N-Docosahexanoylethanolamine Reduces Microglial Activation and Improves Hippocampal Plasticity in a Murine Model of Neuroinflammation


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
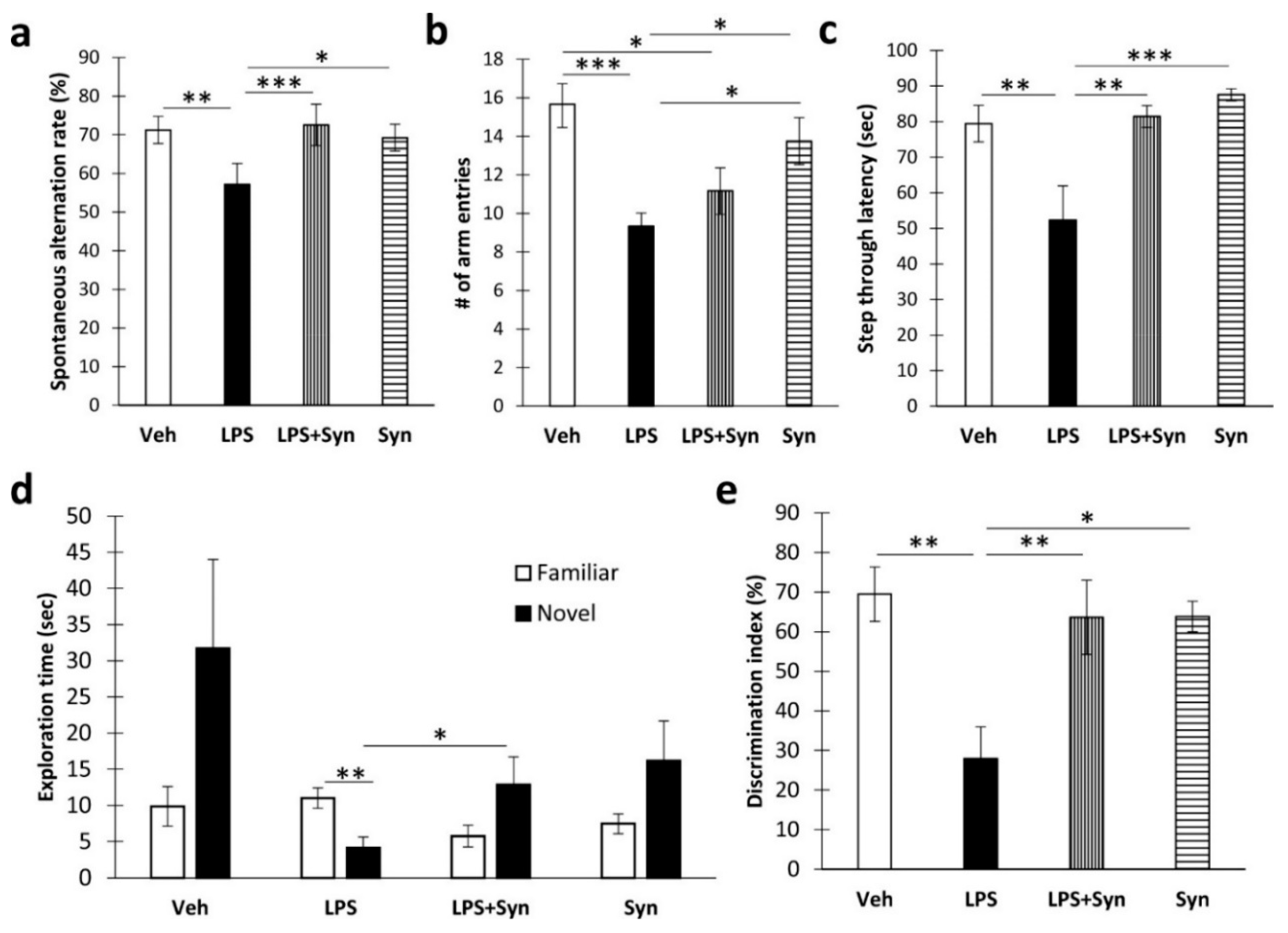
2.1. Synaptamide Treatment Prevents Memory Loss
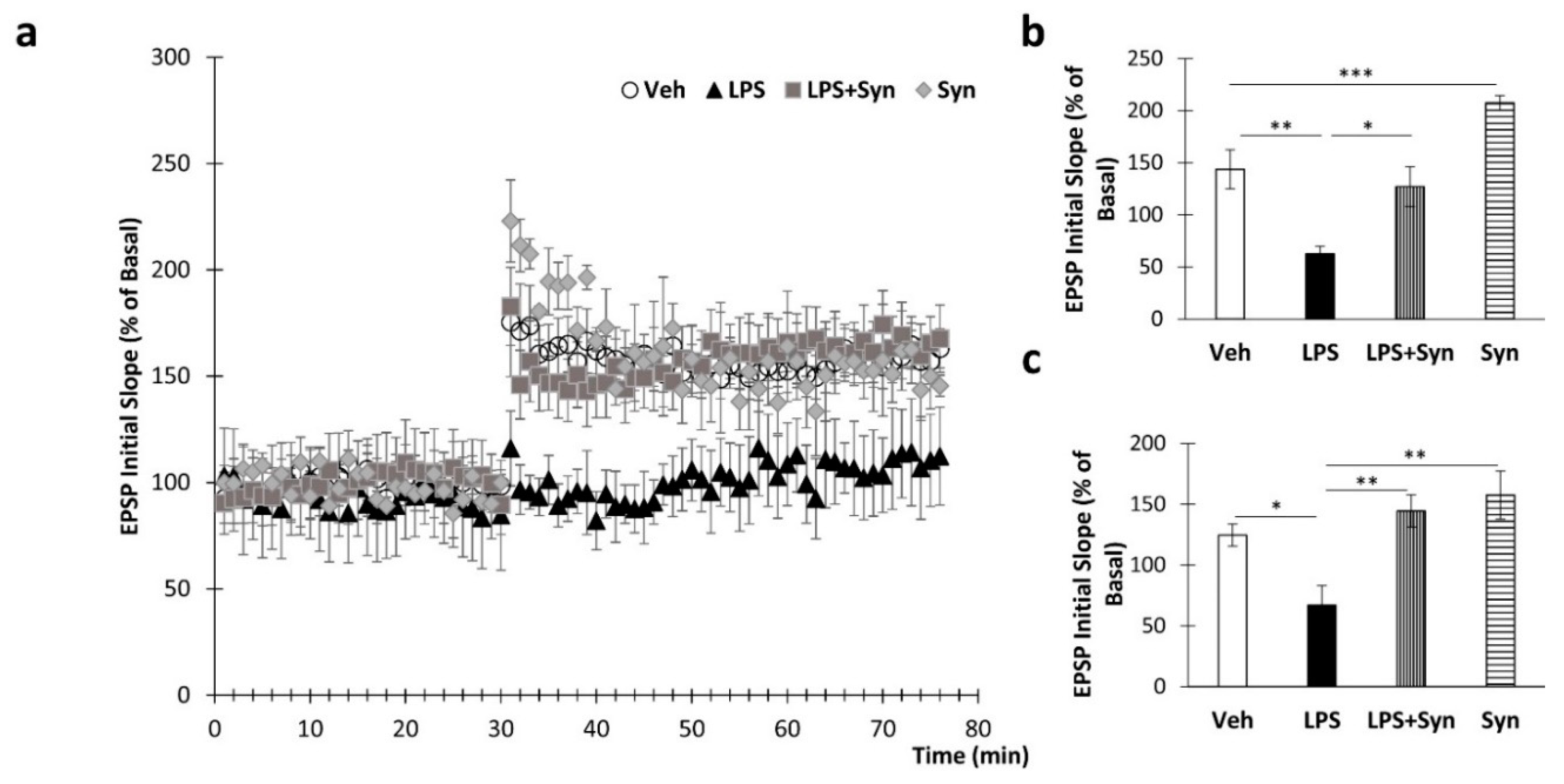
2.2. Synaptamide Prevents Synaptic Plasticity Impairment
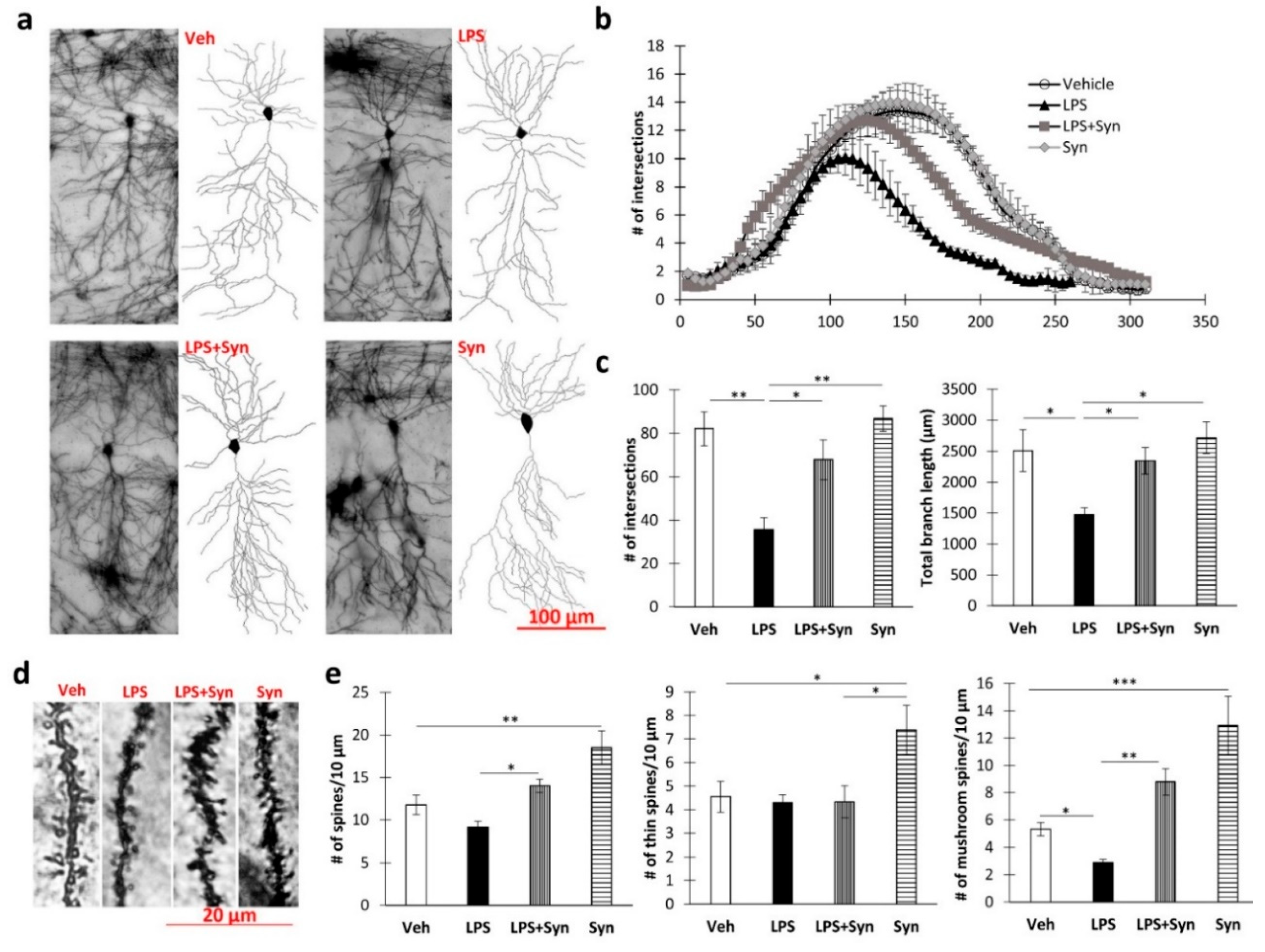
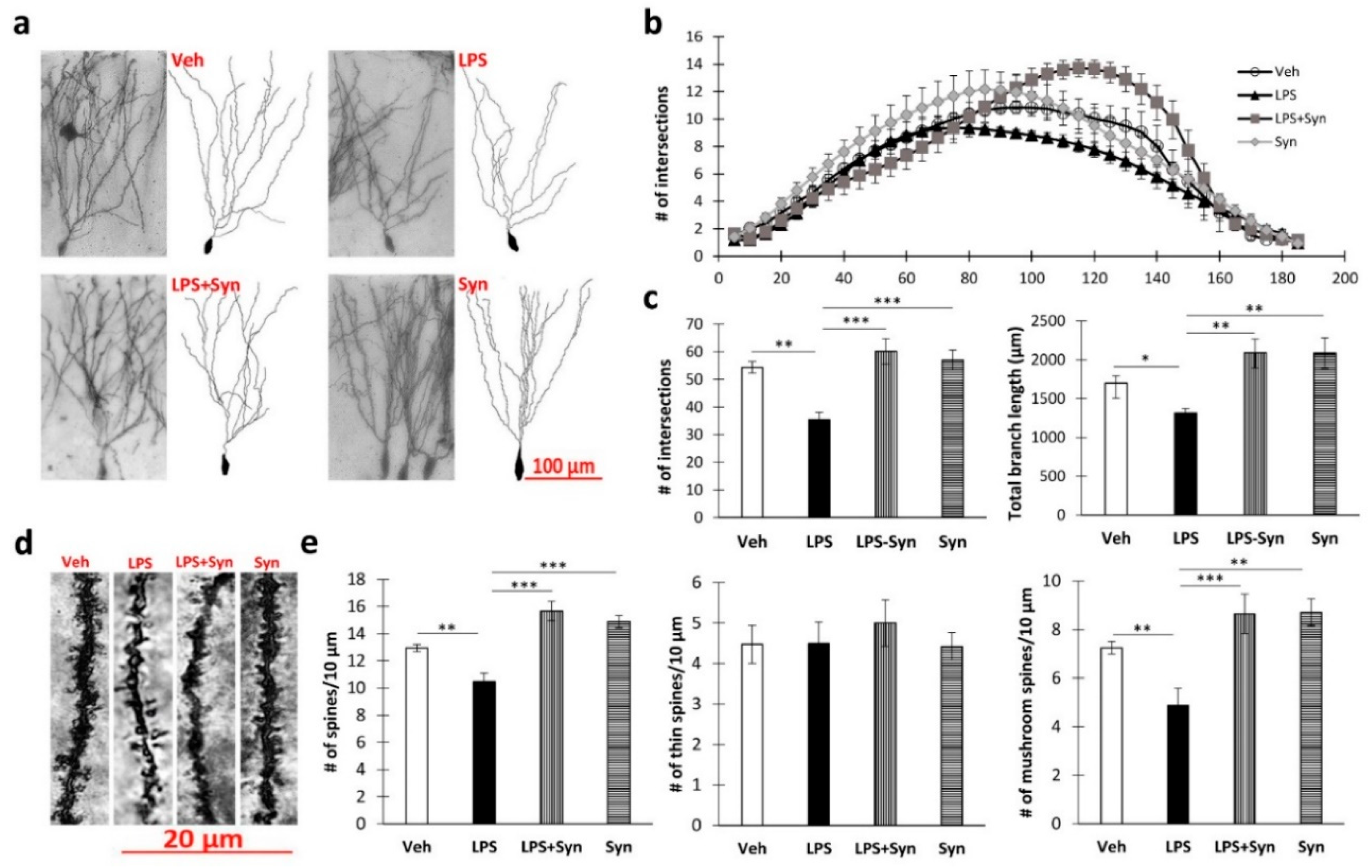
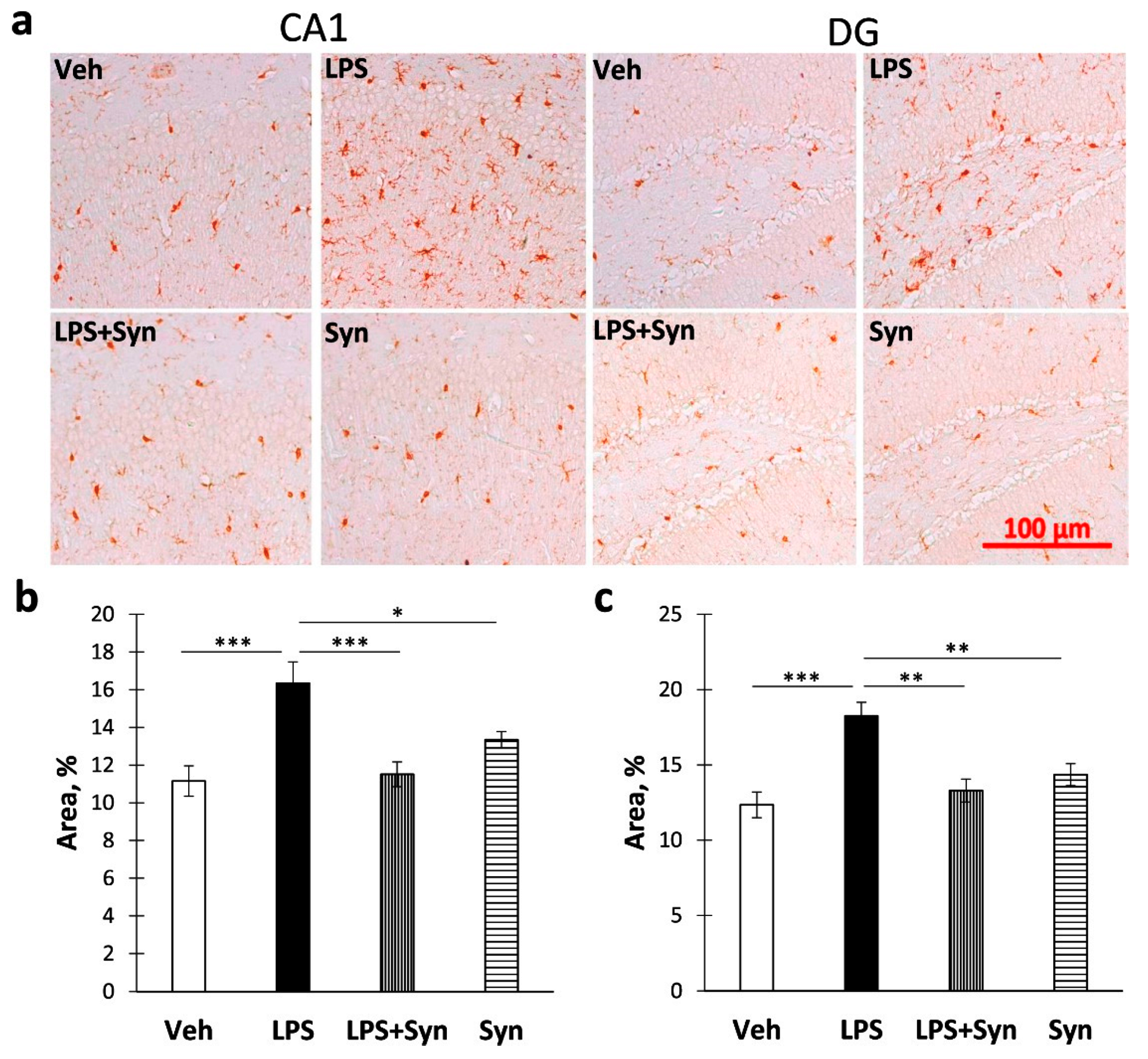
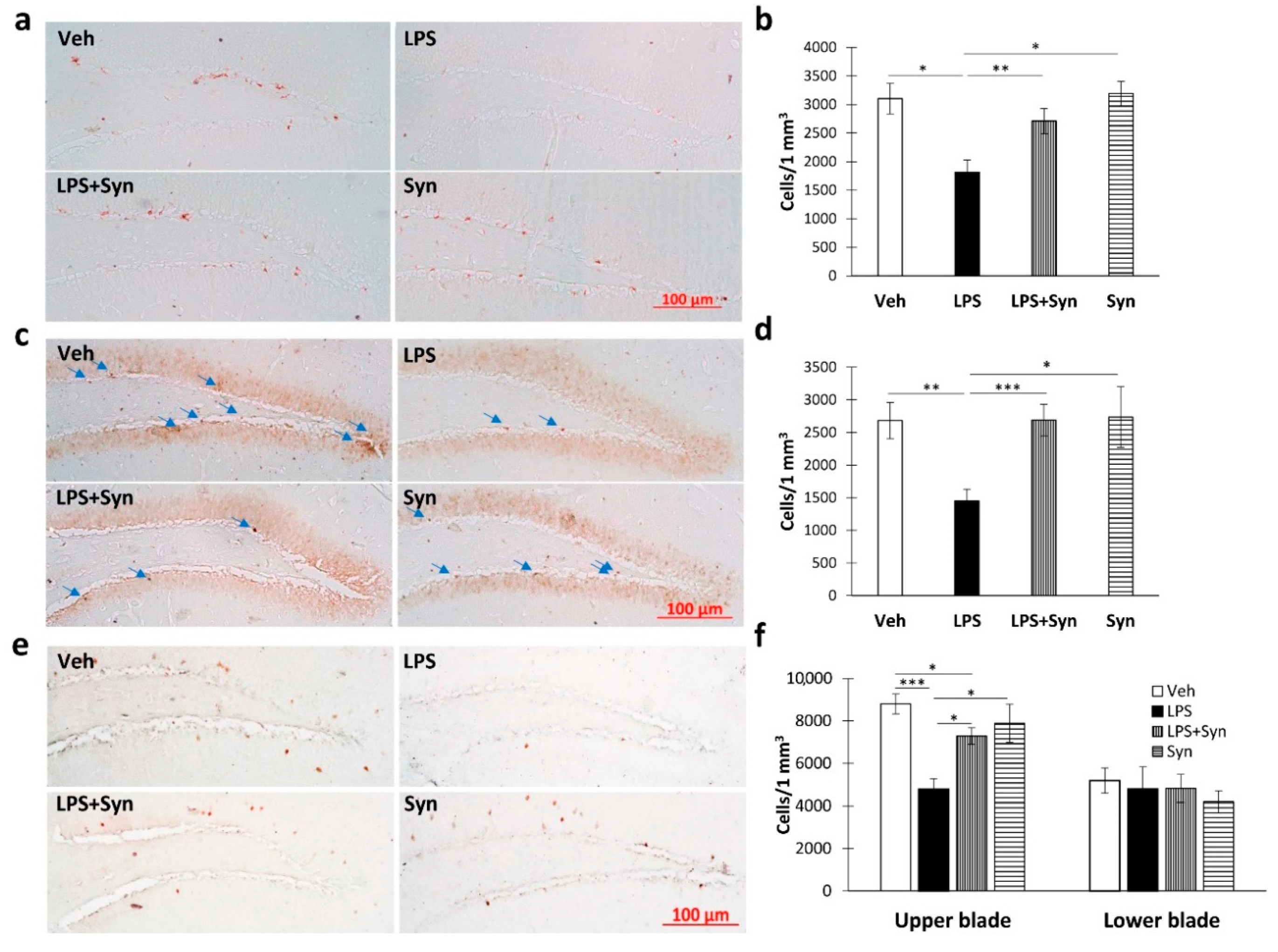
2.3. Effect of Synaptamide Treatment on LPS-Induced Dendrite Degeneration
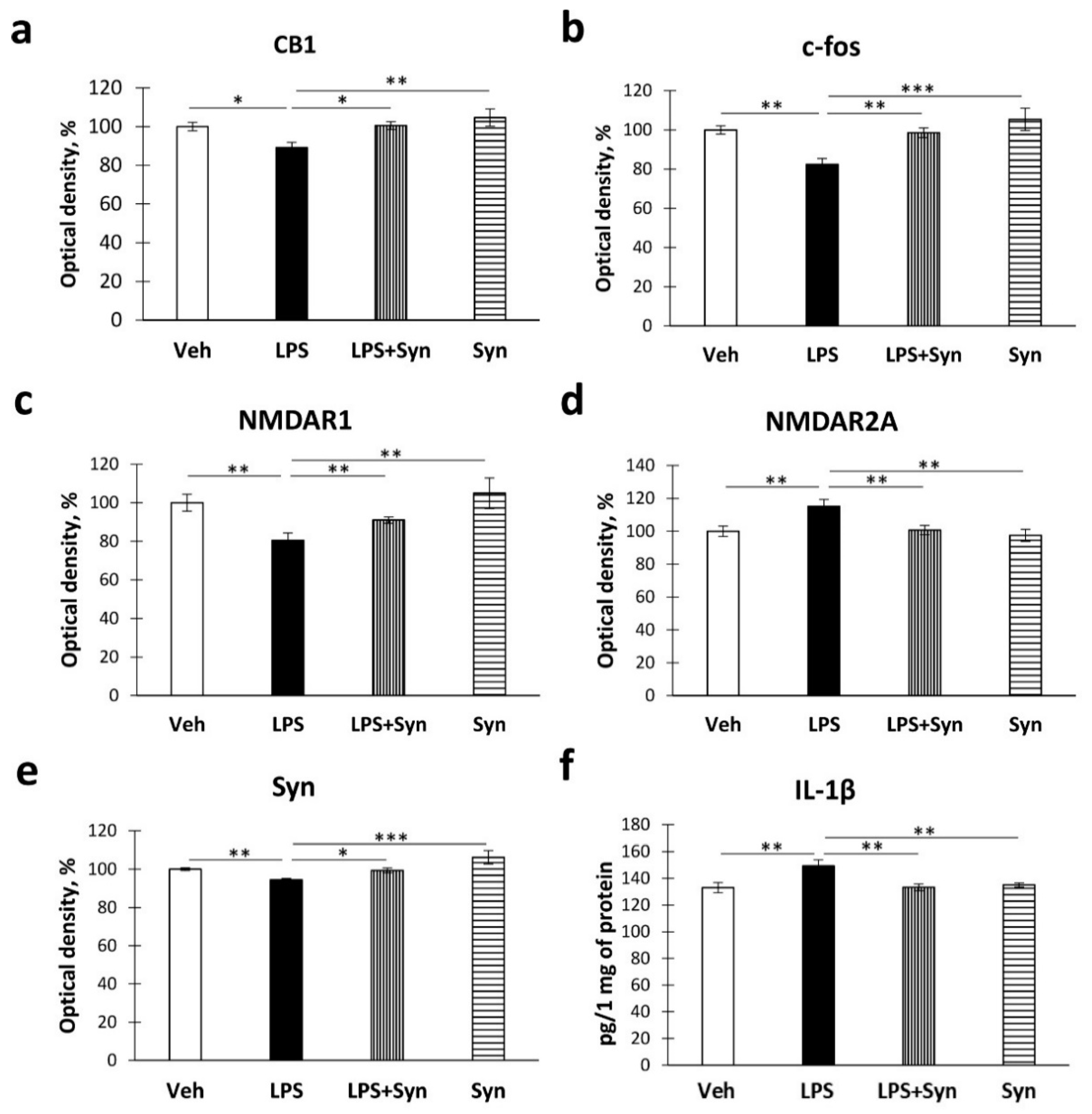
2.4. Effect of Neuroinflammation and Synaptamide Treatment on Hippocampal Protein Expression
3. Discussion
4. Materials and Methods
4.1. Animals and Treatments
4.2. N-Docosahexaenoylethanolamine Preparation
4.3. Working Memory
4.4. Passive Avoidance Test
4.5. Novel Object Recognition
4.6. Electrophysiological Recording
4.7. Golgi–Cox Staining
