The Regulation of Astrocytic Glutamate Transporters in Health and Neurodegenerative Diseases


1
UK Dementia Research Institute at the University of Edinburgh, Chancellor’s Building, Edinburgh Medical School, Edinburgh EH16 4SB, UK
2
Centre for Discovery Brain Sciences, University of Edinburgh, Hugh Robson Building, George Square, Edinburgh EH8 9XD, UK
*
Author to whom correspondence should be addressed.
Int. J. Mol. Sci. 2020, 21(24), 9607; https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21249607
Submission received: 24 October 2020
/
Revised: 4 December 2020
/
Accepted: 11 December 2020
/
Published: 17 December 2020
(This article belongs to the Special Issue Glial Ion Channels and Transporters in Health and Disease)
Abstract
:The astrocytic glutamate transporters excitatory amino acid transporters 1 and 2 (EAAT1 and EAAT2) play a key role in nervous system function to maintain extracellular glutamate levels at low levels. In physiology, this is essential for the rapid uptake of synaptically released glutamate, maintaining the temporal fidelity of synaptic transmission. However, EAAT1/2 hypo-expression or hypo-function are implicated in several disorders, including epilepsy and neurodegenerative diseases, as well as being observed naturally with aging. This not only disrupts synaptic information transmission, but in extremis leads to extracellular glutamate accumulation and excitotoxicity. A key facet of EAAT1/2 expression in astrocytes is a requirement for signals from other brain cell types in order to maintain their expression. Recent evidence has shown a prominent role for contact-dependent neuron-to-astrocyte and/or endothelial cell-to-astrocyte Notch signalling for inducing and maintaining the expression of these astrocytic glutamate transporters. The relevance of this non-cell-autonomous dependence to age- and neurodegenerative disease-associated decline in astrocytic EAAT expression is discussed, plus the implications for disease progression and putative therapeutic strategies.
1. Introduction
Glutamate is the predominant excitatory neurotransmitter in the brain, activating post-synaptic ionotropic N-methyl-D-aspartate (NMDA) and α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA)/kainate receptors. Co-activation of these receptors allows the influx of Na+ ions, which depolarises the cell’s membrane potential, triggering an action potential. Once released, it is important that glutamate is rapidly cleared, since failure of this has two key consequences. Firstly, it will continue to stimulate the post-synaptic receptors after the initial signal has been sent, impairing the detection of the next signal that arrives—akin to a saturation phenomenon, and potentially leading to cell swelling due to ion influx [1]. Secondly, if this glutamate escapes from the synaptic zone it was released into it could activate unintended synapses, triggering activity where it should not. Significantly, if glutamate escapes the synaptic region it can activate extrasynaptic NMDA receptors: too much Ca2+ influx via these extrasynaptic NMDA receptors induces signalling cascades that initiate cell death programs [2,3].
Astrocytes have long been known to be important in promoting the survival of neurons and for counteracting the toxic effects of glutamate [4,5,6,7]. This protection is largely due to their ability to take glutamate up from the extracellular environment via transporters located on astrocytic membranes, preventing excitotoxicity and associated oxidative stress [8,9]. Once inside the astrocytes, glutamate is then either converted into α-ketoglutarate by glutamate dehydrogenase (GDH) or transaminases and shunted into the astrocytic TCA cycle, or else converted into glutamine by the enzyme glutamine synthetase (GS) [10,11]. Glutamine is not toxic to neurons, and is extruded by the SNAT3 glutamine transporter into the extrasynaptic space, which can then be taken up by neurons and converted back into glutamate via the neuronally expressed phosphate-activated glutaminase (PAG), thus replenishing pre-synaptic glutamate stores [12,13,14,15]. This glutamate recycling pathway is referred to as the glutamate-glutamine cycle (see Figure 1).
Astrocytic glutamate uptake and recycling is a vital part of CNS function. Key within this machinery are the two astrocytic glutamate transporters, excitatory amino acid transporters 1 and 2 (EAAT1 and EAAT2), that are responsible for the bulk of glutamate uptake. The astrocytic glutamate transporters EAAT1 and EAAT2 belong to the solute carrier 1A family of transporters (SLC1A), which includes two alanine serine cysteine transporters, ACST1 and ACST2, along with the five excitatory amino acid transporters, EAAT1-5 [16]. The EAATs are electrogenic secondary-active transporters, using the concentration gradients of their co- and counter-transported ions (Na+, H+, and K+) to drive the transport of glutamate against its concentration gradient into the cell [17]. Astrocytes are able to regulate any fluxes in their pH and membrane potential caused by this transport process through their high expression of membrane K+ channels (especially KIR4.1), their Na+/H+ exchangers and Na+/NCO3− cotransporters, as well as dissipating charge and ionic changes throughout the astrocytic network via connexon coupling (see [18] for a recent review on astrocyte physiology).
Unsurprisingly, reductions in the astrocytic glutamate transporters’ expression and function have been implicated in a number of CNS diseases, particularly epilepsy, along with several neurodegenerative diseases, as outlined below. It is therefore of interest to understand how these transporters are functionally regulated, as boosting their expression and function may offer a novel way to reduce severity and progression of neurodegenerative diseases.
2. Glutamate Transporters in the Brain
Glutamate is found at high concentrations in the brain, at a concentration of approximately 10–14 mmol/L depending on region [19,20]. However, most of this glutamate is kept within intracellular compartments, with very low levels maintained in the extracellular fluid (around 3–4 μmol/L in the extracellular space of the hippocampus) [21,22]. Accordingly, there are numerous transporter proteins in the brain that are capable of facilitating glutamate transport to ensure that the right concentration of glutamate is maintained in the right compartment. These transporters fall into two broad categories: those which are found in intracellular compartments, such as the three vesicular glutamate transporters (vGLUT1-3) which package glutamate into synaptic vesicles, and those located on the plasma membrane of cells that can transport glutamate into (or out of) the cell [1,23,24]. The glutamate transporters that are found in the plasma membranes of brain cells consist of five sodium-dependent co-transporters, and one sodium-independent exchanger [17,25,26]. The sodium-independent exchanger, xCT, is found almost exclusively on astrocytes, but preferentially transports cysteine into the cell in exchange for extruding a glutamate molecule out of the astrocyte [27,28,29]. Due to the need to transport glutamate into cells against its electrochemical gradient, it is therefore the sodium-dependent class of transporters that are responsible for quickly sequestering extracellular glutamate back into cells. There are five known members of this family of transporters, excitatory amino acid transporters 1–5 (EAAT1-5).
2.1. Excitatory Amino Acid Transporters
In the early 1970s, a high affinity sodium-dependent uptake system for the negatively charged amino acids l-glutamate and l-Aspartate was first described in synaptosomal preparations, which was hypothesised to be responsible for the accumulation of the putative excitatory neurotransmitter glutamate into cells [30,31]. A few years later Balcar and colleagues went on to show that this glutamate uptake system was also present in glial cells, but not until 1992 were EAATs first purified, with four independent groups cloning three distinct EAAT family members: Glt-1 (EAAT2), GLAST (EAAT1), and EAAC (EAAT3) [32,33,34,35,36]. The final two members were cloned in 1995 (EAAT4) and 1997 (EAAT5) [37,38].
All members of the EAAT family transport l-glutamate into cells under normal conditions using the electrochemical gradients of Na+ and K+ [1,17]. The EAAT family of transporters and the sodium-independent exchanger xCT both display high affinity for glutamate transport compared to the three vesicular glutamate transporters (KM ≈ 2–100 μM for EAAT1-5 and KM ≈ 20–55 μM for xCT versus KM ≈ 1.5–3.5 mM for vGLUT1-3) [37,38,39,40,41,42,43,44,45,46,47,48,49,50]. Although the EAATs have relatively high affinity for glutamate, enabling them to sequester low concentrations of extracellular glutamate to prevent excitotoxicity, they interestingly have relatively slow transport cycle times (see Table 1). This problem may in part be overcome by rapid surface diffusion and transporter trafficking of the EAATs upon glutamate stimulation [51,52].
The different EAAT subtypes are found throughout the body, and within the brain they are found on different cell types and in different brain regions. Although they all transport glutamate into cells, each subtype possesses a different degree of chloride permeability, and it appears the function of each of these subtypes may vary. A summary of the five EAAT transporters is given in Table 1. Note well, for clarity in this review the transporters encoded by SLC1A3/Slc1a3, SLC1A2/Slc1a2, and SLC1A1/Slc1a1 will be consistently referred to as EAAT1, EAAT2, and EAAT3, respectively, independent of species. Rodent EAAT1, EAAT2, and EAAT3 are typically referred to as GLAST (Slc1a3), Glt-1 (Slc1a2), and EAAC (Slc1a1) in other studies. However, there is high amino acid homology between human and rodent proteins, with 96% identity shared between human EAAT1 and rat GLAST, 95% identity between EAAT2 and rat Glt-1, and 92% identity between EAAT3 and rabbit EAAC1 [39].
2.2. Location of Excitatory Amino Acid Transporters
The two subtypes EAAT1 (i.e., GLAST) and EAAT2 (i.e., Glt-1) are referred to as the astrocytic glutamate transporters as they are the only EAAT subtypes expressed on astrocytes, where they are predominantly found on fine astrocytic processes opposed to glutamatergic synapses [53,54,55]. EAAT1 is found in astroglia (including the Bergmann and Müller glia) throughout the brain, on which they are exclusively expressed, and are the primary collectors of glutamate in both the cerebellum and retina (via Bergmann and Müller glia, respectively) [56,57,58,59,60,61,62]. EAAT2 is the primary glutamate transporter in all other brain regions, most prominently in the hippocampus and cortex [54,57]. The location of EAAT2 is less astrocyte-exclusive, with some evidence suggesting it is also found to a small degree in neurons, particularly in the hippocampus and retina (see Zhou and Danbolt, 2013 for discussion). Combined, EAAT1 and EAAT2 make up a significant proportion of the total protein in the brain, representing ~2.1% of protein in the molecular layer of the cerebellum, 1.6% in the hippocampal stratum radiatum, and 1% of protein in forebrain tissue, and are by far the most abundant EAAT subtypes in the CNS [56].
A more recent study using label-free quantitative (LFQ) tandem mass-spectrometry to survey the regional protein expression of postnatal human brain tissue has confirmed that EAAT1 and EAAT2 are abundantly expressed in all regions sampled: the cerebellar cortex (CBC), mediodorsal thalamic nucleus (MD), striatum (STR), amygdala (AMY), hippocampus (HIP), primary visual cortex (V1C), and the dorsal prefrontal cortex (DFC) [63]. Apart from the cerebellar cortex (where EAAT2 was within the 93rd percentile), EAAT2 was the most abundantly expressed of the transporters across all regions, sitting in the 97th (MD), 98th (STR, AMY, HIP, and DFC), and 99th (V1C) percentile of most abundant proteins within each region. EAAT1 on the other hand was more abundantly expressed in the CBC, being in the 99th percentile of expressed proteins, with consistently high, although lower than EAAT2, expression across other regions: 94th percentile in the AMY; 95th in MD, STR, and HIP; and 97th percentile in the V1C and DFC (calculated from supplementary data Table 10 of Carlyle et al., 2017) [63].
The EAAT3 subtype is exclusively neuronal, and is found on neurons throughout the brain [65,66]. It typically localises on dendritic spines and not axon terminals, with its highest expression seen in the hippocampus, at a concentration of 0.013 mg/g [65]. This is 100 times lower than EAAT2 levels in the same region, so even in the hippocampus it may only contribute modestly to glutamate clearance. Consistent with these earlier reports, the more recent LFQ mass spectrometry data showed a relatively low protein abundance of EAAT3 ranging from the 13th percentile in the V1C to the 26th percentile in the MD, and sitting at just the 21st percentile of abundance in the hippocampus (from supplementary Table 10, Carlyle et al., 2017) [63]. This is compared to EAAT1 and EAAT2 which were in the top percentiles of expressed proteins across all regions in these samples (>90th percentile for both EAAT1/2 in all regions) [63].
EAAT4 is another neuronally expressed glutamate transporter, however its expression profile is more restricted than that of EAAT3, being found primarily on Purkinje cells of the cerebellum, with some sparse expression in certain subregions of the forebrain and midbrain [67,68]. It represents about 0.2% of protein in the molecular layer of the cerebellum, which is approximately 10 times less than the predominant astroglial subtype in this region, EAAT1 (which represents 1.8% of total cerebellar protein), and similar to EAAT2 levels (0.3% of total protein) [56,67]. Again, consistent with these earlier findings, Carlyle and colleagues more recent mass spectrometry data revealed EAAT4 to be most abundantly expressed in the cerebellar cortex, in the 89th percentile of proteins. This is less than both EAAT1 and EAAT2 in this region, but significantly higher than EAAT4′s expression across all other regions (8th percentile in HIP, 10th in V1C, 14th in STR and DFC and 19th in MD and AMY), confirming that EAAT4 is primarily restricted to the cerebellum [63]. The final member of the family, EAAT5, has only been found in the eye, where it is located on synaptic terminals of retinal rod bipolar cells as well as rod and cone photoreceptors [38,69]. Again, the astrocytic EAAT1 subtype is expressed more strongly in the retina (located on Müller glia) than EAAT5, and there is evidence that EAAT5 may physiologically act as a chloride channel rather than a glutamate transporter in these retinal neurons [70,71].
2.3. Structure and Function of Excitatory Amino Acid Transporters—The Case for Astrocytic EAAT1 and EAAT2
Given that the glutamate concentration within cells is over 1000-fold higher than that in the extracellular space (≈10 mmol/L compared to 4 μmol/L), combined with the fact that glutamate is an anion carrying −1 charge, transporters responsible for sequestering glutamate must overcome both concentration- and electrochemical gradients. All EAATs support the transport of l-glutamate as well as d and l-Aspartate, displaying a relatively high affinity for inward l-glutamate transport, with reported KM for glutamate ranging from 10–100 μM (see Table 1) [38,39,72]. This high affinity allows the transporters to continue to work efficiently to uptake glutamate under the low concentrations of the seen in the extracellular space. However, the times to complete one transport cycle of a glutamate molecule for the EAATs are relatively low, from 10 ms for EAAT3, 60–70 ms for EAAT1 and 2, >100 ms for EAAT4 and >1000 ms for EAAT5 [40,41,43,44,45]. Although this may seem a limitation as glutamate is required to be quickly cleared, the transporters (especially EAAT1 and EAAT2) are some of the most abundantly expressed proteins in the CNS (see Table 1), and furthermore appear to undergo rapid surface diffusion and shuttling, helping to overcome transporter saturation and slow transport cycle times [51].
The mechanism of EAAT transport was debated for some time, but it has now been established that the EAATs combine the transport of 1 glutamate molecule with the co-transport of 3 Na+ and 1 H+, whilst counter-transporting 1 K+ [32,73,74,75]. This has been supported by more recent studies using the homologous archaeal glutamate transporters GltPh and GltTk whose structures were crystallised in 2004 and 2013 [76,77,78,79]. As a result of this stoichiometry, there is a net +2 charge per molecule of glutamate transported, facilitating the inward movement of the otherwise negatively charged glutamate by using the Na+ and K+ electrochemical gradients to drive transport into the cell. This stoichiometry is estimated to allow the internal glutamate concentration to be in the order of 106 times greater than the external concentration under physiological conditions, ensuring the transporters work to take up rather than extrude glutamate under normal conditions where the concentration differential is only in the order of 103 [1,19,20,22]. The electrogenic nature of EAAT function also enables their function to be measured by both patch clamp and two-electrode voltage clamp electrophysiology [80,81,82]. Although this transporter stoichiometry is believed to be common to all members of the EAAT family, meaning all EAATs could help facilitate glutamate clearance, there is a difference between the EAATs. As well as functioning as a transporter, EAATs can also act as ligand-gated ion channels, with glutamate activation leading to an uncoupled conductance of Cl− through the channel [37,40,41,83]. However, the level of anion conductance varies largely between the different subtypes [84]. EAAT4 and EAAT5 display the largest ion conductance, with their Cl− conductance being greater than that of their glutamate uptake, EAAT1 and EAAT3 have intermediate ion conductance, while EAAT2 displays very little conductance at all [26,37,38,41]. Recent work has suggested that EAAT5 uses this anionic conductance to act as an “inhibitory” glutamate receptor in retinal cells, hyperpolarising these cells’ membrane potentials following glutamate activation [71,85,86]. It is likely that both the EAAT4 and EAAT5 subtypes do not physiologically function as glutamate uptake systems, but instead act as Cl− channels.
The first support for astrocytic glutamate transporters EAAT1 and EAAT2 being primarily responsible for glutamate clearance rather than neuronal subtypes came from studies using autoradiographic localization, which found the bulk of cleared glutamate was seen in glial cells [87,88]. Electrophysiological recordings later went on to show that this astrocytic glutamate clearance was mediated by EAAT1 and EAAT2 [89,90]. Final evidence that it is the astrocytic EAAT1 and EAAT2 subtypes, and not the neuronal EAAT3, that are responsible for glutamate clearance comes from knockout studies.
An EAAT2 knockout animal was generated in 1997, which developed lethal seizures resulting in an 80% death rate by 13 weeks of age, compared to 100% survival in controls [91]. It was found that there was a slower clearance of synaptically released glutamate in knockout animals, with neuronal degeneration appearing specifically in the hippocampal CA1 region [91]. In 1998, the group went on to generate an EAAT1 knockout animal [92]. Unlike the EAAT2 knockout, removal of EAAT1 did not appear to be lethal, and brain development appeared normal. The group focused on the cerebellum, given that is EAAT1′s prominent region of expression, and found that in EAAT1 knockouts glutamate uptake in this region was nearly half that of wild-types. Although finding no difference in basic motor tasks, they found a significant impairment in the knockouts’ ability to complete a more challenging rotor-rod experiment. Furthermore, they found that mutant EAAT1 animals, but not wild-types, were susceptible to cerebellar edema following cold injury [92]. Inevitably, in 2006 the group reported on a double EAAT1/EAAT2 knockout animal. Unlike the single mutants, the double knockout of EAAT1 and EAAT2 was embryonic lethal, with mice dying by E17-18, and brain-wide abnormalities in structure observed [93]. These studies highlight the vital importance of these transporters in the CNS.
In 1997, a second group generated a mouse knockout for the neuronal glutamate uptake transporter, EAAT3 [94]. Contrary to the neurological deficits seen in EAAT1 or EAAT2 knockout animals, removal of EAAT3 had no negative effect on brain formation or function over a period of >12 months. There was no impairment in motor skills, nor in memory, nor in susceptibility to induced seizures [94]. A limitation of all these studies, particularly for the EAAT2 knockout, is that they utilised global knockout models, and not astrocyte specific. As EAAT2 is reportedly expressed on neurons, this does not rule out neuronal EAAT2 glutamate uptake as being an important source of glutamate clearance to prevent excitotoxicity. Addressing this limitation, Rosenberg and colleagues produced conditional neuronal and conditional astrocytic EAAT2 knockout lines [95]. Whilst neuronal knockouts showed no difference in growth and lifespan, astrocytic EAAT2 mutants had lower weight gain and significantly higher mortality rates compared to controls [95]. In proteo-liposome preparations from forebrains they found glutamate uptake in astrocytic mutants was 25% of that in controls, whereas there was no difference in glutamate uptake in these preparations between neuronal knockouts and controls. EEG recordings further showed astrocytic EAAT2 knockouts to have significantly more seizure events than controls, with no difference between the conditional neuronal knockouts and controls [95].
