c-Jun N-Terminal Kinase Inhibitors as Potential Leads for New Therapeutics for Alzheimer’s Diseases

 , , and
, , and
Abstract
:1. Introduction
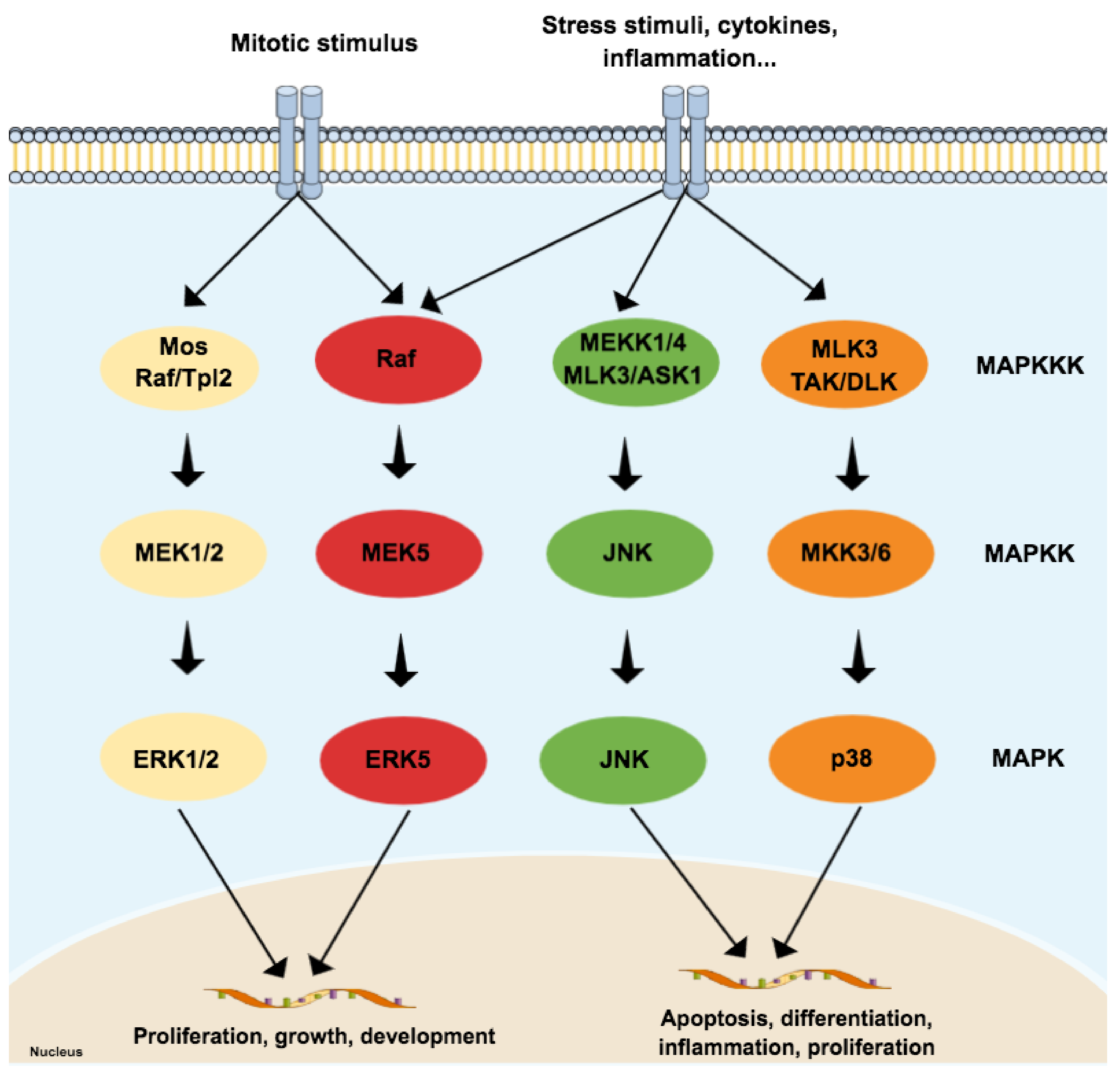
2. General Aspects of Mitogen-Activated Protein Kinases (MAPKs) Family
2.1. JNK3 (and p38), Apoptosis and AD: The Perfect Storm
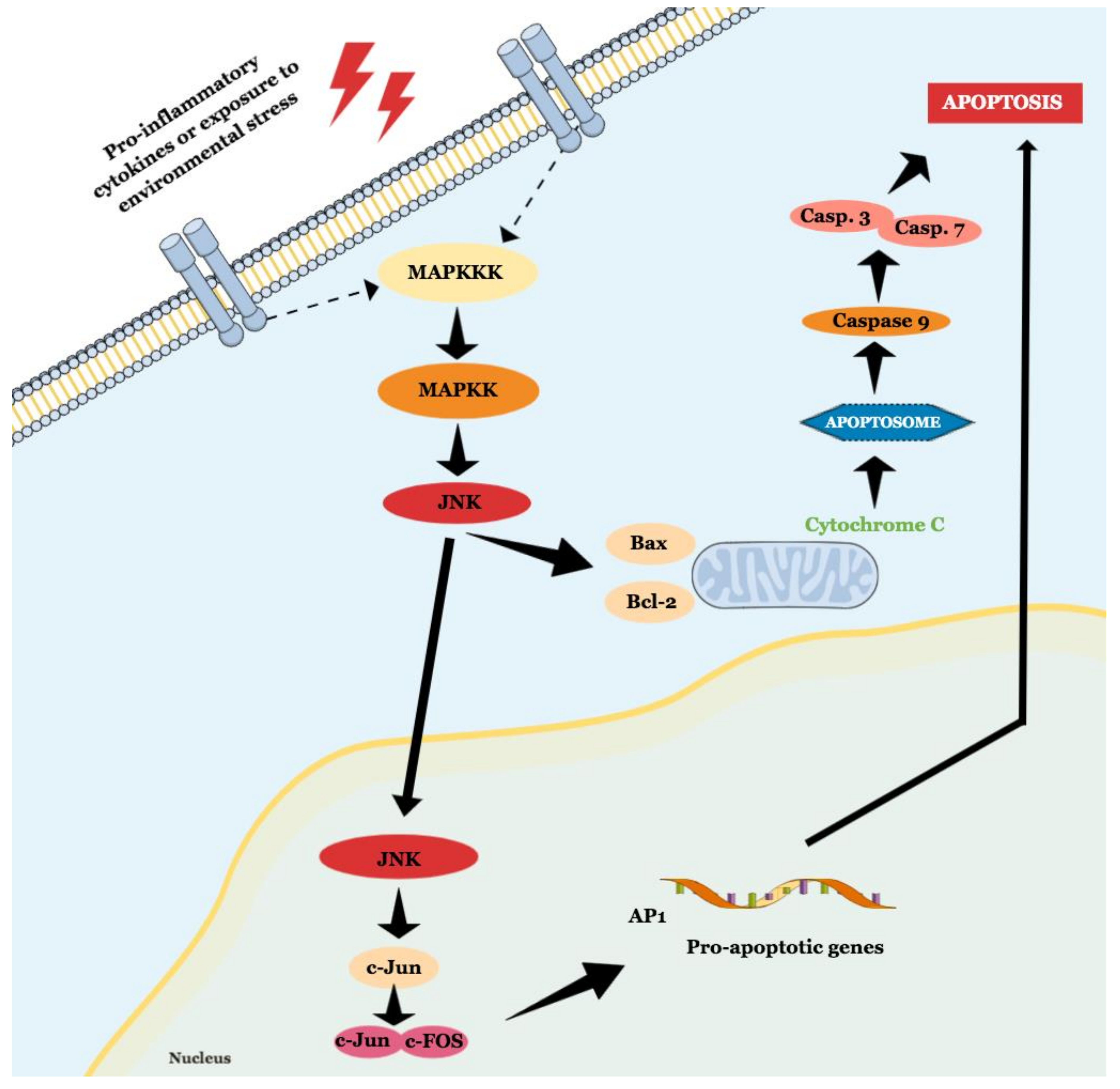
2.1.1. JNK Participates in Both Intrinsic and Extrinsic Pathways of Apoptosis
2.1.2. JNK Isoforms: Are They All the Same Thing?
2.1.3. Brain Regions Normally Affected in AD Are the Same Regions Where JNK3 Expression Was Identified. Is It a Coincidence?
2.1.4. JNK and p38 Working Together towards Chaos
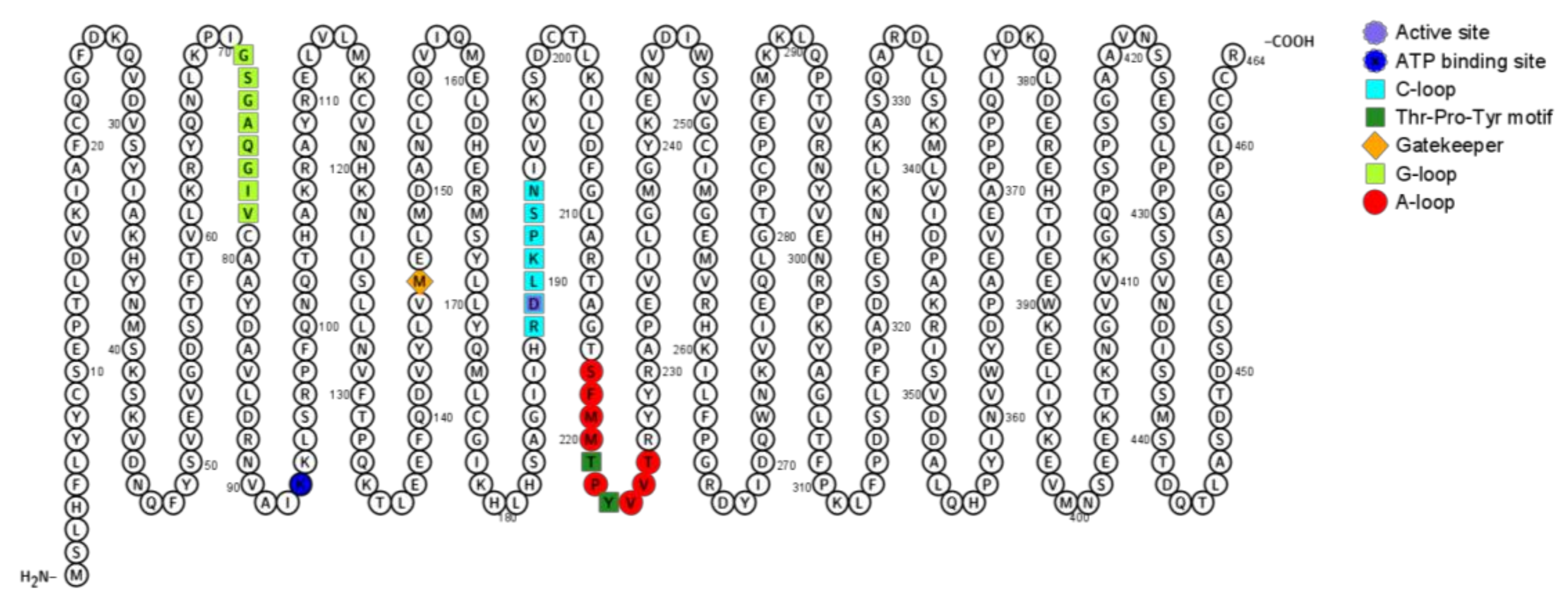
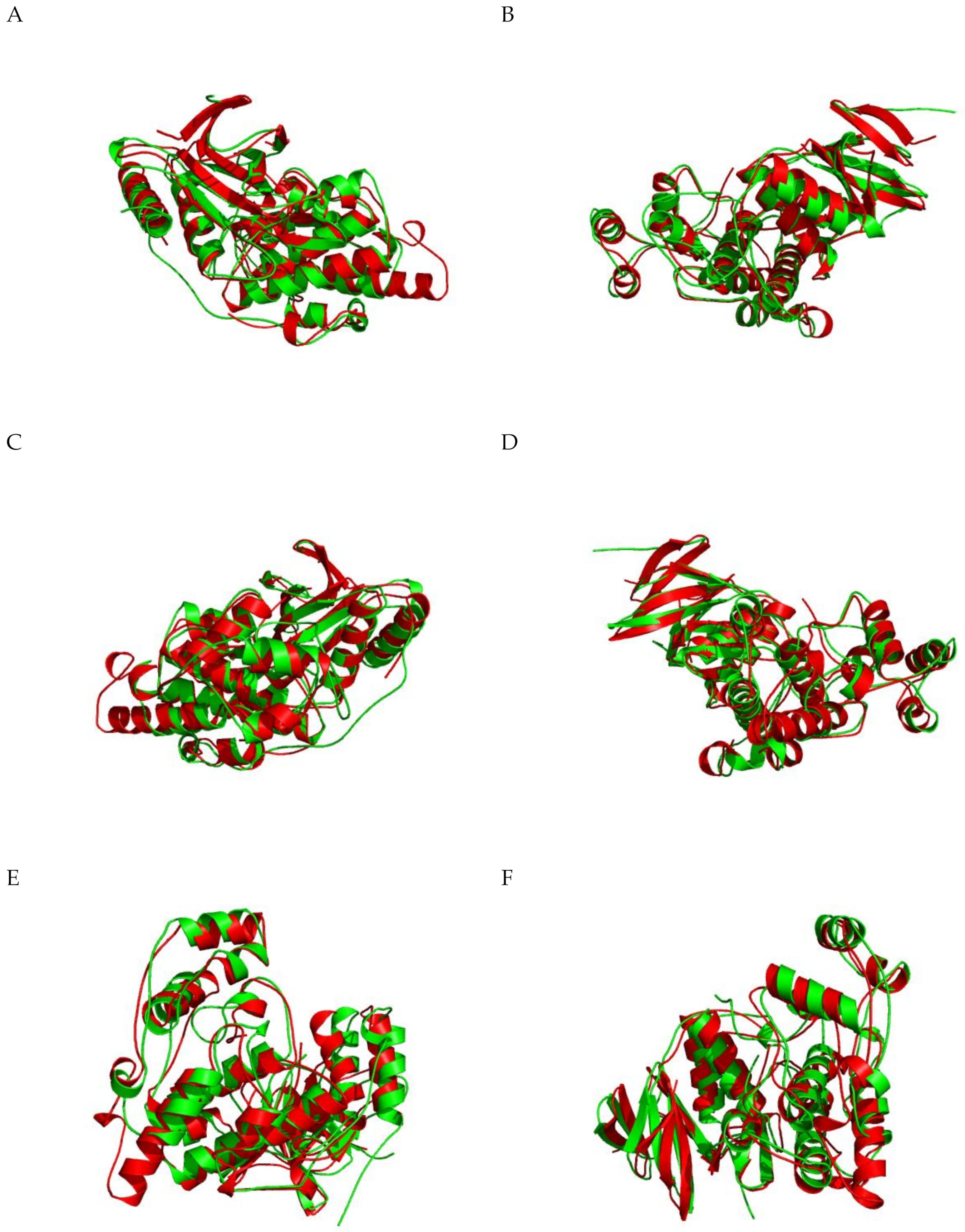
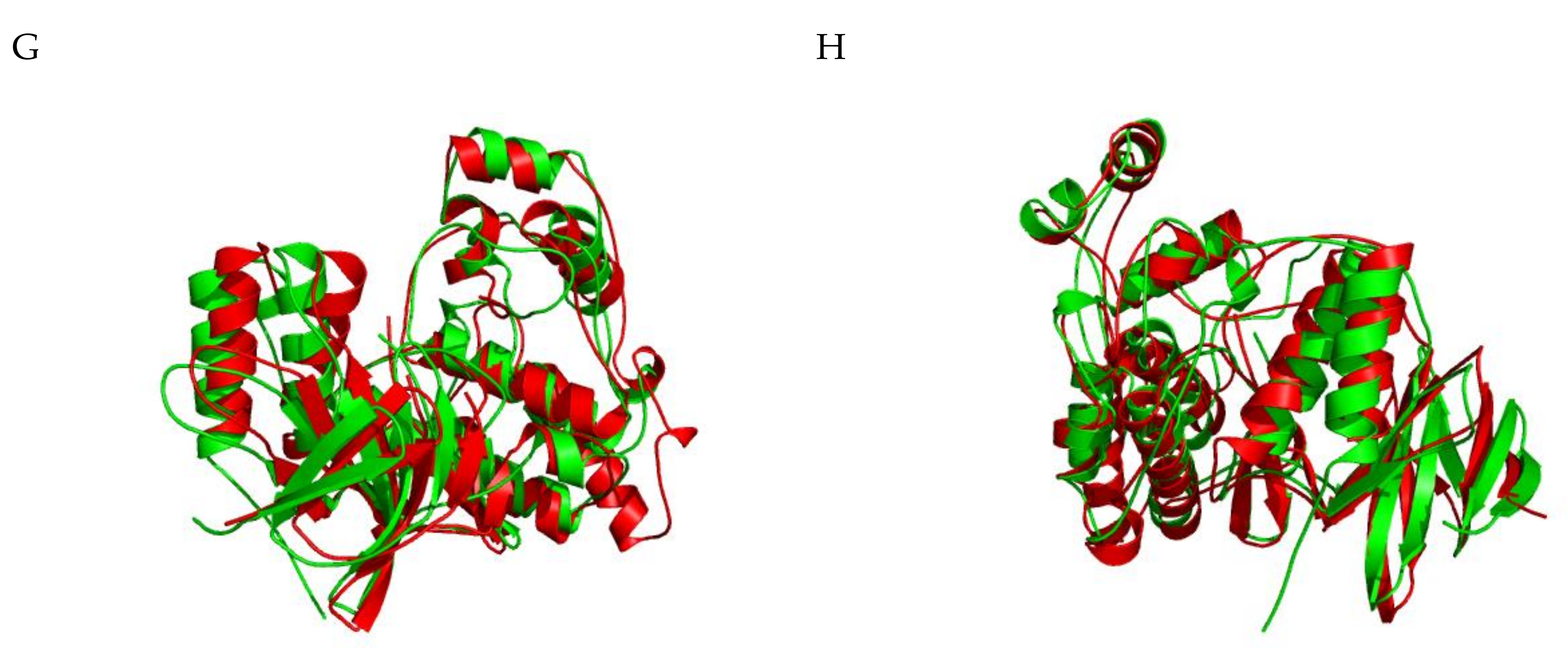
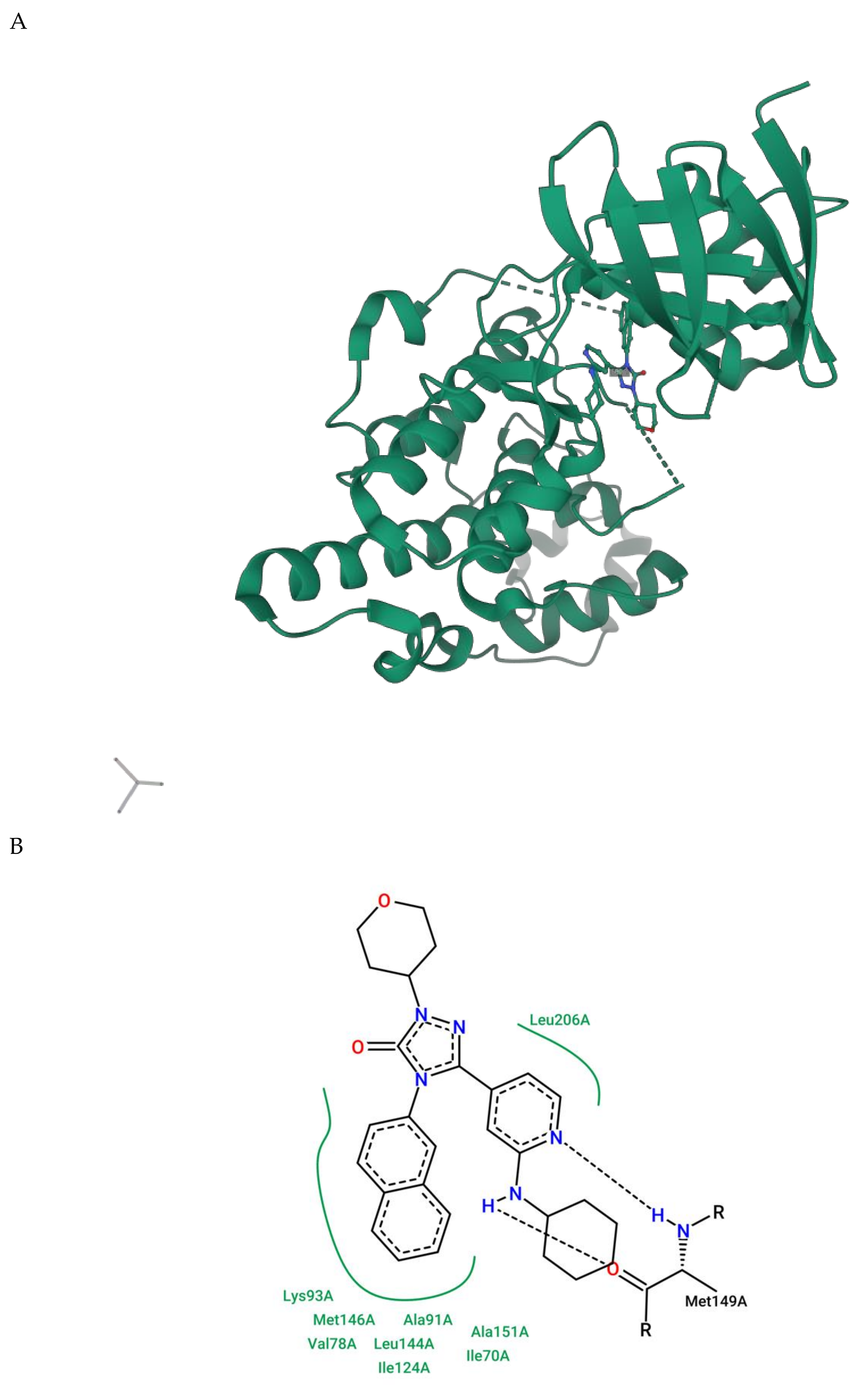
2.1.5. JNK Structure
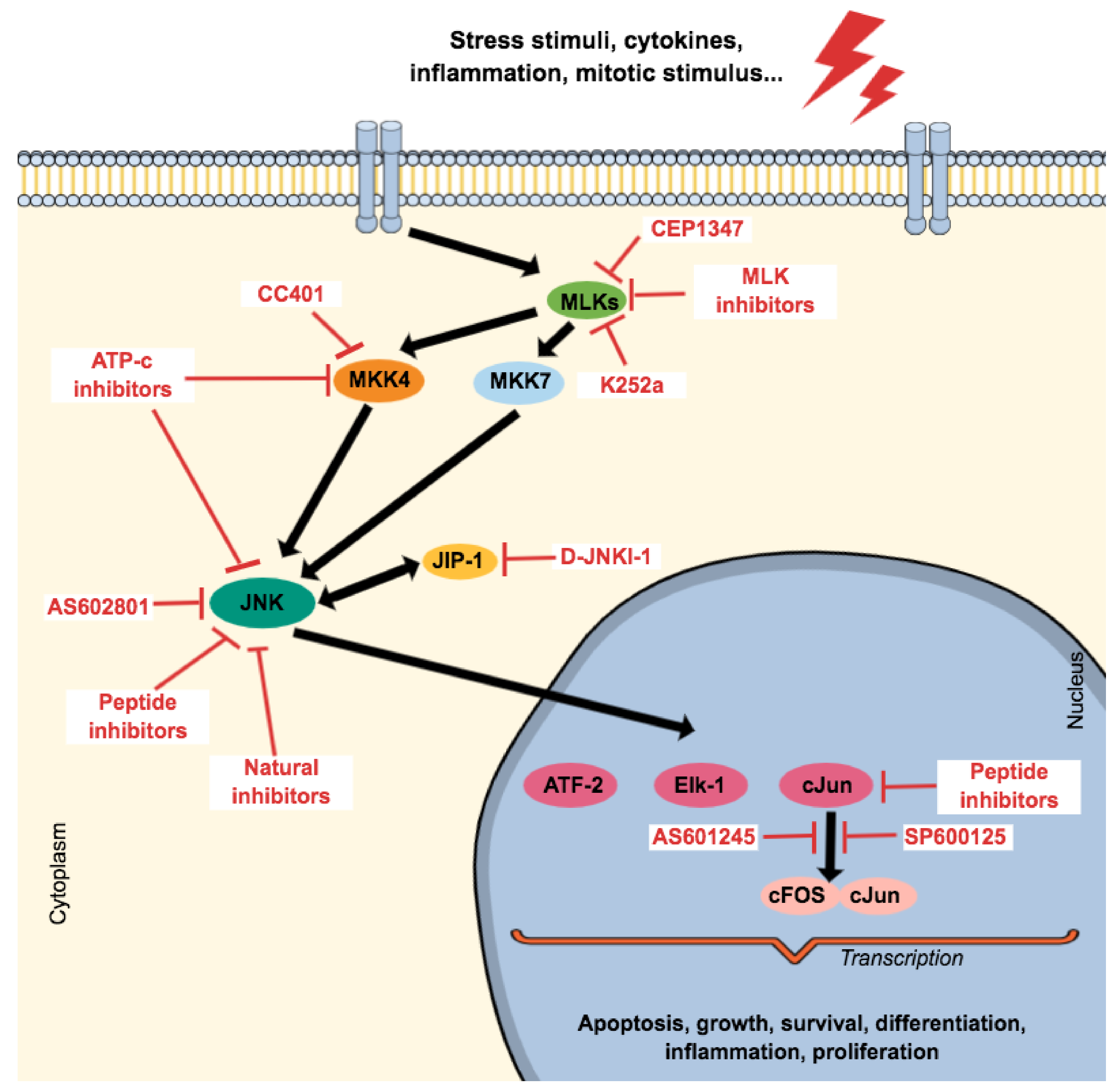
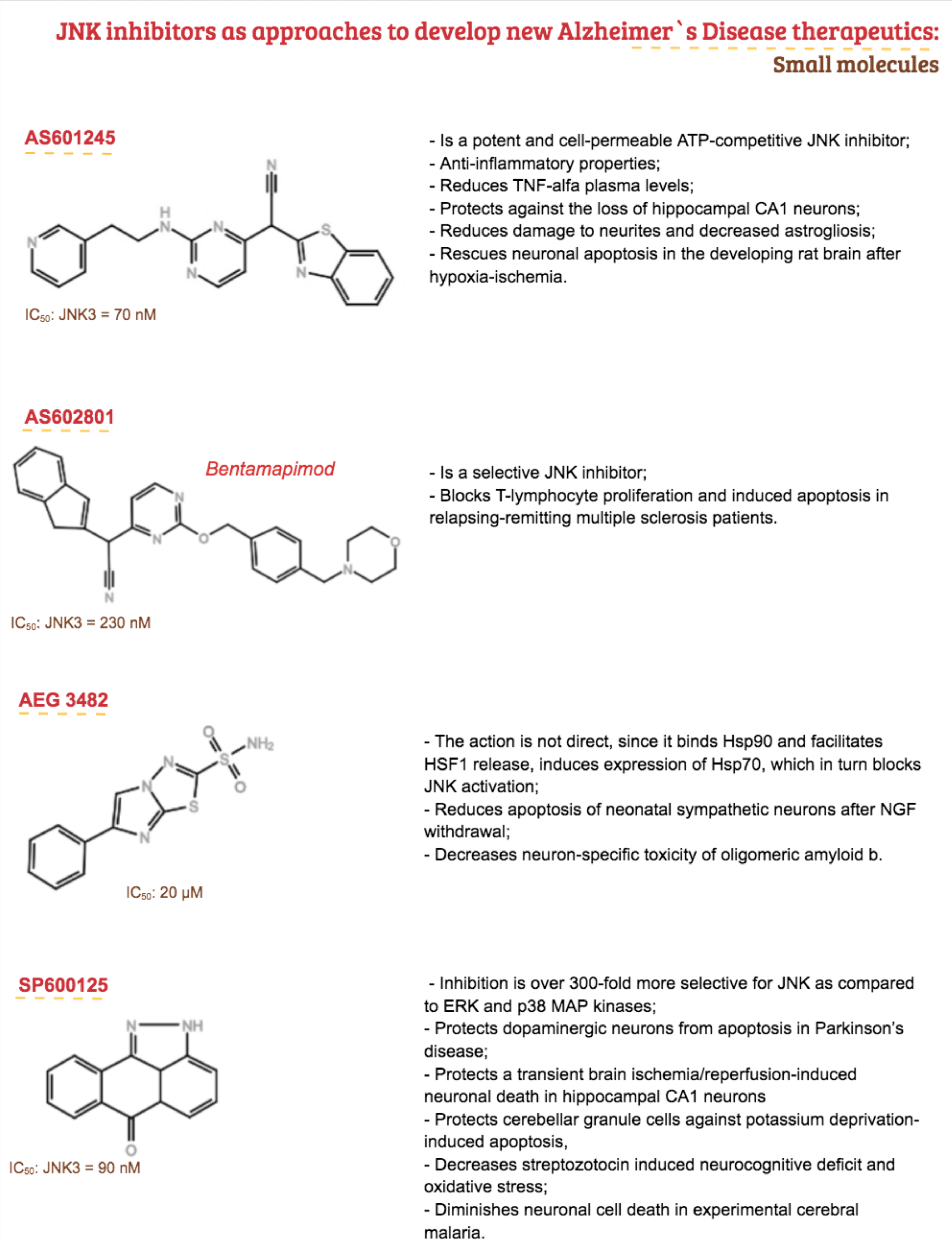
3. Therapeutic Proposal
4. Conclusions and Perspectives
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Appendix A

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Affected Area/System | Major Consequence |
|---|---|
| Entorhinal cortex | Memory impairments |
| Hippocampus | |
| Medial temporal cortex | |
| Neocortex | Deficits in higher cognitive functions, such as language, calculation, problem-solving, and judgement |
| Basal forebrain cholinergic system | Contributes to memory and attention deficits |
| Limbic cortex | Behavioral and emotional disturbances |
| Amygdala | |
| Thalamus | |
| Monoaminergic system |
References
- Small, D.H.; Cappai, R. Alois Alzheimer and Alzheimer’s disease: A centennial perspective. J. Neurochem. 2006, 99, 708–710. [Google Scholar] [CrossRef] [PubMed]
- Alzheimer, A. Über eine eigenartige Erkrankung der Hirnrinde. Allgemeine Zeitschrift fur Psychiatrie und Psychisch-gerichttliche Medizin 1907, 64, 146–148. [Google Scholar]
- Nichols, E.; Szoeke, C.E.I.; Vollset, S.E.; Abbasi, N.; Abd-Allah, F.; Abdela, J.; Aichour, M.T.E.; Akinyemi, R.O.; Alahdab, F.; Asgedom, S.W.; et al. Global, regional, and national burden of Alzheimer’s disease and other dementias, 1990–2016: A systematic analysis for the Global Burden of Disease Study 2016. Lancet Neurol. 2019, 18, 88–106. [Google Scholar] [CrossRef] [Green Version]
- Alzheimer’s Association. Alzheimer’s disease facts and figures. Alzheimer’s Dementia 2017, 13, 325–373. [Google Scholar] [CrossRef]
- Alzheimer’s Association. Alzheimer’s Disease Facts and Figures. Alzheimer’s Dementia 2019, 15, 321–387. [Google Scholar] [CrossRef]