4.8. Sholl Analysis
4.9. ELISA
4.10. Immunohistochemical Studies
4.11. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| Synaptamide | N-docosahexanoylethanolamine |
| LPS | lipopolysaccharides |
| DG SGZ | dentate gyrus subgranular zone |
| DCX | doublecortin |
| PCNA | proliferating cell nuclear antigen |
| LTP | long-term potentiation |
| ELISA | enzyme-linked immunosorbent assay |
| ANOVA | one-way analysis of variance |
| FAAH | fatty acid amide hydrolase |
| DHA | docosahexaenoic acid |
| CB1 | cannabinoid-1 Receptor |
| NMDAR | N-methyl-d-aspartate receptor |
| PBS | phosphate-buffered saline |
| Veh | vehicle |
| Syn | Synaptophysin |
| aCSF | artificial cerebrospinal fluid |
References
- Frank-Cannon, T.C.; Alto, L.T.; E McAlpine, F.; Tansey, M.G. Does neuroinflammation fan the flame in neurodegenerative diseases? Mol. Neurodegener. 2009, 4, 47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fang, F.; Lue, L.-F.; Yan, S.; Xu, H.; Luddy, J.S.; Chen, D.; Walker, D.G.; Stern, D.M.; Yan, S.; Schmidt, A.M.; et al. RAGE-dependent signaling in microglia contributes to neuroinflammation, Aβ accumulation, and impaired learning/memory in a mouse model of Alzheimer’s disease. FASEB J. 2009, 24, 1043–1055. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McGeer, P.L.; McGeer, E.G. NSAIDs and Alzheimer disease: Epidemiological, animal model and clinical studies. Neurobiol. Aging 2007, 28, 639–647. [Google Scholar] [CrossRef] [PubMed]
- Rao, J.S.; Harry, G.J.; Rapoport, S.I.; Kim, H.-W. Increased excitotoxicity and neuroinflammatory markers in postmortem frontal cortex from bipolar disorder patients. Mol. Psychiatry 2009, 15, 384–392. [Google Scholar] [CrossRef]
- Katzman, M.A.; Furtado, M.; Anand, L. Targeting the Endocannabinoid System in Psychiatric Illness. J. Clin. Psychopharmacol. 2016, 36, 691–703. [Google Scholar] [CrossRef]
