Mitophagy and the Brain


1
University of Kansas Alzheimer’s Disease Center, University of Kansas, Kansas City, KS 66160, USA
2
Department of Neurology, University of Kansas Medical Center, Kansas City, KS 66160, USA
3
Department of Biochemistry and Molecular Biology, University of Kansas Medical Center, Kansas City, KS 66160, USA
*
Author to whom correspondence should be addressed.
Int. J. Mol. Sci. 2020, 21(24), 9661; https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21249661
Submission received: 2 December 2020
/
Revised: 16 December 2020
/
Accepted: 17 December 2020
/
Published: 18 December 2020
(This article belongs to the Special Issue Physiological or Pathological Molecular Alterations in Brain Aging)
Abstract
:Stress mechanisms have long been associated with neuronal loss and neurodegenerative diseases. The origin of cell stress and neuronal loss likely stems from multiple pathways. These include (but are not limited to) bioenergetic failure, neuroinflammation, and loss of proteostasis. Cells have adapted compensatory mechanisms to overcome stress and circumvent death. One mechanism is mitophagy. Mitophagy is a form of macroautophagy, were mitochondria and their contents are ubiquitinated, engulfed, and removed through lysosome degradation. Recent studies have implicated mitophagy dysregulation in several neurodegenerative diseases and clinical trials are underway which target mitophagy pathways. Here we review mitophagy pathways, the role of mitophagy in neurodegeneration, potential therapeutics, and the need for further study.
1. Introduction
Mitochondria are essential organelles that regulate energy homeostasis, cell signaling, and cell death [1,2,3]. During threatened cell death or nutrient starvation, mitochondria can be degraded and recycled through mitophagy. Mitophagy is a specific form of autophagy, a process where cell contents are degraded and recycled. In a broad sense, mitophagy involves tagging mitochondria for removal, engulfment of the organelle by an autophagosome, and degradation in a lysosome. There are several pathways which control, initiate, and facilitate mitophagy. The many facets of mitochondrial function contribute to mitophagy pathways.
Mitochondria coordinate and balance energy production through beta oxidation, the citric acid cycle (TCA cycle), and oxidative phosphorylation at the electron transport chain (ETC). Beta oxidation is a catabolic pathway where free fatty acids are converted to acetyl coA, which enter the TCA cycle and ultimately oxidative phosphorylation. In the TCA cycle, either pyruvate (from glycolysis) or acetyl coA are oxidized to generate the high energy electron carriers, NADH and FADH2. NADH and FADH2 enter the ETC at complex I and complex II, respectively. These high energy electron carriers undergo oxidation/reduction reactions in the ETC in order to pump protons into the matrix. These protons ultimately power ATP synthase (or Complex V) for the generation of ATP from ADP. These bioenergetic reactions maintain the mitochondrial electrochemical gradient, or mitochondrial membrane potential. Mitochondrial membrane potential is the main signal which either inhibits or initiates mitophagy.
Mitochondria are double membrane organelles. The outer mitochondrial membrane is imperative for mitophagy function [4,5,6,7]. Transport and signaling proteins localize to the outer mitochondrial membrane to facilitate protein and metabolite import. As discussed in more detail below, aggregation-prone proteins are known to block these import channels on the outer mitochondrial membrane in some neurodegenerative diseases. This could lead to the disruption of mitophagy processes and requires more research efforts to understand. The inner membrane space of mitochondria houses enzymes and allows for proton storage and protein folding. The mitochondrial inner membrane contains the ETC and ATP synthase enzymes and the mitochondrial matrix stores enzymes for the TCA cycle, mitochondrial DNA (mtDNA), and other crucial enzymes for protein folding and maintenance of pH gradients. For more detailed analysis of mitochondrial localized proteins MitoCarta3.0 was recently published [8].
Synaptic loss is strongly correlated with cognitive deficits and motor dysfunction [9,10,11,12]. Mitochondria are essential for synaptic function and neurotransmitter synthesis, release, and uptake [13,14,15]. Accumulation of damaged mitochondria could lead to synaptic dysfunction and neurodegeneration. Mitophagy may play a role in ensuring synaptic mitochondrial integrity by degrading damaged mitochondria.
Mitochondria evolved from a prokaryotic endosymbiont. As such, mitochondria share characteristics with bacteria including a double membrane, circular DNA, formyl-methionine amino acids, and cardiolipin [1]. In certain contexts failure of mitophagy pathways could lead to the release of mitochondrial components into the extracellular space, activation of a damage-associated molecular response (DAMP), and inflammation [1,5]. Mitochondria are also master regulators of cell death pathways (such as apoptosis and necrosis) [4,5,16]. As a by-product of the respiratory chain function superoxide radicals are produced. These free radicals generate multiple species of reactive oxygen species (ROS) and reactive nitrogen species (RNS). During periods of mitochondrial dysfunction and failure of mitochondrial quality control mechanisms (such as mitophagy) ROS/RNS can induce damage to cellular macromolecules and necrotic cell death [4,5,16]. Proper control and coordination of mitophagy pathways are crucial to prevent cell death and inflammation.
Mitophagy is imperative for glial cell function. Signaling between microglia, astrocytes, and neurons are modulated by mitophagy pathways. Novel data show that transcellular mitophagy pathways occur within the brain. Transcellular mitophagy is a process by which cells release mitochondria for engulfment and mitophagy in surrounding cell types. Dysregulation of this process can lead to neuroinflammation and loss of proteostasis [17,18,19].
Disruption of mitophagy is observed with aging and in many neurodegenerative diseases. Recent advances have described novel mechanisms of mitophagy within the central nervous system (CNS). Here, we will review current knowledge of mitophagy regulation, its role in neurodegenerative disease, and therapeutic potential.
For this review article, we used PubMed, clinicaltrials.gov, and Google Scholar to identify studies related to mitophagy, AD, PD, ALS, and MS. We used search terms including mitophagy, mitophagy and neurodegeneration, mitophagy and AD, mitophagy and PD, mitophagy and MS, mitophagy and ALS, mitochondria and neurodegeneration, and autophagy.
2. Mitophagy: Autophagy for Mitochondria
Autophagosomes can be derived from membranes of endoplasmic reticulum (ER), Golgi, mitochondria, or plasma membrane [20,21,22,23,24]. The biogenesis of autophagosomes involves the formation of the isolation membrane (IM), elongation and maturation, closure, and then fusion with the lysosome to form the autolysosome. The first step in autophagosome biogenesis is the activation of the Unc-51-like kinase 1 complex (ULK1; pre-initiation complex), which contains ULK1, autophagy-related proteins 13 and 101 (Atg13, Atg101), and focal adhesion kinase family interacting partner 200 (FIP200) [25,26,27,28]. This complex recruits the class III phosphatidylinositide 3-kinase (PI3K) Vps34 complex (Beclin1, autophagy-related protein 14 (Atg14), autophagy and beclin 1 regulator 1 (Ambra1), and vascular protein sorting 34 and 15 (Vps34 and Vps15)) to produce phosphatidylinositol 3-phosphate (PI3P), also called the initiation complex [25,27,29,30]. PI3P binding proteins, FYVE domain containing proteins (DFCP1), and WD repeat protein interacting with phosphoinositide (WIPIs) localize to the IM [26]. All of this culminates in the formation of the omegasome and IM. Autophagy-related proteins 12, 5, and 16 (Atg12, Atg5, and Atg16) and LC3-phosphatidylethanolamine (PE) facilitate the elongation and closure of the IM, following fusion with the lysosome [25,26,31].
Mitochondrial quality control mechanisms include mitochondrial chaperones, the mitochondrial unfolded protein response (UPRmt), degradation of mitochondrial proteins in the cytoplasm via the proteasome (p97, 26S proteasome), removal of damaged proteins via mitochondrial derived vesicles (MDVs), and mitophagy (Figure 1) [32]. Misfolded mitochondrial proteins can be refolded by mitochondrial chaperone proteins (heat shock proteins 22, 60, and 70) or cleaved/degraded by mitochondrial proteases Lon and Clp through the UPRmt [33,34,35,36,37]. Damaged mitochondrial proteins can also be targeted specifically for proteasome degradation by p97 through the proteasome [38]. MDVs bud off mitochondria after engulfing damaged mitochondrial macromolecules. MDVs are associated with impaired mitochondrial import channels. These MDVs are either degraded by lysosomes, peroxisomes, or exocytosed [39,40]. Mitophagy (reviewed extensively below) functions to remove damaged mitochondria with the goal of preventing cell death.
2.1. Non-Receptor Mediated Mitophagy
Non-receptor mediated mitophagy (Figure 2) or classical mitophagy involves PTEN-induced kinase 1 (PINK1) and Parkin. PINK1 is normally degraded in the inner mitochondrial membrane (IMM) by PARL (presenilin-associated rhomboid-like protease) [41,42]. During mitophagy induction, PINK1 becomes active and accumulates on the outer mitochondrial membrane (OMM), where it recruits Parkin and ubiquitin through phosphorylation [43,44,45,46,47,48,49,50,51]. Parkin accumulates and polyubiquitinates mitochondrial proteins triggering proteasomal degradation. Parkin ubiquitination of OMM proteins leads to more PINK1 activity and phosphorylation of substrates, including the recruitment of additional Parkin proteins at the OMM, creating a positive feedback loop. OMM proteins mitofusins 1 and 2 (MFN1/2), voltage-dependent anion channel 1 (VDAC1), and translocase of the outer mitochondrial membrane 20 (TOM20) are ubiquitinated by Parkin [44,46,47,48,49,50,51,52,53,54,55]. Ubiquitination of MFN1/2 blocks mitochondrial fusion allowing for the isolation of damaged mitochondria and smaller mitochondria facilitate autophagosome targeting [6,32,49,56].
Autophagy adaptor proteins are recruited to the OMM by ubiquitinated proteins. These adaptors include neighbor BRCA1 gene (NBR1), nuclear dot protein 52 (NDP52), optineurin (OPTN), sequestosome-1 (SQSTM1/p62), and Tax1-binding protein (TAX1BP1) [23,52,57,58,59,60,61,62,63,64]. Adaptor proteins recruit and interact with autophagosome proteins; gamma-aminobutyric acid receptor-associated protein (GABARAP) or microtubule-associated protein 1A/1B-light chain 3 (LC3) to mediate the formation of the mitophagosome and lysosomal fusion/degradation. These adaptor proteins interact with LC3 and GABARAP through LC3 interacting regions (LIR) motifs (W/F/YxxL/I) [6,7,23,32]. The importance of different adaptor proteins has been up for debate and often their involvement in mitophagy is context-dependent. The same is true for Parkin, as there are some pathways of mitophagy which are Parkin-independent and these are discussed below [23,44].
2.2. Receptor Mediated Mitophagy
Receptor mediated mitophagy (Figure 2) is driven by mitochondrial receptor proteins which contain LIR motifs (W/F/YxxL/I) [23,50]. Outer mitochondrial membrane proteins, autophagy and Beclin 1 regulator 1 (AMBRA1), Bcl-2 interacting partner 3 (BNIP3), FUN14 domain-containing protein 1 (FUNDC1), Nip3-like protein X (NIX); and inner mitochondrial membrane proteins cardiolipin and prohibitin 2 (PHB2) are the most studied receptors which mediate mitophagy [23,50,65,66,67,68,69,70].
Receptor mediated mitophagy is activated under specific conditions. For example, during hypoxia BNIP3 and NIX transcription are activated by hypoxia inducible factor 1 alpha (HIF1α) [70,71,72,73]. BNIP3 and NIX activity are regulated by phosphorylation where increased phosphorylation increases their binding affinity for LC3 [74,75]. Hypoxia also promotes FUNDC1 binding to LC3 through dephosphorylation via phosphoglycerate mutase family member 5 phosphatase (PGAM5) [65,67,76,77]. Conversely, FUNDC1 phosphorylation by ULK1 is also a mitophagy activating event. Ultimately ubiquitination of FUNDC1 by E3 ubiquitin protein ligase 5 (UBR5) promotes lysosomal degradation of mitochondria [65,67,78]. The inner mitochondrial membrane receptors, PHB2 and cardiolipin have been shown to interact the LC3, especially during times of mitochondrial permeabilization [79,80]. AMBRA1 is sequestered and inhibited by B-cell lymphoma protein 2 (Bcl-2) on the outer mitochondrial membrane but upon mitophagy activated AMBRA1 binds LC3 in a Parkin-dependent or -independent manner [81].
Receptor mediated mitophagy culminates in the elongation of and closure of phagophore membranes, resulting in engulfment of the mitochondria. The elongation and closure of the phagophore is driven by the mitochondrial receptor binding LCR and/or GABARAP, leading to closure of the phagophore by GABARAP [23,50]. Lastly, the autophagosome fuses with a lysosome for degradation.
2.3. Mitoptosis
Separate from mitophagy pathways, damaged mitochondria can be partitioned and removed through mitoptosis. This phenomenon was first proposed in 1992 [82]. Mitoptosis has several proposed definitions. One possible process of mitoptosis occurs when damaged mitochondria gather around the nucleus, are selectively partitioned into lipid membranes and extruded from the cell [83]. A separate definition is when mitochondria undergo condensation with swelling and fragmentation of cristae. This leads to the bursting of the outer mitochondrial membrane and fragmented cristae are extruded into the cytoplasm. In other forms of mitoptosis the outer mitochondrial membrane can remain intact and the cristae deteriorate through refraction and coalescence [6,83,84,85,86,87]. The main benefit of mitoptosis is to prevent opening of the mitochondrial permeability transition pore (MPTP) and apoptosis. Overall, the method of which cells dispose of damaged mitochondria or part of mitochondria can vary and requires further study. Cell-specific pathways which evolved to provide the least devastating consequences based on cell and tissue function likely exist.
Mitoptosis does not require extramitochondrial signaling or protein complexes according to current knowledge. Some evidence suggests PINK1 and Parkin are involved, but no consensus currently exists [6,83,84,85,86,87,88]. Situations which likely activate mitoptosis include mitochondrial membrane depolarization, increased ROS production, and degradation of mtDNA.
2.4. Transcellular Mitophagy
Recent studies have described exocytosis of mitochondria from cells followed by endocytosis or phagocytosis of these extracellular mitochondria. In the brain neurons release mitochondria at synapses, and these extracellular mitochondria were taken up by glial cells for phagocytosis [17,18]. This phenomenon will be referred to as transcellular mitophagy here (Figure 3). A recent study showed that retinal ganglion axons shed mitochondria, which were then then degraded by adjacent astrocytes in mice [18]. Mitochondria might undergo degradation in the axoplasm (cytoplasm of the nerve axon) with the assistance of axonal lysosomes [17]. Essentially instead of being degraded in the soma transcellular mitophagy describes the fact that axonal mitochondria are instead enclosed by axoplasmic membranes that are shed and degraded by neighboring cells.
The notion that transcellular mitophagy occurs is logical given that it is not energetically favorable for neurons to bring mitochondria from dendrites back to the cell body for mitophagy. The process of transcellular mitophagy requires more study and understanding at the basic level and with regards to disease.
In addition to transcellular mitophagy, the observation of glial cells transferring mitochondria to neurons has been documented. In stroke models, glial cells transfer mitochondria to neurons as a likely means to protect neurons from energy stress and hypoxia [89,90]. The specific pathways which facilitate mitochondrial transfer between cells are unknown. However, some studies suggest that in astrocytes, glial acidic fibrillary protein (GFAP), and in neurons, uncoupling protein 2 (UCP2) may play a role [91,92].
In mesenchymal stem cells, connexins (particularly connexin 43) oligomerize to form Gap junctions. These Gap junctions may facilitate the formation of tunneling nanotubules, which allow the exchange of cellular contents, such as mitochondria [93]. Other studies suggest Miro1, a protein which connects cytoskeletal motor proteins to mitochondria is involved in mitochondrial transfer between cells [94]. Finally, S100A4 guides tunneling nanotubule growth [95]. The phenomenon of mitochondrial transfer between cells has been documented in a wide variety of model and tissue types [17,89,90,92,93,94,95,96,97,98,99,100,101]. An important question remains regarding what initiates mitochondrial transfer and the specific mechanisms which control this process.
3. Mitophagy in Aging and Neurodegeneration
Changes in mitophagy flux, signaling, and mitochondrial function are observed with aging and in neurodegenerative diseases. It is imperative to understand the role mitophagy could contribute to brain aging and neurodegeneration. We discuss these implications below.
3.1. Aging
Aging is associated with a loss of proteostasis, mitochondrial dysfunction, genome instability, inflammation, changes to redox balance, and metabolic deficits [32]. Reduced autophagy and mitophagy are observed in models of aging [32,102].
Mitochondrial homeostasis and function are altered in aging. However, the exact mechanisms and findings are not consistent across models and studies. Brain cytochrome oxidase (COX; complex IV) and complex I activity are reduced while ROS production and oxidized proteins are increased in aged rats [103,104]. Calcium homeostasis is changed in aged rat brain mitochondria and synaptosomes; neither were able to take up calcium at a rate equivalent to young rats [105]. Aged mice have altered proteomic expression of glycolytic, TCA, and oxidative phosphorylation pathways in the brain but mitochondrial function is unchanged [106]. This suggests a compensatory mechanism during aging.
Altered brain mitochondrial morphology is observed in aged rats and monkeys [107,108]. Other findings suggest a change to mtDNA epigenetic markers and increased mtDNA deletions in aged mouse brain [109,110]. Oxidative damage to brain mtDNA is related to reduced lifespan across numerous species (birds and mammals) [111,112]. mtDNA in the aged human brain shows increased somatic mutation burden and oxidative damage [113,114,115].
In multiple organisms, mitophagy is associated with longevity and lifespan. Pink1 knockout causes a shorter lifespan, while Parkin overexpression in neurons increases lifespan in D. melanogaster [45,116,117]. C. elegans models exposed to mild mitochondrial stress and upregulated mitophagy have extended lifespan [118,119]. Urolithin A (UA), tomatidine, and catechinic acid induce mitophagy and increase the lifespan of C. elegans models [120,121,122]. In an aging mouse model, stimulation of mitophagy with NAD+ prolongs lifespan [123].