- Cuyvers, E.; Sleegers, K. Genetic variations underlying Alzheimer’s disease: Evidence from genome-wide association studies and beyond. Lancet Neurol. 2016, 15, 857–868. [Google Scholar] [CrossRef]
- Sims, R.; Hill, M.; Williams, J. The multiplex model of the genetics of Alzheimer’s disease. Nat. Neurosci. 2020, 23, 311–322. [Google Scholar] [CrossRef]
- Castello, M.A.; Soriano, S. On the origin of Alzheimer’s disease. Trials and tribulations of the amyloid hypothesis. Ageing Res. Rev. 2014, 13, 10–12. [Google Scholar] [CrossRef]
- Vassar, R. BACE1: The β-secreiase enzyme in Alzheimer’s disease. J. Mol. Neurosci. 2004, 23, 105–113. [Google Scholar] [CrossRef]
- Ashton, N.J.; Schöll, M.; Heurling, K.; Gkanatsiou, E.; Portelius, E.; Höglund, K.; Brinkmalm, G.; Hye, A.; Blennow, K.; Zetterberg, H. Update on biomarkers for amyloid pathology in Alzheimer’s disease. Biomark. Med. 2018, 12, 799–812. [Google Scholar] [CrossRef] [Green Version]
- Braak, E.V.A.; Considerations, A. Staging of Alzheimer’s Disease-Related Neurofibrillary Changes. Neurobiol. Aging 1995, 16, 271–278. [Google Scholar] [CrossRef]
- Iqbal, K.; Zaidi, T.; Wen, G.Y.; Grundke-Iqbal, I.; Merz, P.A.; Shaikh, S.S.; Wisniewski, H.M.; Alafuzoff, I.; Winblad, B. Defective brain microtubule assembly in alzheimer’s disease. Lancet 1986, 328, 421–426. [Google Scholar] [CrossRef]
- Busche, M.A.; Hyman, B.T. Synergy between amyloid-β and tau in Alzheimer’s disease. Nat. Neurosci. 2020, 1–11. [Google Scholar] [CrossRef]
- Zetterberg, H.; Schott, J.M. Biomarkers for Alzheimer’s disease beyond amyloid and tau. Nat. Med. 2019, 25, 201–203. [Google Scholar] [CrossRef]
- Janelidze, S.; Stomrud, E.; Smith, R.; Palmqvist, S.; Mattsson, N.; Airey, D.C.; Proctor, N.K.; Chai, X.; Shcherbinin, S.; Sims, J.R.; et al. Cerebrospinal fluid p-tau217 performs better than p-tau181 as a biomarker of Alzheimer’s disease. Nat. Commun. 2020, 11, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Palmqvist, S.; Janelidze, S.; Quiroz, Y.T.; Zetterberg, H.; Lopera, F.; Stomrud, E.; Su, Y.; Chen, Y.; Serrano, G.E.; Leuzy, A.; et al. Discriminative Accuracy of Plasma Phospho-tau217 for Alzheimer Disease vs. Other Neurodegenerative Disorders. JAMA 2020. [Google Scholar] [CrossRef]
- Fyfe, I. Tau species has potential for Alzheimer disease blood test. Nat. Rev. Neurol. 2020, 16, 521. [Google Scholar] [CrossRef]
- Pillai, J.A.; Bena, J.; Bebek, G.; Bekris, L.M.; Bonner-Jackson, A.; Kou, L.; Pai, A.; Sørensen, L.; Neilsen, M.; Rao, S.M.; et al. Inflammatory pathway analytes predicting rapid cognitive decline in MCI stage of Alzheimer’s disease. Ann. Clin. Transl. Neurol. 2020, 7, 1225–1239. [Google Scholar] [CrossRef]
