA Sequencing Are Complementary Approaches for Identifying Expressed RTEs
4. Materials and Methods
4.1. Plant Material and DNA Isolation
4.2. Pipeline for Genome-Wide Detection of Highly Expressed TEs
4.3. RTE Insertion Time Estimation
4.4. Search of RNA-Binding Motif and GAG ORF Analysis
4.5. Calculation of RTE—Gene Distance
4.6. Mobilome Analysis
4.7. RNA Isolation and RT-PCR
4.8. Extrachromosomal Circular DNA Isolation
4.9. Nanopore Direct RNA Sequencing and ONT Data Analysis
4.10. Statistics and Data Visualization
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Eichler, E.E.; Sankoff, D. Structural dynamics of eukaryotic chromosome evolution. Science 2003, 301, 793–797. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cosby, R.L.; Chang, N.C.; Feschotte, C. Host-transposon interactions: Conflict, cooperation, and cooption. Genes Dev. 2019, 33, 1098–1116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chuong, E.B.; Elde, N.C.; Feschotte, C. Regulatory activities of transposable elements: From conflicts to benefits. Nat. Rev. Genet. 2017, 18, 71–86. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Slotkin, R.K.; Martienssen, R. Transposable elements and the epigenetic regulation of the genome. Nat. Rev. Genet. 2007, 8, 272–285. [Google Scholar] [CrossRef]
- Martinez, G.; Choudury, S.G.; Slotkin, R.K. tRNA-derived small RNAs target transposable element transcripts. Nucleic Acids Res. 2017, 45, 5142–5152. [Google Scholar] [CrossRef]
- Fultz, D.; Slotkin, R.K. Exogenous Transposable Elements Circumvent Identity-Based Silencing, Permitting the Dissection of Expression-Dependent Silencing. Plant Cell 2017, 29, 360–376. [Google Scholar] [CrossRef] [Green Version]
- Panda, K.; Ji, L.; Neumann, D.A.; Daron, J.; Schmitz, R.J.; Slotkin, R.K. Full-length autonomous transposable elements are preferentially targeted by expression-dependent forms of RNA-directed DNA methylation. Genome Biol. 2016, 17, 170. [Google Scholar] [CrossRef] [Green Version]
- Anderson, S.N.; Stitzer, M.C.; Zhou, P.; Ross-Ibarra, J.; Hirsch, C.D.; Springer, N.M. Dynamic patterns of transcript abundance of transposable element families in maize. Genes Genomes Genet. 2019, 9, 3673–3682. [Google Scholar] [CrossRef] [Green Version]
- Guffanti, G.; Bartlett, A.; Klengel, T.; Klengel, C.; Hunter, R.; Glinsky, G.; Macciardi, F. Novel Bioinformatics Approach Identifies Transcriptional Profiles of Lineage-Specific Transposable Elements at Distinct Loci in the Human Dorsolateral Prefrontal Cortex. Mol. Biol. Evol. 2018, 35, 2435–2453. [Google Scholar] [CrossRef]
- Bendall, M.L.; de Mulder, M.; Iniguez, L.P.; Lecanda-Sanchez, A.; Perez-Losada, M.; Ostrowski, M.A.; Jones, R.B.; Mulder, L.C.F.; Reyes-Teran, G.; Crandall, K.A.; et al. Telescope: Characterization of the retrotranscriptome by accurate estimation of transposable element expression. PLoS Comput. Biol. 2019, 15, e1006453. [Google Scholar] [CrossRef] [Green Version]
- Pasquesi, G.I.M.; Perry, B.W.; Vandewege, M.W.; Ruggiero, R.P.; Schield, D.R.; Castoe, T.A. Vertebrate Lineages Exhibit Diverse Patterns of Transposable Element Regulation and Expression across Tissues. Genome Biol. Evol. 2020, 12, 506–521. [Google Scholar] [CrossRef] [PubMed]