Mitochondrial quality control emerges as a central theme in most neurodegenerative diseases, including Alzheimer’ Disease (AD), Parkinson’s Disease (PD, Multiple Sclerosis (MS), and Amyotrophic Lateral Sclerosis (ALS). Mitophagy stimulation has shown positive effects in models of these diseases.
3.2. Alzheimer’s Disease
AD is the most common form of dementia diagnosed upon autopsy with neuropathological examination [124,125]. The pathological hallmarks which lead to AD diagnosis postmortem are considerable Aβ plaques and tau tangles throughout the brain [124,125,126,127]. Recent advances in neuroimaging have allowed the determination of Aβ plaque and tau tangle load in living subjects, showing these proteins accumulate in the brain decades before clinical signs of cognitive decline [126,128,129,130].
One of the earlier observations in AD subjects was reduced glucose uptake/utilization in the brain via fluorodeoxyglucose (FDG)-positron emission tomography (PET) [128,129,130,131,132,133]. Accumulation of evidence supports an overall metabolic deficit in AD subjects both within the brain and systemically [134]. The mitochondrial ETC enzyme, COX (or complex IV), has reduced Vmax in the AD brain, fibroblasts, and blood samples [132,135,136,137,138,139,140,141,142,143,144,145,146]. AD-like changes can be transferred to other cell types when AD patient mitochondria (mtDNA) are transferred [139,147,148,149,150,151]. This process of creating cytoplasmic hybrid cells (cybrids) allows for the determination of the contribution of mtDNA on disease and cell physiological processes [150].
Mitochondria in AD autopsy brain samples have fragmented cristae and vary widely in size compared to age-matched non-demented brain samples [152]. Mitochondria within dendritic spines and presynaptic terminals show the most fragmented and disorganized cristae. Alterations to mitochondrial ultrastructure were observed in areas of the brain with and without Aβ and tau pathology (cerebellar cortex, hypothalamus, cerebellum, and visual cortex). In addition to altered mitochondrial morphology, presynaptic terminals had reduced synaptic vesicles and fragmentation of golgi cisternae was observed [152].
mtDNA inheritance confers risk to AD. Studies show that offspring of maternal AD subjects have a higher risk of AD diagnosis than offspring of paternal AD subjects. Nearly all mitochondria are inherited maternally. Offspring from maternal AD subjects show metabolic and neuroimaging changes earlier in life than offspring of paternal AD subjects [129,153,154,155,156]. Furthermore, inherited mtDNA haplogroups are associated with both increased and decreased AD risk [157,158,159,160,161,162,163,164]. These inherited mtDNA haplogroups also interact with the nuclear DNA encoded risk factor, ApoE (apolipoprotein E) to influence AD risk [159,164,165]. Thus, it is important to understand the role of mitochondria, mitophagy, and metabolism in AD.
AD mouse models have disrupted mitophagy [166]. This is observed in tau transgenic mice and AD postmortem human brains with accumulated tau aggregates [167]. Mutant Amyloid Precursor Protein (APP; Swedish mutant) mice show increased mitochondrial fission proteins and decreased mitochondrial fusion and mitophagy protein expression in hippocampal neurons [168]. APPsw/PS1dE9 transgenic mice show increased LC3, PINK1, and Parkin expression [169]. Cortical neurons derived from AD transgenic mice (J20; Swedish and Indiana APP mutations) also show increased mitophagy protein expression with depolarized mitochondrial membrane potential [170].
In AD postmortem brain samples, accumulation of damaged mitochondria and autophagosome vacuoles is observed [171,172,173]. The UPRmt pathway is upregulated at the gene level, with reduced proteasomal activity through the 26S proteasome. Parkin, SQSTM1/p62, and LCR mitochondrial localization are increased [172,173,174,175,176]. Mitophagy pathways are altered in human postmortem AD brain. Cytosolic Parkin is depleted, and lysosomal deficits are observed. Impaired Parkin recruitment to mitochondria is possibly caused by tau-mediated sequestration of Parkin in the cytosol [170,177]. Defects in the activation of ULK1 and TBK1 lead to impaired mitophagy [177]. In AD, mitophagy increases or decreases depending on the part of the cell observed. Although it is increased in lysosomes, other parts of the cell fail at completing mitophagy.
Mechanisms of altered mitophagy and mitochondrial function warrant further study in AD, specifically given the strong association of mitochondria and mitophagy with synapse health and function.
3.3. Parkinson’s Disease
PD is a neurodegenerative disease with both cognitive and neuromuscular changes. Motor deficits, tremors, rigidity, bradykinesia, dyssomnia, and depression are clinical hallmarks of PD. In some cases, PD can cause cognitive impairment. Within the brain, PD causes degeneration of dopaminergic neurons in the substantia nigra with Lewy body accumulation (composed of aggregating α-synuclein) [178,179,180].
Mitochondrial dysfunction is observed in PD. A complex I deficiency is noted in brain tissue (substantia nigra) but not in skeletal muscle [181,182]. The complex I deficiency might be brain-specific, but some studies suggest deficits in platelets of PD subjects [183,184]. These findings are dependent on the methodology used. Mitochondrial dysfunction is also observed in cybrid cell lines derived from transfer of PD subject mtDNA, suggesting mtDNA may play a role [150,185,186,187]. Furthermore, PD patients have increased rates of mtDNA deletion in the substantia nigra [188,189].
Familial forms of PD are caused by mutations in genes involved in mitophagy. Mutations in PARK6 (encodes for PINK1), PARK2 (encodes for Parkin), PARK1/4 (α-synuclein), PARK7 (DJ1), PARK8 (LRRK2), PARK17 (Vsp35), and PARK9 (ATP13A2) genes are linked to familial PD [177,190,191,192,193,194,195]. The role of PINK1, Parkin, and Vsp35 in mitophagy are well known and reviewed above. DJ1 and α-synuclein have been shown to modulate mitophagy either through direct interactions with PINK1 and Parkin or by causing mitochondrial fragmentation. Loss of function of LRRK2 and ATP13A2 have been shown to impair mitochondrial turnover.
Despite these genetic studies most PD cases are sporadic with no known genetic cause. Inhibition of complex I function with rotenone (a pesticide) or 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) induces PD in rodent and non-human primates [196,197,198,199,200]. Dopaminergic neurons form many synapses (up to one million per neuron), and thus, have a high bioenergetic demand to maintain these unmyelinated synapses [9,201]. In human postmortem brain, mitophagy markers (phosphorylated S65 ubiquitin) increase across age and with PD diagnosis [202]. This mitophagy marker also associated with Lewy Bodies, showing an increase of mitophagy in early PD stages and a decrease in late PD stages [202]. α-synuclein is also associated with an increase in Miro expression in postmortem human brain tissue, human neurons, and fly models of PD [203]. Reducing Miro in the human neuronal and fly models rescued neurodegeneration and mitophagy [203].
Overall, PD patients and disease models consistently show mitochondrial and mitophagy dysfunction.
3.4. Multiple Sclerosis
MS is a neurodegenerative disease marked by an autoimmune response against the myelin sheath. Demyelination of white matter is caused by autoreactive T cells which target the myelin sheath within the central nervous system. This demyelination leads to a secondary loss of neuronal axons and neurodegeneration. MS has no known genetic cause and occurs in young adults with a higher incidence in females (3:1 ratio female to male) [204,205]. Most MS subjects (85%) have a relapse–remission disease course, which includes periods of demyelination followed by neurological recovery. Eventually a secondary progressive course occurs with few remission periods [204,205]. A smaller subset of MS patients (10–15%) show continued progression known as primary progressive MS.
MS pathology begins with the formation of a lesion with acute inflammation, which transitions to a state of chronic inflammation followed by neurodegeneration. Chronic inflammation is believed to allow penetration of the blood brain barrier by activated T cells directed against the myelin sheath. The inflammation observed in MS shows activation of both innate and adaptive immunity pathways [204,205]. In addition to demyelination, loss of oligodendrocytes is observed. Myelin autoreactive T-cells can induce experimental autoimmune encephalomyelitis (EAE) in animal models and human MS subject genome wide association studies (GWAS) have a high representation of immune genes related to T cell differentiation [204,205]. Gray matter lesions and brain atrophy are present before clinical MS onset. These findings highlight the lack of understanding of what ultimately initiates the autoimmune reaction in MS.
Mitochondria and mitophagy are critical for immune signaling. ROS signals from mitochondria are known to modulate inflammatory responses. ROS signals in MS damage myelin and the blood brain barrier, further exacerbating disease. Oxidation of phospholipids and DNA damage are observed during periods of chronic inflammation with disruption of neuronal axons. Chronic inflammation induces damage to macromolecules including mtDNA, ETC protein, and lipids [204]. Systemic mitochondrial changes are observed in MS. Peripheral lymphocytes (mostly T cells) show increased mitochondrial superoxide production, decreased ETC protein expression, increased lactate, and decreased antioxidant capacity in human MS subjects [206]. These findings suggest a mitochondrial and bioenergetic deficit in MS.
Of interest, MS subjects show decreased expression of COX5B [204]. Active lesions from MS subjects show reductions in COX and its catalytic component COX-I; decreased expression was explicitly observed in oligodendrocytes, astrocytes, and neuronal axons [207]. A separate study showed reduced COX activity in lesions from MS patients; they also observed correlation of this endpoint with neurofilament protein (SMI32) expression and with macrophage/microglial density [208]. An axon-specific protein, syntaphilin, which functions as a mitochondrial docking protein, was increased in chronic lesions [208]. Inactive lesion areas in the same MS subjects had elevated COX activity and increased mitochondrial mass [208]. Gene expression analysis of cortex tissue from MS subjects show overall reductions in nuclear encoded mitochondrial genes and ETC complex expression specifically in neurons [209]. Overall, mitochondrial deficits are observed in MS and the role of these deficits requires further study.
Autophagy and mitophagy pathways are altered in MS human subjects and animal models, which mimic MS pathology. Atg5 modulates T cell survival and its expression correlates with clinical disability in mouse models of EAE. In MS brain samples, encephalitogenic T cells appear to be the major source of Atg5 expression and systemic T cells in human MS subjects showed increased Atg5 expression [210]. In MS brain lesions from human subjects, Lamp2 and LC3II/I ratios are decreased, suggesting impaired autophagy [211]. Further studies have shown serum and CSF concentrations of Atg5 are elevated in MS patients. This study also noted increased expression of Parkin in both serum and CSF with higher serum levels of lactate [212]. In addition, blood from MS subjects show altered expression of several autophagy-related genes (these included ATG9A, BCL2, FAS, GAA, HGS, PIK3R1, RAB24, RGS19, ULK1, FOXO1, and HTT) [213]. Autophagy and mitophagy are imperative for immune cell function, differentiation, and adaptive immunity. The role of these pathways in driving autoreactive T cell differentiation in MS needs to be understood.
3.5. Amyotrophic Lateral Sclerosis
ALS is a neurodegenerative disease marked by the loss of alpha motor neurons in the lumbar spinal cord and motor cortex [214,215]. The lifespan post ALS diagnosis is short, often 2–3 years, because progressive muscle wasting leads to lung paralysis. In some cases of ALS, dementia is present [216]. Most ALS cases are sporadic, with a rare subset (less than 5% of total cases) being familial. These familial cases are caused by mutations in genes including Tdp43, Fus, Fig4, Ang, Vapb, and C9orf72 [217,218,219,220]. Mutations in the Optn gene which encodes an autophagy protein, optineurin, were found to be causative of ALS in 2010 [221,222,223]. After the discovery of mutations in Sod1 and Tdp43, transgenic mouse models were developed [224,225,226].
Changes to mitochondrial ultrastructure in human ALS subjects were revealed several decades ago [227,228,229]. Cytoplasmic inclusions that may represent mitochondria-containing autophagic vacuoles are observed in ALS motor neurons [230,231]. Although ALS neurodegeneration is anatomically specific, mitochondrial abnormalities are found systemically [214,215,227,229,232,233]. Mitochondrial dysfunction is present in platelet and muscle mitochondria from ALS subjects [227,229,234,235,236,237,238]. mtDNA may contribute to ALS pathologies, as cybrid cells harboring mtDNA from ALS subjects often show mitochondrial abnormalities and increased cell death [150,229,239,240].
ALS is modeled using rodents that express mutant SOD1 or mutant TDP43 [224,226]. SOD1 is a cytoplasmic enzyme which was identified within mitochondrial membranes [225,229,241,242,243]. This is the case for both mutant SOD1 and to a lesser extent wild type SOD1. Mutant SOD1 ALS transgenic mice have altered mitochondrial morphology and mitochondrial SOD1 accumulation [242,243]. This raises the possibility that mutant SOD1 may drive neurodegeneration by damaging mitochondria. TDP43 mutants are also observed within mitochondria and appear to induce mitochondrial dysfunction [242,243,244,245,246]. Both TDP43 and SOD1 are known to aggregate within motor neurons and muscle; TDP43 interacts with proteins critical to mitophagy in an inhibitory manner [219,244,245,247,248,249,250,251].
Impaired mitophagy was proposed to be involved in the denervation of neuromuscular junctions in an ALS mouse model [252]. Lysosomal dysfunction has also been implicated in ALS. Specifically, lysosomal deficits result in an abnormal accumulation of autophagic vacuoles that engulf damaged mitochondria within the motor neuron axons of G93A SOD1 ALS mice [177]. Impaired mitochondrial turnover along with the accumulation of misfolded proteins and protein aggregates contributes to ALS-linked mitochondrial dysfunction and motor neuron death. Mitochondrial and mitophagy ultrastructure are varied across compartments of motor neurons [253,254]. Parkin, Miro1, and Mfn2 are depleted in an ALS mouse model (G93A SOD1); however, mitochondrial localized p62 is upregulated [255]. Mutant forms of optineurin interfere with Parkin ubiquitin ligase function [256].
In human post-mortem samples, increased autophagic vesicles are observed in lumbar spinal cord motor neurons [257]. Induced pluripotent stem cell (iPSC)-derived motor neurons from familial ALS subjects with C9orf72 mutations or haploinsufficiency have dysfunction of autophagy pathways [258,259].
As discussed above mitochondrial dysfunction and mitophagy alterations are prevalent in human AD, PD, MS, and ALS samples as well as cell and animal models of disease. We discuss below the methods being investigated to modulate mitophagy.
4. Modulating Mitophagy in Neurodegeneration
Increasing mitophagy in transgenic mouse models of neurodegeneration have shown mostly beneficial effects. In AD models (iPSC derived, transgenic mice, and C. elegans), increasing mitophagy using nicotinamide mononucleotide (NMN), UA, or actinonin (AC) reduced Aβ and tau aggregation. In AD transgenic mice, mitophagy induction benefited cognition [260,261]. These compounds are NAD+ precursors, which may drive mitophagy through alterations in redox balance (NAD+/NADH). UA likely drives mitophagy through a PINK1/Parkin/Nix axis.
Broad autophagy induction with Rilmenidine in the G93ASOD1 mouse model of ALS did not change disease progression [262]. The mechanism(s) of Rilmenidine autophagy/mitophagy induction are currently unknown. Rapamaycin (an mTOR inhibitor) treatment of this same mouse model was detrimental unless mature lymphocytes were depleted [263]. These studies highlight the importance of understanding the non-cell autonomous effects of autophagy and mitophagy pathways.
In PD rodent models (MPTP injection), a drug, Salidroside, increased Parkin and PINK1 expression and preserved dopaminergic neurons in the substantia nigra [264]. A cell permeable form of Parkin rescued cells from aggregating α-synuclein, partially restored motor function, and protected dopaminergic neurons in the 6-OHDA PD (6-hydroxydopamine) mouse model [265].
In an MS-related mouse model of EAE administration of rapamycin, an mTOR inhibitor improves outcomes [211]. Further studies of the EAE mouse model show that excessive activation of Drp1 through nitration leads to an overaction of mitophagy [266]. Blocking this pathway alleviated the disease burden in the EAE mouse mode [267]. Genetic ablation of Beclin 1 was also protective in the EAE mouse model [268]. Overall, in MS inhibition of mitophagy specifically in T cells could be beneficial.
UA has been shown to be safe and well-tolerated in elderly adults, with plasma concentrations detectable at a range of doses. Furthermore, UA affected mitochondrial gene expression in muscle [269]. A separate study in healthy adults is registered for UA (NCT04160312), but no results have been posted. Clinical trials for NMN (NCT04228640 safety trial) are recruiting or ongoing (NCT03151239 effects on cardiometabolic health). In Japan, the first human clinical trial of NMN showed no deleterious effects, suggesting NMN is tolerable and safe [270,271]. No clinical trials for these NAD+ precursor mitophagy modulators are currently registered for neurodegenerative diseases.
Lifestyle interventions could be useful tools to boost mitophagy. Exercise and diet have been shown to induce mitophagy [272,273,274,275,276]. In both AD animal models and human clinical trials, exercise has shown cognitive benefit [275,277,278,279,280,281]. The exercise effects in ALS and PD are more controversial, but overall, exercise seems to improve physical and cognitive outcomes [282,283,284,285,286,287]. Intermittent fasting and ketogenic diets have also been shown to induce mitophagy and improve cognition/motor performance [288,289,290,291,292,293,294].
Current clinical trials aimed at increasing autophagy, mitophagy, or mitochondrial function are ongoing or recently completed. For AD, these include treatment with nicotinamide riboside (NR; NCT04430517; NAD+ precursor), Dimebon (NCT00675623, NCT00829374; stimulates mTOR-dependent mitophagy), resveratrol (NCT00678431; mTOR inhibitory), ketogenic diets (NCT03860792), and caloric restriction diets (NCT02460783). In PD, these include nicotinamide supplementation (NCT03568968; NAD+ precursor), ubiquinol/Coenzyme Q10 (NCT03061513; autophagy mechanism unknown), ketogenic diets, and ketone esters (NCT01364545, NCT04477161). In MS, clinical trials include ketogenic diet, dimethyl fumarate, and MitoQ (NCT03740295, NCT04267926, NCT02461069). In ALS, one clinical trial for ubiquinol/Coenzyme Q10 (NCT00243932; autophagy mechanism unknown) was completed. Overall, the clinical trials directly modulating mitophagy are lacking and require more attention. The majority of mitophagy inducers in clinical trials have unknown mechanisms and pleotropic affects.