- Revett, T.J.; Baker, G.B.; Jhamandas, J.; Kar, S. Glutamate system, amyloid? peptides and tau protein: Functional interrelationships and relevance to Alzheimer disease pathology. J. Psychiatry Neurosci. 2013, 38, 6–23. [Google Scholar] [CrossRef] [Green Version]
- Roskoski, R., Jr. A historical overview of protein kinases and their targeted small molecule inhibitors. Pharmacol. Res. 2015, 100, 1–23. [Google Scholar] [CrossRef]
- Serrano-Pozo, A.; Qian, J.; Monsell, S.E.; Frosch, M.P.; Betensky, R.A.; Hyman, B.T. Examination of the Clinicopathologic Continuum of Alzheimer Disease in the Autopsy Cohort of the National Alzheimer Coordinating Center. J. Neuropathol. Exp. Neurol. 2013, 72, 1182–1192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pooler, A.M.; Noble, W.; Hanger, D.P. A role for tau at the synapse in Alzheimer’s disease pathogenesis. Neuropharmacology 2014, 76 Pt A, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Yarza, R.; Vela, S.; Solas, M.; Ramirez, M.J. c-Jun N-terminal Kinase (JNK) Signaling as a Therapeutic Target for Alzheimer’s Disease. Front. Pharmacol. 2016, 6, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takashima, A. Mechanism of neurodegeneration through tau and therapy for Alzheimer’s disease. J. Sport Health Sci. 2016, 5, 391–392. [Google Scholar] [CrossRef] [Green Version]
- Savage, M.J.; Lin, Y.; Ciallella, J.R.; Flood, D.G.; Scott, R.W. Activation of c-Jun N-Terminal Kinase and p38 in an Alzheimer’s Disease Model Is Associated with Amyloid Deposition. J. Neurosci. 2002, 22, 3376–3385. [Google Scholar] [CrossRef] [Green Version]
- Kondoh, K.; Nishida, E. Regulation of MAP kinases by MAP kinase phosphatases. Biochim. Biophys. Acta Mol. Cell Res. 2007, 1773, 1227–1237. [Google Scholar] [CrossRef] [Green Version]
- Galeotti, N.; Ghelardini, C. Regionally selective activation and differential regulation of ERK, JNK and p38 MAP kinase signalling pathway by protein kinase C in mood modulation. Int. J. Neuropsychopharm. 2017, 781–793. [Google Scholar] [CrossRef] [Green Version]
- Tournier, C.; Hess, P.; Yang, D.D.; Xu, J.; Turner, T.K.; Nimnual, A.; Bar-sagi, D.; Jones, S.N.; Flavell, R.A.; Davis, R.J.; et al. Requirement of JNK for stress-induced activation of the cytochrome c-mediated death pathway. Science 2000, 288, 870–874. [Google Scholar] [CrossRef]
- FDA. Drug Safety and Availability. Available online: https://www.fda.gov/drugs/drug-safety-and-availability (accessed on 8 July 2020).
