Anti-Kir4.1 Antibodies in Multiple Sclerosis: Specificity and Pathogenicity

 ,
,
Abstract
:1. Introduction
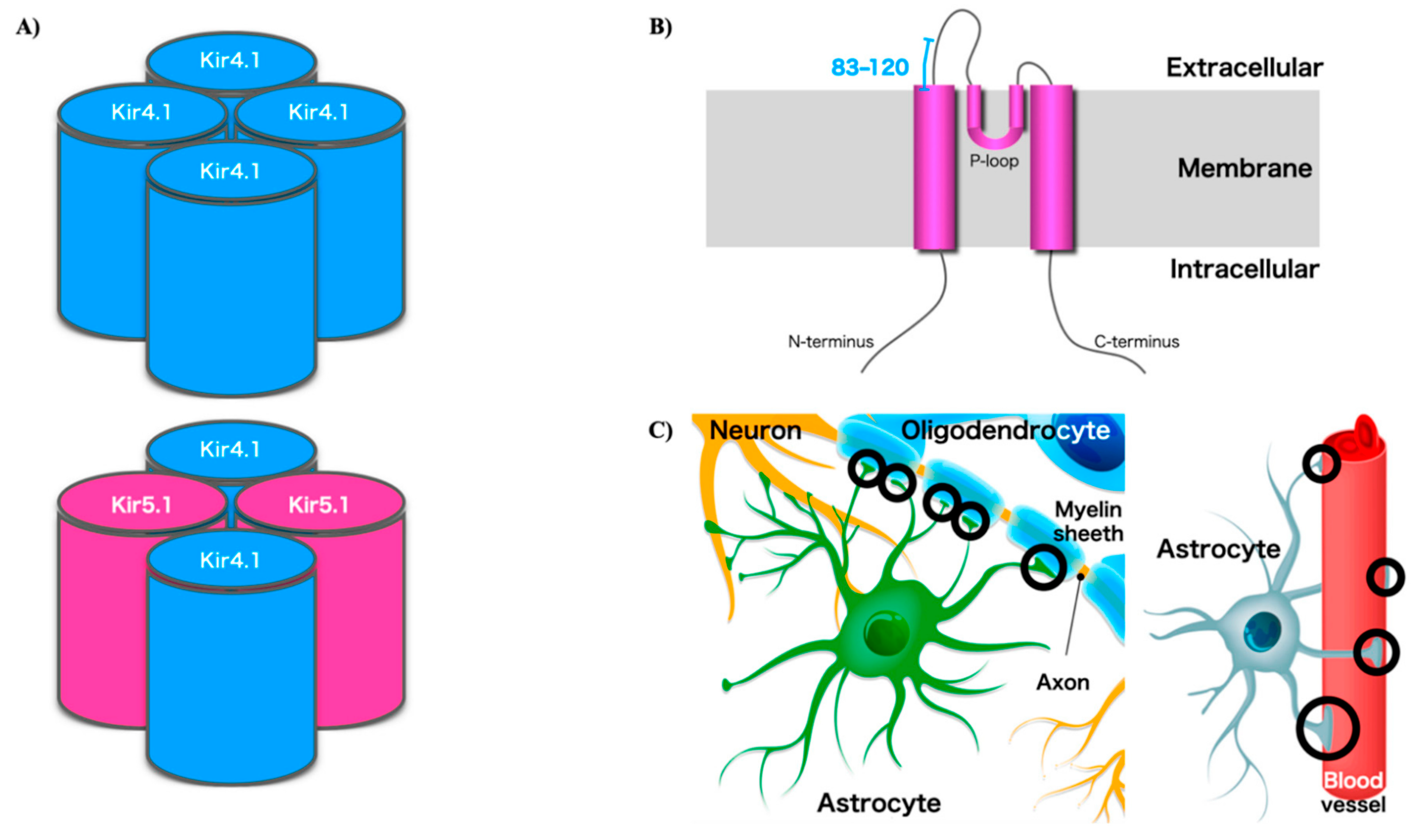
2. Potassium Channels Including Kir4.1
3. Potassium Channels in Neuroimmunology
4. Anti-Kir4.1 Antibodies in MS: Specificity
5. Anti-Kir4.1 Antibodies in MS: Pathogenicity
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| CNS | central nervous system |
| Ig | immunoglobulin |
| CSF | cerebrospinal fluid |
| MS | multiple sclerosis |
| VGKC | voltage-gated potassium channel |
| KCa | calcium-activated potassium |
| Kir | inwardly rectifying potassium |
| K2P | tandem pore domain potassium |
| AQP4 | aquaporin 4 |
| HD | Huntington’s disease |
| SeSAME/EAST | seizures, sensorineural deafness, ataxia, mental retardation and electrolyte imbalance/epilepsy, ataxia, sensorineural deafness and tubulopathy |
| LGI1 | leucine-rich glioma-inactivated protein 1 |
| CASPR2 | contactin-associated protein like 2 |
| OND | other neurological diseases |
| HC | healthy control |
| ELISA | enzyme linked immunosorbent assay |
| NMO | neuromyelitis optica |
| LIPS | luciferase immunoprecipitation systems |
| RRMS | relapsing remitting multiple sclerosis |
| SPMS | secondary progressive multiple sclerosis |
| PPMS | primary progressive multiple sclerosis |
References
- Dobson, R.; Giovannoni, G. Multiple sclerosis—A review. Eur. J. Neurol. 2019, 26, 27–40. [Google Scholar] [CrossRef] [Green Version]
- Oh, J.; Vidal-Jordana, A.; Montalban, X. Multiple sclerosis: Clinical aspects. Curr. Opin. Neurol. 2018, 31, 752–759. [Google Scholar] [CrossRef]
- Reich, D.S.; Lucchinetti, C.F.; Calabresi, P.A. Multiple Sclerosis. N. Engl. J. Med. 2018, 378, 169–180. [Google Scholar] [CrossRef]
- Hauser, S.L.; Bar-Or, A.; Cohen, J.A.; Comi, G.; Correale, J.; Coyle, P.K.; Cross, A.H.; de Seze, J.; Leppert, D.; Montalban, X.; et al. Ofatumumab versus Teriflunomide in Multiple Sclerosis. N. Engl. J. Med. 2020, 383, 546–557. [Google Scholar] [CrossRef]
- Greenfield, A.L.; Hauser, S.L. B-cell Therapy for Multiple Sclerosis: Entering an era. Ann. Neurol. 2018, 83, 13–26. [Google Scholar] [CrossRef]
- Gelfand, J.M.; Cree, B.A.C.; Hauser, S.L. Ocrelizumab and Other CD20(+) B-Cell-Depleting Therapies in Multiple Sclerosis. Neurotherapeutics 2017, 14, 835–841. [Google Scholar] [CrossRef] [Green Version]
- Berger, T.; Reindl, M. Biomarkers in multiple sclerosis: Role of antibodies. Dis. Markers 2006, 22, 207–212. [Google Scholar] [CrossRef] [Green Version]
- McLaughlin, K.A.; Wucherpfennig, K.W. B cells and autoantibodies in the pathogenesis of multiple sclerosis and related inflammatory demyelinating diseases. Adv. Immunol. 2008, 98, 121–149. [Google Scholar]