Targeting specific pathways and tissues could be advantageous in avoiding deleterious or off-target effects. Designing new therapeutic strategies should focus on modulating specific mitophagy targets while also enhancing mitochondrial function and biogenesis.
5. Concluding Remarks
Mitophagy and mitochondrial quality control are important mechanisms which should be further studied in the context of brain aging and neurodegeneration. Novel mechanisms of mitochondrial quality control in neurons and glia have illuminated the knowledge gaps in this field of study. Mitochondria are dynamic and multifaceted organelles at the forefront of pathways associated with aging and neurodegeneration (proteostasis, metabolism, inflammation, and synapse loss). Thus, targeting mitochondrial health and mitochondrial quality control will target the most common pathological mechanisms in neurodegeneration.
Author Contributions
N.S.S. and H.M.W. both contributed to writing the article and research for the article. H.M.W. created the figures and edited the manuscript during the publication process. All authors have read and agreed to the published version of the manuscript.
Funding
Funding for this study was provided by the NIA R00AG056600 to Wilkins and P30AG035982 from the KU Alzheimer’s Disease Center.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Wilkins, H.M.; Swerdlow, R.H. Relationships between Mitochondria and Neuroinflammation: Implications for Alzheimer’s Disease. Curr. Top. Med. Chem. 2016, 16, 849–857. [Google Scholar] [CrossRef] [PubMed]
- Eisner, V.; Picard, M.; Hajnoczky, G. Mitochondrial dynamics in adaptive and maladaptive cellular stress responses. Nat. Cell Biol. 2018, 20, 755–765. [Google Scholar] [CrossRef] [PubMed]
- Swerdlow, R.H.; Koppel, S.; Weidling, I.; Hayley, C.; Ji, Y.; Wilkins, H.M. Mitochondria, Cybrids, Aging, and Alzheimer’s Disease. Prog. Mol. Biol. Transl. Sci. 2017, 146, 259–302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tait, S.W.; Green, D.R. Mitochondria and cell signalling. J. Cell Sci. 2012, 125, 807–815. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Green, D.R.; Galluzzi, L.; Kroemer, G. Mitochondria and the autophagy-inflammation-cell death axis in organismal aging. Science 2011, 333, 1109–1112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mijaljica, D.; Prescott, M.; Devenish, R.J. Mitophagy and mitoptosis in disease processes. Methods Mol. Biol. 2010, 648, 93–106. [Google Scholar] [CrossRef]
- Yoo, S.M.; Jung, Y.K. A Molecular Approach to Mitophagy and Mitochondrial Dynamics. Mol. Cells 2018, 41, 18–26. [Google Scholar] [CrossRef]
- Rath, S.; Sharma, R.; Gupta, R.; Ast, T.; Chan, C.; Durham, T.J.; Goodman, R.P.; Grabarek, Z.; Haas, M.E.; Hung, W.H.W.; et al. MitoCarta3.0: An updated mitochondrial proteome now with sub-organelle localization and pathway annotations. Nucleic Acids Res. 2020. [Google Scholar] [CrossRef]
- Pissadaki, E.K.; Bolam, J.P. The energy cost of action potential propagation in dopamine neurons: Clues to susceptibility in Parkinson’s disease. Front. Comput. Neurosci. 2013, 7, 13. [Google Scholar] [CrossRef] [Green Version]
- Robinson, J.L.; Molina-Porcel, L.; Corrada, M.M.; Raible, K.; Lee, E.B.; Lee, V.M.; Kawas, C.H.; Trojanowski, J.Q. Perforant path synaptic loss correlates with cognitive impairment and Alzheimer’s Disease in the oldest-old. Brain 2014, 137, 2578–2587. [Google Scholar] [CrossRef]
- Kashyap, G.; Bapat, D.; Das, D.; Gowaikar, R.; Amritkar, R.E.; Rangarajan, G.; Ravindranath, V.; Ambika, G. Synapse loss and progress of Alzheimer’s Disease—A network model. Sci. Rep. 2019, 9, 6555. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lepeta, K.; Lourenco, M.V.; Schweitzer, B.C.; Martino Adami, P.V.; Banerjee, P.; Catuara-Solarz, S.; de La Fuente Revenga, M.; Guillem, A.M.; Haidar, M.; Ijomone, O.M.; et al. Synaptopathies: Synaptic dysfunction in neurological disorders—A review from students to students. J. Neurochem. 2016, 138, 785–805. [Google Scholar] [CrossRef] [PubMed]
- Manji, H.; Kato, T.; Di Prospero, N.A.; Ness, S.; Beal, M.F.; Krams, M.; Chen, G. Impaired mitochondrial function in psychiatric disorders. Nat. Rev. Neurosci. 2012, 13, 293–307. [Google Scholar] [CrossRef] [PubMed]
- Sheng, Z.H.; Cai, Q. Mitochondrial transport in neurons: Impact on synaptic homeostasis and neurodegeneration. Nat. Rev. Neurosci. 2012, 13, 77–93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, S.; Jeong, Y.Y.; Sheshadri, P.; Su, X.; Cai, Q. Mitophagy regulates integrity of mitochondria at synapses and is critical for synaptic maintenance. EMBO Rep. 2020, e201949801. [Google Scholar] [CrossRef] [PubMed]
- Green, D.R.; Reed, J.C. Mitochondria and apoptosis. Science 1998, 281, 1309–1312. [Google Scholar] [CrossRef] [PubMed]
- Davis, C.H.; Marsh-Armstrong, N. Discovery and implications of transcellular mitophagy. Autophagy 2014, 10, 2383–2384. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davis, C.H.; Kim, K.Y.; Bushong, E.A.; Mills, E.A.; Boassa, D.; Shih, T.; Kinebuchi, M.; Phan, S.; Zhou, Y.; Bihlmeyer, N.A.; et al. Transcellular degradation of axonal mitochondria. Proc. Natl. Acad. Sci. USA 2014, 111, 9633–9638. [Google Scholar] [CrossRef] [Green Version]
- Joshi, A.U.; Minhas, P.S.; Liddelow, S.A.; Haileselassie, B.; Andreasson, K.I.; Dorn, G.W., 2nd; Mochly-Rosen, D. Fragmented mitochondria released from microglia trigger A1 astrocytic response and propagate inflammatory neurodegeneration. Nat. Neurosci. 2019, 22, 1635–1648. [Google Scholar] [CrossRef]
- Hailey, D.W.; Rambold, A.S.; Satpute-Krishnan, P.; Mitra, K.; Sougrat, R.; Kim, P.K.; Lippincott-Schwartz, J. Mitochondria supply membranes for autophagosome biogenesis during starvation. Cell 2010, 141, 656–667. [Google Scholar] [CrossRef] [Green Version]
- Ravikumar, B.; Moreau, K.; Jahreiss, L.; Puri, C.; Rubinsztein, D.C. Plasma membrane contributes to the formation of pre-autophagosomal structures. Nat. Cell Biol. 2010, 12, 747–757. [Google Scholar] [CrossRef] [PubMed]
- Hayashi-Nishino, M.; Fujita, N.; Noda, T.; Yamaguchi, A.; Yoshimori, T.; Yamamoto, A. A subdomain of the endoplasmic reticulum forms a cradle for autophagosome formation. Nat. Cell Biol. 2009, 11, 1433–1437. [Google Scholar] [CrossRef] [PubMed]
- Feng, D.; Liu, L.; Zhu, Y.; Chen, Q. Molecular signaling toward mitophagy and its physiological significance. Exp. Cell Res. 2013, 319, 1697–1705. [Google Scholar] [CrossRef] [PubMed]
- Ge, L.; Zhang, M.; Kenny, S.J.; Liu, D.; Maeda, M.; Saito, K.; Mathur, A.; Xu, K.; Schekman, R. Remodeling of ER-exit sites initiates a membrane supply pathway for autophagosome biogenesis. EMBO Rep. 2017, 18, 1586–1603. [Google Scholar] [CrossRef]
- Tanida, I. Autophagosome formation and molecular mechanism of autophagy. Antioxid. Redox Signal. 2011, 14, 2201–2214. [Google Scholar] [CrossRef]
- Itakura, E.; Mizushima, N. Characterization of autophagosome formation site by a hierarchical analysis of mammalian Atg proteins. Autophagy 2010, 6, 764–776. [Google Scholar] [CrossRef] [Green Version]
- Hurley, J.H.; Young, L.N. Mechanisms of Autophagy Initiation. Annu. Rev. Biochem. 2017, 86, 225–244. [Google Scholar] [CrossRef]
- Hosokawa, N.; Hara, T.; Kaizuka, T.; Kishi, C.; Takamura, A.; Miura, Y.; Iemura, S.; Natsume, T.; Takehana, K.; Yamada, N.; et al. Nutrient-dependent mTORC1 association with the ULK1-Atg13-FIP200 complex required for autophagy. Mol. Biol. Cell 2009, 20, 1981–1991. [Google Scholar] [CrossRef] [Green Version]
- Funderburk, S.F.; Wang, Q.J.; Yue, Z. The Beclin 1-VPS34 complex—At the crossroads of autophagy and beyond. Trends Cell Biol. 2010, 20, 355–362. [Google Scholar] [CrossRef] [Green Version]
- Tassa, A.; Roux, M.P.; Attaix, D.; Bechet, D.M. Class III phosphoinositide 3-kinase-Beclin1 complex mediates the amino acid-dependent regulation of autophagy in C2C12 myotubes. Biochem. J. 2003, 376, 577–586. [Google Scholar] [CrossRef] [Green Version]
- Romanov, J.; Walczak, M.; Ibiricu, I.; Schuchner, S.; Ogris, E.; Kraft, C.; Martens, S. Mechanism and functions of membrane binding by the Atg5-Atg12/Atg16 complex during autophagosome formation. EMBO J. 2012, 31, 4304–4317. [Google Scholar] [CrossRef] [PubMed]
- Fivenson, E.M.; Lautrup, S.; Sun, N.; Scheibye-Knudsen, M.; Stevnsner, T.; Nilsen, H.; Bohr, V.A.; Fang, E.F. Mitophagy in neurodegeneration and aging. Neurochem. Int. 2017, 109, 202–209. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.F.; Haynes, C.M. Metabolism and the UPR(mt). Mol. Cell 2016, 61, 677–682. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haynes, C.M.; Ron, D. The mitochondrial UPR—Protecting organelle protein homeostasis. J. Cell Sci. 2010, 123, 3849–3855. [Google Scholar] [CrossRef] [Green Version]
- Pellegrino, M.W.; Nargund, A.M.; Haynes, C.M. Signaling the mitochondrial unfolded protein response. Biochim. Biophys. Acta 2013, 1833, 410–416. [Google Scholar] [CrossRef] [Green Version]
- Voos, W.; Rottgers, K. Molecular chaperones as essential mediators of mitochondrial biogenesis. Biochim. Biophys. Acta 2002, 1592, 51–62. [Google Scholar] [CrossRef] [Green Version]
- Ryan, M.T.; Naylor, D.J.; Hoj, P.B.; Clark, M.S.; Hoogenraad, N.J. The role of molecular chaperones in mitochondrial protein import and folding. Int. Rev. Cytol. 1997, 174, 127–193. [Google Scholar] [CrossRef]
- Xu, S.; Peng, G.; Wang, Y.; Fang, S.; Karbowski, M. The AAA-ATPase p97 is essential for outer mitochondrial membrane protein turnover. Mol. Biol. Cell 2011, 22, 291–300. [Google Scholar] [CrossRef]
- Cadete, V.J.; Deschenes, S.; Cuillerier, A.; Brisebois, F.; Sugiura, A.; Vincent, A.; Turnbull, D.; Picard, M.; McBride, H.M.; Burelle, Y. Formation of mitochondrial-derived vesicles is an active and physiologically relevant mitochondrial quality control process in the cardiac system. J. Physiol. 2016, 594, 5343–5362. [Google Scholar] [CrossRef] [Green Version]
- Sugiura, A.; McLelland, G.L.; Fon, E.A.; McBride, H.M. A new pathway for mitochondrial quality control: Mitochondrial-derived vesicles. EMBO J. 2014, 33, 2142–2156. [Google Scholar] [CrossRef] [Green Version]
- Meissner, C.; Lorenz, H.; Weihofen, A.; Selkoe, D.J.; Lemberg, M.K. The mitochondrial intramembrane protease PARL cleaves human Pink1 to regulate Pink1 trafficking. J. Neurochem. 2011, 117, 856–867. [Google Scholar] [CrossRef]
- Jin, S.M.; Lazarou, M.; Wang, C.; Kane, L.A.; Narendra, D.P.; Youle, R.J. Mitochondrial membrane potential regulates PINK1 import and proteolytic destabilization by PARL. J. Cell Biol. 2010, 191, 933–942. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rakovic, A.; Shurkewitsch, K.; Seibler, P.; Grunewald, A.; Zanon, A.; Hagenah, J.; Krainc, D.; Klein, C. Phosphatase and tensin homolog (PTEN)-induced putative kinase 1 (PINK1)-dependent ubiquitination of endogenous Parkin attenuates mitophagy: Study in human primary fibroblasts and induced pluripotent stem cell-derived neurons. J. Biol. Chem. 2013, 288, 2223–2237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, S.M.; Youle, R.J. PINK1- and Parkin-mediated mitophagy at a glance. J. Cell Sci. 2012, 125, 795–799. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Narendra, D.P.; Jin, S.M.; Tanaka, A.; Suen, D.F.; Gautier, C.A.; Shen, J.; Cookson, M.R.; Youle, R.J. PINK1 is selectively stabilized on impaired mitochondria to activate Parkin. PLoS Biol. 2010, 8, e1000298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsuda, N.; Sato, S.; Shiba, K.; Okatsu, K.; Saisho, K.; Gautier, C.A.; Sou, Y.S.; Saiki, S.; Kawajiri, S.; Sato, F.; et al. PINK1 stabilized by mitochondrial depolarization recruits Parkin to damaged mitochondria and activates latent Parkin for mitophagy. J. Cell Biol. 2010, 189, 211–221. [Google Scholar] [CrossRef] [PubMed]
- Geisler, S.; Holmstrom, K.M.; Skujat, D.; Fiesel, F.C.; Rothfuss, O.C.; Kahle, P.J.; Springer, W. PINK1/Parkin-mediated mitophagy is dependent on VDAC1 and p62/SQSTM1. Nat. Cell Biol. 2010, 12, 119–131. [Google Scholar] [CrossRef]
- Vives-Bauza, C.; Zhou, C.; Huang, Y.; Cui, M.; de Vries, R.L.; Kim, J.; May, J.; Tocilescu, M.A.; Liu, W.; Ko, H.S.; et al. PINK1-dependent recruitment of Parkin to mitochondria in mitophagy. Proc. Natl. Acad. Sci. USA 2010, 107, 378–383. [Google Scholar] [CrossRef] [Green Version]
- Gegg, M.E.; Schapira, A.H. PINK1-parkin-dependent mitophagy involves ubiquitination of mitofusins 1 and 2: Implications for Parkinson disease pathogenesis. Autophagy 2011, 7, 243–245. [Google Scholar] [CrossRef] [Green Version]
- Wei, H.; Liu, L.; Chen, Q. Selective removal of mitochondria via mitophagy: Distinct pathways for different mitochondrial stresses. Biochim. Biophys. Acta 2015, 1853, 2784–2790. [Google Scholar] [CrossRef] [Green Version]
- Durcan, T.M.; Fon, E.A. The three ‘P’s of mitophagy: PARKIN, PINK1, and post-translational modifications. Genes Dev. 2015, 29, 989–999. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Narendra, D.; Kane, L.A.; Hauser, D.N.; Fearnley, I.M.; Youle, R.J. p62/SQSTM1 is required for Parkin-induced mitochondrial clustering but not mitophagy; VDAC1 is dispensable for both. Autophagy 2010, 6, 1090–1106. [Google Scholar] [CrossRef] [PubMed]
- Narendra, D.; Tanaka, A.; Suen, D.F.; Youle, R.J. Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J. Cell Biol. 2008, 183, 795–803. [Google Scholar] [CrossRef] [Green Version]
- Yoshii, S.R.; Kishi, C.; Ishihara, N.; Mizushima, N. Parkin mediates proteasome-dependent protein degradation and rupture of the outer mitochondrial membrane. J. Biol. Chem. 2011, 286, 19630–19640. [Google Scholar] [CrossRef] [Green Version]
- Tanaka, A.; Cleland, M.M.; Xu, S.; Narendra, D.P.; Suen, D.F.; Karbowski, M.; Youle, R.J. Proteasome and p97 mediate mitophagy and degradation of mitofusins induced by Parkin. J. Cell Biol. 2010, 191, 1367–1380. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chan, N.C.; Salazar, A.M.; Pham, A.H.; Sweredoski, M.J.; Kolawa, N.J.; Graham, R.L.; Hess, S.; Chan, D.C. Broad activation of the ubiquitin-proteasome system by Parkin is critical for mitophagy. Hum. Mol. Genet. 2011, 20, 1726–1737. [Google Scholar] [CrossRef] [PubMed]
- Mizushima, N.; Komatsu, M. Autophagy: Renovation of cells and tissues. Cell 2011, 147, 728–741. [Google Scholar] [CrossRef] [Green Version]
- Shi, J.; Fung, G.; Deng, H.; Zhang, J.; Fiesel, F.C.; Springer, W.; Li, X.; Luo, H. NBR1 is dispensable for PARK2-mediated mitophagy regardless of the presence or absence of SQSTM1. Cell Death Dis. 2015, 6, e1943. [Google Scholar] [CrossRef] [Green Version]
- Richter, B.; Sliter, D.A.; Herhaus, L.; Stolz, A.; Wang, C.; Beli, P.; Zaffagnini, G.; Wild, P.; Martens, S.; Wagner, S.A.; et al. Phosphorylation of OPTN by TBK1 enhances its binding to Ub chains and promotes selective autophagy of damaged mitochondria. Proc. Natl. Acad. Sci. USA 2016, 113, 4039–4044. [Google Scholar] [CrossRef] [Green Version]