Altogether, the evidence shows that it is EAAT1 and EAAT2 expressed on astrocytes that are primarily responsible for clearing extracellular glutamate to prevent excitotoxicity. The stoichiometry of the EAAT transporters provides one explanation for why astrocytes and not neurons are primarily responsible for glutamate homeostasis: uptake can result in significant depolarisation (up to 2+ charge per molecule). If neurons were required to take up the bulk of released glutamate, this process in itself would cause significant neuronal depolarisation, potentially leading to a hyper-excitable feedback loop. Additionally, uptake would result in a significant increase in internal Na+ concentration, which is counterproductive to the neuron’s need to remove internal Na+ following an action potential and could represent a metabolic strain on the neurons and impede their ability to sustain action potential firing.
3. Astrocytic EAAT Regulation
Before the different EAAT isoforms were isolated, it was observed that culturing cerebellar astrocytes in the presence of cortical neuronal conditioned media increased glutamate uptake [96]. Over a decade later, astrocytic EAAT1 and EAAT2 were first found to be significantly downregulated in the striatum following glutamatergic denervation, and the following year it was reported that EAAT1 was upregulated in cortical astrocyte cultures following activation of astrocytic AMPA and kainate receptors [97,98]. These reports implicated a role for neurons and neuronal activity in regulating astrocytic glutamate transporters. Following on from this work, Swanson and colleagues cultured cortical astrocytes alone or in the presence of cortical neurons, finding that isolated astrocytes expressed very low levels of EAAT1 and EAAT2, that was robustly induced by neuronal co-culture [99]. It was further documented by Gegelashvili and colleagues that physical culture of neurons with cortical astrocytes increased astrocytic EAAT1 expression, as well as inducing EAAT2 expression [100]. Additionally, the group showed that feeding pure cortical astrocytes with neuronal conditioned media was able to induce EAAT2 expression in astrocytes, although they did not find EAAT1 to be affected by conditioned media. They concluded from this work that a neuronally released soluble factor was responsible for the regulation of EAAT2, whereas contact mediated interactions were predominant in the regulation of EAAT1 [100]. Interestingly, this group had earlier reported that glutamate was able to increase cortical astrocytic EAAT1 expression, suggesting soluble factors could play a role in regulating this transporter as well [98]. Work since then has focused on discovering the signalling molecules and pathways behind this neuronal regulation (see Table 2 for overview, references: Hardingham lab unpublished data, [97,98,99,100,101,102,103,104,105,106,107,108,109]).
3.1. Regulation of EAAT Expression by Soluble Factors
3.1.1. Cyclic AMP Signalling
One of the first chemicals shown to induce glutamate transporter function in astrocytes was the cyclic AMP analogue, dibutyryl cyclic AMP (db-cAMP) [110]. Primary astrocytes grown alone showed little response to glutamate and appeared flat, whilst astrocytes fed with db-cAMP became more morphologically complex with significantly greater glutamate uptake [98,110,111]. As a result, many researchers began to treat astrocyte cultures with db-cAMP as standard practice, to make them more reminiscent of their in vivo counterparts. This suggests that one potential mechanism for the neuronal regulation of astrocytic glutamate transporters is through an induction of the astrocytic cAMP signalling pathway.
Further evidence for a role of cAMP signalling in astrocytic EAAT regulation comes from studies showing that application of the adenyl cyclase activator forskolin, to stimulate cAMP production, to both astrocyte cultures and striatal homogenates is able to increase glutamate uptake [102,112]. The mechanism behind this cAMP induced upregulation is less clear, with one group finding inhibition of cAMP’s downstream target of protein kinase A (PKA) to be sufficient to block the effects of forskolin, whilst others found an effect of PKA inhibition in pure astrocyte cultures but no effect on astrocyte glutamate transporter function when grown in the presence of neurons [102,112]. The latter finding suggests that although PKA activation may upregulate EAAT1 and EAAT2 activity in astrocytes in the absence of neurons, in the presence of neurons this pathway is occluded by other mechanisms that are responsible for the observed neuronal regulation of astrocytic EAATs. Also unclear is what neuronally derived factor could be responsible for astrocyte cAMP elevation, since astrocytes express many adenylate cyclase-activating Gs-coupled receptors. Indeed, different ones may play a role in different circumstances.
3.1.2. Glutamatergic Signalling and EAAT Trafficking
Despite early reporting that astrocytic AMPA and kainate receptor activation may upregulate EAAT expression, and that denervation decreases EAAT2 expression, there still lacks a consensus as to whether glutamatergic synaptic activity has a role in astrocytic EAAT expression. Both work from our lab and others have found no effect of pharmacological blockade of neuronal activity on astrocytic EAAT expression [101,102]. Contrary to these findings, it has been reported that in hippocampal astrocyte-neuron co-cultures pharmacological block of synaptic activity does reduce protein levels of both EAAT1 and EAAT2 [113]. Furthermore, acute kainate injections to induce seizure activity in rats were seen to initially cause a significant increase in cortical EAAT2 expression, peaking after 4 h, before ultimately decreasing below baseline levels (following neuronal death) [114].
Although glutamatergic signalling’s role in regulating EAAT expression is not yet clear, it has been observed that glutamate treatment can increase functional astrocytic glutamate clearance, and that this increase in function is mediated directly by the activation of the astrocytic glutamate transporters, rather than glutamate receptors [115]. The mechanism for this glutamate mediated increase in EAAT function was found to be due to an induction of EAAT1 surface expression in mouse astrocytic cultures, with no change in total transporter protein expression [115]. More recently, it has been reported by two groups using quantum dot tracking that glutamate treatment increases astrocytic glutamate clearance via robustly increasing the surface diffusion of EAAT2 in rodent astrocytes from primary culture, organotypic hippocampal slice and acute slice preparations [51,52]. This increase in diffusion, especially in the synaptic region, leads to a faster turnover of unoccupied transporters thereby enabling more efficient clearance of the perisynaptic glutamate. Indeed, immobilising the diffusion of EAAT2 was found to alter synaptic kinetics, increasing both the rise and decay time of spontaneous excitatory postsynaptic currents, although total glutamate clearance was unaltered [51]. As well as surface diffusion, EAATs also undergo surface trafficking, with glutamate triggering Ca+-dependent internalization of EAAT2 through endocytosis [116,117]. On the other hand, surface expression of EAAT1 has been shown to be increased by glutamate application, as well as insulin-like growth factor through phosphatidylinositol-3-kinase signalling, whereas the ubiquitin ligase neuronal developmentally downregulated gene 4, isoform 2 (Nedd4-2) has been shown to decrease EAAT1 surface expression [115,118,119]. These dynamic mechanisms for regulating EAAT surface expression are likely to have a significant impact on the functional glutamate clearance speed and capacity of astrocytes, especially given the EAATs rather slow transport cycle times.
3.1.3. Other Secreted Signals
The EAAT2 transporter is able to be regulated by neuronal secreted factors, and much of the work investigating EAAT regulation has been focused on EAAT2 in particular [100]. Epidermal growth factor application has been shown to upregulate EAAT2 expression through activation of NF-κB signalling, with neuron-dependent induction of astrocytic NF-κB having been shown to upregulate astrocytic EAAT2 expression [103,104,105]. Additionally, enhanced expression of Pax6, a well characterized transcription factor in astrocytes and the CNS [120,121], in pure astrocyte cultures has been recently shown to induce EAAT2 expression, while knockdown of Pax6 in astrocytes grown with neurons was seen to strongly repress neuron-induced EAAT2 expression [106]. However, the authors do not speculate upon the neuronally released factor(s) that may modulate EAAT2 expression through astrocytic Pax6.
3.2. Contact Dependent Regulation: Notch Signalling
In contrast to neuronally released factors, relatively little work had investigated the role of contact-dependent signalling pathways in neuronal control of astrocytic EAAT1 and/or EAAT2 expression. It has been reasonably well established that unlike EAAT2, neuronal upregulation of EAAT1 expression is via a contact-dependent mechanism and not through a soluble factor, but the mechanism remained unclear [122]. Furthermore, it had not been established if contact-dependent signalling also has a role in EAAT2 regulation.
Notch is an important contact dependent signalling pathway present in astrocytes; in fact, it is the interaction of Notch ligands expressed on neuronally committed precursor cells with uncommitted precursors that first initiates the precursors’ development into astrocyte lineage cells [123]. An overview of the Notch signalling pathway is shown in Figure 2. Briefly, when Notch ligands (for example Delta and Jagged1 & 2) contact the receptors (Notch1-4) the receptors undergo cleavage by the enzyme γ-secretase, releasing the Notch intracellular domain (NICD) of the receptor. The NICD then translocates into the cell nucleus, where it associates with the Notch effector (CBF1) and Mastermind-like (MAML) to activate transcription, with the Hes and Hey family of genes being well-established examples of NICD/CBF1 target genes [124,125].
In drosophila only the EAAT1 subtype of high affinity glutamate transporters are found, where it is located on glia cells. Using this model system it was observed that Notch signalling mediated by neuronally expressed Delta ligands induced the expression of EAAT1 in glia cells [107]. If this is a conserved process, these results could suggest a role for Notch signalling not only in allowing astrocyte cell type differentiation, but also in inducing astrocytic EAAT expression.
Strengthening the case for Notch, our lab recently demonstrated using a mixed-species culture model that neuron-to-astrocyte Notch signalling was a major regulator of astrocytic Slc1a2 (EAAT2) and Slc1a3 (EAAT1) expression, functionally boosting glutamate transporter activity in mouse astrocytes [101]. We also found, by expressing a constitutively active form of CBF1, that driving canonical Notch signaling is sufficient to induce glutamate uptake capacity in astrocytes [101].
Around the same time, another group found that endothelial cells were likewise able to induce EAAT1 and EAAT2 expression in mouse astrocytes through contact dependent Notch signalling [108]. The group has now gone on to show that the endothelial Notch ligands responsible for inducing the astrocytic Notch signalling pathway, and downstream increases in astrocytic Slc1a2 and Slc1a3 expression, are the two Delta-like Notch ligands, Dll1 and Dll4 [109].
Interestingly, a link between cAMP signalling and Notch in astrocytes has been reported [126]. Application of db-cAMP was observed to increase the amount of NICD that translocated into the cell nucleus, and that either application of the γ-secretase inhibitor DAPT to block NICD cleavage, or application of the PKA inhibitor H89 to prevent cAMP mediated PKA signalling, was sufficient to prevent this cAMP induced increase [126]. They confirmed that db-cAMP was able to induce Notch transcription, first by showing increased CBF1 activity via a luciferase assay. They then demonstrated that db-cAMP treatment increased both Hes5 gene and Hes5 protein expression, which was also prevented by inhibition of either Notch (via DAPT application) or PKA (via H89) signalling [126]. This suggests the possibility that neuron contact-dependent and -independent signaling may converge on the Notch pathway, with neuron secreted factors that boost cAMP signaling, potentiating Notch signaling.
Is Ongoing Notch Signalling Required to Maintain Glutamate Uptake Capacity?
Having found that Notch signalling was required to functionally increase the activity of the astrocytic glutamate transporters, a key conceptual question remained for us: is this signalling required to maintain expression throughout development, or is EAAT expression set after their initial induction? If constant Notch signalling is required to maintain EAAT activity, then faulty neuronal Notch signalling could be a cause of impaired astrocytic glutamate clearance and increased excitotoxicity.
To investigate this possibility, we set up a mature coculture paradigm, growing established mouse astrocytes in the absence (monoculture, MC) or presence (coculture, CC) of rat neurons for 24 days (to neuron DIV24). The γ-secretase inhibitor DAPT was then applied to some cocultured cells from the point of neuronal plate down (DIV0) or else after two weeks of astrocyte-neuron coculture (DIV14) to block Notch signalling either from the outset or after the establishment of functional astrocytic glutamate transporters had occurred. Astrocyte glutamate transporter function was assessed by electrophysiological recording of astrocytic EAAT currents both on the 13th day of coculture with neurons in untreated astrocytes to confirm EAAT functional establishment, and then again on DIV24 across all conditions.
We found that after two weeks exposure to neurons (DIV13) there was robust induction of astrocytic glutamate transport activity in astrocytes (Figure 3i). As expected, by DIV24 there was significantly greater EAAT activity in astrocytes cultured in the presence of neurons compared to astrocytes cultured without neurons (Figure 3ii,iii). The transport activity of cocultured astrocytes that had Notch inhibited from the outset was (as expected) lower than untreated cocultured cells, although some residual transport activity was seen (Figure 3iv). Most significantly, the EAAT transport activity in cocultured astrocytes that had Notch signalling inhibited on DIV14 of coculture (i.e., after the induction of EAAT function) was indistinguishable from those cells who had Notch signalling inhibited from the outset (Figure 3v,vi, Hardingham lab unpublished data). These data support a model whereby continuous neuron-to-astrocyte Notch signalling is required in order to maintain astrocytic glutamate transporter activity. This is consistent with the observation that in astrocytes isolated ex vivo both EAAT1/2 expression and Notch target genes decline in culture compared to their levels immediately post-isolation [101,127].
3.3. Epigenetic Regulation of EAATs
There is a growing interest into the role of epigenetic regulation of glutamate transporter expression in astrocytes, in particular the regulation of EAAT2. Astrocytes express both a number of epigenetic ‘writers’—enzymes capable of DNA and histone protein modification usually through the processes of methylation or acetylation, and ‘erasers’—enzymes capable of removing the epigenetic marks—i.e., demethylation and deacetylation—allowing for the epigenetic control of astrocytic gene transcription [128,129,130,131,132,133]. With respect to the glutamate transporters, it has been shown that overexpression of the epigenetic ‘erasers’ histone deacetylase (HDAC) 1, 3, 6, and 7 all reduce EAAT2 promotor activity, which can be reversed by treatment with HDAC inhibitors [134]. There are suggestions that altered epigenetic regulation of EAAT expression may be a factor in various diseases. For example it has been observed that in tissue samples from malignant glioma tumors there is a near total absence of astrocytic EAAT2 expression, which was explained by the pronounced hypermethylation of the EAAT2 promotor, resulting in the abolishment of EAAT2 transcription [135,136]. Treatments to inhibit both DNA methyltransferase (an epigenetic ‘writer’ that causes epigenetic transcriptional repression) and HDAC (which likewise leads to transcriptional repression) were successfully able to increase astrocytic EAAT2 expression in these glioma cell lines [135]. These findings raise the possibility of targeting epigenetic mechanisms as a way of controlling astrocytic EAAT expression in the context of disease [137].
4. Astrocytic EAAT in Ageing and Neurodegenerative Disease
As noted above, disruption to glutamate homeostasis has the capacity to disturb synaptic transmission, and by extension, synaptic connectivity and synaptic plasticity of both the classical and homeostatic type. [138,139,140,141], although direct evidence so far extends to Hebbian, spike timing-dependent plasticity [142]. Given the importance of plasticity in shaping circuits in development, one can envisage that even mild disruption to astrocyte-mediated glutamate homeostasis may contribute to deficits in early life brain disorders. Indeed, EAAT2 variants have been associated with cerebral palsy in pre-term infants [143], and with grey matter deficits and working memory in schizophrenia, along with altered EAAT1 and EAAT2 mRNA expression seen in some CNS regions of schizophrenic patients [144,145,146]. Alterations in astrocytic EAAT expression have further been associated with attention deficit hyperactivity disorder [147,148], autism [147,149], and depressive illnesses [150,151,152]. Additionally, given the protective effects of transient, phasic synaptic NMDAR activation, [153,154], perturbations which interfere with this are likely to be deleterious. Moreover, aberrant glutamate homeostasis leads to toxic extrasynaptic NMDAR activation, implicated in the pathophysiology of a number of disorders [155], including stroke [156], epilepsy [157,158], Huntington’s disease [159,160], and Alzheimer’s disease [161,162,163,164]. Though pharmacological inhibition [165], altering synaptic/extrasynaptic signaling balance [166,167], or uncoupling extrasynaptic NMDARs from downstream cascades [168,169] can mitigate these effects, it is important to understand the situations that lead to glutamate dyshomeostasis in the first place. A number of accounts have linked astrocytic glutamate transporter dysfunction both to epilepsy and the neurodegenerative diseases, such as amyotrophic lateral sclerosis (ALS), multiple sclerosis, Alzheimer’s disease, and Parkinson’s disease (see [170] for a recent review of EAATs in diseases of the CNS). An overview of diseases and disorders that aberrant expression and function of the two astrocytic glutamate transporters have been associated with is provided in Table 3. Interestingly, there have been recent suggestions that decreases in astrocytic EAAT expression may also be a natural feature of ageing in humans.
4.1. Epilepsy and EAATs
The first demonstration that increased glutamate concentrations are a feature of epilepsy came from the results of an in vivo microdialysis investigation into the concentrations of GABA and glutamate in the hippocampi of epilepsy patients from 1989 to 1992 [211]. The investigators found that an increase in glutamate concentration appeared in the epileptogenic hippocampus approximately 1.5 min prior to seizure onset, but not in the contralateral hippocampus. At the onset of seizure glutamate levels became further elevated, with concentrations in glutamate also beginning to increase in the opposing hippocampus. Ten minutes post seizure, the non-epileptic hippocampus glutamate concentrations had returned to baseline, whereas the epileptic side had persistently elevated glutamate levels >15 min post seizure [211]. The patients went on to receive surgical resection of the epileptic hippocampus, with microscopy of the removed tissue revealing moderate to severe pyramidal neuron loss throughout the hippocampal tissue along with reactive gliosis [211].
Given the deleterious effects of seizure-induced glutamate elevation in epilepsy, later work investigated the potential role of the glutamate transporters. A reduction in astrocytic EAAT2 expression was found in patients with temporal lobe epilepsy (TLE) that went on to develop hippocampal sclerosis, but no change was found in EAAT2 expression in patients without neuronal loss [188,189]. More recently, it was reported that there was a decrease in both EAAT1 and EAAT2 in epileptic hippocampi of patients with intractable treatment-resistant TLE [190]. It is not known if reduced astrocytic glutamate transporter function initiates some epileptic disorders or exacerbates it, or even if it is simply an outcome of prolonged disease in humans. However, animal models have shown that removal of functional astrocytic EAAT2, and not neuronal EAAT3, is sufficient to cause lethal epilepsy, demonstrating the possible contribution of astrocytic glutamate transporter dysfunction or hypo-expression in the development of certain epileptic disorders [91,212].