- Warman, C.; Panda, K.; Vejlupkova, Z.; Hokin, S.; Unger-Wallace, E.; Cole, R.A.; Chettoor, A.M.; Jiang, D.; Vollbrecht, E.; Evans, M.M.S.; et al. High expression in maize pollen correlates with genetic contributions to pollen fitness as well as with coordinated transcription from neighboring transposable elements. PLoS Genet. 2020, 16, e1008462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Diez, C.M.; Meca, E.; Tenaillon, M.I.; Gaut, B.S. Three groups of transposable elements with contrasting copy number dynamics and host responses in the maize (Zea mays ssp. mays) genome. PLoS Genet. 2014, 10, e1004298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vicient, C.M. Transcriptional activity of transposable elements in maize. BMC Genom. 2010, 11, 601. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Q.-J.; Li, W.; Li, K.; Nan, H.; Shi, C.; Zhang, Y.; Dai, Z.-Y.; Lin, Y.-L.; Yang, X.-L.; Tong, Y.J.M.P. The Chromosome-Level Reference Genome of Tea Tree Unveils Recent Bursts of Non-autonomous LTR Retrotransposons in Driving Genome Size Evolution. Mol. Plant 2020, 13, 935–938. [Google Scholar] [CrossRef]
- Lanciano, S.; Carpentier, M.C.; Llauro, C.; Jobet, E.; Robakowska-Hyzorek, D.; Lasserre, E.; Ghesquiere, A.; Panaud, O.; Mirouze, M. Sequencing the extrachromosomal circular mobilome reveals retrotransposon activity in plants. PLoS Genet. 2017, 13, e1006630. [Google Scholar] [CrossRef] [Green Version]
- Zemach, A.; Kim, M.Y.; Silva, P.; Rodrigues, J.A.; Dotson, B.; Brooks, M.D.; Zilberman, D. Local DNA hypomethylation activates genes in rice endosperm. Proc. Natl. Acad. Sci. USA 2010, 107, 18729–18734. [Google Scholar] [CrossRef] [Green Version]
- Hsieh, T.F.; Ibarra, C.A.; Silva, P.; Zemach, A.; Eshed-Williams, L.; Fischer, R.L.; Zilberman, D. Genome-wide demethylation of Arabidopsis endosperm. Science 2009, 324, 1451–1454. [Google Scholar] [CrossRef] [Green Version]
- Rodrigues, J.A.; Ruan, R.; Nishimura, T.; Sharma, M.K.; Sharma, R.; Ronald, P.C.; Fischer, R.L.; Zilberman, D. Imprinted expression of genes and small RNA is associated with localized hypomethylation of the maternal genome in rice endosperm. Proc. Natl. Acad. Sci. USA 2013, 110, 7934–7939. [Google Scholar] [CrossRef] [Green Version]
- Martínez, G.; Slotkin, R.K. Developmental relaxation of transposable element silencing in plants: Functional or byproduct? Curr. Opin. Plant Biol. 2012, 15, 496–502. [Google Scholar] [CrossRef]
- Hangauer, M.J.; Vaughn, I.W.; McManus, M.T.J.P.G. Pervasive transcription of the human genome produces thousands of previously unidentified long intergenic noncoding RNAs. PLoS Genet. 2013, 9, e1003569. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Jing, Y.; Liu, X.; Li, Q.; Xue, Z.; Cheng, Z.; Wang, D.; He, H.; Qian, W. Heat stress-induced transposon activation correlates with 3D chromatin organization rearrangement in Arabidopsis. Nat. Commun. 2020, 11, 1886. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lisch, D. Epigenetic regulation of transposable elements in plants. Annu. Rev. Plant Biol. 2009, 60, 43–66. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lanciano, S.; Cristofari, G. Measuring and interpreting transposable element expression. Nat. Rev. Genet. 2020, 21, 721–736. [Google Scholar] [CrossRef] [PubMed]
- Panda, K.; Slotkin, R.K. Long-read cDNA Sequencing Enables a’Gene-Like’Transcript Annotation of Transposable Elements. Plant Cell 2020, 32, 2687–2698. [Google Scholar] [CrossRef] [PubMed]
- Ungerer, M.C.; Kawakami, T. Transcriptional dynamics of LTR retrotransposons in early generation and ancient sunflower hybrids. Genome Biol. Evol. 2013, 5, 329–337. [Google Scholar] [CrossRef] [Green Version]