- Tacrine—DrugBank. Available online: https://www.drugbank.ca/drugs/DB00382 (accessed on 8 July 2020).
- Liu, P.-P.; Xie, Y.; Meng, X.-Y.; Kang, J.-S. History and progress of hypotheses and clinical trials for Alzheimer’s disease. Signal Transduct. Target. Ther. 2019, 4, 1–22. [Google Scholar] [CrossRef]
- Parang, K.; Till, J.H.; Ablooglu, A.J.; Kohanski, R.A.; Hubbard, S.R.; Cole, P.A. Mechanism-based design of a protein kinase inhibitor. Nat. Struct. Biol. 2001, 8, 37–41. [Google Scholar] [CrossRef]
- Modi, V.; Dunbrack, R.L. A Structurally-Validated Multiple Sequence Alignment of 497 Human Protein Kinase Domains. Sci. Rep. 2019, 9, 19790. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manning, G. The Protein Kinase Complement of the Human Genome. Science 2002, 298, 1912–1934. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pei, J.J.; Braak, E.; Braak, H.; Grundke-Iqbal, I.; Iqbal, K.; Winblad, B.; Cowburn, R.F. Localization of active forms of C-jun kinase (JNK) and p38 kinase in Alzheimer’s disease brains at different stages of neurofibrillary degeneration. J. Alzheimer’s Dis. 2001, 3, 41–48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kanungo, J. DNA-PK and P38 MAPK: A Kinase Collusion in Alzheimer’s Disease? Brain Disord. Ther. 2017. [Google Scholar] [CrossRef]
- Borroto-Escuela, D.O.; Tarakanov, A.O.; Guidolin, D.; Ciruela, F.; Agnati, L.F.; Fuxe, K. Moonlighting characteristics of G protein-coupled receptors: Focus on receptor heteromers and relevance for neurodegeneration. IUBMB Life 2011, 63, 463–472. [Google Scholar] [CrossRef]
- Shi, Z.; Han, Y.; Han, X.; Zhang, K.; Chang, Y.; Hu, Z.; Qi, H. Journal of the Neurological Sciences Upstream regulators and downstream effectors of NF-κ B in Alzheimer ’ s disease. J. Neurol. Sci. 2016, 366, 127–134. [Google Scholar] [CrossRef]
- Mittal, M.; Siddiqui, M.R.; Tran, K.; Reddy, S.P.; Malik, A.B. Reactive oxygen species in inflammation and tissue injury. Antioxid. Redox Signal. 2014, 20, 1126–1167. [Google Scholar] [CrossRef] [Green Version]
- Shuaib, A.; Hartwell, A.; Kiss-Toth, E.; Holcombe, M. Multi-compartmentalisation in the MAPK signalling pathway contributes to the emergence of oscillatory behaviour and to ultrasensitivity. PLoS ONE 2016, 11. [Google Scholar] [CrossRef] [Green Version]
- Tian, T.; Harding, A. How MAP kinase modules function as robust, yet adaptable, circuits. Cell Cycle 2014, 13, 2379–2390. [Google Scholar] [CrossRef] [Green Version]
- Mishra, P.; Günther, S. New insights into the structural dynamics of the kinase JNK3. Sci. Rep. 2018, 8, 9435. [Google Scholar] [CrossRef] [Green Version]
- Rudalska, R.; Dauch, D.; Longerich, T.; McJunkin, K.; Wuestefeld, T.; Kang, T.-W.; Hohmeyer, A.; Pesic, M.; Leibold, J.; von Thun, A.; et al. In vivo RNAi screening identifies a mechanism of sorafenib resistance in liver cancer. Nat. Med. 2014, 20, 1138–1146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coffey, E.T. Nuclear and cytosolic JNK signalling in neurons. Nat. Rev. Neurosci. 2014, 15, 285–299. [Google Scholar] [CrossRef]
- Yue, J.; López, J.M. Understanding MAPK signaling pathways in apoptosis. Int. J. Mol. Sci. 2020, 21, 2346. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davis, R.J. Signal Transduction by the JNK Group of MAP Kinases. Cell 2000, 103, 239–252. [Google Scholar] [CrossRef] [Green Version]
- Kerr, J.F.R.; Wyllie, A.H.; Currie, A.R. Apoptosis: A basic biological phenomenon with wide-ranging implications in tissue kinetics. Br. J. Cancer 1972, 26, 239–257. [Google Scholar] [CrossRef] [Green Version]
- Hengartner, M.O. The biochemistry of apoptosis. Nature 2000, 407, 770–776. [Google Scholar] [CrossRef]
- Li, H.; Zhu, H.; Xu, C.J.; Yuan, J. Cleavage of BID by caspase 8 mediates the mitochondrial damage in the Fas pathway of apoptosis. Cell 1998, 94, 491–501. [Google Scholar] [CrossRef] [Green Version]
- Kale, J.; Osterlund, E.J.; Andrews, D.W. BCL-2 family proteins: Changing partners in the dance towards death. Cell Death Differ. 2018, 25, 65–80. [Google Scholar] [CrossRef] [Green Version]
- Schroeter, H.; Boyd, C.S.; Ahmed, R.; Spencer, J.P.E.; Duncan, R.F.; Rice-Evans, C.; Cadenas, E. c-Jun N-terminal kinase (JNK)-mediated modulation of brain mitochondria function: New target proteins for JNK signalling in mitochondrion-dependent apoptosis. Biochem. J. 2003, 372, 359–369. [Google Scholar] [CrossRef] [Green Version]
- Galluzzi, L.; Vitale, I.; Aaronson, S.A.; Abrams, J.M.; Adam, D.; Agostinis, P.; Alnemri, E.S.; Altucci, L.; Amelio, I.; Andrews, D.W.; et al. Molecular mechanisms of cell death: Recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ. 2018, 25, 486–541. [Google Scholar] [CrossRef]
- Wajant, H. The Fas Signaling Pathway: More Than a Paradigm. Science 2002, 296, 1635–1636. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Kim, N.; Lee, J.; Kim, S.; Hong, J.; Son, S.; Do Heo, W. Dynamic Fas signaling network regulates neural stem cell proliferation and memory enhancement. Sci. Adv. 2020, 6, eaaz9691. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stavitsky, K.; Brickman, A.M.; Scarmeas, N.; Torgan, R.L.; Tang, M.-X.; Albert, M.; Brandt, J.; Blacker, D.; Stern, Y. The progression of cognition, psychiatric symptoms, and functional abilities in dementia with Lewy bodies and Alzheimer disease. Arch. Neurol. 2006, 63, 1450–1456. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reich, A.; Spering, C.; Schulz, J.B. Death receptor Fas (CD95) signaling in the central nervous system: Tuning neuroplasticity? Trends Neurosci. 2008, 31, 478–486. [Google Scholar] [CrossRef] [PubMed]
- Schulz, E.; Wenzel, P.; Münzel, T.; Daiber, A. Mitochondrial redox signaling: Interaction of mitochondrial reactive oxygen species with other sources of oxidative stress. Antioxid. Redox Signal. 2014, 20, 308–324. [Google Scholar] [CrossRef]
- Kandel, E.R. The molecular biology of memory: cAMP, PKA, CRE, CREB-1, CREB-2, and CPEB. Mol. Brain. 2012, 5, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Raman, M.; Chen, W.; Cobb, M.H. Differential regulation and properties of MAPKs. Oncogene 2007, 3100–3112. [Google Scholar] [CrossRef] [Green Version]
- Dhanasekaran, D.N.; Johnson, G.L. MAPKs: Function, regulation, role in cancer and therapeutic targeting. Oncogene 2007, 3097–3099. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, E.K.; Choi, E. Biochimica et Biophysica Acta Pathological roles of MAPK signaling pathways in human diseases. BBA Mol. Basis Dis. 2010, 1802, 396–405. [Google Scholar] [CrossRef] [Green Version]
- Ardito, F.; Giuliani, M.; Perrone, D.; Troiano, G.; Muzio, L.L.O. The crucial role of protein phosphorylation in cell signaling and its use as targeted therapy (Review). Int. J. Mol. Med. 2017, 271–280. [Google Scholar] [CrossRef] [Green Version]
- Kandel, E.R.; Dudai, Y.; Mayford, M.R. Review The Molecular and Systems Biology of Memory. Cell 2014, 157, 163–186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cohen, P.; Alessi, D.R. Kinase Drug Discovery—What’s Next in the Field? ASC Chem. Biol. 2014, 8, 96–104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, L.; Karin, M. Mammalian MAP kinase signalling cascades. Nature 2001, 410, 37–40. [Google Scholar] [CrossRef]
- Misheva, M.; Bogoyevitch, M.A. JNK Signaling: Regulation and Functions Based on Complex Protein-Protein Partnerships. Microbiol. Mol. Biol. Rev. 2016, 80, 793–835. [Google Scholar] [CrossRef]
- Yasuda, J.U.N.; Whitmarsh, A.J.; Cavanagh, J.; Sharma, M.; Davis, R.J. The JIP Group of Mitogen-Activated Protein Kinase Scaffold Proteins. Mol. Cell. Biol. 1999, 19, 7245–7254. [Google Scholar] [CrossRef] [Green Version]
- Whitmarsh, A.J. The JIP family of MAPK scaffold proteins. Biochem. Soc. Trans. 2006, 34, 828–832. [Google Scholar] [CrossRef]
- Okazawa, H.; Estus, S. The JNK/c-Jun cascade and Alzheimer’s disease. Am. J. Alzheimer’s Dis. Other Dement. 2002, 17, 79–88. [Google Scholar] [CrossRef]
- Xu, Z.; Kukekov, N.V.; Greene, L.A. POSH acts as a scaffold for a multiprotein complex that mediates JNK activation in apoptosis. EMBO J. 2003, 22, 252–261. [Google Scholar] [CrossRef] [Green Version]
- Zhang, F.; Yu, J.; Yang, T.; Xu, D.; Chi, Z.; Xia, Y.; Xu, Z. A Novel c-Jun N-terminal Kinase (JNK) Signaling Complex Involved in Neuronal Migration during Brain Development. J. Biol. Chem. 2016, 291, 11466–11475. [Google Scholar] [CrossRef] [Green Version]
- Zhou, X.; Li, Y.; Shi, X.; Ma, C. An overview on therapeutics attenuating amyloid β level in Alzheimer’s disease: Targeting neurotransmission, inflammation, oxidative stress and enhanced cholesterol levels. Am. J. Transl. Res. 2016, 8, 246–269. [Google Scholar]