4.2. Neurodegenerative Diseases and EAATs
An increasing amount of research has gone into investigating the role of astrocytes and their glutamate clearance in different neurodegenerative diseases, with some links to disease emerging. One example is in amyotrophic lateral sclerosis (ALS), a motor neuron disease characterised by progressive loss of motor neurons in the motor cortex, somatosensory cortex and spinal cord, with around 90% of cases occurring sporadically and 10% with familial linkage. From 1992, it was discovered that impaired glutamate uptake in motor regions was a feature of tissue samples from patients with sporadic ALS, and in 1995 that there was a pronounced reduction specifically in EAAT2 protein levels in these tissue samples [180,181]. Additionally, one patient with sporadic ALS was found to have a mutation in the SLC1A2 gene that resulted in an EAAT2 protein with reduced glutamate transporter activity, suggesting EAAT2 dysfunction may cause some cases of disease [182]. Familial forms of ALS on the other hand were found to be associated with mutations in the superoxide dismutase gene (SOD1), with SOD1 mutant protein being shown to reduce functional EAAT2 protein levels in animal models by initiating the cleavage of EAAT2 by Caspase-3 [183,184,185,186]. Specific deletion of Slc1a2 in the spinal cord of mice was recently shown to be sufficient to lead to motor neuron degeneration by the fifth month of the mice’s lives [187]. Also of note, motor neurons carrying the C9ORF72 expansion are also vulnerable to glutamate excitotoxicity [213], underlying the importance of glutamate homeostasis in this disorder. Finally, in work with our collaborators we have found that in the P301S tauopathy model mouse [214] which results in motor neuron loss of the spinal cord, there is an approximate 35% decrease in Slc1a2 expression in disease mice [101]. It is unclear if it is reduced EAAT function that leads to initial motor neuron death, or if it is the death of neurons that leads to the decrease in functional EAAT (perhaps due to reduced contact-dependent Notch signalling), although a combination of both may be responsible.
Glutamatergic excitotoxicity has been suggested to play a role in PD and other synucleinopathies, with elevated glutamate levels hypothesized as a potential trigger for dopaminergic cell death. Most studies into EAAT dysfunction in PD have focused on animal models, however one human study found that there was reduced glutamate uptake in platelets derived from PD patients [197]. Many rodent models of PD have shown decreases in both EAAT1 and EAAT2 function and expression, including in the PD mutation TJ-1 mouse model, MPTP injection and 6-ODHA lesion models [195,196,198]. However, some animal models have seemingly found the opposite, including one recent study which found that application of pathological α-synuclein oligomers to mice in vivo caused an increase in EAAT1 and EAAT2 expression that persisted into the timepoint when Parkinsonian phenotypes occurred [194]. No functional data for glutamate uptake in this in vivo setting was provided, and it would be interesting for future studies to investigate glutamate transporter function in this scenario, as the increase in expression may not necessarily be accompanied by an increase in functional uptake. One other possibility for these incongruous results is that astrocytes initially upregulate EAATs in response to α-synuclein oligomers (or their effect on neuronal activity) to try and reduce the excitotoxic effects, but over time with disease this upregulation is not sufficient, leading to neuronal loss and eventual reductions in EAAT expression.
As with motor neuron disease, there is interest in the role that glutamate dysregulation may play in the progression of neurodegeneration in Alzheimer’s disease (AD) and other dementias. From the 1990s, it was observed that amyloid β, the main protein found in AD-associated plaques, reduced the function and expression of EAAT1 and EAAT2 in rat hippocampal and cortical astrocytes [171,172,173]. Studies of human tissue samples have found the aberrant expression of both the normally astrocyte-specific EAAT1 transporter and the enzyme glutamine synthetase in subsets of cortical pyramidal neurons of AD patients, suggesting a marked dysfunction in astrocyte glutamate metabolism [174,175]. Reduced expression of both EAAT1 and EAAT2 have further been observed in the hippocampi of patients with AD, alongside a significant decrease in glutamate transporter function in human AD cortices [176,177]. Altogether, accumulated evidence suggests that impaired astrocytic glutamate recycling in the hippocampus and cortex is a feature of dementias and may play a role in the pathological progression of these disorders.
4.3. Ageing and EAATs
It appears that there may be a natural decline in the expression of astrocytic EAAT transporters with age. From a dataset produced by Barres and colleagues in 2016, where they reported the gene expression in astrocytes purified from healthy human CNS tissue (subjects ranging from 8 to 63 years old), the mean expression of the astrocytic glutamate transporters SLC1A2 and SLC1A3 was approximately 35% lower in the six samples from subjects >40 years old compared to the six samples from subjects < 40 years old (see Table 4; data from Zhang et al., 2016, Supplementary Table S6 [129]). A decrease in astrocytic (EAAT1 and EAAT2), but not neuronal (EAAT3) protein has further been observed as a feature in aged rats compared to younger animals, along with a decrease in Slc1a2 expression seen in aged animals [215,216,217]. Thus, reduction in glutamate uptake capacity in ageing may contribute to it being by far the most important risk factor in neurodegenerative diseases.
5. Notch Signalling in Ageing and Disease
If impaired glutamate clearance is a feature of age and disease, and Notch signalling is required to maintain astrocytic EAAT function, then could impaired Notch signalling also be a feature of disease and ageing? A key enzyme in the Notch signalling pathway is the γ-secretase enzyme complex, which is responsible for the cleavage of the Notch intracellular domain, a required step for the induction of the Notch transcriptional pathway. The γ-secretase enzyme complex is itself comprised of four membrane proteins: presenilin (PSEN1 and PSEN2), nicastrin (NCSTN), anterior pharynx-defective 1 (APH1A and APH1B), and presenilin enhancer 2 (PSENEN) [218]. Impairments in γ-secretase function have long been associated with Alzheimer’s disease. As well as γ-secretase’s role in cleaving the NICD, it is also involved in cleavage of the amyloid precursor protein (APP); increased cleavage of APP into toxic Aβ fragments leads to the production of amyloid plaques, a characteristic feature of AD [219]. As it turns out, mutations that cause familial early onset AD have predominantly been found to be in the presenilin component of the γ-secretase complex, PSEN1 and PSEN2 [220,221]. These mutations alter the activity of γ-secretase and cause an increase in production of toxic Aβ fragments.
An early human AD drug trial was run on the broad spectrum γ-secretase inhibitor semagacestat, with the belief that preventing toxic Aβ and amyloid production would improve disease outcome. However, the opposite occurred: inhibiting γ-secretase worsened AD progression, with this worsening eventually attributed to the incidental inhibition of Notch signalling [222,223]. In the light of recent findings, it is a possibility that one of the consequences of Notch inhibition for the CNS was reduced glutamate uptake capacity. Of note, presenilin mutations that give rise to early onset AD and increased pathogenic Aβ production are concurrently found to have a significantly reduced ability to cleave the NICD, suggesting that although mutant presenilin might enhance production of toxic Aβ it also results in loss of Notch activation [224,225,226,227]. Again, this raised the possibility that reduced EAAT1/2 expression may be a feature of these early onset AD variants.
Finally, the same human dataset that saw a reduction in EAAT expression with age likewise found a significant reduction in Notch pathway genes (including HES5 γ-secretase components) in aged human samples, as shown in Table 4 [129]. This raises the possibility that natural reductions in Notch signalling with age in humans could engender vulnerability towards reduced EAAT expression, increased excitotoxicity, and ultimately neurodegeneration.
6. Concluding Remarks
There is emerging evidence that there are reductions in both astrocytic Notch signalling and astrocytic glutamate transporter expression with age in humans, and that this may be further exacerbated in neurodegenerative disease [129,176]. As it appears that Notch signalling is required in order to maintain EAAT expression levels in astrocytes (albeit in a rodent model), it is unsurprising that the reductions in EAAT expression in ageing appear alongside reductions in Notch activity. It is also intuitive that there should be further reductions in both Notch signalling and EAAT expression in astrocytes in neurodegenerative disease, as loss of neurons will lead to loss of contact with astrocytes, and therefore loss of Notch signalling [101]. It is less clear whether this is a downstream symptom of disease, or whether disease can be the outcome of reduced Notch or EAAT function in astrocytes to begin with. Given mutations that alter the function of γ-secretase in such a way that reduces Notch signalling lead to familial AD it is certainly plausible that disease may in part be due to reduced EAAT function leading to excitotoxic synapse loss and neurodegeneration, particularly in patients where increased excitability and glutamate dysregulation are observed [228]. The failed results of the semagacestat drug trial, that worsened disease progression, certainly hint that blocking Notch signalling and perhaps reducing EAAT function exacerbate neurodegenerative disease [222,229].
An involvement of Notch signalling and astrocytic EAAT function would also provide a pleasing explanation of sporadic cases of AD in humans, and why dementia is not a feature of ageing in non-transgenic mice: as reductions in Notch signalling and EAAT are natural features of human (and not so far observed in rodent) ageing, this would mean the aged human brain is particularly vulnerable to the development of dementia, the time when sporadic dementias typically occur. An external factor, such as vascular deficits, reducing bioenergetic status of the brain and thus reduced capacity for energy-expensive glutamate uptake, may exacerbate progression in sporadic neurodegenerative disease where EAATs are under-expressed. A further possibility for the sporadic occurrence of AD and inter-patient variability in EAAT expression could be explained by differences in epigenetic repression of EAAT transcription between individuals. Whether astrocytic Notch, or the modulation of EAAT expression, could be manipulated in a specific enough way to avoid the myriad roles of Notch (or epigenetic regulation) plays elsewhere in the body may however be a major translational hurdle.
Several questions remain to be investigated. Firstly, although we have seen that Notch signalling from neurons to astrocytes is required to maintain EAAT function in a rodent cell model, it needs to be ascertained whether this is the case for human cells. Secondly, investigations into the potential involvement of impaired Notch signalling (and potentially by extension reduced glutamate clearance capacity) have effectively stalled following on from the findings that inhibiting the Notch effector in neurons did not lead to disease in mice [230,231]. Future studies are needed to determine whether impairing Notch in astrocytes contributes to disease, and what the downstream transcriptional results are, for example reduced Slc1a2 and Slc1a3 expression. Finally, although we agree that research into targeting γ-secretases as a treatment for AD should not be abandoned, perhaps chronic partial γ-secretase inhibition is also not the way forward for most cases of AD [222]. More promising are treatments that specifically inhibit γ-secretase’s cleavage of APP without altering γ-secretases ability to cleave the NICD, such as imatinib, although such treatments are still not answering the question of Notch and EAAT involvement in disease [232]. A novel way forward could be to instead investigate the effect of increasing astrocytic γ-secretase activity, or in the case of models based on γ-secretase mutations, specifically boosting Notch signalling in these cells, via direct or indirect mechanisms [233]. The ability to target drugs to specific cell types is still in its infancy (though see [234]), but once a cell type and target have been validated, greater attention and investment will invariably lead to results.
7. Materials and Methods
7.1. Tissue Cultures and Stimulations
Astrocytes and neurons were cultured from E17.5 CD1 mouse and E20.5 Sprague Dawley rat embryos as previously described [235,236]. Cortices or spinal cords were dissected, enzymatically digested with papain and mechanically dissociated using a 5 mL pipette. Mouse cortical and spinal cord astrocytes were obtained by growing cells at low density in DMEM containing 10% fetal bovine serum and were passaged twice, using Trypsin (both Life Technologies), and plated onto coverslips in a 24-well plate. These astrocytes are >99% GFAP positive and <0.1% NeuN positive12. For mixed–species co-cultures, rat neurons were plated on top of a confluent layer of DIV14 mouse astrocytes and both astrocyte monocultures and astrocyte–neuron co-cultures were subsequently kept in Neurobasal-A medium containing B27 (both Life Technologies), but devoid of serum. Cultures were maintained up to DIV24, with regular feeding via 50% media exchanges conducted every three days. During this maintenance the cultured cells were either left untreated, treated with the γ-secretase inhibitor DAPT (10 μM) from DIV0, or treated with DAPT from DIV14.
7.2. Electrophysiological Recordings
Cultures were recorded from at either DIV13 (co-cultured astrocytes prior to DAPT application) or at DIV24 (all conditions), as described [101]. Coverslips containing cortical neurons and astrocytes were transferred to a recording chamber perfused (at a flow rate of 3–5 mL min−1) with an external recording solution composed of (in mM): 150 NaCl, 2.8 KCl, 10 HEPES, 2 CaCl2, 1 MgCl2, and 10 glucose, pH 7.3 (320–330 mOsm). Patch-pipettes were made from thick-walled borosilicate glass (Harvard Apparatus, Kent, UK), and when filled with the internal recording solution had tip resistances of 4–8 MΩ. A KCl-based internal was used, composed of (in mM): KCl 130, glucose 4, HEPES 10, EGTA 0.1, CaCl2 0.025, and sucrose 20; pH 7.2 with KOH. Astrocytes were voltage-clamped at −80 mV and any cells with a resting membrane potential >−60 mV upon break-in were discarded. To determine the maximal induced EAAT transport current, 200 μM l-Aspartate was bath applied followed by the addition of the high affinity EAAT inhibitor TFB-TBOA (20 μM) to ensure that any l-Aspartate-induced current was mediated by the transporter. AP5 (100 μM) was included in the external solution of all astrocyte recordings to block the activation of NMDARs by l-Aspartate. Recordings were at room temperature (21 ± 2 °C) using a Multiclamp 200B amplifier (Molecular Devices, Union City, CA, USA). Recordings were filtered at 5 kHz and digitized online at 20 kHz via a BNC-2090A/PCI-6251 DAQ board interface (National Instruments, Austin, TX, USA) and analysed using WinEDR 3.6 software (Dr John Dempster, University of Strathclyde, Glasgow, UK).
7.3. Statistical Analysis
Results are given as mean ± standard error of mean (SEM), unless otherwise stated. A linear mixed effects (LME) analysis of variance (ANOVA) model was run using R statistical software to assess significance of treatment conditions. This analysis was set up to take variation between the different cultures into consideration. Each data point was coded for the culture batch it was taken from, and the variation in results due to week by week culture variation was then incorporated as a random variable into the statistical analysis.