- Krumbholz, M.; Derfuss, T.; Hohlfeld, R.; Meinl, E. B cells and antibodies in multiple sclerosis pathogenesis and therapy. Nat. Rev. Neurol. 2012, 8, 613–623. [Google Scholar] [CrossRef]
- Levin, M.C.; Lee, S.; Gardner, L.A.; Shin, Y.; Douglas, J.N.; Cooper, C. Autoantibodies to Non-myelin Antigens as Contributors to the Pathogenesis of Multiple Sclerosis. J. Clin. Cell. Immunol. 2013, 4. [Google Scholar] [CrossRef] [Green Version]
- Hemmer, B.; Srivastava, R. Hunting for autoantibodies in multiple sclerosis. Neurology 2013, 81, 944–945. [Google Scholar] [CrossRef] [Green Version]
- Schirmer, L.; Srivastava, R.; Hemmer, B. To look for a needle in a haystack: The search for autoantibodies in multiple sclerosis. Mult. Scler. 2014, 20, 271–279. [Google Scholar] [CrossRef]
- Berger, T.; Reindl, M. Antibody biomarkers in CNS demyelinating diseases—A long and winding road. Eur. J. Neurol. 2015, 22, 1162–1168. [Google Scholar] [CrossRef]
- Srivastava, R.; Aslam, M.; Kalluri, S.R.; Schirmer, L.; Buck, D.; Tackenberg, B.; Rothhammer, V.; Chan, A.; Gold, R.; Berthele, A.; et al. Potassium channel KIR4.1 as an immune target in multiple sclerosis. N. Engl. J. Med. 2012, 367, 115–123. [Google Scholar] [CrossRef] [Green Version]
- Kuang, Q.; Purhonen, P.; Hebert, H. Structure of potassium channels. Cell. Mol. Life Sci. 2015, 72, 3677–3693. [Google Scholar] [CrossRef] [Green Version]
- Tian, C.; Zhu, R.; Zhu, L.; Qiu, T.; Cao, Z.; Kang, T. Potassium channels: Structures, diseases, and modulators. Chem. Biol. Drug Des. 2014, 83, 1–26. [Google Scholar] [CrossRef]
- Kshatri, A.S.; Gonzalez-Hernandez, A.; Giraldez, T. Physiological Roles and Therapeutic Potential of Ca(2+) Activated Potassium Channels in the Nervous System. Front. Mol. Neurosci. 2018, 11, 258. [Google Scholar] [CrossRef]
- Goldstein, S.A.; Bayliss, D.A.; Kim, D.; Lesage, F.; Plant, L.D.; Rajan, S. International Union of Pharmacology. LV. Nomenclature and molecular relationships of two-P potassium channels. Pharmacol. Rev. 2005, 57, 527–540. [Google Scholar] [CrossRef] [Green Version]
- Ohno, Y.; Kinboshi, M.; Shimizu, S. Inwardly Rectifying Potassium Channel Kir4.1 as a Novel Modulator of BDNF Expression in Astrocytes. Int. J. Mol. Sci. 2018, 19, 3313. [Google Scholar] [CrossRef] [Green Version]
- Steinhäuser, C.; Seifert, G.; Bedner, P. Astrocyte dysfunction in temporal lobe epilepsy: K+ channels and gap junction coupling. Glia 2012, 60, 1192–1202. [Google Scholar] [CrossRef]