- Qiu, F.; Ungerer, M.C. Genomic abundance and transcriptional activity of diverse gypsy and copia long terminal repeat retrotransposons in three wild sunflower species. BMC Plant Biol. 2018, 18, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Mascagni, F.; Vangelisti, A.; Usai, G.; Giordani, T.; Cavallini, A.; Natali, L. A computational genome-wide analysis of long terminal repeats retrotransposon expression in sunflower roots (Helianthus annuus L.). Genetica 2020, 148, 13–23. [Google Scholar] [CrossRef]
- Renaut, S.; Rowe, H.C.; Ungerer, M.C.; Rieseberg, L.H. Genomics of homoploid hybrid speciation: Diversity and transcriptional activity of long terminal repeat retrotransposons in hybrid sunflowers. Philos. Trans. R. Soc. B Biol. Sci. 2014, 369, 20130345. [Google Scholar] [CrossRef] [Green Version]
- Vukich, M.; Giordani, T.; Natali, L.; Cavallini, A. Copia and Gypsy retrotransposons activity in sunflower (Helianthus annuus L.). BMC Plant Biol. 2009, 9, 150. [Google Scholar] [CrossRef] [Green Version]
- Santini, S.; Cavallini, A.; Natali, L.; Minelli, S.; Maggini, F.; Cionini, P.G. Ty1 /copia- and Ty3 /gypsy-like DNA sequences in Helianthus species. Chromosoma 2002, 111, 192–200. [Google Scholar] [CrossRef]
- Badouin, H.; Gouzy, J.; Grassa, C.J.; Murat, F.; Staton, S.E.; Cottret, L.; Lelandais-Brière, C.; Owens, G.L.; Carrère, S.; Mayjonade, B. The sunflower genome provides insights into oil metabolism, flowering and Asterid evolution. Nature 2017, 546, 148–152. [Google Scholar] [CrossRef] [PubMed]
- Hübner, S.; Bercovich, N.; Todesco, M.; Mandel, J.R.; Odenheimer, J.; Ziegler, E.; Lee, J.S.; Baute, G.J.; Owens, G.L.; Grassa, C.J.; et al. Sunflower pan-genome analysis shows that hybridization altered gene content and disease resistance. Nat. Plants 2019, 5, 54–62. [Google Scholar] [CrossRef]
- Oberlin, S.; Sarazin, A.; Chevalier, C.; Voinnet, O.; Marí-Ordóñez, A. A genome-wide transcriptome and translatome analysis of Arabidopsis transposons identifies a unique and conserved genome expression strategy for Ty1/Copia retroelements. Genome Res. 2017, 27, 1549–1562. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Faulkner, G.J.; Kimura, Y.; Daub, C.O.; Wani, S.; Plessy, C.; Irvine, K.M.; Schroder, K.; Cloonan, N.; Steptoe, A.L.; Lassmann, T.; et al. The regulated retrotransposon transcriptome of mammalian cells. Nat. Genet. 2009, 41, 563–571. [Google Scholar] [CrossRef] [PubMed]
- Kashkush, K.; Feldman, M.; Levy, A.A. Transcriptional activation of retrotransposons alters the expression of adjacent genes in wheat. Nat. Genet. 2003, 33, 102–106. [Google Scholar] [CrossRef] [PubMed]
- Ito, H.; Gaubert, H.; Bucher, E.; Mirouze, M.; Vaillant, I.; Paszkowski, J. An siRNA pathway prevents transgenerational retrotransposition in plants subjected to stress. Nature 2011, 472, 115–119. [Google Scholar] [CrossRef]
- Atwood, A.; Lin, J.H.; Levin, H.L. The retrotransposon Tf1 assembles virus-like particles that contain excess Gag relative to integrase because of a regulated degradation process. Mol. Cell Biol. 1996, 16, 338–346. [Google Scholar] [CrossRef] [Green Version]
- Chang, W.; Jaaskelainen, M.; Li, S.P.; Schulman, A.H. BARE retrotransposons are translated and replicated via distinct RNA pools. PLoS ONE 2013, 8, e72270. [Google Scholar] [CrossRef] [Green Version]
- Yoshioka, K.; Honma, H.; Zushi, M.; Kondo, S.; Togashi, S.; Miyake, T.; Shiba, T. Virus-like particle formation of Drosophila copia through autocatalytic processing. EMBO J. 1990, 9, 535–541. [Google Scholar] [CrossRef]