Funding
This work was funded by the UK Dementia Research Institute which receives its funding from DRI Ltd., funded by the UK Medical Research Council, Alzheimer’s Society, and Alzheimer’s Research UK.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Danbolt, N.C. Glutamate uptake. Prog. Neurobiol. 2001, 65, 1–105. [Google Scholar] [CrossRef]
- Hardingham, G.E.; Bading, H. Synaptic versus extrasynaptic NMDA receptor signalling: Implications for neurodegenerative disorders. Nat. Rev. Neurosci. 2010, 11, 682–696. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wahl, A.-S.; Buchthal, B.; Rode, F.; Bomholt, S.; Freitag, H.; Hardingham, G.E.; Rønn, L.; Bading, H. Hypoxic/ischemic conditions induce expression of the putative pro-death gene Clca1 via activation of extrasynaptic N-methyl-d-aspartate receptors. Neuroscience 2009, 158, 344–352. [Google Scholar] [CrossRef] [PubMed]
- Lindsay, R.M. Adult rat brain astrocytes support survival of both NGF-dependent and NGF-insensitive neurones. Nat. Cell Biol. 1979, 282, 80–82. [Google Scholar] [CrossRef] [PubMed]
- Banker, G.A. Trophic interactions between astroglial cells and hippocampal neurons in culture. Science 1980, 209, 809–810. [Google Scholar] [CrossRef] [Green Version]
- Rosenberg, P.A.; Amin, S.; Leitner, M. Glutamate uptake disguises neurotoxic potency of glutamate agonists in cerebral cortex in dissociated cell culture. J. Neurosci. 1992, 12, 56–61. [Google Scholar] [CrossRef] [Green Version]
- Rosenberg, P.A.; Aizenman, E. Hundred-fold increase in neuronal vulnerability to glutamate toxicity in astrocyte-poor cultures of rat cerebral cortex. Neurosci. Lett. 1989, 103, 162–168. [Google Scholar] [CrossRef]
- Hardingham, G.E.; Lipton, S.A. Regulation of Neuronal Oxidative and Nitrosative Stress by Endogenous Protective Pathways and Disease Processes. Antioxid. Redox Signal. 2011, 14, 1421–1424. [Google Scholar] [CrossRef]
- Gupta, K.; Hardingham, G.E.; Chandran, S. NMDA receptor-dependent glutamate excitotoxicity in human embryonic stem cell-derived neurons. Neurosci. Lett. 2013, 543, 95–100. [Google Scholar] [CrossRef] [Green Version]
- Hertz, L.; Rodrigues, T.B. Astrocytic—Neuronal—Astrocytic Pathway Selection for Formation and Degradation of Glutamate/GABA. Front. Endocrinol. 2014, 5, 5. [Google Scholar] [CrossRef] [Green Version]
- Sonnewald, U.; Schousboe, A. Introduction to the Glutamate–Glutamine Cycle. In The Glutamate/GABA-Glutamine Cycle: Amino Acid Neurotransmitter Homeostasis; Schousboe, A., Sonnewald, U., Eds.; Springer International Publishing: Cham, Switzerland, 2016; pp. 1–7. [Google Scholar]
- Billups, D.; Marx, M.-C.; Mela, I.; Billups, B. Inducible Presynaptic Glutamine Transport Supports Glutamatergic Transmission at the Calyx of Held Synapse. J. Neurosci. 2013, 33, 17429–17434. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tani, H.; Dulla, C.G.; Farzampour, Z.; Taylor-Weiner, A.; Huguenard, J.R.; Reimer, R.J. A Local Glutamate-Glutamine Cycle Sustains Synaptic Excitatory Transmitter Release. Neuron 2014, 81, 888–900. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Todd, A.C.; Marx, M.-C.; Hulme, S.R.; Bröer, S.; Billups, B. SNAT3-mediated glutamine transport in perisynaptic astrocytesin situis regulated by intracellular sodium. Glia 2017, 65, 900–916. [Google Scholar] [CrossRef] [PubMed]
- Uwechue, N.M.; Marx, M.-C.; Chevy, Q.; Billups, B. Activation of glutamate transport evokes rapid glutamine release from perisynaptic astrocytes. J. Physiol. 2012, 590, 2317–2331. [Google Scholar] [CrossRef] [PubMed]
- Freidman, N.; Chen, I.; Wu, Q.; Briot, C.; Holst, J.; Font, J.; Vandenberg, R.J.; Ryan, R. Amino Acid Transporters and Exchangers from the SLC1A Family: Structure, Mechanism and Roles in Physiology and Cancer. Neurochem. Res. 2020, 45, 1268–1286. [Google Scholar] [CrossRef]
- Grewer, C.; Gameiro, A.; Rauen, T. SLC1 glutamate transporters. Pflügers Arch. Eur. J. Physiol. 2013, 466, 3–24. [Google Scholar] [CrossRef] [Green Version]
- Verkhratsky, A.; Ho, M.S.; Zorec, R.; Parpura, V. Physiology of Astroglia. In Neuroglia in Neurodegenerative Diseases; Springer: Singapore, 2019; pp. 45–91. [Google Scholar]
- Hädel, S.; Wirth, C.; Rapp, M.; Gallinat, J.; Schubert, F. Effects of age and sex on the concentrations of glutamate and glutamine in the human brain. J. Magn. Reson. Imaging 2013, 38, 1480–1487. [Google Scholar] [CrossRef]
- Schubert, F.; Gallinat, J.; Seifert, F.; Rinneberg, H. Glutamate concentrations in human brain using single voxel proton magnetic resonance spectroscopy at 3 Tesla. NeuroImage 2004, 21, 1762–1771. [Google Scholar] [CrossRef]
- Lehmann, A.; Isacsson, H.; Hamberger, A. Effects of In Vivo Administration of Kainic Acid on the Extracellular Amino Acid Pool in the Rabbit Hippocampus. J. Neurochem. 1983, 40, 1314–1320. [Google Scholar] [CrossRef]
- Hamberger, A.; Nyström, B. Extra- and intracellular amino acids in the hippocampus during development of hepatic encephalopathy. Neurochem. Res. 1984, 9, 1181–1192. [Google Scholar] [CrossRef]
- Erecinska, M. Metabolism and role of glutamate in mammalian brain. Prog. Neurobiol. 1990, 35, 245–296. [Google Scholar] [CrossRef]
- Helms, H.C.C.; Nielsen, C.U.; Waagepetersen, H.S.; Brodin, B. Glutamate Transporters in the Blood-Brain Barrier. In Advances in Neurobiology; Ortega, A., Schousboe, A., Eds.; Springer International Publishing: Cham, Switzerland, 2017; pp. 297–314. [Google Scholar]
- Fotiadis, D.; Kanai, Y.; Palacín, M. The SLC3 and SLC7 families of amino acid transporters. Mol. Asp. Med. 2013, 34, 139–158. [Google Scholar] [CrossRef] [PubMed]
- Kanai, Y.; Clémençon, B.; Simonin, A.; Leuenberger, M.; Lochner, M.; Weisstanner, M.; Hediger, M.A. The SLC1 high-affinity glutamate and neutral amino acid transporter family. Mol. Asp. Med. 2013, 34, 108–120. [Google Scholar] [CrossRef] [PubMed]
- Bridges, R.J.; Natale, N.R.; Patel, S.A. System xc- cystine/glutamate antiporter: An update on molecular pharmacology and roles within the CNS. Br. J. Pharmacol. 2011, 165, 20–34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ottestad-Hansen, S.; Hu, Q.X.; Follin-Arbelet, V.V.; Bentea, E.; Sato, H.; Massie, A.; Zhou, Y.; Danbolt, N.C. The cystine-glutamate exchanger (xCT, Slc7a11) is expressed in significant concentrations in a subpopulation of astrocytes in the mouse brain. Glia 2018, 66, 951–970. [Google Scholar] [CrossRef] [Green Version]
- Lewerenz, J.; Baxter, P.; Kassubek, R.; Albrecht, P.; Van Liefferinge, J.; Westhoff, M.-A.; Halatsch, M.-E.; Karpel-Massler, G.; Meakin, P.J.; Hayes, J.D.; et al. Phosphoinositide 3-Kinases Upregulate System xc− via Eukaryotic Initiation Factor 2α and Activating Transcription Factor 4—A Pathway Active in Glioblastomas and Epilepsy. Antioxid. Redox Signal. 2014, 20, 2907–2922. [Google Scholar] [CrossRef] [Green Version]
- Logan, W.J.; Snyder, S.H. Unique High Affinity Uptake Systems for Glycine, Glutamic and Aspartic Acids in Central Nervous Tissue of the Rat. Nature 1971, 234, 297–299. [Google Scholar] [CrossRef]
- Balcar, V.J.; Johnston, G.R. The Structural Specificity of the High Affinity Uptake of l-glutamate and l-Aspartate by Rat Brain Slices. J. Neurochem. 1972, 19, 2657–2666. [Google Scholar] [CrossRef]
- Kanai, Y.; Hediger, M.A. Primary structure and functional characterization of a high-affinity glutamate transporter. Nature 1992, 360, 467–471. [Google Scholar] [CrossRef]
- Pines, G.; Danbolt, N.C.; Bjørås, M.; Zhang, Y.; Bendahan, A.; Eide, L.; Koepsell, H.; Storm-Mathisen, J.; Seeberg, E.; Kanner, B.I. Cloning and expression of a rat brain l-glutamate transporter. Nature 1992, 360, 464–467. [Google Scholar] [CrossRef]
- Storck, T.; Schulte, S.; Hofmann, K.; Stoffel, W. Structure, expression, and functional analysis of a Na(+)-dependent glutamate/aspartate transporter from rat brain. Proc. Natl. Acad. Sci. USA 1992, 89, 10955–10959. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balcar, V.J.; Borg, J.; Mandel, P. High affinity uptake of l-glutamate and l-Aspartate by glial cells. J. Neurochem. 1977, 28, 87–93. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, K. Cloning and expression of a glutamate transporter from mouse brain. Neurosci. Lett. 1993, 159, 183–186. [Google Scholar] [CrossRef]
- Fairman, W.A.; Vandenberg, R.J.; Arriza, J.L.; Kavanaught, M.P.; Amara, S.G. An excitatory amino-acid transporter with properties of a ligand-gated chloride channel. Nature 1995, 375, 599–603. [Google Scholar] [CrossRef] [PubMed]
- Arriza, J.L.; Eliasof, S.; Kavanaugh, M.P.; Amara, S.G. Excitatory amino acid transporter 5, a retinal glutamate transporter coupled to a chloride conductance. Proc. Natl. Acad. Sci. USA 1997, 94, 4155–4160. [Google Scholar] [CrossRef] [Green Version]
- Arriza, J.L.; Fairman, W.A.; Wadiche, J.I.; Murdoch, G.H.; Kavanaugh, M.P.; Amara, S.G. Functional comparisons of three glutamate transporter subtypes cloned from human motor cortex. J. Neurosci. 1994, 14, 5559–5569. [Google Scholar] [CrossRef] [Green Version]
- Wadiche, J.I.; Kavanaugh, M.P. Macroscopic and Microscopic Properties of a Cloned Glutamate Transporter/Chloride Channel. J. Neurosci. 1998, 18, 7650–7661. [Google Scholar] [CrossRef] [Green Version]
- Wadiche, J.I.; Amara, S.G.; Kavanaugh, M.P. Ion fluxes associated with excitatory amino acid transport. Neuron 1995, 15, 721–728. [Google Scholar] [CrossRef] [Green Version]
- Jensen, A.A.; Bräuner-Osborne, H. Pharmacological characterization of human excitatory amino acid transporters EAAT1, EAAT2 and EAAT3 in a fluorescence-based membrane potential assay. Biochem. Pharmacol. 2004, 67, 2115–2127. [Google Scholar] [CrossRef]
- Grewer, C.; Watzke, N.; Wiessner, M.; Rauen, T. Glutamate translocation of the neuronal glutamate transporter EAAC1 occurs within milliseconds. Proc. Natl. Acad. Sci. USA 2000, 97, 9706–9711. [Google Scholar] [CrossRef] [Green Version]
- Gameiro, A.; Braams, S.; Rauen, T.; Grewer, C. The Discovery of Slowness: Low-Capacity Transport and Slow Anion Channel Gating by the Glutamate Transporter EAAT5. Biophys. J. 2011, 100, 2623–2632. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mim, C.; Balani, P.; Rauen, T.; Grewer, C. The Glutamate Transporter Subtypes EAAT4 and EAATs 1-3 Transport Glutamate with Dramatically Different Kinetics and Voltage Dependence but Share a Common Uptake Mechanism. J. Gen. Physiol. 2005, 126, 571–589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bellocchio, E.E.; Reimer, R.J.; Fremeau, R.T.; Edwards, R.H. Uptake of Glutamate into Synaptic Vesicles by an Inorganic Phosphate Transporter. Science 2000, 289, 957–960. [Google Scholar] [CrossRef] [PubMed]
- Herzog, E.; Bellenchi, G.C.; Gras, C.; Bernard, V.; Ravassard, P.; Bedet, C.; Gasnier, B.; Giros, B.; El Mestikawy, S. The Existence of a Second Vesicular Glutamate Transporter Specifies Subpopulations of Glutamatergic Neurons. J. Neurosci. 2001, 21, RC181. [Google Scholar] [CrossRef]
- Fremeau, R.T.; Burman, J.; Qureshi, T.; Tran, C.H.; Proctor, J.; Johnson, J.; Zhang, H.; Sulzer, D.; Copenhagen, D.R.; Storm-Mathisen, J.; et al. The identification of vesicular glutamate transporter 3 suggests novel modes of signaling by glutamate. Proc. Natl. Acad. Sci. USA 2002, 99, 14488–14493. [Google Scholar] [CrossRef] [Green Version]
- Patel, S.A.; Warren, B.A.; Rhoderick, J.F.; Bridges, R.J. Differentiation of substrate and non-substrate inhibitors of transport system xc−: An obligate exchanger of l-glutamate and L-cystine. Neuropharmacology 2004, 46, 273–284. [Google Scholar] [CrossRef]
- Seib, T.M.; Patel, S.A.; Bridges, R.J. Regulation of the System x−C cystine/glutamate exchanger by intracellular glutathione levels in rat astrocyte primary cultures. Glia 2011, 59, 1387–1401. [Google Scholar] [CrossRef]
- Murphy-Royal, C.; Dupuis, J.P.; Varela, J.A.; Panatier, A.; Pinson, B.; Baufreton, J.; Groc, L.; Oliet, S.H.R. Surface diffusion of astrocytic glutamate transporters shapes synaptic transmission. Nat. Neurosci. 2015, 18, 219–226. [Google Scholar] [CrossRef]
- Al Awabdh, S.; Gupta-Agarwal, S.; Sheehan, D.F.; Muir, J.; Norkett, R.; Twelvetrees, A.E.; Griffin, L.D.; Kittler, J.T. Neuronal activity mediated regulation of glutamate transporter GLT-1 surface diffusion in rat astrocytes in dissociated and slice cultures. Glia 2016, 64, 1252–1264. [Google Scholar] [CrossRef] [Green Version]
- Šerý, O.; Sultana, N.; Kashem, M.A.; Pow, D.V.; Balcar, V.J. GLAST But Not Least—Distribution, Function, Genetics and Epigenetics of l-glutamate Transport in Brain—Focus on GLAST/EAAT1. Neurochem. Res. 2015, 40, 2461–2472. [Google Scholar] [CrossRef]
- Zhou, Y.; Danbolt, N.C. GABA and Glutamate Transporters in Brain. Front. Endocrinol. 2013, 4, 165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chaudhry, F.A.; Lehre, K.P.; Campagne, M.V.L.; Ottersen, O.P.; Danbolt, N.C.; Storm-Mathisen, J. Glutamate transporters in glial plasma membranes: Highly differentiated localizations revealed by quantitative ultrastructural immunocytochemistry. Neuron 1995, 15, 711–720. [Google Scholar] [CrossRef] [Green Version]
- Lehre, K.P.; Danbolt, N.C. The Number of Glutamate Transporter Subtype Molecules at Glutamatergic Synapses: Chemical and Stereological Quantification in Young Adult Rat Brain. J. Neurosci. 1998, 18, 8751–8757. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lehre, K.; Levy, L.; Ottersen, O.; Storm-Mathisen, J.; Danbolt, N. Differential expression of two glial glutamate transporters in the rat brain: Quantitative and immunocytochemical observations. J. Neurosci. 1995, 15, 1835–1853. [Google Scholar] [CrossRef] [PubMed]
- Schmitt, A.; Asan, E.; Püschel, B.; Kugler, P. Cellular and Regional Distribution of the Glutamate Transporter GLAST in the CNS of Rats: Nonradioactive In Situ Hybridization and Comparative Immunocytochemistry. J. Neurosci. 1997, 17, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rothstein, J.D.; Martin, L.; Levey, A.I.; Dykes-Hoberg, M.; Jin, L.; Wu, D.; Nash, N.; Kuncl, R.W. Localization of neuronal and glial glutamate transporters. Neuron 1994, 13, 713–725. [Google Scholar] [CrossRef]
- Derouiche, A.; Rauen, T. Coincidence of l-glutamate/l-Aspartate transporter (GLAST) and glutamine synthetase (GS) immunoreactions in retinal glia: Evidence for coupling of GLAST and GS in transmitter clearance. J. Neurosci. Res. 1995, 42, 131–143. [Google Scholar] [CrossRef]
- Lehre, K.P.; Davanger, S.; Danbolt, N.C. Localization of the glutamate transporter protein GLAST in rat retina. Brain Res. 1997, 744, 129–137. [Google Scholar] [CrossRef]
- Pow, D.V.; Barnett, N.L. Changing patterns of spatial buffering of glutamate in developing rat retinae are mediated by the Müller cell glutamate transporter GLAST. Cell Tissue Res. 1999, 297, 57–66. [Google Scholar] [CrossRef]
- Carlyle, B.C.; Kitchen, R.; Kanyo, J.E.; Voss, E.Z.; Pletikos, M.; Sousa, A.M.M.; Lam, T.T.; Gerstein, M.B.; Sestan, N.; Nairn, A.C. A multiregional proteomic survey of the postnatal human brain. Nat. Neurosci. 2017, 20, 1787–1795. [Google Scholar] [CrossRef] [Green Version]
- Rose, C.R.; Ziemens, D.; Untiet, V.; Fahlke, C. Molecular and cellular physiology of sodium-dependent glutamate transporters. Brain Res. Bull. 2018, 136, 3–16. [Google Scholar] [CrossRef]
- Holmseth, S.; Dehnes, Y.; Huang, Y.H.; Follin-Arbelet, V.V.; Grutle, N.J.; Mylonakou, M.N.; Plachez, C.; Zhou, Y.; Furness, D.N.; Bergles, D.E.; et al. The Density of EAAC1 (EAAT3) Glutamate Transporters Expressed by Neurons in the Mammalian CNS. J. Neurosci. 2012, 32, 6000–6013. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shashidharan, P.; Huntley, G.W.; Murray, J.M.; Buku, A.; Moran, T.; Walsh, M.J.; Morrison, J.H.; Plaitakis, A. Immunohistochemical localization of the neuron-specific glutamate transporter EAAC1 (EAAT3) in rat brain and spinal cord revealed by a novel monoclonal antibody. Brain Res. 1997, 773, 139–148. [Google Scholar] [CrossRef]