- Olsen, M.L.; Khakh, B.S.; Skatchkov, S.N.; Zhou, M.; Lee, C.J.; Rouach, N. New Insights on Astrocyte Ion Channels: Critical for Homeostasis and Neuron-Glia Signaling. J. Neurosci. 2015, 35, 13827–13835. [Google Scholar] [CrossRef] [Green Version]
- Gu, C. KIR4.1: K(+) Channel Illusion or Reality in the Autoimmune Pathogenesis of Multiple Sclerosis. Front. Mol. Neurosci. 2016, 9, 90. [Google Scholar] [CrossRef]
- Nwaobi, S.E.; Cuddapah, V.A.; Patterson, K.C.; Randolph, A.C.; Olsen, M.L. The role of glial specific Kir4.1 in normal and pathological states of the CNS. Acta Neuropathol. 2016, 132, 1–21. [Google Scholar] [CrossRef]
- Ferraro, T.N.; Golden, G.T.; Smith, G.G.; Martin, J.F.; Lohoff, F.W.; Gieringer, T.A.; Zamboni, D.; Schwebel, C.L.; Press, D.M.; Kratzer, S.O.; et al. Fine mapping of a seizure susceptibility locus on mouse Chromosome 1: Nomination of Kcnj10 as a causative gene. Mamm. Genome 2004, 15, 239–251. [Google Scholar]
- Scholl, U.I.; Choi, M.; Liu, T.; Ramaekers, V.T.; Häusler, M.G.; Grimmer, J.; Tobe, S.W.; Farhi, A.; Nelson-Williams, C.; Lifton, R.P. Seizures, sensorineural deafness, ataxia, mental retardation, and electrolyte imbalance (SeSAME syndrome) caused by mutations in KCNJ10. Proc. Natl. Acad. Sci. USA 2009, 106, 5842–5847. [Google Scholar] [CrossRef] [Green Version]
- Wilcock, D.M.; Vitek, M.P.; Colton, C.A. Vascular amyloid alters astrocytic water and potassium channels in mouse models and humans with Alzheimer’s disease. Neuroscience 2009, 159, 1055–1069. [Google Scholar] [CrossRef] [Green Version]
- Kaiser, M.; Maletzki, I.; Hülsmann, S.; Holtmann, B.; Schulz-Schaeffer, W.; Kirchhoff, F.; Bähr, M.; Neusch, C. Progressive loss of a glial potassium channel (KCNJ10) in the spinal cord of the SOD1 (G93A) transgenic mouse model of amyotrophic lateral sclerosis. J. Neurochem. 2006, 99, 900–912. [Google Scholar] [CrossRef]
- Magaña, J.J.; Velázquez-Pérez, L.; Cisneros, B. Spinocerebellar ataxia type 2: Clinical presentation, molecular mechanisms, and therapeutic perspectives. Mol. Neurobiol. 2013, 47, 90–104. [Google Scholar] [CrossRef]
- Vit, J.P.; Ohara, P.T.; Bhargava, A.; Kelley, K.; Jasmin, L. Silencing the Kir4.1 potassium channel subunit in satellite glial cells of the rat trigeminal ganglion results in pain-like behavior in the absence of nerve injury. J. Neurosci. 2008, 28, 4161–4171. [Google Scholar] [CrossRef] [Green Version]
- Tang, X.; Hang, D.; Sand, A.; Kofuji, P. Variable loss of Kir4.1 channel function in SeSAME syndrome mutations. Biochem. Biophys. Res. Commun. 2010, 399, 537–541. [Google Scholar] [CrossRef] [Green Version]