- Minowa-Nozawa, A.; Nozawa, T.; Okamoto-Furuta, K.; Kohda, H.; Nakagawa, I. Rab35 GTPase recruits NDP52 to autophagy targets. EMBO J. 2017, 36, 2790–2807. [Google Scholar] [CrossRef]
- Wong, Y.C.; Holzbaur, E.L. Temporal dynamics of PARK2/parkin and OPTN/optineurin recruitment during the mitophagy of damaged mitochondria. Autophagy 2015, 11, 422–424. [Google Scholar] [CrossRef] [Green Version]
- Liu, H.; Dai, C.; Fan, Y.; Guo, B.; Ren, K.; Sun, T.; Wang, W. From autophagy to mitophagy: The roles of P62 in neurodegenerative diseases. J. Bioenerg. Biomembr. 2017, 49, 413–422. [Google Scholar] [CrossRef] [PubMed]
- Yamada, T.; Dawson, T.M.; Yanagawa, T.; Iijima, M.; Sesaki, H. SQSTM1/p62 promotes mitochondrial ubiquitination independently of PINK1 and PRKN/parkin in mitophagy. Autophagy 2019, 15, 2012–2018. [Google Scholar] [CrossRef] [PubMed]
- Whang, M.I.; Tavares, R.M.; Benjamin, D.I.; Kattah, M.G.; Advincula, R.; Nomura, D.K.; Debnath, J.; Malynn, B.A.; Ma, A. The Ubiquitin Binding Protein TAX1BP1 Mediates Autophagasome Induction and the Metabolic Transition of Activated T Cells. Immunity 2017, 46, 405–420. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, L.; Feng, D.; Chen, G.; Chen, M.; Zheng, Q.; Song, P.; Ma, Q.; Zhu, C.; Wang, R.; Qi, W.; et al. Mitochondrial outer-membrane protein FUNDC1 mediates hypoxia-induced mitophagy in mammalian cells. Nat. Cell Biol. 2012, 14, 177–185. [Google Scholar] [CrossRef] [PubMed]
- Novak, I.; Kirkin, V.; McEwan, D.G.; Zhang, J.; Wild, P.; Rozenknop, A.; Rogov, V.; Lohr, F.; Popovic, D.; Occhipinti, A.; et al. Nix is a selective autophagy receptor for mitochondrial clearance. EMBO Rep. 2010, 11, 45–51. [Google Scholar] [CrossRef] [Green Version]
- Wu, W.; Tian, W.; Hu, Z.; Chen, G.; Huang, L.; Li, W.; Zhang, X.; Xue, P.; Zhou, C.; Liu, L.; et al. ULK1 translocates to mitochondria and phosphorylates FUNDC1 to regulate mitophagy. EMBO Rep. 2014, 15, 566–575. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Ney, P.A. NIX induces mitochondrial autophagy in reticulocytes. Autophagy 2008, 4, 354–356. [Google Scholar] [CrossRef]
- Sandoval, H.; Thiagarajan, P.; Dasgupta, S.K.; Schumacher, A.; Prchal, J.T.; Chen, M.; Wang, J. Essential role for Nix in autophagic maturation of erythroid cells. Nature 2008, 454, 232–235. [Google Scholar] [CrossRef]
- Zhang, J.; Ney, P.A. Role of BNIP3 and NIX in cell death, autophagy, and mitophagy. Cell Death Differ. 2009, 16, 939–946. [Google Scholar] [CrossRef] [Green Version]
- Sowter, H.M.; Ratcliffe, P.J.; Watson, P.; Greenberg, A.H.; Harris, A.L. HIF-1-dependent regulation of hypoxic induction of the cell death factors BNIP3 and NIX in human tumors. Cancer Res. 2001, 61, 6669–6673. [Google Scholar] [PubMed]
- Guo, K.; Searfoss, G.; Krolikowski, D.; Pagnoni, M.; Franks, C.; Clark, K.; Yu, K.T.; Jaye, M.; Ivashchenko, Y. Hypoxia induces the expression of the pro-apoptotic gene BNIP3. Cell Death Differ. 2001, 8, 367–376. [Google Scholar] [CrossRef] [Green Version]
- Bellot, G.; Garcia-Medina, R.; Gounon, P.; Chiche, J.; Roux, D.; Pouyssegur, J.; Mazure, N.M. Hypoxia-induced autophagy is mediated through hypoxia-inducible factor induction of BNIP3 and BNIP3L via their BH3 domains. Mol. Cell Biol. 2009, 29, 2570–2581. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, K.E.; Frazier, W.A. Phosphorylation of the BNIP3 C-Terminus Inhibits Mitochondrial Damage and Cell Death without Blocking Autophagy. PLoS ONE 2015, 10, e0129667. [Google Scholar] [CrossRef] [Green Version]
- Rogov, V.V.; Suzuki, H.; Marinkovic, M.; Lang, V.; Kato, R.; Kawasaki, M.; Buljubasic, M.; Sprung, M.; Rogova, N.; Wakatsuki, S.; et al. Phosphorylation of the mitochondrial autophagy receptor Nix enhances its interaction with LC3 proteins. Sci. Rep. 2017, 7, 1131. [Google Scholar] [CrossRef]
- Ma, K.; Zhang, Z.; Chang, R.; Cheng, H.; Mu, C.; Zhao, T.; Chen, L.; Zhang, C.; Luo, Q.; Lin, J.; et al. Dynamic PGAM5 multimers dephosphorylate BCL-xL or FUNDC1 to regulate mitochondrial and cellular fate. Cell Death Differ. 2020, 27, 1036–1051. [Google Scholar] [CrossRef]
- Chen, G.; Han, Z.; Feng, D.; Chen, Y.; Chen, L.; Wu, H.; Huang, L.; Zhou, C.; Cai, X.; Fu, C.; et al. A regulatory signaling loop comprising the PGAM5 phosphatase and CK2 controls receptor-mediated mitophagy. Mol. Cell. 2014, 54, 362–377. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Z.; Liu, L.; Cheng, Q.; Li, Y.; Wu, H.; Zhang, W.; Wang, Y.; Sehgal, S.A.; Siraj, S.; Wang, X.; et al. Mitochondrial E3 ligase MARCH5 regulates FUNDC1 to fine-tune hypoxic mitophagy. EMBO Rep. 2017, 18, 495–509. [Google Scholar] [CrossRef]
- Yan, C.; Gong, L.; Chen, L.; Xu, M.; Abou-Hamdan, H.; Tang, M.; Desaubry, L.; Song, Z. PHB2 (prohibitin 2) promotes PINK1-PRKN/Parkin-dependent mitophagy by the PARL-PGAM5-PINK1 axis. Autophagy 2020, 16, 419–434. [Google Scholar] [CrossRef]
- Li, X.X.; Tsoi, B.; Li, Y.F.; Kurihara, H.; He, R.R. Cardiolipin and its different properties in mitophagy and apoptosis. J. Histochem. Cytochem. 2015, 63, 301–311. [Google Scholar] [CrossRef] [Green Version]
- Strappazzon, F.; Nazio, F.; Corrado, M.; Cianfanelli, V.; Romagnoli, A.; Fimia, G.M.; Campello, S.; Nardacci, R.; Piacentini, M.; Campanella, M.; et al. AMBRA1 is able to induce mitophagy via LC3 binding, regardless of PARKIN and p62/SQSTM1. Cell Death Differ. 2015, 22, 517. [Google Scholar] [CrossRef] [PubMed]
- Zorov, D.B.; Kinnally, K.W.; Tedeschi, H. Voltage activation of heart inner mitochondrial membrane channels. J. Bioenerg. Biomembr. 1992, 24, 119–124. [Google Scholar] [CrossRef] [PubMed]
- Lyamzaev, K.G.; Nepryakhina, O.K.; Saprunova, V.B.; Bakeeva, L.E.; Pletjushkina, O.Y.; Chernyak, B.V.; Skulachev, V.P. Novel mechanism of elimination of malfunctioning mitochondria (mitoptosis): Formation of mitoptotic bodies and extrusion of mitochondrial material from the cell. Biochim. Biophys. Acta 2008, 1777, 817–825. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Skulachev, V.P. Bioenergetic aspects of apoptosis, necrosis and mitoptosis. Apoptosis 2006, 11, 473–485. [Google Scholar] [CrossRef]
- Skulachev, V.P. Mitochondrial physiology and pathology; concepts of programmed death of organelles, cells and organisms. Mol. Aspects Med. 1999, 20, 139–184. [Google Scholar] [CrossRef]
- Jangamreddy, J.R.; Los, M.J. Mitoptosis, a novel mitochondrial death mechanism leading predominantly to activation of autophagy. Hepat. Mon. 2012, 12, e6159. [Google Scholar] [CrossRef] [Green Version]
- Tinari, A.; Garofalo, T.; Sorice, M.; Esposti, M.D.; Malorni, W. Mitoptosis: Different pathways for mitochondrial execution. Autophagy 2007, 3, 282–284. [Google Scholar] [CrossRef] [Green Version]
- Skulachev, V.P. Programmed death phenomena: From organelle to organism. Ann. N. Y. Acad. Sci. 2002, 959, 214–237. [Google Scholar] [CrossRef]
- Hayakawa, K.; Esposito, E.; Wang, X.; Terasaki, Y.; Liu, Y.; Xing, C.; Ji, X.; Lo, E.H. Corrigendum: Transfer of mitochondria from astrocytes to neurons after stroke. Nature 2016, 539, 123. [Google Scholar] [CrossRef] [Green Version]
- Hayakawa, K.; Esposito, E.; Wang, X.; Terasaki, Y.; Liu, Y.; Xing, C.; Ji, X.; Lo, E.H. Transfer of mitochondria from astrocytes to neurons after stroke. Nature 2016, 535, 551–555. [Google Scholar] [CrossRef] [Green Version]
- Hass, D.T.; Barnstable, C.J. Mitochondrial Uncoupling Protein 2 Knock-out Promotes Mitophagy to Decrease Retinal Ganglion Cell Death in a Mouse Model of Glaucoma. J. Neurosci. 2019, 39, 3582–3596. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, L.; Zhang, Z.; Lu, J.; Pei, G. Mitochondria Are Dynamically Transferring Between Human Neural Cells and Alexander Disease-Associated GFAP Mutations Impair the Astrocytic Transfer. Front. Cell Neurosci. 2019, 13, 316. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Islam, M.N.; Das, S.R.; Emin, M.T.; Wei, M.; Sun, L.; Westphalen, K.; Rowlands, D.J.; Quadri, S.K.; Bhattacharya, S.; Bhattacharya, J. Mitochondrial transfer from bone-marrow-derived stromal cells to pulmonary alveoli protects against acute lung injury. Nat. Med. 2012, 18, 759–765. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahmad, T.; Mukherjee, S.; Pattnaik, B.; Kumar, M.; Singh, S.; Kumar, M.; Rehman, R.; Tiwari, B.K.; Jha, K.A.; Barhanpurkar, A.P.; et al. Miro1 regulates intercellular mitochondrial transport & enhances mesenchymal stem cell rescue efficacy. EMBO J. 2014, 33, 994–1010. [Google Scholar] [CrossRef] [PubMed]
- Torralba, D.; Baixauli, F.; Sanchez-Madrid, F. Mitochondria Know No Boundaries: Mechanisms and Functions of Intercellular Mitochondrial Transfer. Front. Cell Dev. Biol. 2016, 4, 107. [Google Scholar] [CrossRef] [Green Version]
- Han, H.; Hu, J.; Yan, Q.; Zhu, J.; Zhu, Z.; Chen, Y.; Sun, J.; Zhang, R. Bone marrow-derived mesenchymal stem cells rescue injured H9c2 cells via transferring intact mitochondria through tunneling nanotubes in an in vitro simulated ischemia/reperfusion model. Mol. Med. Rep. 2016, 13, 1517–1524. [Google Scholar] [CrossRef] [Green Version]
- Liu, K.; Ji, K.; Guo, L.; Wu, W.; Lu, H.; Shan, P.; Yan, C. Mesenchymal stem cells rescue injured endothelial cells in an in vitro ischemia-reperfusion model via tunneling nanotube like structure-mediated mitochondrial transfer. Microvasc. Res. 2014, 92, 10–18. [Google Scholar] [CrossRef]
- Cho, Y.M.; Kim, J.H.; Kim, M.; Park, S.J.; Koh, S.H.; Ahn, H.S.; Kang, G.H.; Lee, J.B.; Park, K.S.; Lee, H.K. Mesenchymal stem cells transfer mitochondria to the cells with virtually no mitochondrial function but not with pathogenic mtDNA mutations. PLoS ONE 2012, 7, e32778. [Google Scholar] [CrossRef] [Green Version]
- Konari, N.; Nagaishi, K.; Kikuchi, S.; Fujimiya, M. Mitochondria transfer from mesenchymal stem cells structurally and functionally repairs renal proximal tubular epithelial cells in diabetic nephropathy in vivo. Sci. Rep. 2019, 9, 5184. [Google Scholar] [CrossRef] [Green Version]
- Spees, J.L.; Olson, S.D.; Whitney, M.J.; Prockop, D.J. Mitochondrial transfer between cells can rescue aerobic respiration. Proc. Natl. Acad. Sci. USA 2006, 103, 1283–1288. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Gerdes, H.H. Transfer of mitochondria via tunneling nanotubes rescues apoptotic PC12 cells. Cell Death Differ. 2015, 22, 1181–1191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hansen, M.; Rubinsztein, D.C.; Walker, D.W. Autophagy as a promoter of longevity: Insights from model organisms. Nat. Rev. Mol. Cell Biol. 2018, 19, 579–593. [Google Scholar] [CrossRef] [PubMed]
- Petrosillo, G.; De Benedictis, V.; Ruggiero, F.M.; Paradies, G. Decline in cytochrome c oxidase activity in rat-brain mitochondria with aging. Role of peroxidized cardiolipin and beneficial effect of melatonin. J. Bioenerg. Biomembr. 2013, 45, 431–440. [Google Scholar] [CrossRef] [PubMed]
- Petrosillo, G.; Matera, M.; Casanova, G.; Ruggiero, F.M.; Paradies, G. Mitochondrial dysfunction in rat brain with aging Involvement of complex I, reactive oxygen species and cardiolipin. Neurochem. Int. 2008, 53, 126–131. [Google Scholar] [CrossRef] [PubMed]
- Leslie, S.W.; Chandler, L.J.; Barr, E.M.; Farrar, R.P. Reduced calcium uptake by rat brain mitochondria and synaptosomes in response to aging. Brain Res. 1985, 329, 177–183. [Google Scholar] [CrossRef]
- Stauch, K.L.; Purnell, P.R.; Villeneuve, L.M.; Fox, H.S. Proteomic analysis and functional characterization of mouse brain mitochondria during aging reveal alterations in energy metabolism. Proteomics 2015, 15, 1574–1586. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bertoni-Freddari, C.; Fattoretti, P.; Casoli, T.; Spagna, C.; Meier-Ruge, W.; Ulrich, J. Morphological plasticity of synaptic mitochondria during aging. Brain Res. 1993, 628, 193–200. [Google Scholar] [CrossRef]
- Bertoni-Freddari, C.; Balietti, M.; Giorgetti, B.; Grossi, Y.; Casoli, T.; Di Stefano, G.; Perretta, G.; Fattoretti, P. Selective decline of the metabolic competence of oversized synaptic mitochondria in the old monkey cerebellum. Rejuvenation Res. 2008, 11, 387–391. [Google Scholar] [CrossRef]
- Tanhauser, S.M.; Laipis, P.J. Multiple deletions are detectable in mitochondrial DNA of aging mice. J. Biol. Chem. 1995, 270, 24769–24775. [Google Scholar] [CrossRef] [Green Version]
- Dzitoyeva, S.; Chen, H.; Manev, H. Effect of aging on 5-hydroxymethylcytosine in brain mitochondria. Neurobiol. Aging 2012, 33, 2881–2891. [Google Scholar] [CrossRef] [Green Version]
- Herrero, A.; Barja, G. 8-oxo-deoxyguanosine levels in heart and brain mitochondrial and nuclear DNA of two mammals and three birds in relation to their different rates of aging. Aging 1999, 11, 294–300. [Google Scholar] [CrossRef] [PubMed]
- Barja, G.; Herrero, A. Oxidative damage to mitochondrial DNA is inversely related to maximum life span in the heart and brain of mammals. FASEB J. 2000, 14, 312–318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mecocci, P.; MacGarvey, U.; Kaufman, A.E.; Koontz, D.; Shoffner, J.M.; Wallace, D.C.; Beal, M.F. Oxidative damage to mitochondrial DNA shows marked age-dependent increases in human brain. Ann. Neurol. 1993, 34, 609–616. [Google Scholar] [CrossRef] [PubMed]
- Wallace, D.C. Mitochondrial DNA in aging and disease. Sci. Am. 1997, 277, 40–47. [Google Scholar] [CrossRef]
- Cortopassi, G.A.; Shibata, D.; Soong, N.W.; Arnheim, N. A pattern of accumulation of a somatic deletion of mitochondrial DNA in aging human tissues. Proc. Natl. Acad. Sci. USA 1992, 89, 7370–7374. [Google Scholar] [CrossRef] [Green Version]
- Clark, I.E.; Dodson, M.W.; Jiang, C.; Cao, J.H.; Huh, J.R.; Seol, J.H.; Yoo, S.J.; Hay, B.A.; Guo, M. Drosophila pink1 is required for mitochondrial function and interacts genetically with parkin. Nature 2006, 441, 1162–1166. [Google Scholar] [CrossRef]
- Rana, A.; Rera, M.; Walker, D.W. Parkin overexpression during aging reduces proteotoxicity, alters mitochondrial dynamics, and extends lifespan. Proc. Natl. Acad. Sci. USA 2013, 110, 8638–8643. [Google Scholar] [CrossRef] [Green Version]
- Palikaras, K.; Lionaki, E.; Tavernarakis, N. Coordination of mitophagy and mitochondrial biogenesis during ageing in C. elegans. Nature 2015, 521, 525–528. [Google Scholar] [CrossRef]
- Schiavi, A.; Maglioni, S.; Palikaras, K.; Shaik, A.; Strappazzon, F.; Brinkmann, V.; Torgovnick, A.; Castelein, N.; De Henau, S.; Braeckman, B.P.; et al. Iron-Starvation-Induced Mitophagy Mediates Lifespan Extension upon Mitochondrial Stress in C. elegans. Curr. Biol. 2015, 25, 1810–1822. [Google Scholar] [CrossRef] [Green Version]