- Kim, S.; Chin, Y.; Cho, J. Protection of Cultured Cortical Neurons by Luteolin against Oxidative Damage through Inhibition of Apoptosis and Induction of Heme. Biol. Pharm. Bull. 2017, 40, 256–265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gupta, S.; Barrett, T.; Whitmarsh, A.J.; Cavanagh, J.; Sluss, H.K.; Dérijard, B.; Davis, R.J. Selective interaction of JNK protein kinase isoforms with transcription factors. EMBO J. 1996, 15, 2760–2770. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sluss, H.K.; Barrett, T.; Dérijard, B.; Davis, R.J. Signal transduction by tumor necrosis factor mediated by JNK protein kinases. Mol. Cell. Biol. 1994, 14, 8376–8384. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kallunki, T.; Su, B.; Tsigelny, I.; Sluss, H.K.; Derijard, B.; Moore, G.; Davis, R.; Karin, M. JNK2 Contains a Specificity-Determining Region Responsible for Efficient c-Jun Binding and Phosphorylation. Genes Dev. 1994, 8, 2996–3007. [Google Scholar] [CrossRef] [Green Version]
- Ota, T.; Suzuki, Y.; Nishikawa, T.; Otsuki, T.; Sugiyama, T.; Irie, R.; Wakamatsu, A.; Hayashi, K.; Sato, H.; Nagai, K.; et al. Complete sequencing and characterization of 21,243 full-length human cDNAs. Nat. Genet. 2004, 36, 40–45. [Google Scholar] [CrossRef] [PubMed]
- Saito, T.; Sadoshima, J. Searching and navigating UniProt databases. Curr. Protoc. Bioinform. 2016, 116, 1477–1490. [Google Scholar] [CrossRef]
- Shoichet, S.A.; Laurence, A.E.; Ae, D.; Hagens, O.; Waetzig, V.; Corinna, A.E.; Ae, M.; Herdegen, T.; Schweiger, S.; Bernard, A.E.; et al. Original investigation Truncation of the CNS-expressed JNK3 in a patient with a severe developmental epileptic encephalopathy. Hum. Genet. 2006. [Google Scholar] [CrossRef]
- Ahmadi, S.; Khaledi, S. Anxiety in rats with bile duct ligation is associated with activation of JNK3 mitogen-activated protein kinase in the hippocampus. Metab. Brain Dis. 2020. [Google Scholar] [CrossRef]
- Haeusgen, W.; Boehm, R.; Zhao, Y.; Herdegen, T.; Waetzig, V. Neuroscience forefront review specific activities of individual c-Jun N-terminal. NSC 2009, 161, 951–959. [Google Scholar] [CrossRef]
- Nakano, R.; Nakayama, T.; Sugiya, H. Biological Properties of JNK3 and Its Function in Neurons, Astrocytes, Pancreatic β-Cells and Cardiovascular Cells. Cells 2020, 9, 1802. [Google Scholar] [CrossRef]
- Mohit, A.A.; Martin, J.H.; Miller, C.A. p493F12 Kinase: A Novel MAP Kinase Expressed in a Subset of Neurons in the Human Nervous System. Neuron 1995, 14, 67–78. [Google Scholar] [CrossRef] [Green Version]
- Arnold, S.E.; Hyman, B.T.; Flory, J.; Damasio, A.R.; Van Hoesen, G.W. The topographical and neuroanatomical distribution of neurofibrillary tangles and neuritic plaques in the cerebral cortex of patients with alzheimer’s disease. Cereb. Cortex 1991, 1, 103–116. [Google Scholar] [CrossRef] [PubMed]
- Firth, N.C.; Primativo, S.; Marinescu, R.-V.; Shakespeare, T.J.; Suarez-Gonzalez, A.; Lehmann, M.; Carton, A.; Ocal, D.; Pavisic, I.; Paterson, R.W.; et al. Longitudinal neuroanatomical and cognitive progression of posterior cortical atrophy. Brain 2019. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bruen, P.D.; Mcgeown, W.J.; Shanks, M.F.; Venneri, A. Neuroanatomical correlates of neuropsychiatric symptoms in Alzheimer’s disease. Brain 2008. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Killick, R.; Ribe, E.M.; Malik, B.; Hooper, C.; Fernandes, C.; Dobson, R.; Nolan, P.M.; Lourdusamy, A.; Furney, S.; Lin, K.; et al. Clusterin regulates b-amyloid toxicity via Dickkopf-1-driven induction of the wnt-PCP-JNK pathway. Mol. Psychiatry 2014, 88–98. [Google Scholar] [CrossRef]
- Neurochemistry, J.O.F. Cell Biology and Physiology Division; CSIR-Indian Institute of Chemical Biology: Kolkata, India, 2015; pp. 1091–1103. [Google Scholar] [CrossRef]
- Zhu, X.; Castellani, R.J.; Takeda, A.; Nunomura, A.; Atwood, C.S.; Perry, G.; Smith, M.A. Differential activation of neuronal ERK, JNK/SAPK and p38 in Alzheimer disease: The ‘two hit’ hypothesis. Mech. Ageing Dev. 2001, 123, 39–46. [Google Scholar] [CrossRef]
- Gourmaud, S.; Paquet, C.; Dumurgier, J.; Pace, C.; Bouras, C.; Gray, F.; Laplanche, J.; Meurs, E.F.; Mouton-liger, F.; Hugon, J. Increased levels of cerebrospinal fluid JNK3 associated with amyloid pathology: Links to cognitive decline. J. Psychiatry Neurosci. JPN 2015, 40, 151–161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takahashi, R.H.; Nagao, T.; Gouras, G.K. Plaque formation and the intraneuronal accumulation of b-amyloid in Alzheimer’s disease. Pathol. Int. 2017, 185–193. [Google Scholar] [CrossRef]
- Yoon, S.O.; Park, D.J.; Ryu, J.C.; Ozer, H.G.; Tep, C.; Shin, Y.J.; Lim, T.H.; Pastorino, L.; Kunwar, A.J.; Walton, J.C.; et al. Article JNK3 Perpetuates Metabolic Stress Induced by A b Peptides. Neuron 2012, 75, 824–837. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aupperle, P.M. Navigating patients and caregivers through the course of Alzheimer’s disease. J. Clin. Psychiatry 2006, 67, 8–14. [Google Scholar]
- Brokaw, D.; Piras, I.; Mastroeni, D.; Weisenberger, D.; Nolz, J.; Delvaux, E.; Serrano, G.; Beach, T.; Huentelman, M.; Coleman, P. Cell Death and Survival Pathways in Alzheimer’s Disease: An Integrative Hypothesis Testing Approach Utilizing-Omic Datasets. Neurobiol. Aging 2020, 95, 15–25. [Google Scholar] [CrossRef] [PubMed]
- Najm, R.; Zalocusky, K.A.; Zilberter, M.; Yoon, S.Y.; Hao, Y.; Koutsodendris, N.; Nelson, M.; Rao, A.; Taubes, A.; Jones, E.A.; et al. In Vivo Chimeric Alzheimer’s Disease Modeling of Apolipoprotein E4 Toxicity in Human Neurons. Cell Rep. 2020, 32, 107962. [Google Scholar] [CrossRef] [PubMed]
- Waetzig, V.; Czeloth, K.; Hidding, U.; Mielke, K.; Kanzow, M.; Brecht, S.; Goetz, M.; Lucius, R.; Herdegen, T.; Hanisch, U.R. c-Jun N-terminal kinases (JNKs) mediate pro-inflammatory actions of microglia. GLIA 2005, 50, 235–246. [Google Scholar] [CrossRef] [PubMed]
- Hidding, U.; Mielke, K.; Waetzig, V.; Brecht, S.; Hanisch, U.; Behrens, A.; Wagner, E.; Herdegen, T. The c-Jun N-terminal kinases in cerebral microglia: Immunological functions in the brain. Biochem. Pharmacol. 2002, 64, 781–788. [Google Scholar] [CrossRef]
- Christensen, D.P.; Ejlerskov, P.; Rasmussen, I.; Vilhardt, F. Reciprocal signals between microglia and neurons regulate α-synuclein secretion by exophagy through a neuronal cJUN-N-terminal kinase-signaling axis. J. Neuroinflamm. 2016, 13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Agarwal, D.; Sandor, C.; Volpato, V.; Caffrey, T.M.; Monzón-Sandoval, J.; Bowden, R.; Alegre-Abarrategui, J.; Wade-Martins, R.; Webber, C. A single-cell atlas of the human substantia nigra reveals cell-specific pathways associated with neurological disorders. Nat. Commun. 2020, 11, 4183. [Google Scholar] [CrossRef]
- Fan, Z.; Brooks, D.J.; Okello, A.; Edison, P. An early and late peak in microglial activation in Alzheimer’s disease trajectory. Brain 2017, 140, 792–803. [Google Scholar] [CrossRef]
- Hamelin, L.; Lagarde, J.; Dorothée, G.; Leroy, C.; Labit, M.; Comley, R.A.; De Souza, L.C.; Corne, H.; Dauphinot, L.; Bertoux, M.; et al. Early and protective microglial activation in Alzheimer’s disease: A prospective study using 18F-DPA-714 PET imaging. Brain 2016, 139, 1252–1264. [Google Scholar] [CrossRef] [Green Version]
- Schiller, M.; Ben-Shaanan, T.L.; Rolls, A. Neuronal regulation of immunity: Why, how and where? Nat. Rev. Immunol. 2020. [Google Scholar] [CrossRef]
- Shi, Y.; Holtzman, D.M. Interplay between innate immunity and Alzheimer disease: APOE and TREM2 in the spotlight. Nat. Rev. Immunol. 2018, 18, 759–772. [Google Scholar] [CrossRef]
- Kracht, L.; Borggrewe, M.; Eskandar, S.; Brouwer, N.; de Sousa, L.S.M.C.; Laman, J.D.; Scherjon, S.A.; Prins, J.R.; Kooistra, S.M.; Eggen, B.J.L. Human fetal microglia acquire homeostatic immune-sensing properties early in development. Science 2020, 369, 530–537. [Google Scholar] [CrossRef] [PubMed]
- Perdiguero, E.G.; Klapproth, K.; Schulz, C.; Busch, K.; Azzoni, E.; Crozet, L.; Garner, H.; Trouillet, C.; De Bruijn, M.F.; Geissmann, F.; et al. Tissue-resident macrophages originate from yolk-sac-derived erythro-myeloid progenitors. Nature 2015, 518, 547–551. [Google Scholar] [CrossRef] [PubMed]
- Keren-shaul, H.; Spinrad, A.; Weiner, A.; Colonna, M.; Schwartz, M.; Amit, I.; Keren-shaul, H.; Spinrad, A.; Weiner, A.; Matcovitch-natan, O.; et al. Article A Unique Microglia Type Associated with Restricting Development of Alzheimer’s Disease. Cell 2017, 169, 1276–1290.e17. [Google Scholar] [CrossRef]
- Maezawa, I.; Zimin, P.I.; Wulff, H.; Jin, L.W. Amyloid-? protein oligomer at low nanomolar concentrations activates microglia and induces microglial neurotoxicity. J. Biol. Chem. 2011, 286, 3693–3706. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maphis, N.; Xu, G.; Kokiko-Cochran, O.N.; Jiang, S.; Cardona, A.; Ransohoff, R.M.; Lamb, B.T.; Bhaskar, K. Reactive microglia drive tau pathology and contribute to the spreading of pathological tau in the brain. Brain 2015, 138, 1738–1755. [Google Scholar] [CrossRef] [PubMed]
- Heneka, M.T.; Carson, M.J.; El Khoury, J. Neuroinflammation in Alzheimer’s disease. Lancet Neurol. 2015, 14, 388–405. [Google Scholar] [CrossRef] [Green Version]
- Subhramanyam, C.S.; Wang, C.; Hu, Q.; Dheen, S.T. Microglia-mediated neuroinflammation in neurodegenerative diseases. Semin. Cell Dev. Biol. 2019, 94, 112–120. [Google Scholar] [CrossRef]