- Dehnes, Y.; Chaudhry, F.A.; Ullensvang, K.; Lehre, K.P.; Storm-Mathisen, J.; Danbolt, N.C. The Glutamate Transporter EAAT4 in Rat Cerebellar Purkinje Cells: A Glutamate-Gated Chloride Channel Concentrated near the Synapse in Parts of the Dendritic Membrane Facing Astroglia. J. Neurosci. 1998, 18, 3606–3619. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Massie, A.; Cnops, L.; Smolders, I.; McCullumsmith, R.; Kooijman, R.; Kwak, S.; Arckens, L.; Michotte, Y. High-affinity Na+/K+-dependent glutamate transporter EAAT4 is expressed throughout the rat fore- and midbrain. J. Comp. Neurol. 2008, 511, 155–172. [Google Scholar] [CrossRef] [PubMed]
- Pow, D.V.; Barnett, N.L. Developmental expression of excitatory amino acid transporter 5: A photoreceptor and bipolar cell glutamate transporter in rat retina. Neurosci. Lett. 2000, 280, 21–24. [Google Scholar] [CrossRef]
- Eliasof, S.; Arriza, J.L.; Leighton, B.H.; Kavanaugh, M.P.; Amara, S.G. Excitatory Amino Acid Transporters of the Salamander Retina: Identification, Localization, and Function. J. Neurosci. 1998, 18, 698–712. [Google Scholar] [CrossRef] [Green Version]
- Wersinger, E.; Schwab, Y.; Sahel, J.-A.; Rendon, A.; Pow, D.V.; Picaud, S.; Roux, M.J. The glutamate transporter EAAT5 works as a presynaptic receptor in mouse rod bipolar cells. J. Physiol. 2006, 577, 221–234. [Google Scholar] [CrossRef]
- Gegelashvili, G.; Schousboe, A. Cellular Distribution and Kinetic Properties of High-Affinity Glutamate Transporters. Brain Res. Bull. 1998, 45, 233–238. [Google Scholar] [CrossRef]
- Levy, L.M.; Warr, O.; Attwell, D. Stoichiometry of the Glial Glutamate Transporter GLT-1 Expressed Inducibly in a Chinese Hamster Ovary Cell Line Selected for Low Endogenous Na+-Dependent Glutamate Uptake. J. Neurosci. 1998, 18, 9620–9628. [Google Scholar] [CrossRef] [Green Version]
- Zerangue, N.; Kavanaugh, M.P. Flux coupling in a neuronal glutamate transporter. Nature 1996, 383, 634–637. [Google Scholar] [CrossRef] [PubMed]
- Kanai, Y.; Nussberger, S.; Romero, M.F.; Boron, W.F.; Hebert, S.C.; Hediger, M.A. Electrogenic Properties of the Epithelial and Neuronal High Affinity Glutamate Transporter. J. Biol. Chem. 1995, 270, 16561–16568. [Google Scholar] [CrossRef] [Green Version]
- Yernool, D.; Boudker, O.; Jin, Y.; Gouaux, E. Structure of a glutamate transporter homologue from Pyrococcus horikoshii. Nature 2004, 431, 811–818. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jensen, S.; Guskov, A.; Rempel, S.; Hänelt, I.; Slotboom, D.J.; Hänelt, I. Crystal structure of a substrate-free aspartate transporter. Nat. Struct. Mol. Biol. 2013, 20, 1224–1226. [Google Scholar] [CrossRef] [Green Version]
- Arkhipova, V.; Trinco, G.; Ettema, T.W.; Jensen, S.; Slotboom, D.J.; Guskov, A. Binding and transport of D-aspartate by the glutamate transporter homolog GltTk. eLife 2019, 8. [Google Scholar] [CrossRef]
- Kortzak, D.; Alleva, C.; Weyand, I.; Ewers, D.; Zimmermann, M.I.; Franzen, A.; Machtens, J.-P.; Fahlke, C. Allosteric gate modulation confers K + coupling in glutamate transporters. EMBO J. 2019, 38, e101468. [Google Scholar] [CrossRef] [PubMed]
- Brew, H.; Attwell, D. Electrogenic glutamate uptake is a major current carrier in the membrane of axolotl retinal glial cells. Nature 1987, 327, 707–709. [Google Scholar] [CrossRef] [PubMed]
- Vandenberg, R.J.; Arriza, J.L.; Amara, S.G.; Kavanaugh, M.P. Constitutive Ion Fluxes and Substrate Binding Domains of Human Glutamate Transporters. J. Biol. Chem. 1995, 270, 17668–17671. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wyllie, D.J.A.; Mathie, A.; Symonds, C.J.; Cull-Candy, S.G. Activation of glutamate receptors and glutamate uptake in identified macroglial cells in rat cerebellar cultures. J. Physiol. 1991, 432, 235–258. [Google Scholar] [CrossRef] [Green Version]
- Machtens, J.-P.; Kortzak, D.; Lansche, C.; Leinenweber, A.; Kilian, P.; Begemann, B.; Zachariae, U.; Ewers, D.; De Groot, B.L.; Briones, R.; et al. Mechanisms of Anion Conduction by Coupled Glutamate Transporters. Cell 2015, 160, 542–553. [Google Scholar] [CrossRef] [Green Version]
- Fahlke, C.; Kortzak, D.; Machtens, J.-P. Molecular physiology of EAAT anion channels. Pflügers Arch. Eur. J. Physiol. 2016, 468, 491–502. [Google Scholar] [CrossRef] [PubMed]
- Schneider, N.; Cordeiro, S.; Machtens, J.-P.; Braams, S.; Rauen, T.; Fahlke, C. Functional Properties of the Retinal Glutamate Transporters GLT-1c and EAAT. J. Biol. Chem. 2013, 289, 1815–1824. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tse, D.Y.; Chung, I.Y.; Wu, S.M. Possible roles of glutamate transporter EAAT5 in mouse cone depolarizing bipolar cell light responses. Vis. Res. 2014, 103, 63–74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McLennan, H. The autoradiographic localization of L-[3H]glutamate in rat brain tissue. Brain Res. 1976, 115, 139–144. [Google Scholar] [CrossRef]
- Wilkin, G.P.; Garthwaite, J.; Bala’zs, R. Putative acidic amino acid transmitters in the cerebellum. II. Electron microscopic localization of transport sites. Brain Res. 1982, 244, 69–80. [Google Scholar] [CrossRef]
- Bergles, D.E.; Jahr, C.E. Glial Contribution to Glutamate Uptake at Schaffer Collateral–Commissural Synapses in the Hippocampus. J. Neurosci. 1998, 18, 7709–7716. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bergles, D.E.; Jahr, C.E. Synaptic Activation of Glutamate Transporters in Hippocampal Astrocytes. Neuron 1997, 19, 1297–1308. [Google Scholar] [CrossRef] [Green Version]
- Tanaka, K.; Watase, K.; Manabe, T.; Yamada, K.; Watanabe, M.; Takahashi, K.; Iwama, H.; Nishikawa, T.; Ichihara, N.; Kikuchi, T.; et al. Epilepsy and Exacerbation of Brain Injury in Mice Lacking the Glutamate Transporter GLT-1. Science 1997, 276, 1699–1702. [Google Scholar] [CrossRef]
- Watase, K.; Hashimoto, K.; Kano, M.; Yamada, K.; Watanabe, M.; Inoue, Y.; Okuyama, S.; Sakagawa, T.; Ogawa, S.-I.; Kawashima, N.; et al. Motor discoordination and increased susceptibility to cerebellar injury in GLAST mutant mice. Eur. J. Neurosci. 1998, 10, 976–988. [Google Scholar] [CrossRef]
- Matsugami, T.R.; Tanemura, K.; Mieda, M.; Nakatomi, R.; Yamada, K.; Kondo, T.; Ogawa, M.; Obata, K.; Watanabe, M.; Hashikawa, T.; et al. Indispensability of the glutamate transporters GLAST and GLT1 to brain development. Proc. Natl. Acad. Sci. USA 2006, 103, 12161–12166. [Google Scholar] [CrossRef] [Green Version]
- Peghini, P.; Janzen, J.; Stoffel, W. Glutamate transporter EAAC-1-deficient mice develop dicarboxylic aminoaciduria and behavioral abnormalities but no neurodegeneration. EMBO J. 1997, 16, 3822–3832. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petr, G.T.; Sun, Y.; Frederick, N.M.; Zhou, Y.; Dhamne, S.C.; Hameed, M.Q.; Miranda, C.; Bedoya, E.A.; Fischer, K.D.; Armsen, W.; et al. Conditional Deletion of the Glutamate Transporter GLT-1 Reveals That Astrocytic GLT-1 Protects against Fatal Epilepsy While Neuronal GLT-1 Contributes Significantly to Glutamate Uptake into Synaptosomes. J. Neurosci. 2015, 35, 5187–5201. [Google Scholar] [CrossRef] [PubMed]
- Drejer, J.; Meier, E.; Schousboe, A. Novel neuron-related regulatory mechanisms for astrocytic glutamate and GABA high affinity uptake. Neurosci. Lett. 1983, 37, 301–306. [Google Scholar] [CrossRef]
- Levy, L.M.; Lehre, K.P.; Walaas, S.I.; Storm-Mathisen, J.; Danbolt, N.C. Down-regulation of Glial Glutamate Transporters after Glutamatergic Denervation in the Rat Brain. Eur. J. Neurosci. 1995, 7, 2036–2041. [Google Scholar] [CrossRef]
- Gegelashvili, G.; Civenni, G.; Racagni, G.; Danbolt, N.C.; Schousboe, I.; Schousboe, A. Glutamate receptor agonists up-regulate glutamate transporter GLAST in astrocytes. NeuroReport 1996, 8, 261–265. [Google Scholar] [CrossRef]
- Swanson, R.A.; Liu, J.; Miller, J.W.; Rothstein, J.D.; Farrell, K.; Stein, B.A.; Longuemare, M.C. Neuronal Regulation of Glutamate Transporter Subtype Expression in Astrocytes. J. Neurosci. 1997, 17, 932–940. [Google Scholar] [CrossRef] [Green Version]
- Gegelashvili, G.; Danbolt, N.C.; Schousboe, A. Neuronal soluble factors differentially regulate the expression of the GLT1 and GLAST glutamate transporters in cultured astroglia. J. Neurochem. 1997, 69, 2612–2615. [Google Scholar] [CrossRef] [Green Version]
- Hasel, P.; Dando, O.R.; Jiwaji, Z.; Baxter, P.; Todd, A.C.; Heron, S.; Márkus, N.M.; McQueen, J.; Hampton, D.W.; Torvell, M.; et al. Neurons and neuronal activity control gene expression in astrocytes to regulate their development and metabolism. Nat. Commun. 2017, 8, 15132. [Google Scholar] [CrossRef]
- Schlag, B.D.; Vondrasek, J.R.; Munir, M.; Kalandadze, A.; Zelenaia, O.A.; Rothstein, J.D.; Robinson, M.B. Regulation of the Glial Na+-Dependent Glutamate Transporters by Cyclic AMP Analogs and Neurons. Mol. Pharmacol. 1998, 53, 355–369. [Google Scholar] [CrossRef]
- Zelenaia, O.; Schlag, B.D.; Gochenauer, G.E.; Ganel, R.; Song, W.; Beesley, J.S.; Grinspan, J.B.; Rothstein, J.D.; Robinson, M.B. Epidermal Growth Factor Receptor Agonists Increase Expression of Glutamate Transporter GLT-1 in Astrocytes through Pathways Dependent on Phosphatidylinositol 3-Kinase and Transcription Factor NF-κB. Mol. Pharmacol. 2000, 57, 667–678. [Google Scholar] [CrossRef]
- Ghosh, M.; Yang, Y.; Rothstein, J.D.; Robinson, M.B. Nuclear Factor- B Contributes to Neuron-Dependent Induction of Glutamate Transporter-1 Expression in Astrocytes. J. Neurosci. 2011, 31, 9159–9169. [Google Scholar] [CrossRef] [PubMed]
- Sitcheran, R.; Gupta, P.; Fisher, P.B.; Baldwin, A.S. Positive and negative regulation of EAAT2 by NF-κB: A role for N-myc in TNFα-controlled repression. EMBO J. 2005, 24, 510–520. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghosh, M.; Lane, M.; Krizman, E.; Sattler, R.; Rothstein, J.D.; Robinson, M.B. The transcription factor Pax6 contributes to the induction of GLT-1 expression in astrocytes through an interaction with a distal enhancer element. J. Neurochem. 2016, 136, 262–275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stacey, S.M.; Muraro, N.I.; Peco, E.; Labbe, A.; Thomas, G.B.; Baines, R.A.; Van Meyel, D.J. Drosophila Glial Glutamate Transporter Eaat1 Is Regulated by Fringe-Mediated Notch Signaling and Is Essential for Larval Locomotion. J. Neurosci. 2010, 30, 14446–14457. [Google Scholar] [CrossRef]
- Lee, M.L.; Martinez-Lozada, Z.; Krizman, E.N.; Robinson, M.B. Brain endothelial cells induce astrocytic expression of the glutamate transporter GLT-1 by a Notch-dependent mechanism. J. Neurochem. 2017, 143, 489–506. [Google Scholar] [CrossRef]
- Martinez-Lozada, Z.; Robinson, M.B. Reciprocal communication between astrocytes and endothelial cells is required for astrocytic glutamate transporter 1 (GLT-1) expression. Neurochem. Int. 2020, 139, 104787. [Google Scholar] [CrossRef]
- Hertz, L.; Bock, E.; Schousboe, A. GFA Content, Glutamate Uptake and Activity of Glutamate Metabolizing Enzymes in Differentiating Mouse Astrocytes in Primary Cultures. Dev. Neurosci. 1978, 1, 226–238. [Google Scholar] [CrossRef]
- Goldman, J.E.; Chiu, F.-C. Dibutyryl cyclic AMP causes intermediate filament accumulation and actin reorganization in astrocytes. Brain Res. 1984, 306, 85–95. [Google Scholar] [CrossRef]
- Pisano, P.; Samuel, D.; Nieoullon, A.; Goff, L.K.-L. Activation of the Adenylate Cyclase-dependent Protein Kinase Pathway Increases High Affinity Glutamate Uptake into Rat Striatal Synaptosomes. Neuropharmacology 1996, 35, 541–547. [Google Scholar] [CrossRef]
- Perego, C.; Vanoni, C.; Bossi, M.; Massari, S.; Basudev, H.; Longhi, R.; Pietrini, G. The GLT-1 and GLAST Glutamate Transporters Are Expressed on Morphologically Distinct Astrocytes and Regulated by Neuronal Activity in Primary Hippocampal Cocultures. J. Neurochem. 2002, 75, 1076–1084. [Google Scholar] [CrossRef]
- Simantov, R.; Crispino, M.; Hoe, W.; Broutman, G.; Tocco, G.; Rothstein, J.D.; Baudry, M. Changes in expression of neuronal and glial glutamate transporters in rat hippocampus following kainate-induced seizure activity. Mol. Brain Res. 1999, 65, 112–123. [Google Scholar] [CrossRef]
- Duan, S.; Anderson, C.M.; Stein, B.A.; Swanson, R.A. Glutamate Induces Rapid Upregulation of Astrocyte Glutamate Transport and Cell-Surface Expression of GLAST. J. Neurosci. 1999, 19, 10193–10200. [Google Scholar] [CrossRef] [PubMed]
- Ibáñez, I.; Díez-Guerra, F.J.; Giménez, C.; Zafra, F. Activity dependent internalization of the glutamate transporter GLT-1 mediated by β-arrestin 1 and ubiquitination. Neuropharmacology 2016, 107, 376–386. [Google Scholar] [CrossRef] [PubMed]
- Ibañez, I.; Bartolomé-Martín, D.; Piniella, D.; Giménez, C.; Zafra, F. Activity dependent internalization of the glutamate transporter GLT-1 requires calcium entry through the NCX sodium/calcium exchanger. Neurochem. Int. 2019, 123, 125–132. [Google Scholar] [CrossRef]
- Gamboa, C.; Ortega, A. Insulin-like growth factor-1 increases activity and surface levels of the GLAST subtype of glutamate transporter. Neurochem. Int. 2002, 40, 397–403. [Google Scholar] [CrossRef]
- Boehmer, C.; Henke, G.; Schniepp, R.; Palmada, M.; Rothstein, J.D.; Bröer, S.; Lang, F. Regulation of the glutamate transporter EAAT1 by the ubiquitin ligase Nedd4-2 and the serum and glucocorticoid-inducible kinase isoforms SGK1/3 and protein kinase B. J. Neurochem. 2003, 86, 1181–1188. [Google Scholar] [CrossRef]
- Götz, M.; Stoykova, A.; Gruss, P. Pax6 Controls Radial Glia Differentiation in the Cerebral Cortex. Neuron 1998, 21, 1031–1044. [Google Scholar] [CrossRef] [Green Version]
- Sakurai, K.; Osumi, N.; Aarts, E.; Roelofs, A.; Van Turennout, M. The Neurogenesis-Controlling Factor, Pax6, Inhibits Proliferation and Promotes Maturation in Murine Astrocytes. J. Neurosci. 2008, 28, 4604–4612. [Google Scholar] [CrossRef]
- Gegelashvili, G. The high-affinity glutamate transporters GLT1, GLAST, and EAAT4 are regulated via different signalling mechanisms. Neurochem. Int. 2000, 37, 163–170. [Google Scholar] [CrossRef]
- Namihira, M.; Kohyama, J.; Semi, K.; Sanosaka, T.; Deneen, B.; Taga, T.; Nakashima, K. Committed Neuronal Precursors Confer Astrocytic Potential on Residual Neural Precursor Cells. Dev. Cell 2009, 16, 245–255. [Google Scholar] [CrossRef] [Green Version]
- Kopan, R.; Ilagan, M.X.G. The Canonical Notch Signaling Pathway: Unfolding the Activation Mechanism. Cell 2009, 137, 216–233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fischer, A.; Gessler, M. Delta Notch and then? Protein interactions and proposed modes of repression by Hes and Hey bHLH factors. Nucleic Acids Res. 2007, 35, 4583–4596. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Angulo-Rojo, C.; Manning-Cela, R.; Aguirre, A.; Ortega, A.; Lopez-Bayghen, E. Involvement of the Notch Pathway in Terminal Astrocytic Differentiation: Role of PKA. ASN Neuro 2013, 5, AN20130023. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cahoy, J.D.; Emery, B.; Kaushal, A.; Foo, L.C.; Zamanian, J.L.; Christopherson, K.S.; Xing, Y.; Lubischer, J.L.; Krieg, P.A.; Krupenko, S.A.; et al. A Transcriptome Database for Astrocytes, Neurons, and Oligodendrocytes: A New Resource for Understanding Brain Development and Function. J. Neurosci. 2008, 28, 264–278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Biswas, S.; Rao, C.M. Epigenetic tools (The Writers, The Readers and The Erasers) and their implications in cancer therapy. Eur. J. Pharmacol. 2018, 837, 8–24. [Google Scholar] [CrossRef]
- Zhang, Y.; Sloan, S.A.; Clarke, L.E.; Caneda, C.; Plaza, C.A.; Blumenthal, P.D.; Vogel, H.; Steinberg, G.K.; Edwards, M.S.B.; Li, G.; et al. Purification and Characterization of Progenitor and Mature Human Astrocytes Reveals Transcriptional and Functional Differences with Mouse. Neuron 2016, 89, 37–53. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Chen, K.; Sloan, S.A.; Bennett, M.L.; Scholze, A.R.; O’Keeffe, S.; Phatnani, H.P.; Guarnieri, P.; Caneda, C.; Ruderisch, N.; et al. An RNA-Sequencing Transcriptome and Splicing Database of Glia, Neurons, and Vascular Cells of the Cerebral Cortex. J. Neurosci. 2014, 34, 11929–11947. [Google Scholar] [CrossRef]
- Zhang, X.; Kusumo, H.; Sakharkar, A.J.; Pandey, S.C.; Guizzetti, M. Regulation of DNA methylation by ethanol induces tissue plasminogen activator expression in astrocytes. J. Neurochem. 2014, 128, 344–349. [Google Scholar] [CrossRef] [Green Version]
- Broide, R.S.; Redwine, J.M.; Aftahi, N.; Young, W.; Bloom, F.E.; Winrow, C.J. Distribution of histone deacetylases 1–11 in the rat brain. J. Mol. Neurosci. 2007, 31, 47–58. [Google Scholar] [CrossRef]