- Shin, J.Y.; Fang, Z.H.; Yu, Z.X.; Wang, C.E.; Li, S.H.; Li, X.J. Expression of mutant huntingtin in glial cells contributes to neuronal excitotoxicity. J. Cell Biol. 2005, 171, 1001–1012. [Google Scholar] [CrossRef]
- Faideau, M.; Kim, J.; Cormier, K.; Gilmore, R.; Welch, M.; Auregan, G.; Dufour, N.; Guillermier, M.; Brouillet, E.; Hantraye, P.; et al. In vivo expression of polyglutamine-expanded huntingtin by mouse striatal astrocytes impairs glutamate transport: A correlation with Huntington’s disease subjects. Hum. Mol. Genet. 2010, 19, 3053–3067. [Google Scholar] [CrossRef]
- Tong, X.; Ao, Y.; Faas, G.C.; Nwaobi, S.E.; Xu, J.; Haustein, M.D.; Anderson, M.A.; Mody, I.; Olsen, M.L.; Sofroniew, M.V.; et al. Astrocyte Kir4.1 ion channel deficits contribute to neuronal dysfunction in Huntington’s disease model mice. Nat. Neurosci. 2014, 17, 694–703. [Google Scholar] [CrossRef] [Green Version]
- Irani, S.R.; Vincent, A. Autoimmune encephalitis—New awareness, challenging questions. Discov. Med. 2011, 11, 449–458. [Google Scholar]
- Irani, S.R.; Vincent, A. Voltage-gated potassium channel-complex autoimmunity and associated clinical syndromes. Handb. Clin. Neurol. 2016, 133, 185–197. [Google Scholar]
- Irani, S.R.; Michell, A.W.; Lang, B.; Pettingill, P.; Waters, P.; Johnson, M.R.; Schott, J.M.; Armstrong, R.J.; Zagami, A.S.; Bleasel, A.; et al. Faciobrachial dystonic seizures precede LGI1 antibody limbic encephalitis. Ann. Neurol. 2011, 69, 892–900. [Google Scholar] [CrossRef]
- Binks, S.N.M.; Klein, C.J.; Waters, P.; Pittock, S.J.; Irani, S.R. LGI1, CASPR2 and related antibodies: A molecular evolution of the phenotypes. J. Neurol. Neurosurg. Psychiatry 2018, 89, 526–534. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, S.; Utsugisawa, K.; Yoshikawa, H.; Motomura, M.; Matsubara, S.; Yokoyama, K.; Nagane, Y.; Maruta, T.; Satoh, T.; Sato, H.; et al. Autoimmune targets of heart and skeletal muscles in myasthenia gravis. Arch. Neurol. 2009, 66, 1334–1338. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, S.; Baba, A.; Kaida, K.; Utsugisawa, K.; Kita, Y.; Tsugawa, J.; Ogawa, G.; Nagane, Y.; Kuwana, M.; Suzuki, N. Cardiac involvements in myasthenia gravis associated with anti-Kv1.4 antibodies. Eur. J. Neurol. 2014, 21, 223–230. [Google Scholar] [CrossRef]
- Kraus, V.; Srivastava, R.; Kalluri, S.R.; Seidel, U.; Schuelke, M.; Schimmel, M.; Rostasy, K.; Leiz, S.; Hosie, S.; Grummel, V.; et al. Potassium channel KIR4.1-specific antibodies in children with acquired demyelinating CNS disease. Neurology 2014, 82, 470–473. [Google Scholar] [CrossRef]