- Wu, X.; Al-Amin, M.; Zhao, C.; An, F.; Wang, Y.; Huang, Q.; Teng, H.; Song, H. Catechinic acid, a natural polyphenol compound, extends the lifespan of Caenorhabditis elegans via mitophagy pathways. Food Funct. 2020, 11, 5621–5634. [Google Scholar] [CrossRef]
- Fang, E.F.; Waltz, T.B.; Kassahun, H.; Lu, Q.; Kerr, J.S.; Morevati, M.; Fivenson, E.M.; Wollman, B.N.; Marosi, K.; Wilson, M.A.; et al. Tomatidine enhances lifespan and healthspan in C. elegans through mitophagy induction via the SKN-1/Nrf2 pathway. Sci. Rep. 2017, 7, 46208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ryu, D.; Mouchiroud, L.; Andreux, P.A.; Katsyuba, E.; Moullan, N.; Nicolet-Dit-Felix, A.A.; Williams, E.G.; Jha, P.; Lo Sasso, G.; Huzard, D.; et al. Urolithin A induces mitophagy and prolongs lifespan in C. elegans and increases muscle function in rodents. Nat. Med. 2016, 22, 879–888. [Google Scholar] [CrossRef] [PubMed]
- Fang, E.F.; Hou, Y.; Lautrup, S.; Jensen, M.B.; Yang, B.; SenGupta, T.; Caponio, D.; Khezri, R.; Demarest, T.G.; Aman, Y.; et al. NAD(+) augmentation restores mitophagy and limits accelerated aging in Werner syndrome. Nat. Commun. 2019, 10, 5284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Braak, H.; Braak, E. Diagnostic criteria for neuropathologic assessment of Alzheimer’s Disease. Neurobiol. Aging 1997, 18, S85–S88. [Google Scholar] [CrossRef]
- Powers, J.M. Diagnostic criteria for the neuropathologic assessment of Alzheimer’s Disease. Neurobiol. Aging 1997, 18, S53–S54. [Google Scholar] [CrossRef]
- Jack, C.R., Jr.; Barrio, J.R.; Kepe, V. Cerebral amyloid PET imaging in Alzheimer’s Disease. Acta Neuropathol. 2013, 126, 643–657. [Google Scholar] [CrossRef] [Green Version]
- Dubois, B.; Feldman, H.H.; Jacova, C.; Dekosky, S.T.; Barberger-Gateau, P.; Cummings, J.; Delacourte, A.; Galasko, D.; Gauthier, S.; Jicha, G.; et al. Research criteria for the diagnosis of Alzheimer’s Disease: Revising the NINCDS-ADRDA criteria. Lancet Neurol. 2007, 6, 734–746. [Google Scholar] [CrossRef]
- Marcus, C.; Mena, E.; Subramaniam, R.M. Brain PET in the diagnosis of Alzheimer’s Disease. Clin. Nucl. Med. 2014, 39, e413–e422. [Google Scholar] [CrossRef] [Green Version]
- Mosconi, L.; McHugh, P.F. FDG- and amyloid-PET in Alzheimer’s Disease: Is the whole greater than the sum of the parts? Q. J. Nucl. Med. Mol. Imaging 2011, 55, 250–264. [Google Scholar]
- Suppiah, S.; Didier, M.A.; Vinjamuri, S. The Who, When, Why, and How of PET Amyloid Imaging in Management of Alzheimer’s Disease-Review of Literature and Interesting Images. Diagnostics 2019, 9, 65. [Google Scholar] [CrossRef] [Green Version]
- Herholz, K.; Salmon, E.; Perani, D.; Baron, J.C.; Holthoff, V.; Frolich, L.; Schonknecht, P.; Ito, K.; Mielke, R.; Kalbe, E.; et al. Discrimination between Alzheimer dementia and controls by automated analysis of multicenter FDG PET. Neuroimage 2002, 17, 302–316. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valla, J.; Berndt, J.D.; Gonzalez-Lima, F. Energy hypometabolism in posterior cingulate cortex of Alzheimer’s patients: Superficial laminar cytochrome oxidase associated with disease duration. J. Neurosci. 2001, 21, 4923–4930. [Google Scholar] [CrossRef] [PubMed]
- Messa, C.; Perani, D.; Lucignani, G.; Zenorini, A.; Zito, F.; Rizzo, G.; Grassi, F.; Del Sole, A.; Franceschi, M.; Gilardi, M.C.; et al. High-resolution technetium-99m-HMPAO SPECT in patients with probable Alzheimer’s Disease: Comparison with fluorine-18-FDG PET. J. Nucl. Med. 1994, 35, 210–216. [Google Scholar] [PubMed]
- Morris, J.K.; Honea, R.A.; Vidoni, E.D.; Swerdlow, R.H.; Burns, J.M. Is Alzheimer’s Disease a systemic disease? Biochim. Biophys. Acta 2014, 1842, 1340–1349. [Google Scholar] [CrossRef] [Green Version]
- Bosetti, F.; Brizzi, F.; Barogi, S.; Mancuso, M.; Siciliano, G.; Tendi, E.A.; Murri, L.; Rapoport, S.I.; Solaini, G. Cytochrome c oxidase and mitochondrial F1F0-ATPase (ATP synthase) activities in platelets and brain from patients with Alzheimer’s Disease. Neurobiol. Aging 2002, 23, 371–376. [Google Scholar] [CrossRef]
- Fukui, H.; Diaz, F.; Garcia, S.; Moraes, C.T. Cytochrome c oxidase deficiency in neurons decreases both oxidative stress and amyloid formation in a mouse model of Alzheimer’s Disease. Proc. Natl. Acad. Sci. USA 2007, 104, 14163–14168. [Google Scholar] [CrossRef] [Green Version]
- Parker, W.D., Jr.; Parks, J.K. Cytochrome c oxidase in Alzheimer’s Disease brain: Purification and characterization. Neurology 1995, 45, 482–486. [Google Scholar] [CrossRef]
- Cardoso, S.M.; Proenca, M.T.; Santos, S.; Santana, I.; Oliveira, C.R. Cytochrome c oxidase is decreased in Alzheimer’s Disease platelets. Neurobiol. Aging 2004, 25, 105–110. [Google Scholar] [CrossRef]
- Khan, S.M.; Cassarino, D.S.; Abramova, N.N.; Keeney, P.M.; Borland, M.K.; Trimmer, P.A.; Krebs, C.T.; Bennett, J.C.; Parks, J.K.; Swerdlow, R.H.; et al. Alzheimer’s Disease cybrids replicate beta-amyloid abnormalities through cell death pathways. Ann. Neurol. 2000, 48, 148–155. [Google Scholar] [CrossRef]
- Kish, S.J.; Bergeron, C.; Rajput, A.; Dozic, S.; Mastrogiacomo, F.; Chang, L.J.; Wilson, J.M.; DiStefano, L.M.; Nobrega, J.N. Brain cytochrome oxidase in Alzheimer’s Disease. J. Neurochem. 1992, 59, 776–779. [Google Scholar] [CrossRef] [PubMed]
- Kish, S.J. Brain energy metabolizing enzymes in Alzheimer’s Disease: Alpha-ketoglutarate dehydrogenase complex and cytochrome oxidase. Ann. N. Y. Acad. Sci. 1997, 826, 218–228. [Google Scholar] [CrossRef] [PubMed]
- Mutisya, E.M.; Bowling, A.C.; Beal, M.F. Cortical cytochrome oxidase activity is reduced in Alzheimer’s Disease. J. Neurochem. 1994, 63, 2179–2184. [Google Scholar] [CrossRef]
- Parker, W.D., Jr. Cytochrome oxidase deficiency in Alzheimer’s Disease. Ann. N. Y. Acad. Sci. 1991, 640, 59–64. [Google Scholar] [CrossRef] [PubMed]
- Parker, W.D., Jr.; Filley, C.M.; Parks, J.K. Cytochrome oxidase deficiency in Alzheimer’s Disease. Neurology 1990, 40, 1302–1303. [Google Scholar] [CrossRef] [PubMed]
- Alberts, M.J.; Ioannou, P.; Deucher, R.; Gilbert, J.; Lee, J.; Middleton, L.; Roses, A.D. Isolation of a cytochrome oxidase gene overexpressed in Alzheimer’s Disease brain. Mol. Cell Neurosci. 1992, 3, 461–470. [Google Scholar] [CrossRef]
- Curti, D.; Rognoni, F.; Gasparini, L.; Cattaneo, A.; Paolillo, M.; Racchi, M.; Zani, L.; Bianchetti, A.; Trabucchi, M.; Bergamaschi, S.; et al. Oxidative metabolism in cultured fibroblasts derived from sporadic Alzheimer’s Disease (AD) patients. Neurosci. Lett. 1997, 236, 13–16. [Google Scholar] [CrossRef]
- Silva, D.F.; Selfridge, J.E.; Lu, J.; Lezi, E.; Roy, N.; Hutfles, L.; Burns, J.M.; Michaelis, E.K.; Yan, S.; Cardoso, S.M.; et al. Bioenergetic flux, mitochondrial mass and mitochondrial morphology dynamics in AD and MCI cybrid cell lines. Hum. Mol. Genet. 2013, 22, 3931–3946. [Google Scholar] [CrossRef] [PubMed]
- Sheehan, J.P.; Swerdlow, R.H.; Miller, S.W.; Davis, R.E.; Parks, J.K.; Parker, W.D.; Tuttle, J.B. Calcium homeostasis and reactive oxygen species production in cells transformed by mitochondria from individuals with sporadic Alzheimer’s Disease. J. Neurosci. 1997, 17, 4612–4622. [Google Scholar] [CrossRef] [PubMed]
- Cardoso, S.M.; Santana, I.; Swerdlow, R.H.; Oliveira, C.R. Mitochondria dysfunction of Alzheimer’s Disease cybrids enhances Abeta toxicity. J. Neurochem. 2004, 89, 1417–1426. [Google Scholar] [CrossRef] [Green Version]
- Swerdlow, R.H. Mitochondria in cybrids containing mtDNA from persons with mitochondriopathies. J. Neurosci. Res. 2007, 85, 3416–3428. [Google Scholar] [CrossRef]
- Trimmer, P.A.; Keeney, P.M.; Borland, M.K.; Simon, F.A.; Almeida, J.; Swerdlow, R.H.; Parks, J.P.; Parker, W.D., Jr.; Bennett, J.P., Jr. Mitochondrial abnormalities in cybrid cell models of sporadic Alzheimer’s Disease worsen with passage in culture. Neurobiol. Dis. 2004, 15, 29–39. [Google Scholar] [CrossRef] [PubMed]
- Baloyannis, S.J. Mitochondria in Alzheimer’s Disease: An Electron Microscopy Study. Alzheimer Dis. Treat. 2019. [Google Scholar] [CrossRef] [Green Version]
- Berti, V.; Mosconi, L.; Glodzik, L.; Li, Y.; Murray, J.; De Santi, S.; Pupi, A.; Tsui, W.; De Leon, M.J. Structural brain changes in normal individuals with a maternal history of Alzheimer’s. Neurobiol. Aging 2011, 32, 2325-e17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mosconi, L. Glucose metabolism in normal aging and Alzheimer’s Disease: Methodological and physiological considerations for PET studies. Clin. Transl. Imaging 2013, 1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mosconi, L.; Mistur, R.; Switalski, R.; Brys, M.; Glodzik, L.; Rich, K.; Pirraglia, E.; Tsui, W.; De Santi, S.; de Leon, M.J. Declining brain glucose metabolism in normal individuals with a maternal history of Alzheimer disease. Neurology 2009, 72, 513–520. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mosconi, L.; Rinne, J.O.; Tsui, W.H.; Murray, J.; Li, Y.; Glodzik, L.; McHugh, P.; Williams, S.; Cummings, M.; Pirraglia, E.; et al. Amyloid and metabolic positron emission tomography imaging of cognitively normal adults with Alzheimer’s parents. Neurobiol. Aging 2013, 34, 22–34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Swerdlow, R.H.; Hui, D.; Chalise, P.; Sharma, P.; Wang, X.; Andrews, S.J.; Pa, J.; Mahnken, J.D.; Morris, J.; Wilkins, H.M.; et al. Exploratory analysis of mtDNA haplogroups in two Alzheimer’s longitudinal cohorts. Alzheimers Dement. 2020, 16, 1164–1172. [Google Scholar] [CrossRef]
- Andrews, S.J.; Fulton-Howard, B.; Patterson, C.; McFall, G.P.; Gross, A.; Michaelis, E.K.; Goate, A.; Swerdlow, R.H.; Pa, J. Mitochondrial haplogroups & a nuclear encoded mitochondrial polygenic risk score interact to influence dementia risk. Neurobiol. Aging 2020, 87, 138-e7. [Google Scholar]
- Carrieri, G.; Bonafe, M.; De Luca, M.; Rose, G.; Varcasia, O.; Bruni, A.; Maletta, R.; Nacmias, B.; Sorbi, S.; Corsonello, F.; et al. Mitochondrial DNA haplogroups and APOE4 allele are non-independent variables in sporadic Alzheimer’s Disease. Hum. Genet. 2001, 108, 194–198. [Google Scholar] [CrossRef]
- Coto, E.; Gomez, J.; Alonso, B.; Corao, A.I.; Diaz, M.; Menendez, M.; Martinez, C.; Calatayud, M.T.; Moris, G.; Alvarez, V. Late-onset Alzheimer’s Disease is associated with mitochondrial DNA 7028C/haplogroup H and D310 poly-C tract heteroplasmy. Neurogenetics 2011, 12, 345–346. [Google Scholar] [CrossRef]
- Fesahat, F.; Houshmand, M.; Panahi, M.S.; Gharagozli, K.; Mirzajani, F. Do haplogroups H and U act to increase the penetrance of Alzheimer’s Disease? Cell Mol. Neurobiol. 2007, 27, 329–334. [Google Scholar] [CrossRef] [PubMed]
- Takasaki, S. Mitochondrial haplogroups associated with Japanese Alzheimer’s patients. J. Bioenerg. Biomembr. 2009, 41, 407–410. [Google Scholar] [CrossRef] [PubMed]
- van der Walt, J.M.; Dementieva, Y.A.; Martin, E.R.; Scott, W.K.; Nicodemus, K.K.; Kroner, C.C.; Welsh-Bohmer, K.A.; Saunders, A.M.; Roses, A.D.; Small, G.W.; et al. Analysis of European mitochondrial haplogroups with Alzheimer disease risk. Neurosci. Lett. 2004, 365, 28–32. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Chen, Y.; Mok, K.Y.; Kwok, T.C.Y.; Mok, V.C.T.; Guo, Q.; Ip, F.C.; Chen, Y.; Mullapudi, N.; Alzheimer’s Disease Neuroimaging, I.; et al. Non-coding variability at the APOE locus contributes to the Alzheimer’s risk. Nat. Commun. 2019, 10, 3310. [Google Scholar] [CrossRef] [Green Version]
- Lakatos, A.; Derbeneva, O.; Younes, D.; Keator, D.; Bakken, T.; Lvova, M.; Brandon, M.; Guffanti, G.; Reglodi, D.; Saykin, A.; et al. Association between mitochondrial DNA variations and Alzheimer’s Disease in the ADNI cohort. Neurobiol. Aging 2010, 31, 1355–1363. [Google Scholar] [CrossRef] [Green Version]
- Reddy, P.H.; Oliver, D.M. Amyloid Beta and Phosphorylated Tau-Induced Defective Autophagy and Mitophagy in Alzheimer’s Disease. Cells 2019, 8, 488. [Google Scholar] [CrossRef] [Green Version]
- Hu, Y.; Li, X.C.; Wang, Z.H.; Luo, Y.; Zhang, X.; Liu, X.P.; Feng, Q.; Wang, Q.; Yue, Z.; Chen, Z.; et al. Tau accumulation impairs mitophagy via increasing mitochondrial membrane potential and reducing mitochondrial Parkin. Oncotarget 2016, 7, 17356–17368. [Google Scholar] [CrossRef] [Green Version]
- Manczak, M.; Kandimalla, R.; Yin, X.; Reddy, P.H. Hippocampal mutant APP and amyloid beta-induced cognitive decline, dendritic spine loss, defective autophagy, mitophagy and mitochondrial abnormalities in a mouse model of Alzheimer’s Disease. Hum. Mol. Genet. 2018, 27, 1332–1342. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Zhao, X.L.; Xu, L.L.; Wang, C.F.; Wei, L.F.; Liu, Z.; Yang, H.; Wang, P.; Xie, Z.H.; Bi, J.Z. Mitophagy in APPsw/PS1dE9 transgenic mice and APPsw stably expressing in HEK293 cells. Eur. Rev. Med. Pharmacol. Sci. 2015, 19, 4595–4602. [Google Scholar]
- Ye, X.; Sun, X.; Starovoytov, V.; Cai, Q. Parkin-mediated mitophagy in mutant hAPP neurons and Alzheimer’s Disease patient brains. Hum. Mol. Genet. 2015, 24, 2938–2951. [Google Scholar] [CrossRef]
- Bordi, M.; Berg, M.J.; Mohan, P.S.; Peterhoff, C.M.; Alldred, M.J.; Che, S.; Ginsberg, S.D.; Nixon, R.A. Autophagy flux in CA1 neurons of Alzheimer hippocampus: Increased induction overburdens failing lysosomes to propel neuritic dystrophy. Autophagy 2016, 12, 2467–2483. [Google Scholar] [CrossRef]
- Kerr, J.S.; Adriaanse, B.A.; Greig, N.H.; Mattson, M.P.; Cader, M.Z.; Bohr, V.A.; Fang, E.F. Mitophagy and Alzheimer’s Disease: Cellular and Molecular Mechanisms. Trends Neurosci. 2017, 40, 151–166. [Google Scholar] [CrossRef] [Green Version]
- Nixon, R.A. The role of autophagy in neurodegenerative disease. Nat. Med. 2013, 19, 983–997. [Google Scholar] [CrossRef]
- Kurosawa, M.; Matsumoto, G.; Sumikura, H.; Hatsuta, H.; Murayama, S.; Sakurai, T.; Shimogori, T.; Hattori, N.; Nukina, N. Serine 403-phosphorylated p62/SQSTM1 immunoreactivity in inclusions of neurodegenerative diseases. Neurosci. Res. 2016, 103, 64–70. [Google Scholar] [CrossRef] [PubMed]
- Bharadwaj, P.; Martins, R.N. PRKAG2 Gene Expression Is Elevated and its Protein Levels Are Associated with Increased Amyloid-beta Accumulation in the Alzheimer’s Disease Brain. J. Alzheimers Dis. 2020, 74, 441–448. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lonskaya, I.; Shekoyan, A.R.; Hebron, M.L.; Desforges, N.; Algarzae, N.K.; Moussa, C.E. Diminished parkin solubility and co-localization with intraneuronal amyloid-beta are associated with autophagic defects in Alzheimer’s Disease. J. Alzheimers Dis. 2013, 33, 231–247. [Google Scholar] [CrossRef]
- Yan, X.; Wang, B.; Hu, Y.; Wang, S.; Zhang, X. Abnormal Mitochondrial Quality Control in Neurodegenerative Diseases. Front. Cell Neurosci. 2020, 14, 138. [Google Scholar] [CrossRef]
- Poewe, W.; Seppi, K.; Tanner, C.M.; Halliday, G.M.; Brundin, P.; Volkmann, J.; Schrag, A.E.; Lang, A.E. Parkinson disease. Nat. Rev. Dis. Primers 2017, 3, 17013. [Google Scholar] [CrossRef] [PubMed]