- Zhang, Z.; Wang, Y.; Chen, J.; Tan, Q.; Xie, C.; Li, C.; Zhan, W.; Wang, M. Silencing of histone deacetylase 2 suppresses malignancy for proliferation, migration, and invasion of glioblastoma cells and enhances temozolomide sensitivity. Cancer Chemother. Pharmacol. 2016, 78, 1289–1296. [Google Scholar] [CrossRef]
- Karki, P.; Webb, A.; Smith, K.; Johnson, J.; Lee, K.; Son, D.-S.; Aschner, M.; Lee, E. Yin Yang 1 Is a Repressor of Glutamate Transporter EAAT2, and It Mediates Manganese-Induced Decrease of EAAT2 Expression in Astrocytes. Mol. Cell. Biol. 2014, 34, 1280–1289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zschocke, J.; Allritz, C.; Engele, J.; Rein, T. DNA methylation dependent silencing of the human glutamate transporterEAAT2 gene in glial cells. Glia 2007, 55, 663–674. [Google Scholar] [CrossRef] [PubMed]
- De Groot, J.F.; Liu, T.J.; Fuller, G.N.; Yung, W.A. The Excitatory Amino Acid Transporter-2 Induces Apoptosis and Decreases Glioma GrowthIn vitroandIn vivo. Cancer Res. 2005, 65, 1934–1940. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alam, M.A.; Datta, P.K. Epigenetic Regulation of Excitatory Amino Acid Transporter 2 in Neurological Disorders. Front. Pharmacol. 2019, 10, 1510. [Google Scholar] [CrossRef] [Green Version]
- Hardingham, N.R.; Hardingham, G.E.; Fox, K.D.; Jack, J.J.B. Presynaptic Efficacy Directs Normalization of Synaptic Strength in Layer 2/3 Rat Neocortex After Paired Activity. J. Neurophysiol. 2007, 97, 2965–2975. [Google Scholar] [CrossRef] [Green Version]
- Turrigiano, G. Homeostatic Synaptic Plasticity: Local and Global Mechanisms for Stabilizing Neuronal Function. Cold Spring Harb. Perspect. Biol. 2012, 4, a005736. [Google Scholar] [CrossRef] [Green Version]
- Larkman, A.U.; Jack, J.J.B. Synaptic plasticity: Hippocampal LTP. Curr. Opin. Neurobiol. 1995, 5, 324–334. [Google Scholar] [CrossRef]
- Hardingham, N.; Read, J.C.A.; Trevelyan, A.J.; Nelson, J.C.; Jack, J.J.B.; Bannister, N.J. Quantal analysis reveals a functional correlation between presynaptic and postsynaptic efficacy in excitatory connections from rat neocortex. J. Neurosci. 2010, 30, 1441–1451. [Google Scholar] [CrossRef] [Green Version]
- Valtcheva, S.; Venance, L. Astrocytes gate Hebbian synaptic plasticity in the striatum. Nat. Commun. 2016, 7, 13845. [Google Scholar] [CrossRef] [Green Version]
- Rajatileka, S.; Odd, D.; Robinson, M.T.; Spittle, A.C.; Dwomoh, L.; Williams, M.; Harding, D.; Wagstaff, M.; Owen, M.; Crosby, C.; et al. Variants of the EAAT2 Glutamate Transporter Gene Promoter Are Associated with Cerebral Palsy in Preterm Infants. Mol. Neurobiol. 2017, 55, 2013–2024. [Google Scholar] [CrossRef] [Green Version]
- Poletti, S.; Radaelli, D.; Bosia, M.; Buonocore, M.; Pirovano, A.; Lorenzi, C.; Cavallaro, R.; Smeraldi, E.; Benedetti, F. Effect of glutamate transporter EAAT2 gene variants and gray matter deficits on working memory in schizophrenia. Eur. Psychiatry 2014, 29, 219–225. [Google Scholar] [CrossRef] [PubMed]
- Parkin, G.M.; Gibbons, A.; Udawela, M.; Dean, B. Excitatory amino acid transporter (EAAT)1 and EAAT2 mRNA levels are altered in the prefrontal cortex of subjects with schizophrenia. J. Psychiatr. Res. 2020, 123, 151–158. [Google Scholar] [CrossRef] [PubMed]
- Bauer, D.; Gupta, D.S.; Harotunian, V.; Meador-Woodruff, J.H.; McCullumsmith, R.E. Abnormal expression of glutamate transporter and transporter interacting molecules in prefrontal cortex in elderly patients with schizophrenia. Schizophr. Res. 2008, 104, 108–120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Amen-Hellebrekers, C.J.; Jansen, S.; Pfundt, R.; Schuurs-Hoeijmakers, J.H.; Koolen, D.; Marcelis, C.L.; De Leeuw, N.; De Vries, B.B. Duplications of SLC1A3: Associated with ADHD and autism. Eur. J. Med Genet. 2016, 59, 373–376. [Google Scholar] [CrossRef]
- Huang, X.; Zhang, Q.; Chen, X.; Gu, X.; Wang, M.; Wu, J. A functional variant in SLC1A3 influences ADHD risk by disrupting a hsa-miR-3171 binding site: A two-stage association study. Genes Brain Behav. 2019, 18, e12574. [Google Scholar] [CrossRef]
- Higashimori, H.; Schin, C.S.; Chiang, M.S.R.; Morel, L.; Shoneye, T.A.; Nelson, D.L.; Yang, Y. Selective Deletion of Astroglial FMRP Dysregulates Glutamate Transporter GLT1 and Contributes to Fragile X Syndrome Phenotypes In Vivo. J. Neurosci. 2016, 36, 7079–7094. [Google Scholar] [CrossRef]
- Blacker, C.J.; Millischer, V.; Webb, L.M.; Ho, A.M.C.; Schalling, M.; Frye, M.A.; Veldic, M. EAAT2 as a Research Target in Bipolar Disorder and Unipolar Depression: A Systematic Review. Mol. Neuropsychiatry 2019, 5, 1–16. [Google Scholar] [CrossRef]
- Bernard, R.; Kerman, I.A.; Thompson, R.C.; Jones, E.G.; Bunney, W.E.; Barchas, J.D.; Schatzberg, A.F.; Myers, R.M.; Akil, H.; Watson, S.J. Altered expression of glutamate signaling, growth factor, and glia genes in the locus coeruleus of patients with major depression. Mol. Psychiatry 2011, 16, 634–646. [Google Scholar] [CrossRef] [Green Version]
- Choudary, P.V.; Molnar, M.; Evans, S.J.; Tomita, H.; Li, J.Z.; Vawter, M.P.; Myers, R.M.; Bunney, W.E.; Akil, H.; Watson, S.J.; et al. Altered cortical glutamatergic and GABAergic signal transmission with glial involvement in depression. Proc. Natl. Acad. Sci. USA 2005, 102, 15653–15658. [Google Scholar] [CrossRef] [Green Version]
- Soriano, F.X.; Hardingham, G.E. Compartmentalized NMDA receptor signalling to survival and death. J. Physiol. 2007, 584, 381–387. [Google Scholar] [CrossRef]
- Bell, K.F.S.; Hardingham, G.E. The influence of synaptic activity on neuronal health. Curr. Opin. Neurobiol. 2011, 21, 299–305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parsons, M.P.; Raymond, L.A. Extrasynaptic NMDA Receptor Involvement in Central Nervous System Disorders. Neuron 2014, 82, 279–293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soria, F.N.; Pérez-Samartín, A.; Martin, A.; Gona, K.B.; Llop, J.; Szczupak, B.; Chara, J.C.; Matute, C.; Domercq, M. Extrasynaptic glutamate release through cystine/glutamate antiporter contributes to ischemic damage. J. Clin. Investig. 2014, 124, 3645–3655. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frasca, A.; Aalbers, M.; Frigerio, F.; Fiordaliso, F.; Salio, M.; Gobbi, M.; Cagnotto, A.; Gardoni, F.; Battaglia, G.S.; Hoogland, G.; et al. Misplaced NMDA receptors in epileptogenesis contribute to excitotoxicity. Neurobiol. Dis. 2011, 43, 507–515. [Google Scholar] [CrossRef]
- Stark, D.T.; Bazan, N.G. Synaptic and Extrasynaptic NMDA Receptors Differentially Modulate Neuronal Cyclooxygenase-2 Function, Lipid Peroxidation, and Neuroprotection. J. Neurosci. 2011, 31, 13710–13721. [Google Scholar] [CrossRef]
- Milnerwood, A.; Gladding, C.; Pouladi, M.; Kaufman, A.; Hines, R.; Boyd, J.; Ko, R.; Vasuta, O.; Graham, R.; Hayden, M.; et al. Early increase in extrasynaptic NMDA receptor signalling and expression contributes to phenotype onset in Huntington’s disease mice. Neuron 2010, 65, 178–190. [Google Scholar] [CrossRef] [Green Version]
- Okamoto, S.-I.; Pouladi, M.A.; Talantova, M.; Yao, D.; Xia, P.; Ehrnhoefer, D.E.; Zaidi, R.; Clemente, A.; Kaul, M.; Graham, R.K.; et al. Balance between synaptic versus extrasynaptic NMDA receptor activity influences inclusions and neurotoxicity of mutant huntingtin. Nat. Med. 2009, 15, 1407–1413. [Google Scholar] [CrossRef] [Green Version]
- Talantova, M.; Sanz-Blasco, S.; Zhang, X.; Xia, P.; Akhtar, M.W.; Okamoto, S.-I.; Dziewczapolski, G.; Nakamura, T.; Cao, G.; Pratt, A.E.; et al. A induces astrocytic glutamate release, extrasynaptic NMDA receptor activation, and synaptic loss. Proc. Natl. Acad. Sci. USA 2013, 110, E2518–E2527. [Google Scholar] [CrossRef] [Green Version]
- Bordji, K.; Becerril-Ortega, J.; Nicole, O.; Buisson, A. Activation of Extrasynaptic, But Not Synaptic, NMDA Receptors Modifies Amyloid Precursor Protein Expression Pattern and Increases Amyloid-β Production. J. Neurosci. 2010, 30, 15927–15942. [Google Scholar] [CrossRef]
- Molokanova, E.; Akhtar, M.W.; Sanz-Blasco, S.; Tu, S.; Piña-Crespo, J.C.; McKercher, S.R.; Lipton, S.A. Differential Effects of Synaptic and Extrasynaptic NMDA Receptors on Aβ-Induced Nitric Oxide Production in Cerebrocortical Neurons. J. Neurosci. 2014, 34, 5023–5028. [Google Scholar] [CrossRef] [Green Version]
- Li, S.; Jin, M.; Koeglsperger, T.; Shepardson, N.E.; Shankar, G.M.; Selkoe, D. Soluble A Oligomers Inhibit Long-Term Potentiation through a Mechanism Involving Excessive Activation of Extrasynaptic NR2B-Containing NMDA Receptors. J. Neurosci. 2011, 31, 6627–6638. [Google Scholar] [CrossRef] [PubMed]
- Xia, P.; Chen, H.-S.V.; Zhang, D.; Lipton, S.A. Memantine Preferentially Blocks Extrasynaptic over Synaptic NMDA Receptor Currents in Hippocampal Autapses. J. Neurosci. 2010, 30, 11246–11250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lau, D.; Bengtson, C.P.; Buchthal, B.; Bading, H. BDNF Reduces Toxic Extrasynaptic NMDA Receptor Signaling via Synaptic NMDA Receptors and Nuclear-Calcium-Induced Transcription of inhba/Activin A. Cell Rep. 2015, 12, 1353–1366. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Puddifoot, C.; Martel, M.-A.; Soriano, F.X.; Camacho, A.; Vidal-Puig, A.; Wyllie, D.J.A.; Hardingham, G.E. PGC-1 Negatively Regulates Extrasynaptic NMDAR Activity and Excitotoxicity. J. Neurosci. 2012, 32, 6995–7000. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karpova, A.; Emikhaylova, M.; Bera, S.; Bär, J.; Reddy, P.P.; Behnisch, T.; Rankovic, V.; Spilker, C.; Bethge, P.; Sahin, J.; et al. Encoding and Transducing the Synaptic or Extrasynaptic Origin of NMDA Receptor Signals to the Nucleus. Cell 2013, 152, 1119–1133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, J.; Bengtson, C.P.; Buchthal, B.; Hagenston, A.M.; Bading, H. Coupling of NMDA receptors and TRPM4 guides discovery of unconventional neuroprotectants. Science 2020, 370, eaay3302. [Google Scholar] [CrossRef] [PubMed]
- Malik, A.R.; Willnow, T.E. Excitatory Amino Acid Transporters in Physiology and Disorders of the Central Nervous System. Int. J. Mol. Sci. 2019, 20, 5671. [Google Scholar] [CrossRef] [Green Version]
- Harris, M.E.; Wang, Y.; Pedigo, N.W.; Hensley, K.; Butterfield, D.A.; Carney, J.M. Amyloid ? Peptide (25–35) Inhibits Na+?Dependent Glutamate Uptake in Rat Hippocampal Astrocyte Cultures. J. Neurochem. 1996, 67, 277–286. [Google Scholar] [CrossRef]
- Parpura-Gill, A.; Beitz, D.; Uemura, E. The inhibitory effects of β-amyloid on glutamate and glucose uptakes by cultured astrocytes. Brain Res. 1997, 754, 65–71. [Google Scholar] [CrossRef]
- Matos, M.; Augusto, E.; Oliveira, C.; Agostinho, P. Amyloid-beta peptide decreases glutamate uptake in cultured astrocytes: Involvement of oxidative stress and mitogen-activated protein kinase cascades. Neuroscience 2008, 156, 898–910. [Google Scholar] [CrossRef]
- Robinson, S.R. Neuronal expression of glutamine synthetase in Alzheimer’s disease indicates a profound impairment of metabolic interactions with astrocytes. Neurochem. Int. 2000, 36, 471–482. [Google Scholar] [CrossRef]
- Scott, H.L.; Pow, D.V.; Tannenberg, A.E.; Dodd, P.R. Aberrant expression of the glutamate transporter excitatory amino acid transporter 1 (EAAT1) in Alzheimer’s disease. J. Neurosci. 2002, 22, 201–205. [Google Scholar] [CrossRef] [Green Version]
- Jacob, C.P.; Koutsilieri, E.; Bartl, J.; Neuen-Jacob, E.; Arzberger, T.; Zander, N.; Ravid, R.; Roggendorf, W.; Riederer, P.; Grunblatt, E. Alterations in expression of glutamatergic transporters and receptors in sporadic Alzheimer’s disease. J. Alzheimer’s Dis. 2007, 11, 97–116. [Google Scholar] [CrossRef] [PubMed]
- Masliah, E.; Hansen, L.; Alford, M.; DeTeresa, R.; Mallory, M. Deficient glutamate tranport is associated with neurodegeneration in Alzheimer’s disease. Ann. Neurol. 1996, 40, 759–766. [Google Scholar] [CrossRef]
- Hoshi, A.; Tsunoda, A.; Yamamoto, T.; Tada, M.; Kakita, A.; Ugawa, Y. Altered expression of glutamate transporter-1 and water channel protein aquaporin-4 in human temporal cortex with Alzheimer’s disease. Neuropathol. Appl. Neurobiol. 2018, 44, 628–638. [Google Scholar] [CrossRef]
- Scimemi, A.; Meabon, J.S.; Woltjer, R.L.; Sullivan, J.M.; Diamond, J.S.; Cook, D.G. Amyloid- 1-42 Slows Clearance of Synaptically Released Glutamate by Mislocalizing Astrocytic GLT-1. J. Neurosci. 2013, 33, 5312–5318. [Google Scholar] [CrossRef]
- Rothstein, J.D.; Ba, M.V.K.; Levey, A.I.; Martin, L.J.; Kuncl, R.W. Selective loss of glial glutamate transporter GLT-1 in amyotrophic lateral sclerosis. Ann. Neurol. 1995, 38, 73–84. [Google Scholar] [CrossRef]
- Rothstein, J.D.; Martin, L.J.; Kuncl, R.W. Decreased Glutamate Transport by the Brain and Spinal Cord in Amyotrophic Lateral Sclerosis. N. Engl. J. Med. 1992, 326, 1464–1468. [Google Scholar] [CrossRef]
- Trotti, D.; Aoki, M.; Pasinelli, P.; Berger, U.V.; Danbolt, N.C.; Brown, R.H.; Hediger, M.A. Amyotrophic Lateral Sclerosis-linked Glutamate Transporter Mutant Has Impaired Glutamate Clearance Capacity. J. Biol. Chem. 2001, 276, 576–582. [Google Scholar] [CrossRef] [Green Version]
- Gibb, S.L.; Boston-Howes, W.; Lavina, S.Z.; Gustincich, S.; Brown, R.H.; Pasinelli, P.; Trotti, D. A Caspase-3-cleaved Fragment of the Glial Glutamate Transporter EAAT2 Is Sumoylated and Targeted to Promyelocytic Leukemia Nuclear Bodies in Mutant SOD1-linked Amyotrophic Lateral Sclerosis. J. Biol. Chem. 2007, 282, 32480–32490. [Google Scholar] [CrossRef] [Green Version]
- Boston-Howes, W.; Gibb, S.L.; Williams, E.O.; Pasinelli, P.; Brown, R.H.; Trotti, D. Caspase-3 Cleaves and Inactivates the Glutamate Transporter EAAT. J. Biol. Chem. 2006, 281, 14076–14084. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosen, D.R.; Siddique, T.; Patterson, D.; Figlewicz, D.A.; Sapp, P.C.; Hentati, A.; Donaldson, D.H.; Goto, J.; O’Regan, J.P.; Deng, H.-X.; et al. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature 1993, 362, 59–62. [Google Scholar] [CrossRef] [PubMed]
- Deng, H.X.; Hentati, A.; Tainer, J.A.; Iqbal, Z.; Cayabyab, A.; Hung, W.Y.; Getzoff, E.D.; Hu, P.; Herzfeldt, B.; Roos, R.P.; et al. Amyotrophic lateral sclerosis and structural defects in Cu,Zn superoxide dismutase. Science 1993, 261, 1047–1051. [Google Scholar] [CrossRef] [PubMed]
- Sugiyama, K.; Tanaka, K. Spinal cord-specific deletion of the glutamate transporter GLT1 causes motor neuron death in mice. Biochem. Biophys. Res. Commun. 2018, 497, 689–693. [Google Scholar] [CrossRef] [PubMed]
- Proper, E.A.; Hoogland, G.; Kappen, S.M.; Jansen, G.H.; Rensen, M.G.A.; Schrama, L.H.; Van Veelen, C.W.M.; Van Rijen, P.C.; Van Nieuwenhuizen, O.; Gispen, W.H.; et al. Distribution of glutamate transporters in the hippocampus of patients with pharmaco-resistant temporal lobe epilepsy. Brain 2002, 125, 32–43. [Google Scholar] [CrossRef] [PubMed]
- Mathern, G.W.; Mendoza, D.; Lozada, A.; Pretorius, J.K.; Dehnes, Y.; Danbolt, N.C.; Nelson, N.; Leite, J.P.; Chimelli, L.; Born, D.E.; et al. Hippocampal GABA and glutamate transporter immunoreactivity in patients with temporal lobe epilepsy. Neurology 1999, 52, 453. [Google Scholar] [CrossRef]
- Sarac, S.; Afzal, S.; Broholm, H.; Madsen, F.F.; Ploug, T.; Laursen, H. Excitatory amino acid transporters EAAT-1 and EAAT-2 in temporal lobe and hippocampus in intractable temporal lobe epilepsy. APMIS 2009, 117, 291–301. [Google Scholar] [CrossRef]
- Vallejo-Illarramendi, A.; Domercq, M.; Pérez-Cerdá, F.; Ravid, R.; Matute, C. Increased expression and function of glutamate transporters in multiple sclerosis. Neurobiol. Dis. 2006, 21, 154–164. [Google Scholar] [CrossRef]
- Mitosek-Szewczyk, K.; Sulkowski, G.; Stelmasiak, Z.; Strużyńska, L. Expression of glutamate transporters GLT-1 and GLAST in different regions of rat brain during the course of experimental autoimmune encephalomyelitis. Neurosci. 2008, 155, 45–52. [Google Scholar] [CrossRef]
- Vercellino, M.; Merola, A.; Piacentino, C.; Votta, B.; Capello, E.; Mancardi, G.L.; Mutani, R.; Giordana, M.T.; Cavalla, P. Altered Glutamate Reuptake in Relapsing-Remitting and Secondary Progressive Multiple Sclerosis Cortex: Correlation With Microglia Infiltration, Demyelination, and Neuronal and Synaptic Damage. J. Neuropathol. Exp. Neurol. 2007, 66, 732–739. [Google Scholar] [CrossRef] [Green Version]