- Brickshawana, A.; Hinson, S.R.; Romero, M.F.; Lucchinetti, C.F.; Guo, Y.; Buttmann, M.; McKeon, A.; Pittock, S.J.; Chang, M.H.; Chen, A.P.; et al. Investigation of the KIR4.1 potassium channel as a putative antigen in patients with multiple sclerosis: A comparative study. Lancet Neurol. 2014, 13, 795–806. [Google Scholar] [CrossRef] [Green Version]
- Malyavantham, K.; Weinstock-Guttman, B.; Suresh, L.; Zivadinov, R.; Shanahan, T.; Badgett, D.; Ramanathan, M. Humoral Responses to Diverse Autoimmune Disease-Associated Antigens in Multiple Sclerosis. PLoS ONE 2015, 10, e0129503. [Google Scholar] [CrossRef] [Green Version]
- Chastre, A.; Hafler, D.A.; O’Connor, K.C. Evaluation of KIR4.1 as an Immune Target in Multiple Sclerosis. N. Engl. J. Med. 2016, 374, 1495–1496. [Google Scholar] [CrossRef] [Green Version]
- Zhong, R.; Liang, J.; Tao, A.; Wu, L.; Yang, X.; Xu, H.; Huang, Q.; Zhuang, S.; Long, Y.; Gao, C. Anti-KIR4.1 antibodies in Chinese patients with central nervous system inflammatory demyelinating disorders. Neuroimmunomodulation 2016, 23, 295–300. [Google Scholar] [CrossRef]
- Nerrant, E.; Salsac, C.; Charif, M.; Ayrignac, X.; Carra-Dalliere, C.; Castelnovo, G.; Goulabchand, R.; Tisseyre, J.; Raoul, C.; Eliaou, J.F.; et al. Lack of confirmation of anti-inward rectifying potassium channel 4.1 antibodies as reliable markers of multiple sclerosis. Mult. Scler. 2014, 20, 1699–1703. [Google Scholar] [CrossRef]
- Brill, L.; Goldberg, L.; Karni, A.; Petrou, P.; Abramsky, O.; Ovadia, H.; Ben-Hur, T.; Karussis, D.; Vaknin-Dembinsky, A. Increased anti-KIR4.1 antibodies in multiple sclerosis: Could it be a marker of disease relapse? Mult. Scler. 2015, 21, 572–579. [Google Scholar] [CrossRef]
- Marnetto, F.; Valentino, P.; Caldano, M.; Bertolotto, A. Detection of potassium channel KIR4.1 antibodies in Multiple Sclerosis patients. J. Immunol. Methods 2017, 445, 53–58. [Google Scholar] [CrossRef]
- Marino, M.; Frisullo, G.; Di Sante, G.; Samengo, D.M.; Provenzano, C.; Mirabella, M.; Pani, G.; Ria, F.; Bartoccioni, E. Low reliability of anti-KIR4.1(83-120) peptide auto-antibodies in multiple sclerosis patients. Mult. Scler. 2018, 24, 910–918. [Google Scholar] [CrossRef]
- Watanabe, M.; Yamasaki, R.; Kawano, Y.; Imamura, S.; Kira, J.-I. Anti-KIR4.1 antibodies in Japanese patients with idiopathic central nervous system demyelinating diseases. Clin. Exp. Neuroimmunol. 2013, 4, 241–242. [Google Scholar] [CrossRef]
- Higuchi, O.; Nakane, S.; Sakai, W.; Maeda, Y.; Niino, M.; Takahashi, T.; Fukazawa, T.; Kikuchi, S.; Fujihara, K.; Matsuo, H. Lack of KIR4.1 autoantibodies in Japanese patients with MS and NMO. Neurol. Neuroimmunol. Neuroinflamm. 2016, 3, e263. [Google Scholar] [CrossRef] [Green Version]