- Dodonova, S.O.; Prinz, S.; Bilanchone, V.; Sandmeyer, S.; Briggs, J.A.G. Structure of the Ty3/Gypsy retrotransposon capsid and the evolution of retroviruses. Proc. Natl. Acad. Sci. USA 2019, 116, 10048–10057. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saha, A.; Mitchell, J.A.; Nishida, Y.; Hildreth, J.E.; Ariberre, J.A.; Gilbert, W.V.; Garfinkel, D.J. A trans-dominant form of Gag restricts Ty1 retrotransposition and mediates copy number control. J. Virol. 2015, 89, 3922–3938. [Google Scholar] [CrossRef] [Green Version]
- Chaparro, C.; Gayraud, T.; de Souza, R.F.; Domingues, D.S.; Akaffou, S.; Laforga Vanzela, A.L.; Kochko, A.; Rigoreau, M.; Crouzillat, D.; Hamon, S.; et al. Terminal-repeat retrotransposons with GAG domain in plant genomes: A new testimony on the complex world of transposable elements. Genome Biol. Evol. 2015, 7, 493–504. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mirouze, M.; Reinders, J.; Bucher, E.; Nishimura, T.; Schneeberger, K.; Ossowski, S.; Cao, J.; Weigel, D.; Paszkowski, J.; Mathieu, O. Selective epigenetic control of retrotransposition in Arabidopsis. Nature 2009, 461, 427–430. [Google Scholar] [CrossRef]
- Masuta, Y.; Kawabe, A.; Nozawa, K.; Naito, K.; Kato, A.; Ito, H. Characterization of a heat-activated retrotransposon in Vigna angularis. Breed. Sci. 2018, 68, 168–176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McCue, A.D.; Nuthikattu, S.; Slotkin, R.K. Genome-wide identification of genes regulated in trans by transposable element small interfering RNAs. RNA Biol. 2013, 10, 1379–1395. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cho, J. Transposon-Derived Non-coding RNAs and Their Function in Plants. Front. Plant Sci. 2018, 9. [Google Scholar] [CrossRef] [PubMed]
- Slotkin, R.K. The case for not masking away repetitive DNA. Mob. DNA 2018, 9, 15. [Google Scholar] [CrossRef]
- Parker, M.T.; Knop, K.; Sherwood, A.V.; Schurch, N.J.; Mackinnon, K.; Gould, P.D.; Hall, A.J.; Barton, G.J.; Simpson, G.G. Nanopore direct RNA sequencing maps the complexity of Arabidopsis mRNA processing and m6A modification. eLife 2020, 9, e49658. [Google Scholar] [CrossRef]
- Knyazev, A.; Glushkevich, A.; Fesenko, I. Direct RNA sequencing dataset of SMG1 KO mutant Physcomitrella (Physcomitrium patens). Data Brief 2020, 106602. [Google Scholar] [CrossRef]
- Workman, R.E.; Tang, A.D.; Tang, P.S.; Jain, M.; Tyson, J.R.; Razaghi, R.; Zuzarte, P.C.; Gilpatrick, T.; Payne, A.; Quick, J.; et al. Nanopore native RNA sequencing of a human poly(A) transcriptome. Nat. Methods 2019, 16, 1297–1305. [Google Scholar] [CrossRef] [PubMed]
- Sessegolo, C.; Cruaud, C.; Da Silva, C.; Cologne, A.; Dubarry, M.; Derrien, T.; Lacroix, V.; Aury, J.-M.J. Transcriptome profiling of mouse samples using nanopore sequencing of cDNA and RNA molecules. Sci. Rep. 2019, 9, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rogers, S.O.; Bendich, A.J. Extraction of DNA from milligram amounts of fresh, herbarium and mummified plant tissues. Plant Mol. Biol. 1985, 5, 69–76. [Google Scholar] [CrossRef] [PubMed]
- Ellinghaus, D.; Kurtz, S.; Willhoeft, U. LTRharvest, an efficient and flexible software for de novo detection of LTR retrotransposons. BMC Bioinform. 2008, 9, 18. [Google Scholar] [CrossRef] [Green Version]
- Steinbiss, S.; Willhoeft, U.; Gremme, G.; Kurtz, S. Fine-grained annotation and classification of de novo predicted LTR retrotransposons. Nucleic Acids Res. 2009, 37, 7002–7013. [Google Scholar] [CrossRef]