- Chen, J.; Buchanan, J.B.; Sparkman, N.L.; Godbout, J.P.; Freund, G.G.; Johnson, R.W. Neuroinflammation and disruption in working memory in aged mice after acute stimulation of the peripheral innate immune system. Brain Behav. Immun. 2008, 22, 301–311. [Google Scholar] [CrossRef] [Green Version]
- Murray, C.; Sanderson, D.J.; Barkus, C.; Deacon, R.M.; Rawlins, J.N.P.; Bannerman, D.M.; Cunningham, C. Systemic inflammation induces acute working memory deficits in the primed brain: Relevance for delirium. Neurobiol. Aging 2012, 33, 603–616. [Google Scholar] [CrossRef] [Green Version]
- Czerniawski, J.; Miyashita, T.; Lewandowski, G.; Guzowski, J.F. Systemic lipopolysaccharide administration impairs retrieval of context–object discrimination, but not spatial, memory: Evidence for selective disruption of specific hippocampus-dependent memory functions during acute neuroinflammation. Brain Behav. Immun. 2015, 44, 159–166. [Google Scholar] [CrossRef] [Green Version]
- Frühauf, P.K.S.; Ineu, R.P.; Tomazi, L.; Duarte, T.; Mello, C.F.; Rubin, M.A. Spermine reverses lipopolysaccharide-induced memory deficit in mice. J. Neuroinflamm. 2015, 12, 3–11. [Google Scholar] [CrossRef] [Green Version]
- Di Filippo, M.; Chiasserini, D.; Tozzi, A.; Picconi, B.; Calabresi, P. Mitochondria and the Link Between Neuroinflammation and Neurodegeneration. J. Alzheimer’s Dis. 2010, 20, S369–S379. [Google Scholar] [CrossRef] [Green Version]
- Dayton, E.T.; Major, E.O. Recombinant human interleukin 1β induces production of prostaglandins in primary human fetal astrocytes and immortalized human fetal astrocyte cultures. J. Neuroimmunol. 1996, 71, 11–18. [Google Scholar] [CrossRef]
- Stella, N.; Estellés, A.; Siciliano, J.; Tencé, M.; Desagher, S.; Piomelli, D.; Glowinski, J.; Prémont, J. Interleukin-1 Enhances the ATP-Evoked Release of Arachidonic Acid from Mouse Astrocytes. J. Neurosci. 1997, 17, 2939–2946. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McCann, S.M.; Licinio, J.; Wong, M.-L.; Yu, W.H.; Karanth, S.; Rettorri, V. The nitric oxide hypothesis of aging. Exp. Gerontol. 1999, 33, 813–826. [Google Scholar] [CrossRef]