- Diniz, L.P.; Araujo, A.P.B.; Matias, I.; Garcia, M.N.; Barros-Aragão, F.G.; Reis, R.A.D.M.; Foguel, D.; Braga, C.; Figueiredo, C.P.; Romão, L.; et al. Astrocyte glutamate transporters are increased in an early sporadic model of synucleinopathy. Neurochem. Int. 2020, 138, 104758. [Google Scholar] [CrossRef] [PubMed]
- Chung, E.; Chen, L.; Chan, Y.; Yung, K.K.L. Downregulation of glial glutamate transporters after dopamine denervation in the striatum of 6-hydroxydopamine-lesioned rats. J. Comp. Neurol. 2008, 511, 421–437. [Google Scholar] [CrossRef] [PubMed]
- El Arfani, A.; Albertini, G.; Bentea, E.; Demuyser, T.; Van Eeckhaut, A.; Smolders, I.; Massie, A. Alterations in the motor cortical and striatal glutamatergic system and d-serine levels in the bilateral 6-hydroxydopamine rat model for Parkinson’s disease. Neurochem. Int. 2015, 88, 88–96. [Google Scholar] [CrossRef] [PubMed]
- Ferrarese, C.; Tremolizzo, L.; Rigoldi, M.; Sala, G.; Brighina, L.; Ricci, G.; Albizzati, M.G.; Piolti, R.; Crosti, F.; Frattola, L.; et al. Decreased platelet glutamate uptake and genetic risk factors in patients with Parkinson’s disease. Neurol. Sci. 2001, 22, 65–66. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.-M.; Cha, S.-H.; Choi, Y.R.; Jou, I.; Joe, E.-H.; Park, S.M. DJ-1 deficiency impairs glutamate uptake into astrocytes via the regulation of flotillin-1 and caveolin-1 expression. Sci. Rep. 2016, 6, 28823. [Google Scholar] [CrossRef] [PubMed]
- Arzberger, T.; Krampfl, K.; Leimgruber, S.; Weindl, A. Changes of NMDA Receptor Subunit (NR1, NR2B) and Glutamate Transporter (GLT1) mRNA Expression in Huntington’s Disease—An In Situ Hybridization Study. J. Neuropathol. Exp. Neurol. 1997, 56, 440–454. [Google Scholar] [CrossRef] [Green Version]
- Behrens, P.F.; Franz, P.; Woodman, B.; Lindenberg, K.S.; Landwehrmeyer, G.B. Impaired glutamate transport and glutamate-glutamine cycling: Downstream effects of the Huntington mutation. Brain 2002, 125, 1908–1922. [Google Scholar] [CrossRef] [Green Version]
- Scarr, E.; Udawela, M.; Thomas, E.A.; Dean, B. Changed gene expression in subjects with schizophrenia and low cortical muscarinic M1 receptors predicts disrupted upstream pathways interacting with that receptor. Mol. Psychiatry 2018, 23, 295–303. [Google Scholar] [CrossRef] [Green Version]
- Vallejo-Illarramendi, A.; Melone, M.; Conti, F.; Matute, C. Clozapine reduces GLT-1 expression and glutamate uptake in astrocyte cultures. Glia 2005, 50, 276–279. [Google Scholar] [CrossRef] [Green Version]
- Medina, A.; Burke, S.; Thompson, R.C.; Bunney, W.; Myers, R.M.; Schatzberg, A.; Akil, H.; Watson, S.J. Glutamate transporters: A key piece in the glutamate puzzle of major depressive disorder. J. Psychiatr. Res. 2013, 47, 1150–1156. [Google Scholar] [CrossRef]
- Purcell, A.E.; Jeon, O.H.; Zimmerman, A.W.; Blue, M.E.; Pevsner, J. Postmortem brain abnormalities of the glutamate neurotransmitter system in autism. Neurology 2001, 57, 1618–1628. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, W.; Imai, S.; Zou, S.; Yang, J.; Watanabe, M.; Wang, J.; Dubner, R.; Wei, F.; Ren, K. Altered glial glutamate transporter expression in descending circuitry and the emergence of pain chronicity. Mol. Pain 2019, 15, 1744806918825044. [Google Scholar] [CrossRef] [PubMed]
- Cirillo, G.; Bianco, M.R.; Colangelo, A.M.; Cavaliere, C.; Daniele, D.L.; Zaccaro, L.; Alberghina, L.; Papa, M. Reactive astrocytosis-induced perturbation of synaptic homeostasis is restored by nerve growth factor. Neurobiol. Dis. 2011, 41, 630–639. [Google Scholar] [CrossRef] [PubMed]
- Cavaliere, C.; Cirillo, G.; Bianco, M.R.; Rossi, F.; De Novellis, V.; Maione, S.; Papa, M. Gliosis alters expression and uptake of spinal glial amino acid transporters in a mouse neuropathic pain model. Neuron Glia Biol. 2007, 3, 141–153. [Google Scholar] [CrossRef]
- Aguirre, G.; Rosas, S.; Lopez-Bayghen, E.; Ortega, A. Valproate-dependent transcriptional regulation of GLAST/EAAT1 expression: Involvement of Ying-Yang. Neurochem. Int. 2008, 52, 1322–1331. [Google Scholar] [CrossRef]
- Gegelashvili, G.; Bjerrum, O.J. Glutamate Transport System as a Novel Therapeutic Target in Chronic Pain: Molecular Mechanisms and Pharmacology. In Advances in Neurobiology; Ortega, A., Schousboe, A., Eds.; Springer: Cham, Switzerland, 2017; pp. 225–253. [Google Scholar]
- Chivukula, A.S.; Suslova, M.; Kortzak, D.; Kovermann, P.; Fahlke, C. Functional consequences of SLC1A3 mutations associated with episodic ataxia 6. Hum. Mutat. 2020, 41, 1892–1905. [Google Scholar] [CrossRef]
- During, M. Extracellular hippocampal glutamate and spontaneous seizure in the conscious human brain. Lancet 1993, 341, 1607–1610. [Google Scholar] [CrossRef]
- Binder, D.K.; Steinhäuser, C. Functional changes in astroglial cells in epilepsy. Glia 2006, 54, 358–368. [Google Scholar] [CrossRef]
- Selvaraj, B.T.; Livesey, M.R.; Zhao, C.; Gregory, J.M.; James, O.T.; Cleary, E.M.; Chouhan, A.K.; Gane, A.B.; Perkins, E.M.; Dando, O.R.; et al. C9ORF72 repeat expansion causes vulnerability of motor neurons to Ca2+-permeable AMPA receptor-mediated excitotoxicity. Nat. Commun. 2018, 9, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Allen, B.; Ingram, E.; Takao, M.; Smith, M.J.; Jakes, R.; Virdee, K.; Yoshida, H.; Holzer, M.; Craxton, M.; Emson, P.C.; et al. Abundant Tau Filaments and Nonapoptotic Neurodegeneration in Transgenic Mice Expressing Human P301S Tau Protein. J. Neurosci. 2002, 22, 9340–9351. [Google Scholar] [CrossRef] [Green Version]
- Potier, B.; Billard, J.-M.; Rivière, S.; Sinet, P.-M.; Denis, I.; Champeil-Potokar, G.; Grintal, B.; Jouvenceau, A.; Kollen, M.; Dutar, P. Reduction in glutamate uptake is associated with extrasynaptic NMDA and metabotropic glutamate receptor activation at the hippocampal CA1 synapse of aged rats. Aging Cell 2010, 9, 722–735. [Google Scholar] [CrossRef] [PubMed]
- Brothers, H.M.; Bardou, I.; Hopp, S.C.; Kaercher, R.M.; Corona, A.W.; Fenn, A.M.; Godbout, J.P.; Wenk, G.L. Riluzole Partially Rescues Age-Associated, but not LPS-Induced, Loss of Glutamate Transporters and Spatial Memory. J. Neuroimmune Pharmacol. 2013, 8, 1098–1105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pereira, A.C.; Gray, J.D.; Kogan, J.F.; Davidson, R.L.; Rubin, T.G.; Okamoto, M.; Morrison, J.H.; McEwen, B.S. Age and Alzheimer’s disease gene expression profiles reversed by the glutamate modulator riluzole. Mol. Psychiatry 2017, 22, 296–305. [Google Scholar] [CrossRef] [PubMed]
- Wolfe, M.S. Structure and Function of the γ-Secretase Complex. Biochemistry 2019, 58, 2953–2966. [Google Scholar] [CrossRef]
- Jurisch-Yaksi, N.; Sannerud, R.; Annaert, W. A fast growing spectrum of biological functions of γ-secretase in development and disease. Biochim. Biophys. Acta (BBA) Biomembr. 2013, 1828, 2815–2827. [Google Scholar] [CrossRef] [Green Version]
- Hutton, M. The presenilins and Alzheimer’s disease. Hum. Mol. Genet. 1997, 6, 1639–1646. [Google Scholar] [CrossRef]
- Sassi, C.; Guerreiro, R.; Gibbs, R.; Ding, J.; Lupton, M.K.; Troakes, C.; Lunnon, K.; Al-Sarraj, S.; Pickering-Brown, S.; Medway, C.; et al. Exome sequencing identifies 2 novel presenilin 1 mutations (p.L166V and p.S230R) in British early-onset Alzheimer’s disease. Neurobiol. Aging 2014, 35, 2422.e13–2422.e16. [Google Scholar] [CrossRef]
- De Strooper, B. Lessons from a Failed γ-Secretase Alzheimer Trial. Cell 2014, 159, 721–726. [Google Scholar] [CrossRef] [Green Version]
- Henley, D.B.; Sundell, K.L.; Sethuraman, G.; Dowsett, S.A.; May, P.C. Safety profile of semagacestat, a gamma-secretase inhibitor: IDENTITY trial findings. Curr. Med Res. Opin. 2014, 30, 2021–2032. [Google Scholar] [CrossRef]
- Moehlmann, T.; Winkler, E.; Xia, X.; Edbauer, D.; Murrell, J.; Capell, A.; Kaether, C.; Zheng, H.; Ghetti, B.; Haass, C.; et al. Presenilin-1 mutations of leucine 166 equally affect the generation of the Notch and APP intracellular domains independent of their effect on A 42 production. Proc. Natl. Acad. Sci. USA 2002, 99, 8025–8030. [Google Scholar] [CrossRef] [Green Version]
- Brai, E.; Raio, N.A.; Auber, L.A. Notch1 hallmarks fibrillary depositions in sporadic Alzheimer’s disease. Acta Neuropathol. Commun. 2016, 4, 64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, W.; Nadeau, P.; Yuan, M.; Yang, X.; Shen, J.; Yankner, B.A. Proteolytic release and nuclear translocation of Notch-1 are induced by presenilin-1 and impaired by pathogenic presenilin-1 mutations. Proc. Natl. Acad. Sci. USA 1999, 96, 6959–6963. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, F.; Gu, Y.; Hasegawa, H.; Ruan, X.; Arawaka, S.; Fraser, P.E.; Westaway, D.; Mount, H.; George-Hyslop, P.S. Presenilin 1 Mutations Activate γ42-Secretase but Reciprocally Inhibit ε-Secretase Cleavage of Amyloid Precursor Protein (APP) and S3-Cleavage of Notch. J. Biol. Chem. 2002, 277, 36521–36526. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amatniek, J.C.; Hauser, W.A.; DelCastillo-Castaneda, C.; Jacobs, D.M.; Marder, K.; Bell, K.; Albert, M.; Brandt, J.; Stern, Y. Incidence and Predictors of Seizures in Patients with Alzheimer’s Disease. Epilepsia 2006, 47, 867–872. [Google Scholar] [CrossRef] [PubMed]
- Doody, R.S.; Raman, R.; Farlow, M.; Iwatsubo, T.; Vellas, B.; Joffe, S.; Kieburtz, K.; Sun, X.; Thomas, R.G.; Aisen, P.S.; et al. A Phase 3 Trial of Semagacestat for Treatment of Alzheimer’s Disease. N. Engl. J. Med. 2013, 369, 341–350. [Google Scholar] [CrossRef] [PubMed]
- Zheng, J.; Watanabe, H.; Wines-Samuelson, M.; Zhao, H.; Gridley, T.; Kopan, R.; Shen, J. Conditional Deletion ofNotch1andNotch2Genes in Excitatory Neurons of Postnatal Forebrain Does Not Cause Neurodegeneration or Reduction of Notch mRNAs and Proteins. J. Biol. Chem. 2012, 287, 20356–20368. [Google Scholar] [CrossRef] [Green Version]
- Sato, C.; Turkoz, M.; Dearborn, J.T.; Wozniak, D.F.; Kopan, R.; Hass, M.R. Loss of RBPj in Postnatal Excitatory Neurons Does Not Cause Neurodegeneration or Memory Impairments in Aged Mice. PLoS ONE 2012, 7, e48180. [Google Scholar] [CrossRef]
- He, G.; Luo, W.; Li, P.; Remmers, C.; Netzer, W.J.; Hendrick, J.P.; Bettayeb, K.; Flajolet, M.; Gorelick, F.S.; Wennogle, L.P.; et al. Gamma-secretase activating protein is a therapeutic target for Alzheimer’s disease. Nature 2010, 467, 95–98. [Google Scholar] [CrossRef] [Green Version]
- McKenzie, G.J.; Stevenson, P.; Ward, G.; Papadia, S.; Bading, H.; Chawla, S.; Privalsky, M.; Hardingham, G.E. Nuclear Ca2+ and CaM kinase IV specify hormonal- and Notch-responsiveness. J. Neurochem. 2005, 93, 171–185. [Google Scholar] [CrossRef]
- Needham, L.A.; Davidson, A.H.; Bawden, L.J.; Belfield, A.; Bone, E.A.; Brotherton, D.H.; Bryant, S.; Charlton, M.H.; Clark, V.L.; Davies, S.J.; et al. Drug Targeting to Monocytes and Macrophages Using Esterase-Sensitive Chemical Motifs. J. Pharmacol. Exp. Ther. 2011, 339, 132–142. [Google Scholar] [CrossRef] [Green Version]
- Baxter, P.S.; Bell, K.F.S.; Hasel, P.; Kaindl, A.M.; Fricker, M.; Thomson, D.; Cregan, S.P.; Gillingwater, T.H.; Hardingham, G.E. Synaptic NMDA receptor activity is coupled to the transcriptional control of the glutathione system. Nat. Commun. 2015, 6, 6761. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Al-Mubarak, B.; Soriano, F.X.; Hardingham, G.E. Synaptic NMDAR activity suppresses FOXO1 expression via a cis-acting FOXO binding site: FOXO1 is a FOXO target gene. Channels 2009, 3, 233–239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
Figure 1.
Glutamate-glutamine cycle. Glutamate (Glu−) released after excitatory transmission is collected by astrocytic EAAT transporters 1 and 2. The glutamate is then either converted into α-ketoglutarate (α-KG) via glutamate dehydrogenase (GDH) or transaminase reaction and enters the TCA cycle, or else is converted into glutamine (Gln) by glutamine synthetase (GS). Astrocytes excrete Gln back into the extracellular environment via the Na+ driven SNAT3 transporter, which is then taken up by an as yet unconfirmed neuronal Gln transporter. Neurons then convert Gln back to Glu− via a phosphate-activated glutaminase (PAG) reaction to replenish their vesicular Glu− stores.
Figure 1.
Glutamate-glutamine cycle. Glutamate (Glu−) released after excitatory transmission is collected by astrocytic EAAT transporters 1 and 2. The glutamate is then either converted into α-ketoglutarate (α-KG) via glutamate dehydrogenase (GDH) or transaminase reaction and enters the TCA cycle, or else is converted into glutamine (Gln) by glutamine synthetase (GS). Astrocytes excrete Gln back into the extracellular environment via the Na+ driven SNAT3 transporter, which is then taken up by an as yet unconfirmed neuronal Gln transporter. Neurons then convert Gln back to Glu− via a phosphate-activated glutaminase (PAG) reaction to replenish their vesicular Glu− stores.

Figure 2.
The Notch signalling pathway. Notch is a contact dependent signalling pathway. When a Notch ligand contacts a Notch receptor this initiates a cleavage event through the enzyme γ-secretase, releasing the Notch intracellular domain (NICD). The NICD then translocates into the cell nucleus, where it pulls down various proteins, such as MAML, and associates with the Notch effector, CBF1. This association turns on transcription, with the Hes and Hey family of genes prominent examples of genes transcribed by this cascade. The γ-secretase inhibitor DAPT is able to prevent activation of the Notch signalling pathway as the NICD is unable to be cleaved.
Figure 2.
The Notch signalling pathway. Notch is a contact dependent signalling pathway. When a Notch ligand contacts a Notch receptor this initiates a cleavage event through the enzyme γ-secretase, releasing the Notch intracellular domain (NICD). The NICD then translocates into the cell nucleus, where it pulls down various proteins, such as MAML, and associates with the Notch effector, CBF1. This association turns on transcription, with the Hes and Hey family of genes prominent examples of genes transcribed by this cascade. The γ-secretase inhibitor DAPT is able to prevent activation of the Notch signalling pathway as the NICD is unable to be cleaved.

Figure 3.
Notch signalling is needed to maintain astrocytic EAAT function. Example traces of EAAT mediated currents in response to 200 μM L-Asp in (i) DIV13 neuronal cocultured (CC) astrocyte, (ii) DIV24 CC astrocyte, (iii) DIV24 monocultured (MC) astrocyte, (iv) DIV24 CC astrocyte +DAPT from DIV0, and (v) DIV24 astrocyte +DAPT from DIV14. Cells were voltage-clamped at −80 mV and all recordings were done in the presence of 100 μM AP-5. (vi) At DIV24 there was a significantly larger EAAT mediated response in control CC astrocytes compared to MC astrocytes (p = 0.009, LME ANOVE, df = 29), as well as CC astrocytes treated with DAPT from either DIV0 or DIV14 (p = 0.028 & 0.027, for DIV0 and DIV14 respectively, LME ANOVA, df = 29). There was no difference in EAAT response in CC astrocytes treated with DAPT from DIV0 or from DIV14 after currents had been established. Recordings were taken from cells across at least three independent culture batches. * p < 0.05, ** p < 0.001.
Figure 3.
Notch signalling is needed to maintain astrocytic EAAT function. Example traces of EAAT mediated currents in response to 200 μM L-Asp in (i) DIV13 neuronal cocultured (CC) astrocyte, (ii) DIV24 CC astrocyte, (iii) DIV24 monocultured (MC) astrocyte, (iv) DIV24 CC astrocyte +DAPT from DIV0, and (v) DIV24 astrocyte +DAPT from DIV14. Cells were voltage-clamped at −80 mV and all recordings were done in the presence of 100 μM AP-5. (vi) At DIV24 there was a significantly larger EAAT mediated response in control CC astrocytes compared to MC astrocytes (p = 0.009, LME ANOVE, df = 29), as well as CC astrocytes treated with DAPT from either DIV0 or DIV14 (p = 0.028 & 0.027, for DIV0 and DIV14 respectively, LME ANOVA, df = 29). There was no difference in EAAT response in CC astrocytes treated with DAPT from DIV0 or from DIV14 after currents had been established. Recordings were taken from cells across at least three independent culture batches. * p < 0.05, ** p < 0.001.