- Llorens, C.; Futami, R.; Covelli, L.; Dominguez-Escriba, L.; Viu, J.M.; Tamarit, D.; Aguilar-Rodriguez, J.; Vicente-Ripolles, M.; Fuster, G.; Bernet, G.P.; et al. The Gypsy Database (GyDB) of mobile genetic elements: Release 2.0. Nucleic Acids Res. 2011, 39, D70–D74. [Google Scholar] [CrossRef] [Green Version]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [Green Version]
- Kim, D.; Paggi, J.M.; Park, C.; Bennett, C.; Salzberg, S.L. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat. Biotechnol. 2019, 37, 907–915. [Google Scholar] [CrossRef]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R.; Genome Project Data Processing, S. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [Green Version]
- Barnett, D.W.; Garrison, E.K.; Quinlan, A.R.; Stromberg, M.P.; Marth, G.T. BamTools: A C++ API and toolkit for analyzing and managing BAM files. Bioinformatics 2011, 27, 1691–1692. [Google Scholar] [CrossRef]
- Kovaka, S.; Zimin, A.V.; Pertea, G.M.; Razaghi, R.; Salzberg, S.L.; Pertea, M. Transcriptome assembly from long-read RNA-seq alignments with StringTie2. Genome Biol. 2019, 20, 278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quinlan, A.R. BEDTools: The Swiss-Army Tool for Genome Feature Analysis. Curr. Protoc. Bioinform. 2014, 47. [Google Scholar] [CrossRef] [PubMed]
- Kimura, M. A simple method for estimating evolutionary rates of base substitutions through comparative studies of nucleotide sequences. J. Mol. Evol. 1980, 16, 111–120. [Google Scholar] [CrossRef]
- Ma, J.; Bennetzen, J.L. Rapid recent growth and divergence of rice nuclear genomes. Proc. Natl. Acad. Sci. USA 2004, 101, 12404–12410. [Google Scholar] [CrossRef] [Green Version]
- Rice, P.; Longden, I.; Bleasby, A. EMBOSS: The European Molecular Biology Open Software Suite. Trends Genet. 2000, 16, 276–277. [Google Scholar] [CrossRef]
- Neumann, P.; Novák, P.; Hoštáková, N.; Macas, J. Systematic survey of plant LTR-retrotransposons elucidates phylogenetic relationships of their polyprotein domains and provides a reference for element classification. Mob. DNA 2019, 10, 1. [Google Scholar] [CrossRef]
- Li, H. Minimap2: Pairwise alignment for nucleotide sequences. Bioinformatics 2018, 34, 3094–3100. [Google Scholar] [CrossRef]
- Pertea, G.; Pertea, M. GFF Utilities: GffRead and GffCompare. F1000Research 2020, 9. [Google Scholar] [CrossRef]
- Buels, R.; Yao, E.; Diesh, C.M.; Hayes, R.D.; Munoz-Torres, M.; Helt, G.; Goodstein, D.M.; Elsik, C.G.; Lewis, S.E.; Stein, L.; et al. JBrowse: A dynamic web platform for genome visualization and analysis. Genome Biol. 2016, 17, 66. [Google Scholar] [CrossRef] [Green Version]
- Wickham, H. ggplot2. Wiley Interdiscip. Rev. Comput. Stat. 2011, 3, 180–185. [Google Scholar] [CrossRef]
- Gu, Z.; Eils, R.; Schlesner, M. Complex heatmaps reveal patterns and correlations in multidimensional genomic data. Bioinformatics 2016, 32, 2847–2849. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, H.; Boutros, P.C. VennDiagram: A package for the generation of highly-customizable Venn and Euler diagrams in R. BMC Bioinform. 2011, 12, 35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wagih, O. ggseqlogo: A versatile R package for drawing sequence logos. Bioinformatics 2017, 33, 3645–3647. [Google Scholar] [CrossRef] [PubMed] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Accession Number in NCBI | Description | Number of Reads after Quality Filtering |