- Sung, C.-S.; Wen, Z.-H.; Chang, W.-K.; Ho, S.-T.; Tsai, S.-K.; Chang, Y.-C.; Wong, C.-S. Intrathecal interleukin-1β administration induces thermal hyperalgesia by activating inducible nitric oxide synthase expression in the rat spinal cord. Brain Res. 2004, 1015, 145–153. [Google Scholar] [CrossRef] [PubMed]
- Ye, L.; Huang, Y.; Zhao, L.; Li, Y.; Sun, L.; Zhou, Y.; Qian, G.; Zheng, J.C. IL-1β and TNF-α induce neurotoxicity through glutamate production: A potential role for neuronal glutaminase. J. Neurochem. 2013, 125, 897–908. [Google Scholar] [CrossRef] [PubMed]
- Mozrzymas, J.W. Dynamism of GABAA receptor activation shapes the “personality” of inhibitory synapses. Neuropharmacol. 2004, 47, 945–960. [Google Scholar] [CrossRef] [PubMed]
- Dravid, S.M.; Erreger, K.; Yuan, H.; Nicholson, K.; Le, P.; Lyuboslavsky, P.; Almonte, A.; Murray, E.; Mosley, C.; Barber, J.; et al. Subunit-specific mechanisms and proton sensitivity of NMDA receptor channel block. J. Physiol. 2007, 581, 107–128. [Google Scholar] [CrossRef]