- Gibb, W.R.; Lees, A.J. The relevance of the Lewy body to the pathogenesis of idiopathic Parkinson’s disease. J. Neurol. Neurosurg. Psychiatry 1988, 51, 745–752. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clarke, C.E. Parkinson’s disease. BMJ 2007, 335, 441–445. [Google Scholar] [CrossRef]
- Mann, V.M.; Cooper, J.M.; Krige, D.; Daniel, S.E.; Schapira, A.H.; Marsden, C.D. Brain, skeletal muscle and platelet homogenate mitochondrial function in Parkinson’s disease. Brain 1992, 115 Pt 2, 333–342. [Google Scholar] [CrossRef]
- Gatt, A.P.; Duncan, O.F.; Attems, J.; Francis, P.T.; Ballard, C.G.; Bateman, J.M. Dementia in Parkinson’s disease is associated with enhanced mitochondrial complex I deficiency. Mov. Disord. 2016, 31, 352–359. [Google Scholar] [CrossRef] [PubMed]
- Blake, C.I.; Spitz, E.; Leehey, M.; Hoffer, B.J.; Boyson, S.J. Platelet mitochondrial respiratory chain function in Parkinson’s disease. Mov. Disord. 1997, 12, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Krige, D.; Carroll, M.T.; Cooper, J.M.; Marsden, C.D.; Schapira, A.H. Platelet mitochondrial function in Parkinson’s disease. The Royal Kings and Queens Parkinson Disease Research Group. Ann. Neurol. 1992, 32, 782–788. [Google Scholar] [CrossRef] [PubMed]
- Arduino, D.M.; Esteves, A.R.; Swerdlow, R.H.; Cardoso, S.M. A cybrid cell model for the assessment of the link between mitochondrial deficits and sporadic Parkinson’s disease. Methods Mol. Biol. 2015, 1265, 415–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Esteves, A.R.; Domingues, A.F.; Ferreira, I.L.; Januario, C.; Swerdlow, R.H.; Oliveira, C.R.; Cardoso, S.M. Mitochondrial function in Parkinson’s disease cybrids containing an nt2 neuron-like nuclear background. Mitochondrion 2008, 8, 219–228. [Google Scholar] [CrossRef]
- Esteves, A.R.; Lu, J.; Rodova, M.; Onyango, I.; Lezi, E.; Dubinsky, R.; Lyons, K.E.; Pahwa, R.; Burns, J.M.; Cardoso, S.M.; et al. Mitochondrial respiration and respiration-associated proteins in cell lines created through Parkinson’s subject mitochondrial transfer. J. Neurochem. 2010, 113, 674–682. [Google Scholar] [CrossRef]
- Dolle, C.; Flones, I.; Nido, G.S.; Miletic, H.; Osuagwu, N.; Kristoffersen, S.; Lilleng, P.K.; Larsen, J.P.; Tysnes, O.B.; Haugarvoll, K.; et al. Defective mitochondrial DNA homeostasis in the substantia nigra in Parkinson disease. Nat. Commun. 2016, 7, 13548. [Google Scholar] [CrossRef]
- Gu, G.; Reyes, P.E.; Golden, G.T.; Woltjer, R.L.; Hulette, C.; Montine, T.J.; Zhang, J. Mitochondrial DNA deletions/rearrangements in parkinson disease and related neurodegenerative disorders. J. Neuropathol. Exp. Neurol. 2002, 61, 634–639. [Google Scholar] [CrossRef] [Green Version]
- Valente, E.M.; Abou-Sleiman, P.M.; Caputo, V.; Muqit, M.M.; Harvey, K.; Gispert, S.; Ali, Z.; Del Turco, D.; Bentivoglio, A.R.; Healy, D.G.; et al. Hereditary early-onset Parkinson’s disease caused by mutations in PINK1. Science 2004, 304, 1158–1160. [Google Scholar] [CrossRef] [Green Version]
- Polymeropoulos, M.H.; Lavedan, C.; Leroy, E.; Ide, S.E.; Dehejia, A.; Dutra, A.; Pike, B.; Root, H.; Rubenstein, J.; Boyer, R.; et al. Mutation in the alpha-synuclein gene identified in families with Parkinson’s disease. Science 1997, 276, 2045–2047. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.Y.; Nagano, Y.; Taylor, J.P.; Lim, K.L.; Yao, T.P. Disease-causing mutations in parkin impair mitochondrial ubiquitination, aggregation, and HDAC6-dependent mitophagy. J. Cell Biol. 2010, 189, 671–679. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lesage, S.; Condroyer, C.; Klebe, S.; Honore, A.; Tison, F.; Brefel-Courbon, C.; Durr, A.; Brice, A.; French Parkinson’s Disease Genetics Study, G. Identification of VPS35 mutations replicated in French families with Parkinson disease. Neurology 2012, 78, 1449–1450. [Google Scholar] [CrossRef] [PubMed]
- Kitada, T.; Asakawa, S.; Hattori, N.; Matsumine, H.; Yamamura, Y.; Minoshima, S.; Yokochi, M.; Mizuno, Y.; Shimizu, N. Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature 1998, 392, 605–608. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Chen, K.; Song, W.; Chen, X.; Cao, B.; Huang, R.; Zhao, B.; Guo, X.; Burgunder, J.; Li, J.; et al. VPS35 Asp620Asn and EIF4G1 Arg1205His mutations are rare in Parkinson disease from southwest China. Neurobiol. Aging 2013, 34, 1709-e7. [Google Scholar] [CrossRef] [PubMed]
- Saravanan, K.S.; Sindhu, K.M.; Mohanakumar, K.P. Acute intranigral infusion of rotenone in rats causes progressive biochemical lesions in the striatum similar to Parkinson’s disease. Brain Res. 2005, 1049, 147–155. [Google Scholar] [CrossRef] [PubMed]
- Duty, S.; Jenner, P. Animal models of Parkinson’s disease: A source of novel treatments and clues to the cause of the disease. Br. J. Pharmacol. 2011, 164, 1357–1391. [Google Scholar] [CrossRef] [Green Version]
- Betarbet, R.; Sherer, T.B.; MacKenzie, G.; Garcia-Osuna, M.; Panov, A.V.; Greenamyre, J.T. Chronic systemic pesticide exposure reproduces features of Parkinson’s disease. Nat. Neurosci. 2000, 3, 1301–1306. [Google Scholar] [CrossRef]
- Zeng, X.S.; Geng, W.S.; Jia, J.J. Neurotoxin-Induced Animal Models of Parkinson Disease: Pathogenic Mechanism and Assessment. ASN Neuro 2018, 10, 1759091418777438. [Google Scholar] [CrossRef]
- von Wrangel, C.; Schwabe, K.; John, N.; Krauss, J.K.; Alam, M. The rotenone-induced rat model of Parkinson’s disease: Behavioral and electrophysiological findings. Behav. Brain Res. 2015, 279, 52–61. [Google Scholar] [CrossRef]
- Bolam, J.P.; Pissadaki, E.K. Living on the edge with too many mouths to feed: Why dopamine neurons die. Mov. Disord. 2012, 27, 1478–1483. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hou, X.; Fiesel, F.C.; Truban, D.; Castanedes Casey, M.; Lin, W.L.; Soto, A.I.; Tacik, P.; Rousseau, L.G.; Diehl, N.N.; Heckman, M.G.; et al. Age- and disease-dependent increase of the mitophagy marker phospho-ubiquitin in normal aging and Lewy body disease. Autophagy 2018, 14, 1404–1418. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hsieh, C.H.; Shaltouki, A.; Gonzalez, A.E.; Bettencourt da Cruz, A.; Burbulla, L.F.; St Lawrence, E.; Schule, B.; Krainc, D.; Palmer, T.D.; Wang, X. Functional Impairment in Miro Degradation and Mitophagy Is a Shared Feature in Familial and Sporadic Parkinson’s Disease. Cell Stem Cell 2016, 19, 709–724. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barcelos, I.P.; Troxell, R.M.; Graves, J.S. Mitochondrial Dysfunction and Multiple Sclerosis. Biology 2019, 8, 37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vaughn, C.B.; Jakimovski, D.; Kavak, K.S.; Ramanathan, M.; Benedict, R.H.B.; Zivadinov, R.; Weinstock-Guttman, B. Epidemiology and treatment of multiple sclerosis in elderly populations. Nat. Rev. Neurol. 2019, 15, 329–342. [Google Scholar] [CrossRef] [PubMed]
- Gonzalo, H.; Nogueras, L.; Gil-Sanchez, A.; Hervas, J.V.; Valcheva, P.; Gonzalez-Mingot, C.; Martin-Gari, M.; Canudes, M.; Peralta, S.; Solana, M.J.; et al. Impairment of Mitochondrial Redox Status in Peripheral Lymphocytes of Multiple Sclerosis Patients. Front. Neurosci. 2019, 13, 938. [Google Scholar] [CrossRef]
- Mahad, D.; Ziabreva, I.; Lassmann, H.; Turnbull, D. Mitochondrial defects in acute multiple sclerosis lesions. Brain 2008, 131, 1722–1735. [Google Scholar] [CrossRef]
- Mahad, D.J.; Ziabreva, I.; Campbell, G.; Lax, N.; White, K.; Hanson, P.S.; Lassmann, H.; Turnbull, D.M. Mitochondrial changes within axons in multiple sclerosis. Brain 2009, 132, 1161–1174. [Google Scholar] [CrossRef] [Green Version]
- Dutta, R.; McDonough, J.; Yin, X.; Peterson, J.; Chang, A.; Torres, T.; Gudz, T.; Macklin, W.B.; Lewis, D.A.; Fox, R.J.; et al. Mitochondrial dysfunction as a cause of axonal degeneration in multiple sclerosis patients. Ann. Neurol. 2006, 59, 478–489. [Google Scholar] [CrossRef]
- Alirezaei, M.; Fox, H.S.; Flynn, C.T.; Moore, C.S.; Hebb, A.L.; Frausto, R.F.; Bhan, V.; Kiosses, W.B.; Whitton, J.L.; Robertson, G.S.; et al. Elevated ATG5 expression in autoimmune demyelination and multiple sclerosis. Autophagy 2009, 5, 152–158. [Google Scholar] [CrossRef] [Green Version]
- Liang, P.; Le, W. Role of autophagy in the pathogenesis of multiple sclerosis. Neurosci. Bull. 2015, 31, 435–444. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castellazzi, M.; Patergnani, S.; Donadio, M.; Giorgi, C.; Bonora, M.; Fainardi, E.; Casetta, I.; Granieri, E.; Pugliatti, M.; Pinton, P. Correlation between auto/mitophagic processes and magnetic resonance imaging activity in multiple sclerosis patients. J. Neuroinflamm. 2019, 16, 131. [Google Scholar] [CrossRef] [PubMed]
- Igci, M.; Baysan, M.; Yigiter, R.; Ulasli, M.; Geyik, S.; Bayraktar, R.; Bozgeyik, I.; Bozgeyik, E.; Bayram, A.; Cakmak, E.A. Gene expression profiles of autophagy-related genes in multiple sclerosis. Gene 2016, 588, 38–46. [Google Scholar] [CrossRef] [PubMed]
- Smith, E.F.; Shaw, P.J.; De Vos, K.J. The role of mitochondria in amyotrophic lateral sclerosis. Neurosci. Lett. 2019, 710, 132933. [Google Scholar] [CrossRef] [PubMed]
- Swerdlow, R.H.; Parks, J.K.; Pattee, G.; Parker, W.D., Jr. Role of mitochondria in amyotrophic lateral sclerosis. Amyotroph. Lateral Scler. Other Motor Neuron Disord. 2000, 1, 185–190. [Google Scholar] [CrossRef]
- Beeldman, E.; Raaphorst, J.; Klein Twennaar, M.; de Visser, M.; Schmand, B.A.; de Haan, R.J. The cognitive profile of ALS: A systematic review and meta-analysis update. J. Neurol. Neurosurg. Psychiatry 2016, 87, 611–619. [Google Scholar] [CrossRef] [Green Version]
- Gubbay, S.S.; Kahana, E.; Zilber, N.; Cooper, G.; Pintov, S.; Leibowitz, Y. Amyotrophic lateral sclerosis. A study of its presentation and prognosis. J. Neurol. 1985, 232, 295–300. [Google Scholar] [CrossRef]
- Norris, F.; Shepherd, R.; Denys, E.; Kwei, U.; Mukai, E.; Elias, L.; Holden, D.; Norris, H. Onset, natural history and outcome in idiopathic adult motor neuron disease. J. Neurol. Sci. 1993, 118, 48–55. [Google Scholar] [CrossRef]
- Rothstein, J.D. Current hypotheses for the underlying biology of amyotrophic lateral sclerosis. Ann. Neurol. 2009, 65 (Suppl. 1), S3–S9. [Google Scholar] [CrossRef]
- Rowland, L.P.; Shneider, N.A. Amyotrophic lateral sclerosis. N. Engl. J. Med. 2001, 344, 1688–1700. [Google Scholar] [CrossRef]
- Deng, H.X.; Bigio, E.H.; Zhai, H.; Fecto, F.; Ajroud, K.; Shi, Y.; Yan, J.; Mishra, M.; Ajroud-Driss, S.; Heller, S.; et al. Differential involvement of optineurin in amyotrophic lateral sclerosis with or without SOD1 mutations. Arch. Neurol. 2011, 68, 1057–1061. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van Blitterswijk, M.; van Vught, P.W.; van Es, M.A.; Schelhaas, H.J.; van der Kooi, A.J.; de Visser, M.; Veldink, J.H.; van den Berg, L.H. Novel optineurin mutations in sporadic amyotrophic lateral sclerosis patients. Neurobiol. Aging 2012, 33, 1016-e1. [Google Scholar] [CrossRef] [PubMed]
- Sakaguchi, T.; Irie, T.; Kawabata, R.; Yoshida, A.; Maruyama, H.; Kawakami, H. Optineurin with amyotrophic lateral sclerosis-related mutations abrogates inhibition of interferon regulatory factor-3 activation. Neurosci. Lett. 2011, 505, 279–281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gurney, M.E.; Pu, H.; Chiu, A.Y.; Dal Canto, M.C.; Polchow, C.Y.; Alexander, D.D.; Caliendo, J.; Hentati, A.; Kwon, Y.W.; Deng, H.X.; et al. Motor neuron degeneration in mice that express a human Cu,Zn superoxide dismutase mutation. Science 1994, 264, 1772–1775. [Google Scholar] [CrossRef]
- Kong, J.; Xu, Z. Massive mitochondrial degeneration in motor neurons triggers the onset of amyotrophic lateral sclerosis in mice expressing a mutant SOD1. J. Neurosci. 1998, 18, 3241–3250. [Google Scholar] [CrossRef] [PubMed]
- Wegorzewska, I.; Bell, S.; Cairns, N.J.; Miller, T.M.; Baloh, R.H. TDP-43 mutant transgenic mice develop features of ALS and frontotemporal lobar degeneration. Proc. Natl. Acad. Sci. USA 2009, 106, 18809–18814. [Google Scholar] [CrossRef] [Green Version]
- Vielhaber, S.; Kunz, D.; Winkler, K.; Wiedemann, F.R.; Kirches, E.; Feistner, H.; Heinze, H.J.; Elger, C.E.; Schubert, W.; Kunz, W.S. Mitochondrial DNA abnormalities in skeletal muscle of patients with sporadic amyotrophic lateral sclerosis. Brain 2000, 123 Pt 7, 1339–1348. [Google Scholar] [CrossRef] [Green Version]
- Duffy, L.M.; Chapman, A.L.; Shaw, P.J.; Grierson, A.J. Review: The role of mitochondria in the pathogenesis of amyotrophic lateral sclerosis. Neuropathol. Appl. Neurobiol. 2011, 37, 336–352. [Google Scholar] [CrossRef]
- Swerdlow, R.H.; Parks, J.K.; Cassarino, D.S.; Trimmer, P.A.; Miller, S.W.; Maguire, D.J.; Sheehan, J.P.; Maguire, R.S.; Pattee, G.; Juel, V.C.; et al. Mitochondria in sporadic amyotrophic lateral sclerosis. Exp. Neurol. 1998, 153, 135–142. [Google Scholar] [CrossRef]
- Hart, M.N.; Cancilla, P.A.; Frommes, S.; Hirano, A. Anterior horn cell degeneration and Bunina-type inclusions associated with dementia. Acta Neuropathol. 1977, 38, 225–228. [Google Scholar] [CrossRef]
- Mizuno, Y.; Amari, M.; Takatama, M.; Aizawa, H.; Mihara, B.; Okamoto, K. Immunoreactivities of p62, an ubiqutin-binding protein, in the spinal anterior horn cells of patients with amyotrophic lateral sclerosis. J. Neurol. Sci. 2006, 249, 13–18. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, G.E.; Gonzalez, D.M.; Monachelli, G.M.; Costa, J.J.; Nicola, A.F.; Sica, R.E. Morphological abnormalities in mitochondria of the skin of patients with sporadic amyotrophic lateral sclerosis. Arq. Neuropsiquiatr. 2012, 70, 40–44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Swerdlow, R.H. The neurodegenerative mitochondriopathies. J. Alzheimers Dis. 2009, 17, 737–751. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Catalan-Garcia, M.; Garrabou, G.; Moren, C.; Guitart-Mampel, M.; Hernando, A.; Diaz-Ramos, A.; Gonzalez-Casacuberta, I.; Juarez, D.L.; Bano, M.; Enrich-Bengoa, J.; et al. Mitochondrial DNA disturbances and deregulated expression of oxidative phosphorylation and mitochondrial fusion proteins in sporadic inclusion body myositis. Clin. Sci. 2016, 130, 1741–1751. [Google Scholar] [CrossRef] [Green Version]
- Horvath, R.; Fu, K.; Johns, T.; Genge, A.; Karpati, G.; Shoubridge, E.A. Characterization of the mitochondrial DNA abnormalities in the skeletal muscle of patients with inclusion body myositis. J. Neuropathol. Exp. Neurol. 1998, 57, 396–403. [Google Scholar] [CrossRef] [Green Version]
- Wiedemann, F.R.; Manfredi, G.; Mawrin, C.; Beal, M.F.; Schon, E.A. Mitochondrial DNA and respiratory chain function in spinal cords of ALS patients. J. Neurochem. 2002, 80, 616–625. [Google Scholar] [CrossRef] [Green Version]