- Navas-Madroñal, M.; Valero-Mut, A.; Martínez-Zapata, M.J.; Simón-Talero, M.J.; Figueroa, S.; Vidal-Fernández, N.; López-Góngora, M.; Escartín, A.; Querol, L. Absence of antibodies against KIR4.1 in multiple sclerosis: A three-technique approach and systematic review. PLoS ONE 2017, 12, e0175538. [Google Scholar] [CrossRef]
- Pröbstel, A.K.; Kuhle, J.; Lecourt, A.C.; Vock, I.; Sanderson, N.S.; Kappos, L.; Derfuss, T. Multiple Sclerosis and Antibodies against KIR4.1. N. Engl. J. Med. 2016, 374, 1496–1498. [Google Scholar] [CrossRef]
- Hemmer, B. Antibodies to the inward rectifying potassium channel 4.1 in multiple sclerosis: Different methodologies—Conflicting results? Mult. Scler. 2015, 21, 537–539. [Google Scholar] [CrossRef] [Green Version]
- Filippi, M.; Rocca, M.A.; Lassmann, H. KIR4.1: Another misleading expectation in multiple sclerosis? Lancet Neurol. 2014, 13, 753–755. [Google Scholar] [CrossRef]
- López-Chiriboga, A.S.; Clardy, S.L. Emerging subspecialties in Neurology: Autoimmune neurology. Neurology 2017, 89, e129–e133. [Google Scholar] [CrossRef] [Green Version]
- Dalmau, J. NMDA receptor encephalitis and other antibody-mediated disorders of the synapse: The 2016 Cotzias Lecture. Neurology 2016, 87, 2471–2482. [Google Scholar] [CrossRef] [Green Version]
- Crisp, S.J.; Kullmann, D.M.; Vincent, A. Autoimmune synaptopathies. Nat. Rev. Neurosci. 2016, 17, 103–117. [Google Scholar] [CrossRef]
- Rodriguez, M. Have We Finally Identified an Autoimmune Demyelinating Disease? Ann. Neurol. 2009, 66, 572–573. [Google Scholar] [CrossRef]
- Schwartz, R.; Datta, S. Autoimmunity and autoimmune diseases. In Fundamental Immunology, 2nd ed.; Paul, W., Ed.; Raven Press: New York, NY, USA, 1989; pp. 819–866. [Google Scholar]
- Drachman, D.B. Autonomic “myasthenia”: The case for an autoimmune pathogenesis. J. Clin. Investig. 2003, 111, 797–799. [Google Scholar] [CrossRef]
- Nakajima, M.; Kawamura, T.; Tokui, R.; Furuta, K.; Sugino, M.; Nakanishi, M.; Okuyama, S.; Furukawa, Y. Enhanced accumulation of Kir4.1 protein, but not mRNA, in a murine model of cuprizone-induced demyelination. Brain Res. 2013, 1537, 340–349. [Google Scholar] [CrossRef]
- Schirmer, L.; Srivastava, R.; Kalluri, S.R.; Böttinger, S.; Herwerth, M.; Carassiti, D.; Srivastava, B.; Gempt, J.; Schlegel, J.; Kuhlmann, T.; et al. Differential loss of KIR4.1 immunoreactivity in multiple sclerosis lesions. Ann. Neurol. 2014, 75, 810–828. [Google Scholar] [CrossRef]
- Sato, J.-I.; Tabunoki, H.; Ishida, T.; Saito, Y.; Konno, H.; Arima, K. Reactive astrocytes express the potassium channel Kir4.1 in active multiple sclerosis lesions. Clin. Exp. Neuroimmunol. 2013, 4, 19–28. [Google Scholar] [CrossRef]