- Ledo, J.H.; Liebmann, T.; Zhang, R.; Chang, J.C.; Azevedo, E.P.; Wong, E.; Silva, H.M.; Troyanskaya, O.G.; Bustos, V.; Greengard, P. Presenilin 1 phosphorylation regulates amyloid-β degradation by microglia. Mol. Psychiatry 2020, 1–16. [Google Scholar] [CrossRef]
- Harry, G.J. Microglia during development and aging. Pharmacol. Ther. 2013, 139, 313–326. [Google Scholar] [CrossRef]
- Hickman, S.E.; Allison, E.K.; El Khoury, J. Microglial dysfunction and defective β-amyloid clearance pathways in aging alzheimer’s disease mice. J. Neurosci. 2008, 28, 8354–8360. [Google Scholar] [CrossRef]
- Tang, Y.; Le, W. Differential Roles of M1 and M2 Microglia in Neurodegenerative Diseases. Mol. Neurobiol. 2016, 53, 1181–1194. [Google Scholar] [CrossRef] [PubMed]
- Ransohoff, R.M. A polarizing question: Do M1 and M2 microglia exist. Nat. Neurosci. 2016, 19, 987–991. [Google Scholar] [CrossRef] [PubMed]
- Lue, L.F.; Beach, T.G. Walker Alzheimer’s Disease Research Using Human Microglia. Cells 2019, 8, 838. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodríguez-Gómez, J.A.; Kavanagh, E.; Engskog-Vlachos, P.; Engskog, M.K.R.; Herrera, A.J.; Espinosa-Oliva, A.M.; Joseph, B.; Hajji, N.; Venero, J.L.; Burguillos, M.A. Microglia: Agents of the CNS Pro-Inflammatory Response. Cells 2020, 9, 1717. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Liu, L.; Barger, S.W.; Griffin, W.S. Interleukin-1 mediates pathological effects of microglia on tau phosphorylation and on synaptophysin synthesis in cortical neurons through a p38-MAPK pathway. J. Neurosci. 2003, 23, 1605–1611. [Google Scholar] [CrossRef] [Green Version]
- Zarubin, T.; Han, J. Activation and signaling of the p38 MAP kinase pathway. Cell Res. 2005, 15, 11–18. [Google Scholar] [CrossRef] [Green Version]
- Enslen, H.; Brancho, D.M.; Davis, R.J. Molecular determinants that mediate selective activation of p38 MAP kinase isoforms. EMBO J. 2000, 19, 1301–1311. [Google Scholar] [CrossRef] [Green Version]
- Sun, A.; Liu, M.; Nguyen, X.V.; Bing, G. p38 MAP kinase is activated at early stages in Alzheimer’s disease brain. Exp. Neurol. 2003, 183, 394–405. [Google Scholar] [CrossRef]
- Hensley, K.; Floyd, R.A.; Zheng, N.Y.; Nael, R.; Robinson, K.A.; Nguyen, X.; Pye, Q.N.; Stewart, C.A.; Geddes, J.; Markesbery, W.R.; et al. p38 Kinase is activated in the Alzheimer’s disease brain. J. Neurochem. 1999, 72, 2053–2058. [Google Scholar] [CrossRef]
- Zhu, X.; Rottkamp, C.A.; Boux, H.; Takeda, A.; Perry, G.; Smith, M.A. Activation of p38 kinase links tau phosphorylation, oxidative stress, and cell cycle-related events in Alzheimer disease. J. Neuropathol. Exp. Neurol. 2000, 59, 880–888. [Google Scholar] [CrossRef] [Green Version]
- Ugbode, C.; Fort-Aznar, L.; Evans, G.; Chawla, S.; Sweeney, S. JNK Signalling Mediates Context-Dependent Responses to Reactive Oxygen Species in Neurons. SSRN Electron. J. 2019. [Google Scholar] [CrossRef]
- Kheiri, G.; Dolatshahi, M.; Rahmani, F.; Rezaei, N. Role of p38/MAPKs in Alzheimer’s disease: Implications for amyloid beta toxicity targeted therapy. Rev. Neurosci. 2019, 30, 9–30. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.W.; Qin, Z.H.; Ren, M.; Kanai, H.; Chalecka-Franaszek, E.; Leeds, P.; Chuang, D.M. Regulation of c-Jun N-terminal kinase, p38 kinase and AP-1 DNA binding in cultured brain neurons: Roles in glutamate excitotoxicity and lithium neuroprotection. J. Neurochem. 2003, 84, 566–575. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Zhang, H.; Wu, J.; Yin, L.; Yan, L.-J.; Zhang, C. Humanin Attenuates NMDA-Induced Excitotoxicity by Inhibiting ROS-dependent JNK/p38 MAPK Pathway. Int. J. Mol. Sci. 2018, 19, 2982. [Google Scholar] [CrossRef] [Green Version]
- Gee, M.S.; Kim, S.W.; Kim, N.; Lee, S.J.; Oh, M.S.; Jin, H.K.; Bae, J.S.; Inn, K.S.; Kim, N.J.; Lee, J.K. A Novel and Selective p38 Mitogen-Activated Protein Kinase Inhibitor Attenuates LPS-Induced Neuroinflammation in BV2 Microglia and a Mouse Model. Neurochem. Res. 2018, 43, 2362–2371. [Google Scholar] [CrossRef]
- Gee, M.S.; Son, S.H.; Jeon, S.H.; Do, J.; Kim, N.; Ju, Y.J.; Lee, S.J.; Chung, E.K.; Inn, K.S.; Kim, N.J.; et al. A selective p38α/β MAPK inhibitor alleviates neuropathology and cognitive impairment, and modulates microglia function in 5XFAD mouse. Alzheimer’s Res. Ther. 2020, 12. [Google Scholar] [CrossRef] [Green Version]
- Munoz, L.; Ammit, A.J. Targeting p38 MAPK pathway for the treatment of Alzheimer’s disease. Neuropharmacol. 2010, 58, 561–568. [Google Scholar] [CrossRef]
- Dafsari, F.S.; Jessen, F. Depression—An underrecognized target for prevention of dementia in Alzheimer’s disease. Transl. Psychiatry 2020, 10, 1–13. [Google Scholar] [CrossRef]
- Snowden, M.B.; Atkins, D.C.; Steinman, L.E.; Bell, J.F.; Bryant, L.L.; Copeland, C.; Fitzpatrick, A.L. Longitudinal association of dementia and depression. Am. J. Geriatr. Psychiatry 2015, 23, 897–905. [Google Scholar] [CrossRef] [Green Version]
- Lim, G.Y.; Tam, W.W.; Lu, Y.; Ho, C.S.; Zhang, M.W.; Ho, R.C. Prevalence of Depression in the Community from 30 Countries between 1994 and 2014. Sci. Rep. 2018, 8, 1–10. [Google Scholar] [CrossRef]
- Zhu, C.B.; Blakely, R.D.; Hewlett, W.A. The proinflammatory cytokines interleukin-1beta and tumor necrosis factor-alpha activate serotonin transporters. Neuropsychopharmacology 2006, 31, 2121–2131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, C.B.; Lindler, K.M.; Owens, A.W.; Daws, L.C.; Blakely, R.D.; Hewlett, W.A. Interleukin-1 receptor activation by systemic lipopolysaccharide induces behavioral despair linked to MAPK regulation of CNS serotonin transporters. Neuropsychopharmacology 2010, 35, 2510–2520. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stefanoska, K.; Bertz, J.; Volkerling, A.M.; van der Hoven, J.; Ittner, L.M.; Ittner, A. Neuronal MAP kinase p38α inhibits c-Jun N-terminal kinase to modulate anxiety-related behaviour. Sci. Rep. 2018, 8, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanks, S.; Quinn, A.; Hunter, T. The protein kinase family: Conserved features and deduced phylogeny of the catalytic domains. Science 1988, 241, 42–52. [Google Scholar] [CrossRef] [PubMed]
- McClendon, C.L.; Kornev, A.P.; Gilson, M.K.; Taylor, S.S. Dynamic architecture of a protein kinase. Proc. Natl. Acad. Sci. USA 2014, 111, E4623–E4631. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zuccotto, F.; Ardini, E.; Casale, E.; Angiolini, M. Through the “Gatekeeper Door”: Exploiting the Active Kinase Conformation. J. Med. Chem. 2010, 53, 2681–2694. [Google Scholar] [CrossRef] [PubMed]
- Treiber, D.K.; Shah, N.P. Ins and Outs of Kinase DFG Motifs. Chem. Biol. 2013, 20, 745–746. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, X.; Gu, Y.; Fox, T.; Coll, J.T.; Fleming, M.A.; Markland, W.; Caron, P.R.; Wilson, K.P.; Su, M.S. Crystal structure of JNK3: A kinase implicated in neuronal apoptosis. Structure 1998, 6, 983–991. [Google Scholar] [CrossRef] [Green Version]
- Scapin, G.; Patel, S.B.; Lisnock, J.M.; Becker, J.W.; LoGrasso, P.V. The structure of JNK3 in complex with small molecule inhibitors: Structural basis for potency and selectivity. Chem. Biol. 2003, 10, 705–712. [Google Scholar] [CrossRef] [Green Version]
- Macalino, S.J.Y.; Gosu, V.; Hong, S.; Choi, S. Role of computer-aided drug design in modern drug discovery. Arch. Pharm. Res. 2015, 38, 1686–1701. [Google Scholar] [CrossRef]
- Park, H.J.; Iqbal, S.; Hernandez, P.; Mora, R.; Zheng, K.; Feng, Y.; LoGrasso, P. Structural basis and biological consequences for JNK2/3 isoform selective aminopyrazoles. Sci. Rep. 2015, 5, 8047. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Probst, G.D.; Bowers, S.; Sealy, J.M.; Truong, A.P.; Hom, R.K.; Galemmo, R.A.; Konradi, A.W.; Sham, H.L.; Quincy, D.A.; Pan, H.; et al. Highly selective c-Jun N-terminal kinase (JNK) 2 and 3 inhibitors with in vitro CNS-like pharmacokinetic properties prevent neurodegeneration. Bioorganic Med. Chem. Lett. 2011, 21, 315–319. [Google Scholar] [CrossRef] [PubMed]
- Hay, M.; Thomas, D.W.; Craighead, J.L.; Economides, C.; Rosenthal, J. Clinical development success rates for investigational drugs. Nat. Biotechnol. 2014, 32, 40–51. [Google Scholar] [CrossRef] [PubMed]
- Daina, A.; Michielin, O.; Zoete, V. SwissTargetPrediction: Updated data and new features for efficient prediction of protein targets of small molecules. Nucleic Acids Res. 2019, 47, 357–364. [Google Scholar] [CrossRef] [Green Version]
- Drachman, D.A.; Leavitt, J. Human memory and the cholinergic system. A relationship to aging? Arch. Neurol. 1974, 30, 113–121. [Google Scholar] [CrossRef]
- Bowen, D.M.; Smith, C.B.; White, P.; Davison, A.N. Neurotransmitter-related enzymes and indices of hypoxia in senile dementia and other abiotrophies. Brain 1976, 99, 459–496. [Google Scholar] [CrossRef] [Green Version]
- Davies, P.; Maloney, A.J.F. Selective Loss of Central Cholinergic Neurons in Alzheimer’S Disease. Lancet 1976, 308, 1403. [Google Scholar] [CrossRef]