- Borthwick, G.M.; Johnson, M.A.; Ince, P.G.; Shaw, P.J.; Turnbull, D.M. Mitochondrial enzyme activity in amyotrophic lateral sclerosis: Implications for the role of mitochondria in neuronal cell death. Ann. Neurol. 1999, 46, 787–790. [Google Scholar] [CrossRef]
- Fujita, K.; Yamauchi, M.; Shibayama, K.; Ando, M.; Honda, M.; Nagata, Y. Decreased cytochrome c oxidase activity but unchanged superoxide dismutase and glutathione peroxidase activities in the spinal cords of patients with amyotrophic lateral sclerosis. J. Neurosci. Res. 1996, 45, 276–281. [Google Scholar] [CrossRef]
- Shrivastava, M.; Subbiah, V. Elevated caspase 3 activity and cytosolic cytochrome c in NT2 cybrids containing amyotrophic lateral sclerosis subject mtDNA. Int. J. Neurosci. 2016, 126, 839–849. [Google Scholar] [CrossRef]
- Wilkins, H.M.; Carl, S.M.; Swerdlow, R.H. Cytoplasmic hybrid (cybrid) cell lines as a practical model for mitochondriopathies. Redox Biol. 2014, 2, 619–631. [Google Scholar] [CrossRef] [Green Version]
- Wong, P.C.; Pardo, C.A.; Borchelt, D.R.; Lee, M.K.; Copeland, N.G.; Jenkins, N.A.; Sisodia, S.S.; Cleveland, D.W.; Price, D.L. An adverse property of a familial ALS-linked SOD1 mutation causes motor neuron disease characterized by vacuolar degeneration of mitochondria. Neuron 1995, 14, 1105–1116. [Google Scholar] [CrossRef] [Green Version]
- Mattiazzi, M.; D’Aurelio, M.; Gajewski, C.D.; Martushova, K.; Kiaei, M.; Beal, M.F.; Manfredi, G. Mutated human SOD1 causes dysfunction of oxidative phosphorylation in mitochondria of transgenic mice. J. Biol. Chem. 2002, 277, 29626–29633. [Google Scholar] [CrossRef] [Green Version]
- Tafuri, F.; Ronchi, D.; Magri, F.; Comi, G.P.; Corti, S. SOD1 misplacing and mitochondrial dysfunction in amyotrophic lateral sclerosis pathogenesis. Front. Cell Neurosci. 2015, 9, 336. [Google Scholar] [CrossRef] [Green Version]
- Davis, S.A.; Itaman, S.; Khalid-Janney, C.M.; Sherard, J.A.; Dowell, J.A.; Cairns, N.J.; Gitcho, M.A. TDP-43 interacts with mitochondrial proteins critical for mitophagy and mitochondrial dynamics. Neurosci. Lett. 2018, 678, 8–15. [Google Scholar] [CrossRef]
- Huntley, M.L.; Gao, J.; Termsarasab, P.; Wang, L.; Zeng, S.; Thammongkolchai, T.; Liu, Y.; Cohen, M.L.; Wang, X. Association between TDP-43 and mitochondria in inclusion body myositis. Lab. Investig. 2019, 99, 1041–1048. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Deng, J.; Dong, J.; Liu, J.; Bigio, E.H.; Mesulam, M.; Wang, T.; Sun, L.; Wang, L.; Lee, A.Y.; et al. TDP-43 induces mitochondrial damage and activates the mitochondrial unfolded protein response. PLoS Genet. 2019, 15, e1007947. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blokhuis, A.M.; Groen, E.J.; Koppers, M.; van den Berg, L.H.; Pasterkamp, R.J. Protein aggregation in amyotrophic lateral sclerosis. Acta Neuropathol. 2013, 125, 777–794. [Google Scholar] [CrossRef] [Green Version]
- Johnson, B.S.; Snead, D.; Lee, J.J.; McCaffery, J.M.; Shorter, J.; Gitler, A.D. TDP-43 is intrinsically aggregation-prone, and amyotrophic lateral sclerosis-linked mutations accelerate aggregation and increase toxicity. J. Biol. Chem. 2009, 284, 20329–20339. [Google Scholar] [CrossRef] [Green Version]
- Nogalska, A.; D’Agostino, C.; Terracciano, C.; Engel, W.K.; Askanas, V. Impaired autophagy in sporadic inclusion-body myositis and in endoplasmic reticulum stress-provoked cultured human muscle fibers. Am. J. Pathol. 2010, 177, 1377–1387. [Google Scholar] [CrossRef]
- Vattemi, G.; Nogalska, A.; King Engel, W.; D’Agostino, C.; Checler, F.; Askanas, V. Amyloid-beta42 is preferentially accumulated in muscle fibers of patients with sporadic inclusion-body myositis. Acta Neuropathol. 2009, 117, 569–574. [Google Scholar] [CrossRef]
- Watanabe, M.; Dykes-Hoberg, M.; Culotta, V.C.; Price, D.L.; Wong, P.C.; Rothstein, J.D. Histological evidence of protein aggregation in mutant SOD1 transgenic mice and in amyotrophic lateral sclerosis neural tissues. Neurobiol. Dis. 2001, 8, 933–941. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rogers, R.S.; Tungtur, S.; Tanaka, T.; Nadeau, L.L.; Badawi, Y.; Wang, H.; Ni, H.M.; Ding, W.X.; Nishimune, H. Impaired Mitophagy Plays a Role in Denervation of Neuromuscular Junctions in ALS Mice. Front. Neurosci. 2017, 11, 473. [Google Scholar] [CrossRef] [PubMed]
- Natale, G.; Lenzi, P.; Lazzeri, G.; Falleni, A.; Biagioni, F.; Ryskalin, L.; Fornai, F. Compartment-dependent mitochondrial alterations in experimental ALS, the effects of mitophagy and mitochondriogenesis. Front. Cell Neurosci. 2015, 9, 434. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruffoli, R.; Bartalucci, A.; Frati, A.; Fornai, F. Ultrastructural studies of ALS mitochondria connect altered function and permeability with defects of mitophagy and mitochondriogenesis. Front. Cell Neurosci. 2015, 9, 341. [Google Scholar] [CrossRef] [Green Version]
- Palomo, G.M.; Granatiero, V.; Kawamata, H.; Konrad, C.; Kim, M.; Arreguin, A.J.; Zhao, D.; Milner, T.A.; Manfredi, G. Parkin is a disease modifier in the mutant SOD1 mouse model of ALS. EMBO Mol. Med. 2018, 10. [Google Scholar] [CrossRef]
- Wong, Y.C.; Holzbaur, E.L. Optineurin is an autophagy receptor for damaged mitochondria in parkin-mediated mitophagy that is disrupted by an ALS-linked mutation. Proc. Natl. Acad. Sci. USA 2014, 111, E4439–E4448. [Google Scholar] [CrossRef] [Green Version]
- Sasaki, S. Autophagy in spinal cord motor neurons in sporadic amyotrophic lateral sclerosis. J. Neuropathol. Exp. Neurol. 2011, 70, 349–359. [Google Scholar] [CrossRef]
- Almeida, S.; Gascon, E.; Tran, H.; Chou, H.J.; Gendron, T.F.; Degroot, S.; Tapper, A.R.; Sellier, C.; Charlet-Berguerand, N.; Karydas, A.; et al. Modeling key pathological features of frontotemporal dementia with C9ORF72 repeat expansion in iPSC-derived human neurons. Acta Neuropathol. 2013, 126, 385–399. [Google Scholar] [CrossRef] [Green Version]
- Webster, C.P.; Smith, E.F.; Bauer, C.S.; Moller, A.; Hautbergue, G.M.; Ferraiuolo, L.; Myszczynska, M.A.; Higginbottom, A.; Walsh, M.J.; Whitworth, A.J.; et al. The C9orf72 protein interacts with Rab1a and the ULK1 complex to regulate initiation of autophagy. EMBO J. 2016, 35, 1656–1676. [Google Scholar] [CrossRef]
- Fang, E.F. Mitophagy and NAD(+) inhibit Alzheimer disease. Autophagy 2019, 15, 1112–1114. [Google Scholar] [CrossRef] [Green Version]
- Fang, E.F.; Hou, Y.; Palikaras, K.; Adriaanse, B.A.; Kerr, J.S.; Yang, B.; Lautrup, S.; Hasan-Olive, M.M.; Caponio, D.; Dan, X.; et al. Mitophagy inhibits amyloid-beta and tau pathology and reverses cognitive deficits in models of Alzheimer’s Disease. Nat. Neurosci. 2019, 22, 401–412. [Google Scholar] [CrossRef]
- Perera, N.D.; Sheean, R.K.; Lau, C.L.; Shin, Y.S.; Beart, P.M.; Horne, M.K.; Turner, B.J. Rilmenidine promotes MTOR-independent autophagy in the mutant SOD1 mouse model of amyotrophic lateral sclerosis without slowing disease progression. Autophagy 2018, 14, 534–551. [Google Scholar] [CrossRef] [PubMed]
- Staats, K.A.; Hernandez, S.; Schonefeldt, S.; Bento-Abreu, A.; Dooley, J.; Van Damme, P.; Liston, A.; Robberecht, W.; Van Den Bosch, L. Rapamycin increases survival in ALS mice lacking mature lymphocytes. Mol. Neurodegener. 2013, 8, 31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, R.; Chen, J. Salidroside Protects Dopaminergic Neurons by Enhancing PINK1/Parkin-Mediated Mitophagy. Oxid. Med. Cell Longev. 2019, 2019, 9341018. [Google Scholar] [CrossRef] [PubMed]
- Chung, E.; Choi, Y.; Park, J.; Nah, W.; Park, J.; Jung, Y.; Lee, J.; Lee, H.; Park, S.; Hwang, S.; et al. Intracellular delivery of Parkin rescues neurons from accumulation of damaged mitochondria and pathological alpha-synuclein. Sci. Adv. 2020, 6, eaba1193. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Feng, J.; Gao, C.; Wu, M.; Du, Q.; Tsoi, B.; Wang, Q.; Yang, D.; Shen, J. Nitration of Drp1 provokes mitophagy activation mediating neuronal injury in experimental autoimmune encephalomyelitis. Free Radic. Biol. Med. 2019, 143, 70–83. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Deng, R.; Jing, X.; Chen, J.; Yang, D.; Shen, J. Acteoside ameliorates experimental autoimmune encephalomyelitis through inhibiting peroxynitrite-mediated mitophagy activation. Free Radic. Biol. Med. 2020, 146, 79–91. [Google Scholar] [CrossRef]
- Kovacs, J.R.; Li, C.; Yang, Q.; Li, G.; Garcia, I.G.; Ju, S.; Roodman, D.G.; Windle, J.J.; Zhang, X.; Lu, B. Autophagy promotes T-cell survival through degradation of proteins of the cell death machinery. Cell Death Differ. 2012, 19, 144–152. [Google Scholar] [CrossRef]
- Andreux, P.A.; Blanco-Bose, W.; Ryu, D.; Burdet, F.; Ibberson, M.; Aebischer, P.; Auwerx, J.; Singh, A.; Rinsch, C. The mitophagy activator urolithin A is safe and induces a molecular signature of improved mitochondrial and cellular health in humans. Nat. Metab. 2019, 1, 595–603. [Google Scholar] [CrossRef]
- Irie, J.; Inagaki, E.; Fujita, M.; Nakaya, H.; Mitsuishi, M.; Yamaguchi, S.; Yamashita, K.; Shigaki, S.; Ono, T.; Yukioka, H.; et al. Effect of oral administration of nicotinamide mononucleotide on clinical parameters and nicotinamide metabolite levels in healthy Japanese men. Endocr. J. 2020, 67, 153–160. [Google Scholar] [CrossRef] [Green Version]
- Tsubota, K. The first human clinical study for NMN has started in Japan. NPJ Aging Mech. Dis. 2016, 2, 16021. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoo, S.Z.; No, M.H.; Heo, J.W.; Park, D.H.; Kang, J.H.; Kim, J.H.; Seo, D.Y.; Han, J.; Jung, S.J.; Kwak, H.B. Effects of Acute Exercise on Mitochondrial Function, Dynamics, and Mitophagy in Rat Cardiac and Skeletal Muscles. Int. Neurourol. J. 2019, 23, S22–S31. [Google Scholar] [CrossRef]
- Kwon, I.; Jang, Y.; Lee, Y. Endurance Exercise-Induced Autophagy/Mitophagy Coincides with a Reinforced Anabolic State and Increased Mitochondrial Turnover in the Cortex of Young Male Mouse Brain. J. Mol. Neurosci. 2020. [Google Scholar] [CrossRef] [PubMed]
- Guan, Y.; Drake, J.C.; Yan, Z. Exercise-Induced Mitophagy in Skeletal Muscle and Heart. Exerc. Sport Sci. Rev. 2019, 47, 151–156. [Google Scholar] [CrossRef] [PubMed]
- Bernardo, T.C.; Marques-Aleixo, I.; Beleza, J.; Oliveira, P.J.; Ascensao, A.; Magalhaes, J. Physical Exercise and Brain Mitochondrial Fitness: The Possible Role Against Alzheimer’s Disease. Brain Pathol. 2016, 26, 648–663. [Google Scholar] [CrossRef] [PubMed]
- Vainshtein, A.; Tryon, L.D.; Pauly, M.; Hood, D.A. Role of PGC-1alpha during acute exercise-induced autophagy and mitophagy in skeletal muscle. Am. J. Physiol. Cell Physiol. 2015, 308, C710–C719. [Google Scholar] [CrossRef] [Green Version]
- Morris, J.K.; Vidoni, E.D.; Johnson, D.K.; Van Sciver, A.; Mahnken, J.D.; Honea, R.A.; Wilkins, H.M.; Brooks, W.M.; Billinger, S.A.; Swerdlow, R.H.; et al. Aerobic exercise for Alzheimer’s Disease: A randomized controlled pilot trial. PLoS ONE 2017, 12, e0170547. [Google Scholar] [CrossRef]
- Baker, L.D.; Frank, L.L.; Foster-Schubert, K.; Green, P.S.; Wilkinson, C.W.; McTiernan, A.; Cholerton, B.A.; Plymate, S.R.; Fishel, M.A.; Watson, G.S.; et al. Aerobic exercise improves cognition for older adults with glucose intolerance, a risk factor for Alzheimer’s Disease. J. Alzheimers Dis. 2010, 22, 569–579. [Google Scholar] [CrossRef]
- Vidoni, E.D.; Johnson, D.K.; Morris, J.K.; Van Sciver, A.; Greer, C.S.; Billinger, S.A.; Donnelly, J.E.; Burns, J.M. Dose-Response of Aerobic Exercise on Cognition: A Community-Based, Pilot Randomized Controlled Trial. PLoS ONE 2015, 10, e0131647. [Google Scholar] [CrossRef] [Green Version]
- Tomporowski, P.D. Effects of acute bouts of exercise on cognition. Acta Psychol. 2003, 112, 297–324. [Google Scholar] [CrossRef]
- Nichol, K.; Deeny, S.P.; Seif, J.; Camaclang, K.; Cotman, C.W. Exercise improves cognition and hippocampal plasticity in APOE epsilon4 mice. Alzheimers Dement. 2009, 5, 287–294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kiernan, M.C. Amyotrophic lateral sclerosis and the neuroprotective potential of exercise. J. Physiol. 2009, 587, 3759–3760. [Google Scholar] [CrossRef] [PubMed]
- De Almeida, J.P.; Silvestre, R.; Pinto, A.C.; de Carvalho, M. Exercise and amyotrophic lateral sclerosis. Neurol. Sci. 2012, 33, 9–15. [Google Scholar] [CrossRef] [PubMed]
- David, F.J.; Robichaud, J.A.; Leurgans, S.E.; Poon, C.; Kohrt, W.M.; Goldman, J.G.; Comella, C.L.; Vaillancourt, D.E.; Corcos, D.M. Exercise improves cognition in Parkinson’s disease: The PRET-PD randomized, clinical trial. Mov. Disord. 2015, 30, 1657–1663. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Armon, C. A randomized controlled trial of resistance exercise in individuals with ALS. Neurology 2008, 71, 865–866. [Google Scholar] [CrossRef] [PubMed]
- Bello-Haas, V.D.; Florence, J.M.; Kloos, A.D.; Scheirbecker, J.; Lopate, G.; Hayes, S.M.; Pioro, E.P.; Mitsumoto, H. A randomized controlled trial of resistance exercise in individuals with ALS. Neurology 2007, 68, 2003–2007. [Google Scholar] [CrossRef]
- Dalbello-Haas, V.; Florence, J.M.; Krivickas, L.S. Therapeutic exercise for people with amyotrophic lateral sclerosis or motor neuron disease. Cochrane Database Syst. Rev. 2008, CD005229. [Google Scholar] [CrossRef] [Green Version]
- Wing, R.R.; Vazquez, J.A.; Ryan, C.M. Cognitive effects of ketogenic weight-reducing diets. Int. J. Obes. Relat. Metab. Disord. 1995, 19, 811–816. [Google Scholar]
- Ari, C.; Murdun, C.; Goldhagen, C.; Koutnik, A.P.; Bharwani, S.R.; Diamond, D.M.; Kindy, M.; D’Agostino, D.P.; Kovacs, Z. Exogenous Ketone Supplements Improved Motor Performance in Preclinical Rodent Models. Nutrients 2020, 12, 2459. [Google Scholar] [CrossRef]
- Hernandez, A.R.; Hernandez, C.M.; Campos, K.; Truckenbrod, L.; Federico, Q.; Moon, B.; McQuail, J.A.; Maurer, A.P.; Bizon, J.L.; Burke, S.N. A Ketogenic Diet Improves Cognition and Has Biochemical Effects in Prefrontal Cortex That Are Dissociable From Hippocampus. Front. Aging Neurosci. 2018, 10, 391. [Google Scholar] [CrossRef]
- Brownlow, M.L.; Benner, L.; D’Agostino, D.; Gordon, M.N.; Morgan, D. Ketogenic diet improves motor performance but not cognition in two mouse models of Alzheimer’s pathology. PLoS ONE 2013, 8, e75713. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koppel, S.J.; Swerdlow, R.H. Neuroketotherapeutics: A modern review of a century-old therapy. Neurochem. Int. 2018, 117, 114–125. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Wang, Z.; Zuo, Z. Chronic intermittent fasting improves cognitive functions and brain structures in mice. PLoS ONE 2013, 8, e66069. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krikorian, R.; Shidler, M.D.; Dangelo, K.; Couch, S.C.; Benoit, S.C.; Clegg, D.J. Dietary ketosis enhances memory in mild cognitive impairment. Neurobiol. Aging 2012, 33, 425.e19–425.e27. [Google Scholar] [CrossRef] [Green Version]
Figure 1.