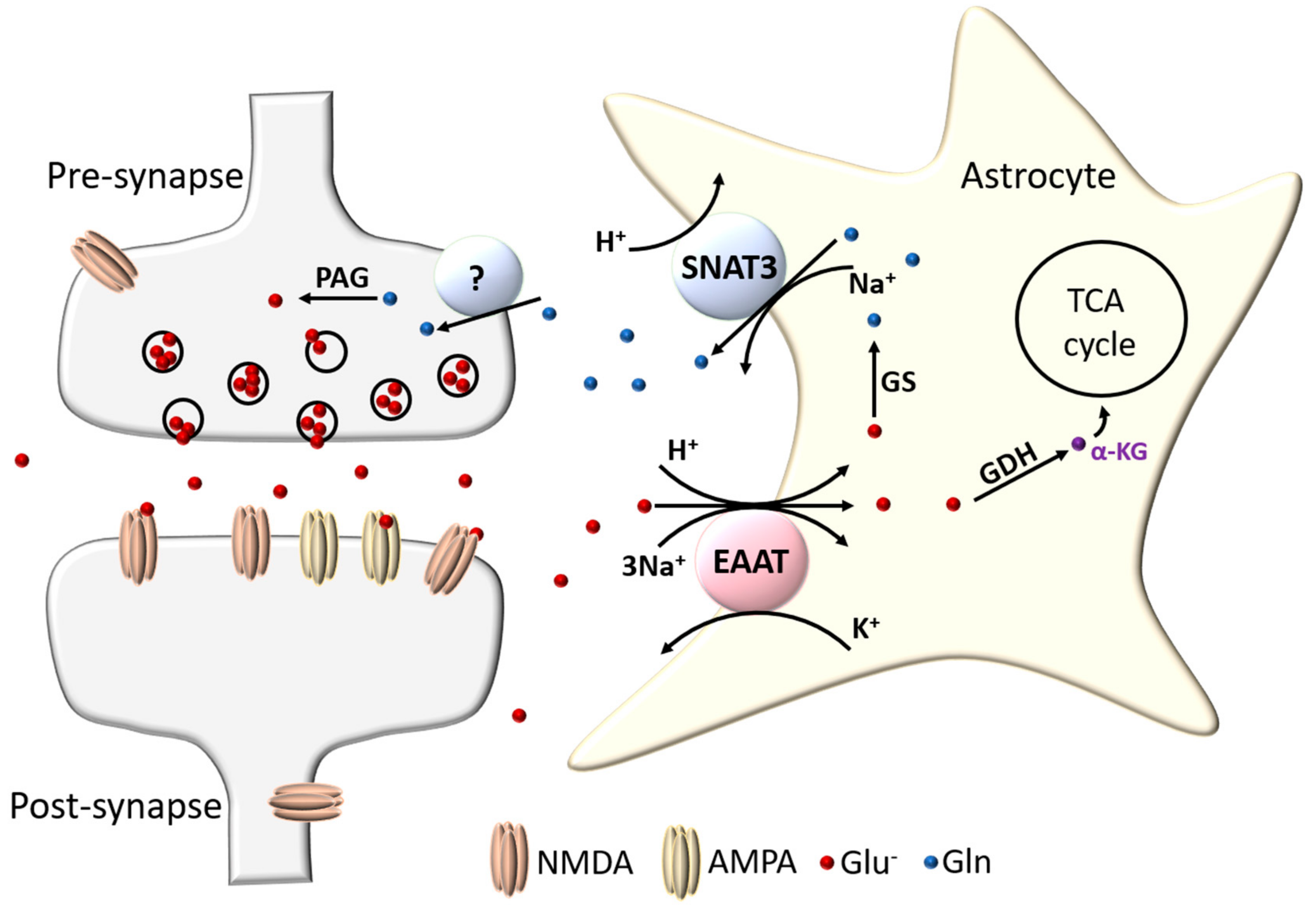{kind=link}
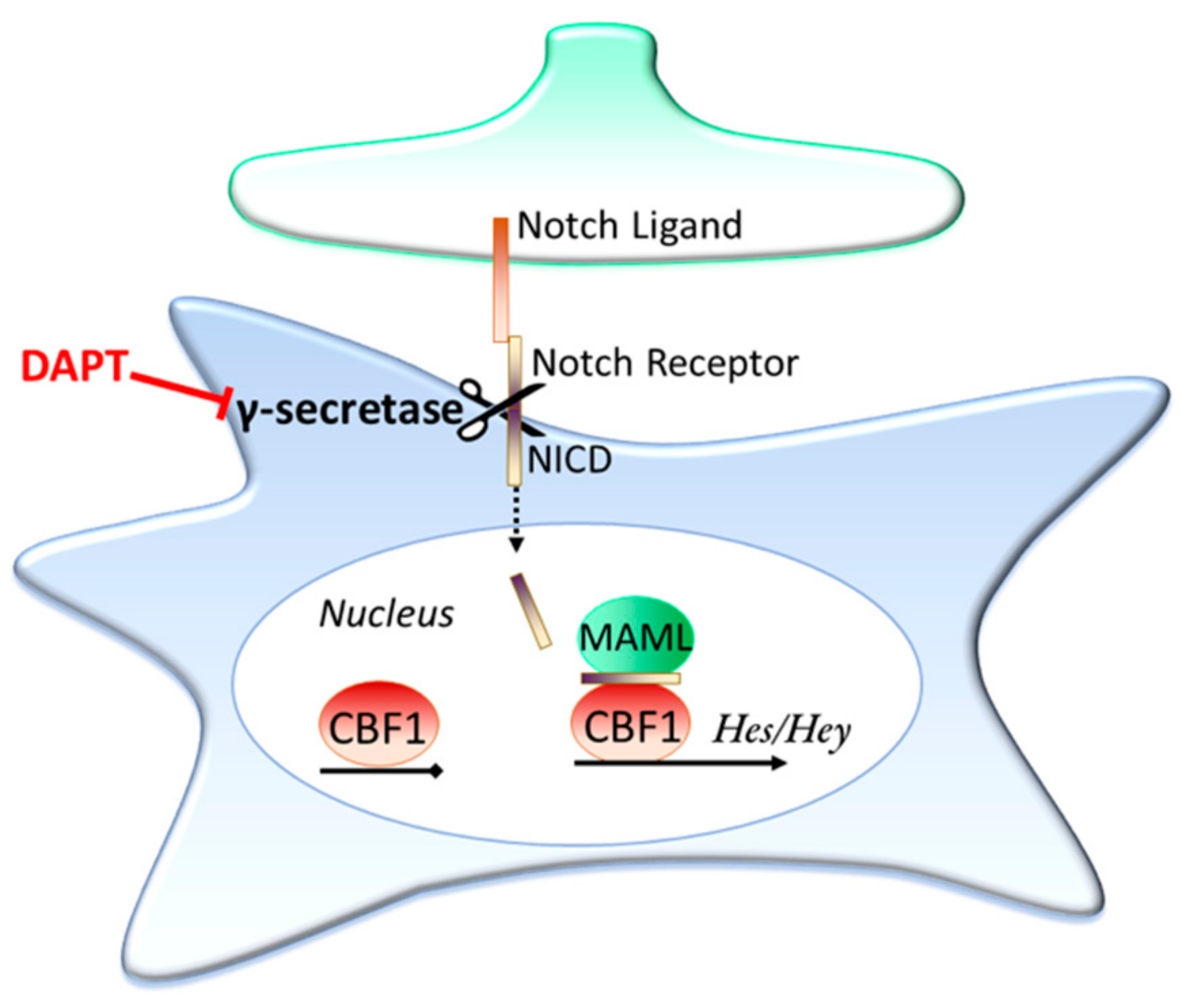{kind=link}
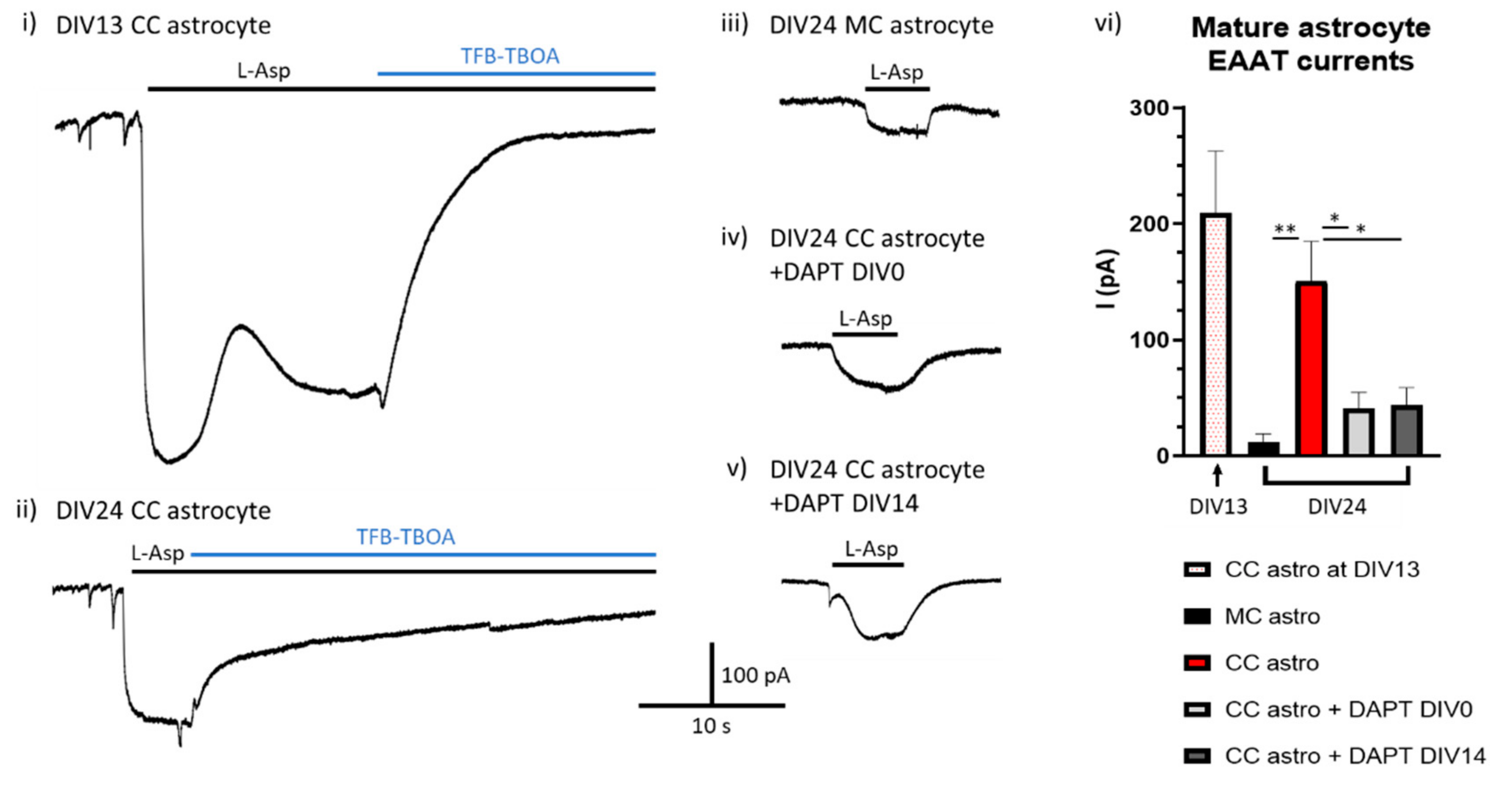{kind=link}
Table 1.
Excitatory amino acid transporter family overview.
| Protein | Gene | Cl− Conduct. | Kinetics | Location | Protein Abundance |
|---|---|---|---|---|---|
| EAAT1 | SLC1A3 | Mod | KM = 22–48 μM Cycle time = 62 ms | Astrocytes (incl. Bergmann & Müller glia); Predominant EAAT subtype in cerebellum (1.8 mg/g of protein) and retina | 99th percentile of protein found in human CBC; ≥95th in V1C, DFC, MD, STR and HIP; F794th in AMY |
| EAAT2 | SLC1A2 | Low | KM = 25–97 μM Cycle time = 70 ms | Astrocytes (and some sparse neurons); Predominant EAAT subtype in hippocampus (1.3 mg/g of protein) and cortex (0.8 mg/g) | 99th percentile of protein found in human V1C; ≥95th in DFC, HIP, AMY, STR and MD; 93rd in CBC |
| EAAT3 | SLC1A1 | Mod | KM = 42–62 μM Cycle time = 10 ms | Neurons (typically on spines); Highest concentration in hippocampus (0.013 mg/g of protein) | 26th percentile of protein found in human MD; 21st in HIP; ≤20th in AMY, STR, CBC, DFC and V1C |
| EAAT4 | SLC1A6 | High | KM = 2.5 μM Cycle time >166 ms | Cerebellar Purkinje cells (0.2 mg/g of protein in cerebellar molecular layer) | 89th percentile of protein in human CBC; 9th in MD and AMY; <15th in STR, DFC and HIP |
| EAAT5 | SLC1A7 | High | KM = 61–62 μM Cycle time >1000 ms | Retina (rod photo receptors, bipolar cells) |
Abbreviations: CBC = cerebellar cortex; V1C = primary visual cortex; DFC = dorsal prefrontal cortex; MD = mediodorsal thalamic nucleus; STR = striatum; HIP = hippocampus and AMY = amygdala. See ref [64] for references.
Table 2.
Regulation of astrocytic glutamate transporters.
| Treatment | Slc1a3/EAAT1 | Slc1a2/EAAT2 | Species |
|---|---|---|---|
| Neuronal coculture | Increased expression | Robust induction of expression | Mouse and rat; in vitro |
| Neuronal Conditioned Media | No | Yes | Mouse; in vitro |
| cAMP | Increases expression and function | Robust increases in expression and function | Mouse; in vitro |
| Glutamate | Downregulated by glutamaterigic denervation; Upregulated by AMPA receptor activation | Downregulated by glutamaterigic denervation | Mouse and Rat; in situ |
| Epidermal growth factor/NF-κB | No | Increased expression. Overlap with NCM and cAMP pathway | Mouse, rat and human; in vitro |
| Pax6 | No | Induces expression in pure astrocytes; knockdown represses neuron coculture induction | Mouse; in vitro |
| Notch (Neuron/endothelial cell to astrocyte contact dependant) | Increases expression; inhibition decreases expression | Increased expression; inhibition decreases expression | Mouse, rat and drosophila; in vitro and in vivo |
Table 3.
CNS diseases and disorders associated with astrocytic glutamate transporter dysfunction.
| Disease | Transporter | Observed Association | Refs |
|---|---|---|---|
| Alzheimer’s Disease (AD) | EAAT1, EAAT2 | EAAT1/2 function and expression reduced by amyloid β; Aberrant EAAT1 expression in AD patient neurons; Reduced function and expression of EAAT1 and 2 in hippocampal and cortical AD tissue | [171,172,173,174,175,176,177,178,179] |
| Amyotrophic lateral sclerosis (ALS)/motor neuron disease | EAAT2 | Impaired Glu uptake in patients with sporadic ALS; Reduced EAAT2 protein in tissue from motor regions; One reported case of a patient with a mutation in SLC1A2 causing reduced EAAT2 activity; Familial ALS with SOD1 mutations expected to reduce functional EAAT2 protein; Deletion of slc1a2 in mice spinal cord leads to motor neuron degeneration; Reduction in slc1a2 in P301S tauopathy mouse model | [101,180,181,182,183,184,185,186,187] |
| Epilepsy/temporal lobe epilepsy (TLE) | EAAT1, EAAT2 | Reduced EAAT2 in TLE patients with hippocampal sclerosis; Reduced EAAT1 & 2 in treatment resistant TLE patients; Mouse EAAT2 KO → lethal epilepsy | [91,188,189,190] |
| Multiple sclerosis (MS) | EAAT1, EAAT2 | Increased EAAT1 and EAAT2 mRNA and protein in MS optic nerve, with increased glutamate uptake; Loss of EAAT1 and EAAT2 in areas surrounding cortical lesions of MS patients; In rat EAE model cortex, increased EAAT2 mRNA and protein, increased EAAT1 mRNA but decreased protein. In rat EAE model cerebellum, increased EAAT1 and EAAT2 mRNA, but decreased EAAT1 and EAAT2 protein. | [191,192,193] |
| Synucleinop-athies (including Parkinson’s disease -PD) | EAAT1, EAAT2 | Increased EAAT1 and EAAT2 expression following injection of α-synuclein oligomers in mouse striatum; Decreased EAAT1 and EAAT2 in rat striatum following dopaminergic denervation via MPTP treatment or 6-ODHA induced lesion rat models; Reduced glutamate uptake in platelets from PD patients; PD-related mutation DJ-1 mouse model showed reduced EAAT2 function | [194,195,196,197,198] |
| Huntington’s disease (HD) | EAAT2 | Decrease in EAAT2 mRNA expression in neostriatum of HD patients, decrease corresponding to disease severity; Decease in EAAT2 mRNA and protein in mice expressing mutant huntingtin; | [199,200] |
| Schizophrenia (SCZ) | EAAT1, EAAT2 | Increased mRNA expression of EAAT1 in Brodmann’s area (BA)9; Increased mRNA expression of EAAT1 and EAAT2 in BA10; Increased EAAT1 mRNA and decreased protein in post mortem SCZ CNS tissue; Clozapine (used to treat SCZ) decreased EAAT2 expression; | [145,146,201,202] |
| Major depressive disorder (MDD) | EAAT1, EAAT2 | Reduced mRNA expression of EAAT1 and EAAT2 in anterior cingulate, dorsolateral prefrontal cortex, locus coeruleus and hippocampus of human MDD patients; Decreased protein in orbitofrontal cortex of MDD patients; | [150,151,152,203] |
| Autism | EAAT1,F7EAAT2 | EAAT1 mRNA expression upregulated; Decreased functional EAAT2 in conditional Fmr1 KO mouse astrocytes (mouse model of fragile-X) | [149,204] |
| Attention deficit hyperactivity disorder (ADHD) | EAAT1 | Duplication of SLC1A3 gene observed in clinical case of ADHD; SLC1A3 rs1049522 allele significantly associated with ADHD; Increased EAAT1 mRNA expression in cerebellar cortex | [147,148] |
| Chronic pain | EAAT2 | Decreased EAAT2 mRNA in rostral ventromedial medulla and spinal cord in rodent chronic pain models; Administration of EAAT2 antagonist alleviates hyperalgesia in rats; Analgesic effects of valproic acid suggested to be due to increasing EAAT1 expression. | [205,206,207,208,209] |
| Episodic ataxia type 6 | EAAT1 | Caused by mutations in SLC1A3 altering properties of EAAT1 | [210] |
Table 4.
Notch pathway and astrocytic EAAT expression in human astrocytes with age. There is a reduction in gene expression for both astrocytic EAAT transporters and Notch pathway associated genes with age in astrocytes isolated from human tissue.
Table 4.
Notch pathway and astrocytic EAAT expression in human astrocytes with age. There is a reduction in gene expression for both astrocytic EAAT transporters and Notch pathway associated genes with age in astrocytes isolated from human tissue.
| Gene | Mean Human Expression in <40 y.o. (FPKM) | Mean Human Expression in >40 y.o. (FPKM) | Relative Expression with Older Age |
|---|---|---|---|
| Notch genes | |||
| HES1 | 9.31 | 6.57 | 0.71 |
| HES6 | 2.91 | 1.28 | 0.44 |
| HES5 | 3.47 | 0.92 | 0.26 |
| HEY2 | 1.40 | 1.57 | 1.12 |
| HEY1 | 10.43 | 9.76 | 0.94 |
| BCL2 | 5.41 | 4.75 | 0.88 |
| Total FPKM | 32.93 | 24.84 | 0.75 |
| Notch receptors | |||
| NOTCH2 | 22.36 | 14.39 | 0.64 |
| NOTCH1 | 0.84 | 0.41 | 0.49 |
| NOTCH3 | 0.53 | 0.20 | 0.38 |
| NOTCH4 | 0.11 | 0.13 | 1.22 |
| Total FPKM | 23.83 | 15.14 | 0.64 |
| γ-secretase genes | |||
| PSEN1 | 7.87 | 6.99 | 0.89 |
| PSEN2 | 1.23 | 0.67 | 0.55 |
| NCSTN | 24.59 | 11.58 | 0.47 |
| APH1A | 1.63 | 1.27 | 0.78 |
| APH1B | 5.07 | 4.72 | 0.93 |
| PSENEN | 0.26 | 0.65 | 2.51 |
| Total FPKM | 40.64 | 25.89 | 0.64 |
| Notch effectors/activators | |||
| MAML1 | 1.83 | 0.90 | 0.49 |
| MED8 | 4.42 | 2.77 | 0.63 |
| RBPJ | 6.52 | 7.22 | 1.11 |
| FURIN | 0.46 | 0.17 | 0.38 |
| Total FPKM | 13.23 | 11.07 | 0.84 |
| Sum Notch related genes (FPKM) | 110.64 | 76.93 | 0.70 |
| Glutamate transporters | |||
| SLC1A2 | 2454.47 | 1521.31 | 0.62 |
| SLC1A3 | 1146.57 | 797.77 | 0.70 |
| Total EAAT (FPKM) | 3601.05 | 2319.07 | 0.64 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Todd, A.C.; Hardingham, G.E. The Regulation of Astrocytic Glutamate Transporters in Health and Neurodegenerative Diseases. Int. J. Mol. Sci. 2020, 21, 9607. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21249607
AMA Style
Todd AC, Hardingham GE. The Regulation of Astrocytic Glutamate Transporters in Health and Neurodegenerative Diseases. International Journal of Molecular Sciences. 2020; 21(24):9607. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21249607
Chicago/Turabian StyleTodd, Alison C., and Giles E. Hardingham. 2020. "The Regulation of Astrocytic Glutamate Transporters in Health and Neurodegenerative Diseases" International Journal of Molecular Sciences 21, no. 24: 9607. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21249607
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.