- Burke, J.F.; Langa, K.M.; Hayward, R.A.; Albin, R.L. Modeling test and treatment strategies for presymptomatic alzheimer disease. PLoS ONE 2014, 9, 1–20. [Google Scholar] [CrossRef] [Green Version]
- Schneider, L.S.; Mangialasche, F.; Andreasen, N.; Feldman, H.; Giacobini, E.; Jones, R.; Mantua, V.; Mecocci, P.; Pani, L. Disease: An Appraisal From 1984 To 2014. J. Intern. Med. 2014, 275, 251–283. [Google Scholar] [CrossRef]
- Fuster-Matanzo, A.; Llorens-Martin, M.; Hernandez, F.; Avila, J. Role of neuroinflammation in adult neurogenesis and Alzheimer disease: Therapeutic approaches. Mediat. Inflamm. 2013, 2013, 260925. [Google Scholar] [CrossRef] [Green Version]
- Muller, F.L.; Lustgarten, M.S.; Jang, Y.; Richardson, A.; Van Remmen, H. Trends in oxidative aging theories. Free Radic. Biol. Med. 2007, 43, 477–503. [Google Scholar] [CrossRef]
- Chilmonczyk, Z.; Bojarski, A.J.; Pilc, A.; Sylte, I. Pharmacological Reports Serotonin transporter and receptor ligands with antidepressant activity as neuroprotective and proapoptotic agents. Pharmacol. Rep. 2017, 69, 469–478. [Google Scholar] [CrossRef] [PubMed]
- Clarke, M.; Pentz, R.; Bobyn, J.; Hayley, S. Stressor-Like Effects of c-Jun N-Terminal Kinase (JNK) Inhibition. PLoS ONE 2012, 7. [Google Scholar] [CrossRef]
- Cicenas, J. JNK inhibitors: Is there a future? MAP Kinase 2015, 4. [Google Scholar] [CrossRef] [Green Version]
- Hunot, S.; Vila, M.; Teismann, P.; Davis, R.J.; Hirsch, E.C.; Przedborski, S.; Rakic, P.; Flavell, R.A. JNK-mediated induction of cyclooxygenase 2 is required for neurodegeneration in a mouse model of Parkinson’s disease. Proc. Natl. Acad. Sci. USA 2004, 101, 665–670. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mehan, S.; Meena, H.; Sharma, D.; Sankhla, R. JNK: A stress-activated protein kinase therapeutic strategies and involvement in Alzheimer’s and various neurodegenerative abnormalities. J. Mol. Neurosci. 2011, 43, 376–390. [Google Scholar] [CrossRef]
- Hirosumi, J.; Tuncman, G.; Chang, L.; Görgün, C.Z.; Uysal, K.T.; Maeda, K.; Karin, M.; Hotamisligil, G.S. A central, role for JNK in obesity and insulin resistance. Nature 2002, 420, 333–336. [Google Scholar] [CrossRef]
- Sabio, G.; Kennedy, N.J.; Cavanagh-Kyros, J.; Jung, D.Y.; Ko, H.J.; Ong, H.; Barrett, T.; Kim, J.K.; Davis, R.J. Role of Muscle c-Jun NH2-Terminal Kinase 1 in Obesity-Induced Insulin Resistance. Mol. Cell. Biol. 2010, 30, 106–115. [Google Scholar] [CrossRef] [Green Version]
- Han, Z.; Chang, L.; Yamanishi, Y.; Karin, M.; Firestein, G.S. Joint damage and inflammation in c-Jun N-terminal kinase 2 knockout mice with passive murine collagen-induced arthritis. Arthritis Rheum. 2002, 46, 818–823. [Google Scholar] [CrossRef]
- Pelaia, G.; Cuda, G.; Vatrella, A.; Gallelli, L.; Caraglia, M.; Marra, M.; Abbruzzese, A.; Caputi, M.; Maselli, R.; Costanzo, F.S.; et al. Mitogen-activated protein kinases and asthma. J. Cell. Physiol. 2005, 202, 642–653. [Google Scholar] [CrossRef]
- Blease, K.; Lewis, A.; Raymon, H.K. Emerging treatments for asthma. Expert Opin. Emerg. Drugs 2003, 8, 71–81. [Google Scholar] [CrossRef] [PubMed]
- Chialda, L.; Zhang, M.; Brune, K.; Pahl, A. Inhibitors of mitogen-activated protein kinases differentially regulate costimulated T cell cytokine production and mouse airway eosinophilia. Respir. Res. 2005, 6, 36. [Google Scholar] [CrossRef] [Green Version]
- Osto, E.; Matter, C.M.; Kouroedov, A.; Malinski, T.; Bachschmid, M.; Camici, G.G.; Kilic, U.; Stallmach, T.; Boren, J.; Iliceto, S.; et al. c-Jun N-Terminal kinase 2 deficiency protects against hypercholesterolemia-induced endothelial dysfunction and oxidative stress. Circulation 2008, 118, 2073–2080. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Noble, M.E.M.; Endicott, J.A.; Johnson, L.N. Protein Kinase Inhibitors: Insights into Drug Design from Structure. Science 2004, 303, 1800–1805. [Google Scholar] [CrossRef] [PubMed]
- Comess, K.M.; Sun, C.; Abad-Zapatero, C.; Goedken, E.R.; Gum, R.J.; Borhani, D.W.; Argiriadi, M.; Groebe, D.R.; Jia, Y.; Clampit, J.E.; et al. Discovery and Characterization of Non-ATP Site Inhibitors of the Mitogen Activated Protein (MAP) Kinases. ACS Chem. Biol. 2011, 6, 234–244. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Gray, N.S. Rational design of inhibitors that bind to inactive kinase conformations. Nat. Chem. Biol. 2006, 2, 358–364. [Google Scholar] [CrossRef] [PubMed]
- Roskoski, R. Classification of small molecule protein kinase inhibitors based upon the structures of their drug-enzyme complexes. Pharmacol. Res. 2016, 103, 26–48. [Google Scholar] [CrossRef]
- Huang, D.; Zhou, T.; Lafleur, K.; Nevado, C.; Caflisch, A. Kinase selectivity potential for inhibitors targeting the ATP binding site: A network analysis. Bioinformatics 2010, 26, 198–204. [Google Scholar] [CrossRef] [Green Version]
- Kufareva, I.; Abagyan, R. Type-II Kinase Inhibitor Docking, Screening, and Profiling Using Modified Structures of Active Kinase States. J. Med. Chem. 2008, 51, 7921–7932. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Z.; Wu, H.; Wang, L.; Liu, Y.; Knapp, S.; Liu, Q.; Gray, N.S. Exploration of Type II Binding Mode: A Privileged Approach for Kinase Inhibitor Focused Drug Discovery? ACS Chem. Biol. 2014, 9, 1230–1241. [Google Scholar] [CrossRef]
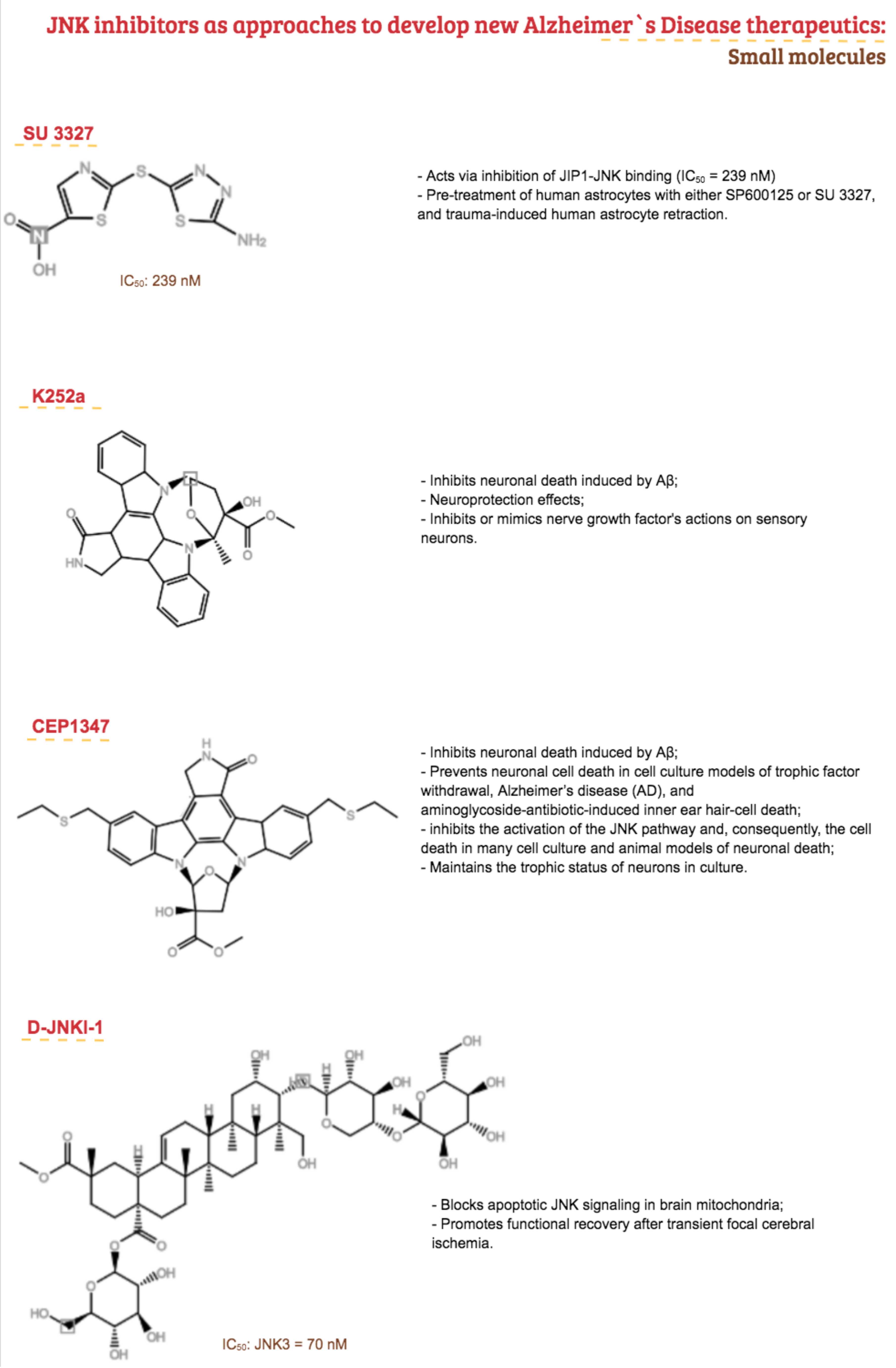
- LoGrasso, P.; Kamenecka, T. Inhibitors of c-jun-N-Terminal Kinase (JNK). Mini-Rev. Med. Chem. 2008, 8, 755–766. [Google Scholar] [CrossRef] [PubMed]
- Bain, J.; Plater, L.; Elliott, M.; Shpiro, N.; Hastie, C.J.; McLauchlan, H.; Klevernic, I.; Arthur, J.S.C.; Alessi, D.R.; Cohen, P. The selectivity of protein kinase inhibitors: A further update. Biochem. J. 2007, 408, 297–315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonny, C.; Oberson, A.; Negri, S.; Sauser, C.; Schorderet, D.F. Cell-Permeable Peptide Inhibitors of JNK: Novel Blockers of -Cell Death. Diabetes 2001, 50, 77–82. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krenitsky, V.P.; Nadolny, L.; Delgado, M.; Ayala, L.; Clareen, S.S.; Hilgraf, R.; Albers, R.; Hegde, S.; D’Sidocky, N.; Sapienza, J.; et al. Discovery of CC-930, an orally active anti-fibrotic JNK inhibitor. Bioorg. Med. Chem. Lett. 2012, 22, 1433–1438. [Google Scholar] [CrossRef]
- Atsriku, C.; Hoffmann, M.; Ye, Y.; Kumar, G.; Surapaneni, S. Metabolism and disposition of a potent and selective JNK inhibitor [14C]tanzisertib following oral administration to rats, dogs and humans. Xenobiotica 2015, 45, 428–441. [Google Scholar] [CrossRef]
- van der Velden, J.L.J.; Ye, Y.; Nolin, J.D.; Hoffman, S.M.; Chapman, D.G.; Lahue, K.G.; Abdalla, S.; Chen, P.; Liu, Y.; Bennett, B.; et al. JNK inhibition reduces lung remodeling and pulmonary fibrotic systemic markers. Clin. Transl. Med. 2016, 5, 36. [Google Scholar] [CrossRef] [Green Version]
- Hussein, M.; Chai, D.C.; Kyama, C.M.; Mwenda, J.M.; Palmer, S.S.; Gotteland, J.-P.; D’Hooghe, T.M. c-Jun NH2-terminal kinase inhibitor bentamapimod reduces induced endometriosis in baboons: An assessor-blind placebo-controlled randomized study. Fertil. Steril. 2016, 105, 815–824.e5. [Google Scholar] [CrossRef] [Green Version]
- Palmer, S.S.; Altan, M.; Denis, D.; Tos, E.G.; Gotteland, J.-P.; Osteen, K.G.; Bruner-Tran, K.L.; Nataraja, S.G. Bentamapimod (JNK Inhibitor AS602801) Induces Regression of Endometriotic Lesions in Animal Models. Reprod. Sci. 2016, 23, 11–23. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.; Sun, C.; Tao, S.; Osunkoya, A.O.; Arnold, R.S.; Petros, J.A.; Zu, X.; Moreno, C.S. The JNK inhibitor AS602801 Synergizes with Enzalutamide to Kill Prostate Cancer Cells In Vitro and In Vivo and Inhibit Androgen Receptor Expression. Transl. Oncol. 2020, 13, 100751. [Google Scholar] [CrossRef]
- Antoniou, X.; Falconi, M.; Di Marino, D.; Borsello, T. JNK3 as a therapeutic target for neurodegenerative diseases. J. Alzheimer’s Dis. 2011, 24, 633–642. [Google Scholar] [CrossRef]
- Bogoyevitch, M.A.; Boehm, I.; Oakley, A.; Ketterman, A.J.; Barr, R.K. Targeting the JNK MAPK cascade for inhibition: Basic science and therapeutic potential. Biochim. Biophys. Acta Proteins Proteom. 2004, 1697, 89–101. [Google Scholar] [CrossRef] [PubMed]
- Colombo, A.; Repici, M.; Pesaresi, M.; Santambrogio, S.; Forloni, G.; Borsello, T. The TAT-JNK inhibitor peptide interferes with beta amyloid protein stability. Cell Death Differ. 2007, 14, 1845–1848. [Google Scholar] [CrossRef] [PubMed]
- Colombo, A.; Bastone, A.; Ploia, C.; Sclip, A.; Salmona, M.; Forloni, G.; Borsello, T. JNK regulates APP cleavage and degradation in a model of Alzheimer’s disease. Neurobiol. Dis. 2009, 33, 518–525. [Google Scholar] [CrossRef]
- Gourmaud, S.; Thomas, P.; Thomasseau, S.; Tible, M.; Abadie, C.; Paquet, C.; Hugon, J. Brimapitide Reduced Neuronal Stress Markers and Cognitive Deficits in 5XFAD Transgenic Mice. J. Alzheimer’s Dis. 2018, 63, 665–674. [Google Scholar] [CrossRef] [PubMed]
- Guo, C.; Whitmarsh, A.J. The β-arrestin-2 scaffold protein promotes c-Jun N-terminal kinase-3 activation by binding to its nonconserved N terminus. J. Biol. Chem. 2008, 283, 15903–15911. [Google Scholar] [CrossRef] [Green Version]