Mitochondrial Quality Control Mechanisms. Unfolded protein response, mitochondrial derived vesicles, and mitophagy [23,33] Created with BioRender.com.

Figure 2.
Mitophagy. (A) Non-Receptor Mediated Mitophagy or Classical Mitophagy. Activation of PINK1 leads to recruitment of ubiquitin and Parkin. Parkin ubiquitinates and phosphorylates mitochondrial proteins (such as VDAC, MFN1/2, and TOM20) and this initiates receptor adaptor protein recruitment (p62, NDP52, OPTN, TAX1BP1, and NBR1). These adaptor proteins interact with LC3 to form the autophagosome. The Vps34 and Atg5/12/16 complex facilitate autophagosome maturation, closure, and lysosome fusion [44,50] (B) Receptor Mediated Mitophagy. Mitochondrial receptor proteins (BNIP3, NIX, FUNC1, AMBRA1, PHB2, or cardiolipin) are ubiquitinated and phosphorylated. This facilitates their interaction with LC3 and GABARAP for autophagosome formation. The Vps34 and Atg5/12/16 complex facilitate autophagosome maturation, closure, and lysosome fusion [44,50]. Created with BioRender.com.
Figure 2.
Mitophagy. (A) Non-Receptor Mediated Mitophagy or Classical Mitophagy. Activation of PINK1 leads to recruitment of ubiquitin and Parkin. Parkin ubiquitinates and phosphorylates mitochondrial proteins (such as VDAC, MFN1/2, and TOM20) and this initiates receptor adaptor protein recruitment (p62, NDP52, OPTN, TAX1BP1, and NBR1). These adaptor proteins interact with LC3 to form the autophagosome. The Vps34 and Atg5/12/16 complex facilitate autophagosome maturation, closure, and lysosome fusion [44,50] (B) Receptor Mediated Mitophagy. Mitochondrial receptor proteins (BNIP3, NIX, FUNC1, AMBRA1, PHB2, or cardiolipin) are ubiquitinated and phosphorylated. This facilitates their interaction with LC3 and GABARAP for autophagosome formation. The Vps34 and Atg5/12/16 complex facilitate autophagosome maturation, closure, and lysosome fusion [44,50]. Created with BioRender.com.


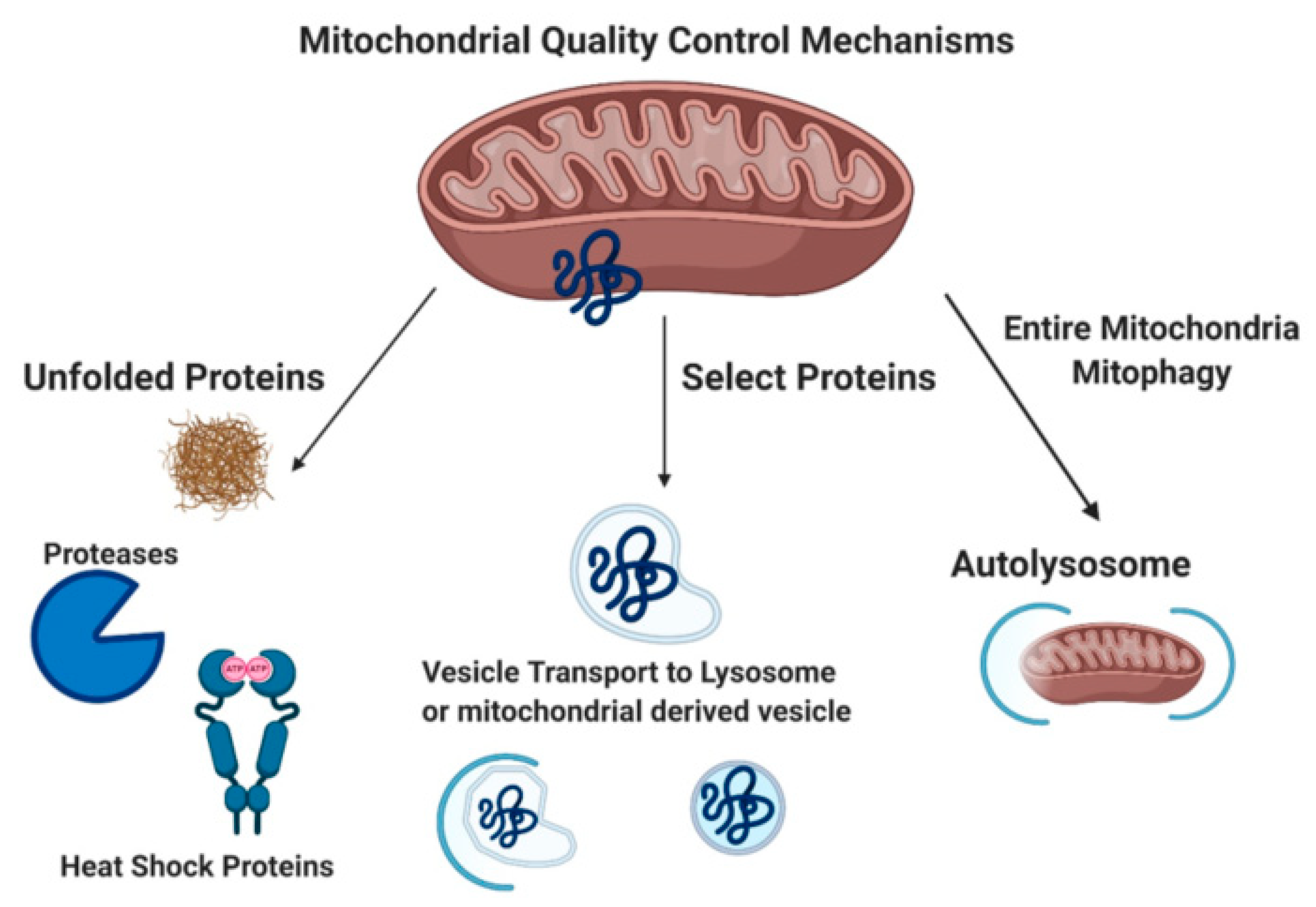{kind=link}
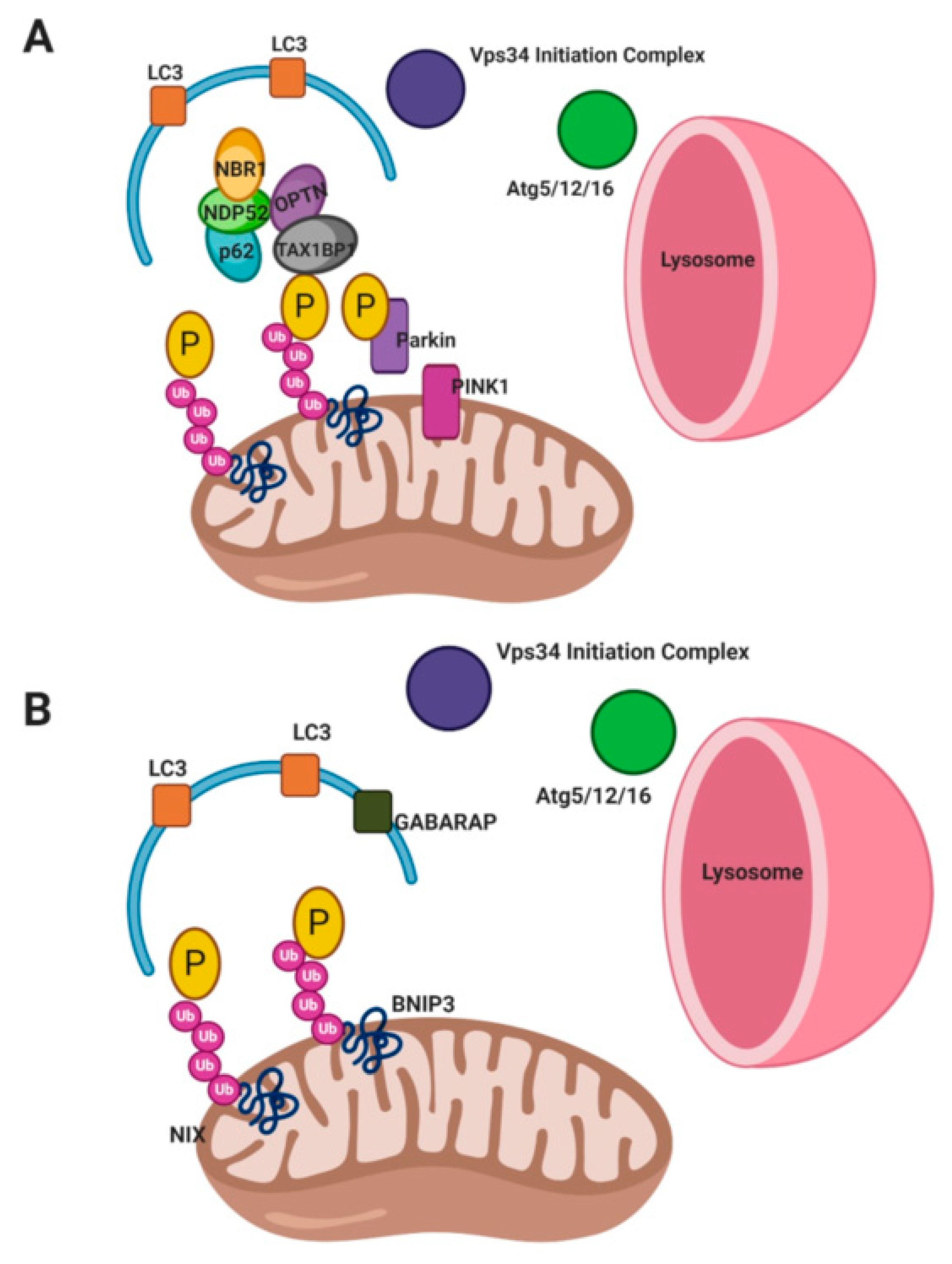{kind=link}
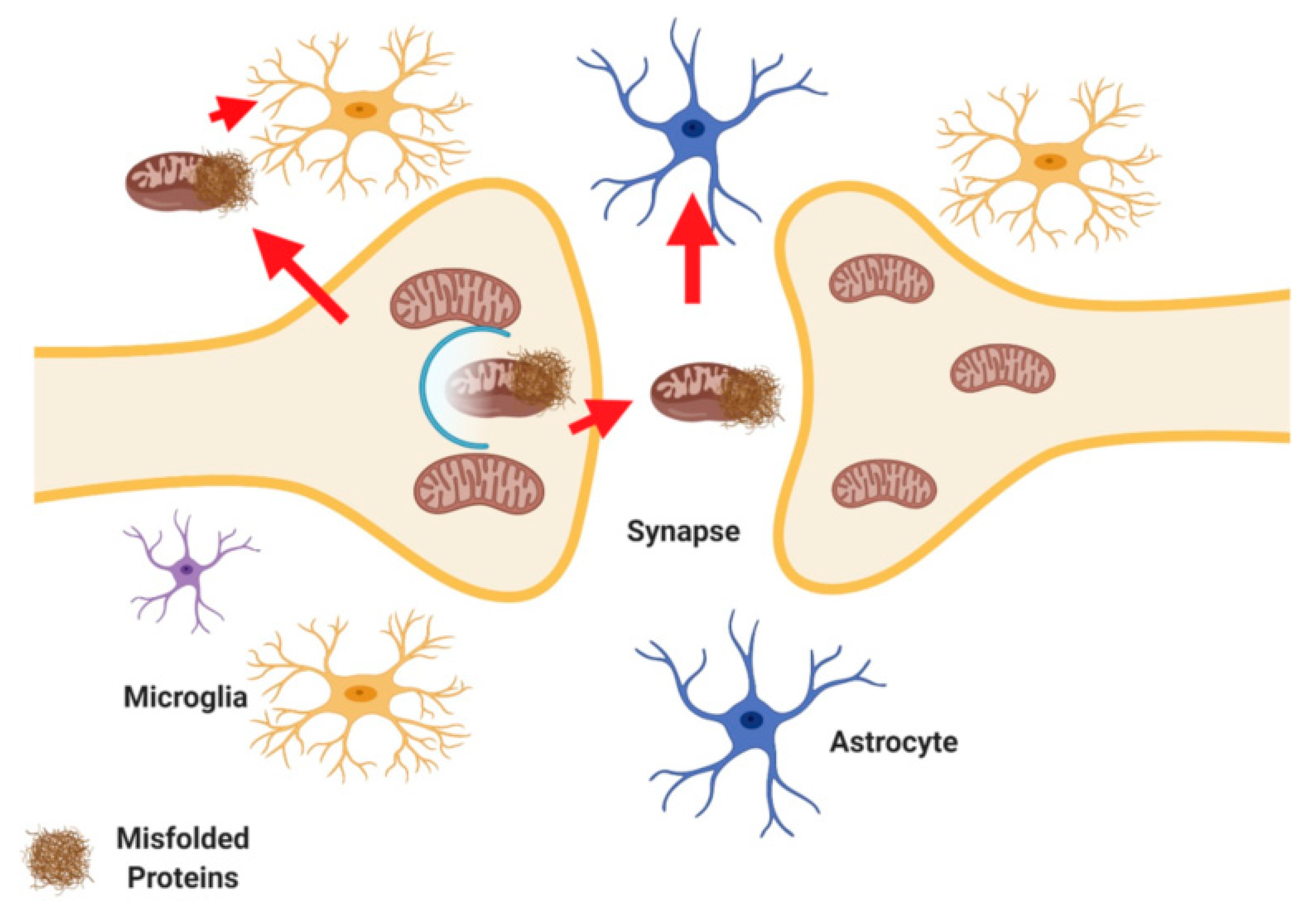{kind=link}
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Swerdlow, N.S.; Wilkins, H.M. Mitophagy and the Brain. Int. J. Mol. Sci. 2020, 21, 9661. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21249661
AMA Style
Swerdlow NS, Wilkins HM. Mitophagy and the Brain. International Journal of Molecular Sciences. 2020; 21(24):9661. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21249661
Chicago/Turabian StyleSwerdlow, Natalie S., and Heather M. Wilkins. 2020. "Mitophagy and the Brain" International Journal of Molecular Sciences 21, no. 24: 9661. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21249661
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.