|---|---|---|
| SRR7691052 | Pistil | 19,498,261 |
| SRR7691053 | Stamen | 21,945,535 |
| SRR7691054 | Ligule | 21,602,172 |
| SRR7691055 | Leaf | 20,908,077 |
| SRR7691059 | NaCl 12 h | 23,081,013 |
| SRR7691051 | NaCl 3 h | 21,727,606 |
| SRR7691057 | Seeds | 22,697,764 |
| SRR7691056 | Roots | 18,190,537 |
| SRR7691047 | PEG 12 h | 20,937,047 |
| SRR7691048 | PEG 6 h | 23,482,045 |
| SRR4996808 | Ovary | 13,636,857 |
| SRR4996851 | ABA leaves | 31,312,818 |
| SRR4996849 | Meja leaves | 20,970,863 |
| SRA | Cultivar |
|---|---|
| SRR10484607 | SAM227 |
| SRR10484608 | SAM060 |
| SRR10484609 | SAM167 |
| SRR10484610 | SAM175 |
| SRR10737894 | SAM210 |
| SRR5140325 | SAM012 |
| SRR5140331 | SAM011 |
| SRR5140336 | SAM010 |
| SRR5140395 | SAM006 |
| SRR5907847 | ann04-nwAR |
| SRR5907848 | ann05-ccNM |
| SRR5907869 | ann01-cwIA |
| SRR5912489 | SAM009 |
| Gene/TE ids | Primers |
|---|---|
| Actin | TTCAACGTTCCCGCCATGTA; GTTCGGCAGTGGTTGTGAAC |
| TE01s125448413 | ATTGGCTTCGATCCATCTCGACG; AGATGTAGGGAAACGGGTGGAGT |
| TE04s156439376 | CACTGTGACTTGTGGACATCCCC; GACGAATCATGCGCTCGGATTTC |
| TE05s178342634 | CCGGGTCAACCTGTCATGGATTT; TGGGCATCCTAAATTGTGTGGCA |
| TE13s189833316 | ACCACTTAGCAGCACAAACTCGT; GGTAACCGACATGCCTTCCTTCA |
| TE05s252167574 | AGCCGTACAGAGACGAAGAGACA; TTTGCCCACCAGGTTGATGCATA |
| TE09s40344039 | GATCTGGAGCATGCGTATGGAGG; GTGGCCGCCTTAGAAGCAATAGA |
| TE04s45676682 | TACCAGCAAGAATTTGAGCGGCT; GGCGGTCACGTATTTCTGCACTA; |
| TE03s54222821 | TAGAACTCTTGCTAGGGCGTGGA; TCTGGGAAGATTTGGTGCAAGCA |
| TE08s71329455 | GATGGGTGATGGTTCGGGTGAAA; CGGACCAAACTTCTGCTGCCTAT |
| Int1/2 (Tyran) | CCAGTCACCAGGATTCTCCC; GATTCGAAATCAGGGAGAATC |
| ORF (Tyran) |
AGGGTGATAGTTCTGGGTCCT; GGAACACAGGGTTAGCTGCT |
| gRNA (Varan) | CTGTTTCAGCCCATACAGCGACT; GGTCCTCTAGAACTTCTGTTGCTCC |
| Gene/TE Id | Primers |
|---|---|
| Tyran | TCACTTGCTTGGAGATATGGGT; TCCTCACTACCCCGACTTCA |
| Gagarin | CGAAGAGGCTACTTGGGAGA; CGGACTGGATTTCTTGCATT |
| TE13s189833316 | CAAAACCCGCTTCAAAGAAA; CAGCCCCTTGTGTCTTCCTA |
| TE05s252167574 | GGTGAGGTTGACGGTGGTAT; GGAGTCGAAACGGAATGTGT |
| TE04s45676682 | GGATTTGTTTGTTTTAATGTGATG; TGGAATTCAGCATTGGTACG |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kirov, I.; Omarov, M.; Merkulov, P.; Dudnikov, M.; Gvaramiya, S.; Kolganova, E.; Komakhin, R.; Karlov, G.; Soloviev, A. Genomic and Transcriptomic Survey Provides New Insight into the Organization and Transposition Activity of Highly Expressed LTR Retrotransposons of Sunflower (Helianthus annuus L.). Int. J. Mol. Sci. 2020, 21, 9331. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21239331
Kirov I, Omarov M, Merkulov P, Dudnikov M, Gvaramiya S, Kolganova E, Komakhin R, Karlov G, Soloviev A. Genomic and Transcriptomic Survey Provides New Insight into the Organization and Transposition Activity of Highly Expressed LTR Retrotransposons of Sunflower (Helianthus annuus L.). International Journal of Molecular Sciences. 2020; 21(23):9331. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21239331
Chicago/Turabian StyleKirov, Ilya, Murad Omarov, Pavel Merkulov, Maxim Dudnikov, Sofya Gvaramiya, Elizaveta Kolganova, Roman Komakhin, Gennady Karlov, and Alexander Soloviev. 2020. "Genomic and Transcriptomic Survey Provides New Insight into the Organization and Transposition Activity of Highly Expressed LTR Retrotransposons of Sunflower (Helianthus annuus L.)" International Journal of Molecular Sciences 21, no. 23: 9331. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21239331