- Schirmer, L.; Möbius, W.; Zhao, C.; Cruz-Herranz, A.; Ben Haim, L.; Cordano, C.; Shiow, L.R.; Kelley, K.W.; Sadowski, B.; Timmons, G.; et al. Oligodendrocyte-encoded Kir4.1 function is required for axonal integrity. Elife 2018, 7, e36428. [Google Scholar] [CrossRef]
- Schneider, R. Autoantibodies to Potassium Channel KIR4.1 in Multiple Sclerosis. Front. Neurol. 2013, 4, 125. [Google Scholar] [CrossRef] [Green Version]


{kind=link}
| Class | Name | Class | Name |
|---|---|---|---|
| Voltage-gated potassium channel | Kv1.1–1.3 | Inwardly rectifying potassium channel | Kir1.1 |
| Kv1.4 | Kir2.1–2.4 | ||
| Kv1.5 | Kir3.1–3.4 | ||
| Kv1.6 | Kir4.1–4.2 | ||
| Kv1.7 | Kir5.1 | ||
| Kv1.8 | Kir6.1 | ||
| Kv2.1, 2.2 | Kir6.2 | ||
| Kv3.1, 3.2 | Kir7.1 | ||
| Kv3.3, 3.4 | Tandem pore domain potassium channel | K2P1.1 | |
| Kv4.1–4.3 | K2P2.1 | ||
| Kv5.1 | K2P3.1 | ||
| Kv6.1–6.4 | K2P4.1 | ||
| Kv7.1 | K2P5.1 | ||
| Kv7.2–7.5 | K2P6.1 | ||
| Kv8.1, 8.2 | K2P7.1 | ||
| Kv9.1–9.3 | K2P8.1 | ||
| Kv10.1, 10.2 | K2P9.1 | ||
| Kv11.1–11.3 | K2P10.1 | ||
| Kv12.1–12.3 | K2P11.1 | ||
| Calcium-activated potassium channel | KCa1.1 | K2P12.1 | |
| KCa2.1–2.3 | K2P13.1 | ||
| KCa3.1 | K2P14.1 | ||
| KCa4.1, 4.2 | K2P15.1 | ||
| KCa5.1 | K2P16.1 | ||
| K2P17.1 | |||
| K2P18.1 |
| Author | Year | Countries | Assay | Sample | Frequency in MS | Frequency in NMO | Frequency in OND | Frequency in HC |
|---|---|---|---|---|---|---|---|---|
| Srivastava et al. [14] | 2012 | Germany | ELISA | Serum | 46.9% (186/397) | 0.9% (3/329) | 0.0% (0/59) | |
| Watanabe et al. [49] | 2013 | Japan | ELISA | Serum | 3.9% (7/180) | 1.3% (1/75) | 9.5% (2/21) | 0.0% (1/49) |
| Kraus et al. * [40] | 2013 | Germany | ELISA | Serum | 57.4% (27/47) | 0.0% (0/62) | ||
| Nerrant et al. [45] | 2014 | France | ELISA | Serum | 7.5% (20/268) | 4.3% (2/46) | 4.4% (2/45) | |
| Brickshawana et al. [41] | 2014 | USA | ELISA | Serum | 1.0% (3/286) | 0.9% (2/208) | ||
| ELISA | CSF | 0.0% (0/50) | ||||||
| Brill et al. [46] | 2015 | Israel | ELISA | Serum | 26.3% (21/80) | 22.2% (10/45) | 6.2% (2/32) | |
| Malyavantham et al. ** [42] | 2015 | USA | ELISA | Serum | 4.9% and 7.5% (20 and 31/411: RRMS) 8.6% and 8.6% (11 and 11/128: SPMS) 6.1% and 6.1% (2 and 2/33: PPMS) | 8.5% and 12.2% (7 and 12/82) | 9.8% and 11.4% (31 and 36/315) | |
| Chastre et al. [43] | 2015 | USA | ELISA | Serum | 0.0% (0/86) | 0.0% (0/51) | ||
| Higuchi et al. [50] | 2016 | Japan | ELISA | Serum | 0.0% (0/57) | 0.0% (0/40) | 0.0% (0/50) | |