- Shenoy, S.K.; Lefkowitz, R.J. Seven-transmembrane receptor signaling through beta-arrestin. Sci. Signal. 2005, 2005, cm10. [Google Scholar] [CrossRef]
- Gower, C.M.; Chang, M.E.K.; Maly, D.J. Bivalent inhibitors of protein kinases. Crit. Rev. Biochem. Mol. Biol. 2014, 49, 102–115. [Google Scholar] [CrossRef] [Green Version]
- Muth, F.; El-gokha, A.; Ansideri, F.; Eitel, M.; Doering, E.; Sievers, A.; Lange, A.; Boeckler, F.M.; Lämmerhofer, M.; Koch, P.; et al. Tri- and Tetra-substituted Pyridinylimidazoles as Covalent Inhibitors of c-Jun N-Terminal Kinase 3 Tri- and Tetra-substituted Pyridinylimidazoles as Covalent Inhibitors of c-Jun N-Terminal Kinase 3. J. Med. Chem. 2016, 60, 594–607. [Google Scholar] [CrossRef]
- Zhang, T.; Inesta-Vaquera, F.; Niepel, M.; Zhang, J.; Ficarro, S.B.; Machleidt, T.; Xie, T.; Marto, J.A.; Kim, N.; Sim, T.; et al. Discovery of Potent and Selective Covalent Inhibitors of JNK. Chem. Biol. 2012, 19, 140–154. [Google Scholar] [CrossRef] [Green Version]
- Guan, Q.; Pei, D.; Liu, X.; Wang, X.; Xu, T.; Zhang, G. Neuroprotection against ischemic brain injury by SP600125 via suppressing the extrinsic and intrinsic pathways of apoptosis. Brain Res. 2006, 92. [Google Scholar] [CrossRef]
- Bogoyevitch, M.A.; Ngoei, K.R.W.; Zhao, T.T.; Yeap, Y.Y.C.; Ng, D.C.H. Biochimica et Biophysica Acta c-Jun N-terminal kinase (JNK) signaling: Recent advances and challenges. BBA Proteins Proteom. 2010, 1804, 463–475. [Google Scholar] [CrossRef]
- Guan, Q.; Pei, D.; Zhang, Q.; Hao, Z. The neuroprotective action of SP600125, a new inhibitor of JNK, on transient brain ischemia/reperfusion-induced neuronal death in rat hippocampal CA1 via nuclear and non-nuclear pathways. Brain Res. 2005, 1035, 51–59. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Shi, L.; Xie, Y.; Ma, C.; Li, W.; Su, X.; Huang, S.; Chen, R.; Zhu, Z.; Mao, Z.; et al. SP600125, a new JNK inhibitor, protects dopaminergic neurons in the MPTP model of Parkinson’s disease. Neurosci. Res. 2004, 48, 195–202. [Google Scholar] [CrossRef] [PubMed]
- Mu, F.; Haas, E.; Schwenzer, R.; Schubert, G.; Grell, M.; Smith, C.; Scheurich, P.; Wajant, H. TRAIL/Apo2L Activates c-Jun NH2-terminal Kinase (JNK) via Caspase-dependent and Caspase-independent Pathways. J. Biol. Chem. 1998, 273, 33091–33098. [Google Scholar]
- Prause, M.; Adrian, L.; Ibarra, A.; Hyldgaard, M.; Billestrup, N.; Mandrup-poulsen, T.; Størling, J. Molecular and Cellular Endocrinology TRAF2 mediates JNK and STAT3 activation in response to IL-1 b and IFN g and facilitates apoptotic death of insulin-producing b-cells. Mol. Cell. Endocrinol. 2016, 420, 24–36. [Google Scholar] [CrossRef] [PubMed]
- Tan, Y.; Dourdin, N.; Wu, C.; Veyra, T.; De Elce, J.S.; Greer, P.A. Ubiquitous Calpains Promote Caspase-12 and JNK Activation during Endoplasmic Reticulum Stress-induced Apoptosis. J. Biol. Chem. 2006, 281, 16016–16024. [Google Scholar] [CrossRef] [Green Version]
- Choi, W.; Yoon, S.; Oh, T.H.; Choi, E.; Malley, K.L.O. Two Distinct Mechanisms Are Involved in 6-Hydroxydopamine- and MPP-Induced Dopaminergic Neuronal Cell Death: Role of Caspases, ROS, and JNK. J. Neurosci. Res. 1999, 94, 86–94. [Google Scholar] [CrossRef]
- Martinez, R.; Defnet, A.; Shapiro, P. Avoiding or Co-Opting ATP Inhibition: Overview of Type III, IV, V, and VI Kinase Inhibitors. In Next Generation Kinase Inhibitors; Springer International Publishing: Berlin/Heidelberg, Germany, 2020; pp. 29–59. [Google Scholar]
- Vagnoni, A.; Glennon, E.B.C.; Perkinton, M.S.; Gray, E.H.; Noble, W.; Miller, C.C.J. Loss of c-Jun N-terminal kinase-interacting protein-1 does not affect axonal transport of the amyloid precursor protein or Aβ production. Hum. Mol. Genet. 2013, 22, 4646–4652. [Google Scholar] [CrossRef] [Green Version]
- Matsuda, S.; Yasukawa, T.; Homma, Y.; Ito, Y.; Niikura, T.; Hiraki, T.; Hirai, S.; Ohno, S.; Kita, Y.; Kawasumi, M.; et al. c-Jun N-terminal kinase (JNK)-interacting protein-1b/islet-brain-1 scaffolds Alzheimer’s amyloid precursor protein with JNK. J. Neurosci. 2001, 21, 6597–6607. [Google Scholar] [CrossRef] [Green Version]
- Harding, T.C.; Xue, L.; Bienemann, A.; Haywood, D.; Dickens, M.; Tolkovsky, A.M.; Uney, J.B. Inhibition of JNK by Overexpression of the JNK Binding Domain of JIP-1 Prevents Apoptosis in Sympathetic Neurons. J. Biol. Chem. 2001, 276, 4531–4534. [Google Scholar] [CrossRef] [Green Version]
- Morel, C.; Sherrin, T.; Kennedy, N.J.; Forest, K.H.; Barutcu, S.A.; Robles, M.; Carpenter-Hyland, E.; Alfulaij, N.; Standen, C.L.; Nichols, R.A.; et al. JIP1-mediated JNK activation negatively regulates synaptic plasticity and spatial memory. J. Neurosci. 2018, 38, 3708–3728. [Google Scholar] [CrossRef] [PubMed]
- Omotehara, Y.; Hakuba, N.; Hato, N.; Okada, M.; Gyo, K. Protection against ischemic cochlear damage by intratympanic administration of AM-111. Otol. Neurotol. 2011, 32, 1422–1427. [Google Scholar] [CrossRef] [PubMed]
- Barkdull, G.C.; Hondarrague, Y.; Meyer, T.; Harris, J.P.; Keithley, E.M. AM-111 reduces hearing loss in a guinea pig model of acute labyrinthitis. Laryngoscope 2007, 117, 2174–2182. [Google Scholar] [CrossRef] [PubMed]
- Reinecke, K.; Eminel, S.; Dierck, F.; Roessner, W.; Kersting, S.; Chromik, A.M.; Gavrilova, O.; Laukevicience, A.; Leuschner, I.; Waetzig, V.; et al. The JNK inhibitor XG-102 protects against TNBS-induced colitis. PLoS ONE 2012, 7, e30985. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suckfuell, M.; Lisowska, G.; Domka, W.; Kabacinska, A.; Morawski, K.; Bodlaj, R.; Klimak, P.; Kostrica, R.; Meyer, T. Efficacy and safety of AM-111 in the treatment of acute sensorineural hearing loss: A double-blind, randomized, placebo-controlled phase II study. Otol. Neurotol. 2014, 35, 1317–1326. [Google Scholar] [CrossRef] [PubMed]
- Staecker, H.; Jokovic, G.; Karpishchenko, S.; Kienle-Gogolok, A.; Krzyzaniak, A.; Lin, C.-D.; Navratil, P.; Tzvetkov, V.; Wright, N.; Meyer, T. Efficacy and Safety of AM-111 in the Treatment of Acute Unilateral Sudden Deafness—A Double-blind, Randomized, Placebo-controlled Phase 3 Study. Otol. Neurotol. 2019, 40, 584–594. [Google Scholar] [CrossRef]
- Beydoun, T.; Deloche, C.; Perino, J.; Kirwan, B.-A.; Combette, J.-M.; Behar-Cohen, F. Subconjunctival Injection of XG-102, a JNK Inhibitor Peptide, in Patients with Intraocular Inflammation: A Safety and Tolerability Study. J. Ocul. Pharmacol. Ther. 2015, 31, 93–99. [Google Scholar] [CrossRef] [Green Version]
- Chiquet, C.; Aptel, F.; Creuzot-Garcher, C.; Berrod, J.-P.; Kodjikian, L.; Massin, P.; Deloche, C.; Perino, J.; Kirwan, B.-A.; de Brouwer, S.; et al. Postoperative Ocular Inflammation: A Single Subconjunctival Injection of XG-102 Compared to Dexamethasone Drops in a Randomized Trial. Am. J. Ophthalmol. 2017, 174, 76–84. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.; Spigolon, G.; Bonny, C.; Culman, J.; Vercelli, A.; Herdegen, T. The JNK inhibitor D-JNKI-1 blocks apoptotic JNK signaling in brain mitochondria. Mol. Cell Neurosci. 2012, 49, 300–310. [Google Scholar] [CrossRef]
- Esneault, E.; Castagne, V.; Moser, P.; Bonny, C.; Bernaudin, M. D-JNKi, a peptide inhibitor of c-Jun N-terminal kinase, promotes functional recovery after transient focal cerebral ischemia in rats. Neuroscience 2008, 152, 308–320. [Google Scholar] [CrossRef]
- Wang, L.H.; Besirli, C.G.; Johnson, E.M., Jr. Mixed-lineage kinases: A target for the prevention of neurodegeneration. Annu. Rev. Pharmacol. Toxicol. 2004, 44, 451–474. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ylikoski, J.; Xing-Qun, L.; Virkkala, J.; Pirvola, U. Blockade of c-Jun N-terminal kinase pathway attenuates gentamicin-induced cochlear and vestibular hair cell death. Heart Res. 2002, 163, 71–81. [Google Scholar] [CrossRef]
- Buck, H.; Winter, J. K252a modulates the expression of nerve growth factor-dependent capsaicin sensitivity and substance P levels in cultured adult rat dorsal root ganglion neurones. J. Neurochem. 1996, 67, 345–351. [Google Scholar] [CrossRef] [PubMed]









| JNK1 | JNK2 | JNK3 2 |
|---|---|---|
| 1α1 | 2α1 | 3α1 |
| 1α2 1 | 2α2 1 | 3α2 1 |
| 1β1 | 2β1 | |
| 1β2 | 2β2 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hepp Rehfeldt, S.C.; Majolo, F.; Goettert, M.I.; Laufer, S. c-Jun N-Terminal Kinase Inhibitors as Potential Leads for New Therapeutics for Alzheimer’s Diseases. Int. J. Mol. Sci. 2020, 21, 9677. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21249677
Hepp Rehfeldt SC, Majolo F, Goettert MI, Laufer S. c-Jun N-Terminal Kinase Inhibitors as Potential Leads for New Therapeutics for Alzheimer’s Diseases. International Journal of Molecular Sciences. 2020; 21(24):9677. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21249677
Chicago/Turabian StyleHepp Rehfeldt, Stephanie Cristine, Fernanda Majolo, Márcia Inês Goettert, and Stefan Laufer. 2020. "c-Jun N-Terminal Kinase Inhibitors as Potential Leads for New Therapeutics for Alzheimer’s Diseases" International Journal of Molecular Sciences 21, no. 24: 9677. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21249677