| LIPS | Serum | 3.5% (2/57) | 0.0% (0/40) | 0.0% (0/50) | ||||
| Marnetto et al. [47] | 2017 | Italy | ELISA | Serum | 27.5% (8/29) | 4.5% (1/22) | ||
| Zhong et al. [44] | 2017 | China | CBA | Serum | 12.2% (23/188) | 15.9% (42/264) | 11.8% (24/203) | 5.0% (2/40) |
| Marino et al. [48] | 2017 | Italy | ELISA | Serum | 16.6% (13/78) | |||
| Flow cytometry | Serum | 2.6% (2/78) | ||||||
| Navas-Madronal et al. [51] | 2017 | Spain | ELISA | Serum | 0.0% (0/108) | 0.0% (0/77) |
| Criteria for determining a disease as autoimmune [58] | |
|---|---|
| 1 | Demonstration of an immune response to a precise autoantigen in all patients with the disease. |
| 2 | Reproduction of the lesion by administration of autoantibody or T cells into a normal animal. |
| 3 | Induction of lesion by immunizing an animal with relevant purified autoantigen. |
| 4 | Isolation or presence of autoantibody or autoreactive T cell from lesion (or serum). |
| 5 | Correlation of autoantibody or autoreactive T cell with disease activity. |
| 6 | Presence of other autoimmune disorders or autoantigens associated with disease. |
| 7 | Immune absorption with purified autoantigen abrogates pathogenic autoantibody or autoreactive T cell. |
| 8 | Reduction of pathogenic autoantibody or T cell associated with clinical improvement. |
| Five criteria for recognizing antibody-mediated autoimmune disease [59] | |
| 1 | Autoantibodies are present in patients with the disease. |
| 2 | Antibody interacts with the target antigen. |
| 3 | Passive transfer of antibody reproduces features of disease. |
| 4 | Immunization with antigen produces a model disease. |
| 5 | Reduction of antibody levels ameliorates the disease. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Imamura, M.; Higuchi, O.; Maeda, Y.; Mukaino, A.; Ueda, M.; Matsuo, H.; Nakane, S. Anti-Kir4.1 Antibodies in Multiple Sclerosis: Specificity and Pathogenicity. Int. J. Mol. Sci. 2020, 21, 9632. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21249632
Imamura M, Higuchi O, Maeda Y, Mukaino A, Ueda M, Matsuo H, Nakane S. Anti-Kir4.1 Antibodies in Multiple Sclerosis: Specificity and Pathogenicity. International Journal of Molecular Sciences. 2020; 21(24):9632. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21249632
Chicago/Turabian StyleImamura, Michie, Osamu Higuchi, Yasuhiro Maeda, Akihiro Mukaino, Mitsuharu Ueda, Hidenori Matsuo, and Shunya Nakane. 2020. "Anti-Kir4.1 Antibodies in Multiple Sclerosis: Specificity and Pathogenicity" International Journal of Molecular Sciences 21, no. 24: 9632. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21249632