- Tyrtyshnaia, A.A.; Lysenko, L.V.; Madamba, F.; Manzhulo, I.V.; Khotimchenko, M.Y.; Kleschevnikov, A.M. Acute neuroinflammation provokes intracellular acidification in mouse hippocampus. J. Neuroinflamm. 2016, 13, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.-Y.; Spector, A.A.; Xiong, Z.-M. A synaptogenic amide N-docosahexaenoylethanolamide promotes hippocampal development. Prostaglandins Other Lipid Mediat. 2011, 96, 114–120. [Google Scholar] [CrossRef] [Green Version]
- Meijerink, J.; Balvers, M.; Witkamp, R.F. N-acyl amines of docosahexaenoic acid and other n–3 polyunsatured fatty acids – from fishy endocannabinoids to potential leads. Br. J. Pharmacol. 2013, 169, 772–783. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.-Y.; Spector, A.A. N-Docosahexaenoylethanolamine: A neurotrophic and neuroprotective metabolite of docosahexaenoic acid. Mol. Asp. Med. 2018, 64, 34–44. [Google Scholar] [CrossRef] [PubMed]
- Rashid, M.A.; Katakura, M.; Kharebava, G.; Kevala, K.; Kim, H.-Y. N-Docosahexaenoylethanolamine is a potent neurogenic factor for neural stem cell differentiation. J. Neurochem. 2013, 125, 869–884. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, T.; Chen, H.; Kevala, K.; Lee, J.-W.; Kim, H.-Y. N-Docosahexaenoylethanolamine ameliorates LPS-induced neuroinflammation via cAMP/PKA-dependent signaling. J. Neuroinflamm. 2016, 13, 1–15. [Google Scholar] [CrossRef] [Green Version]
- McDougle, D.R.; Watson, J.E.; Abdeen, A.A.; Adili, R.; Caputo, M.P.; Krapf, J.E.; Johnson, R.W.; Kilian, K.A.; Holinstat, M.; Das, A. Anti-inflammatory ω-3 endocannabinoid epoxides. Proc. Natl. Acad. Sci. USA 2017, 114, E6034–E6043. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, H.-Y.; Spector, A.A. Synaptamide, endocannabinoid-like derivative of docosahexaenoic acid with cannabinoid-independent function. Prostaglandins, Leukot. Essent. Fat. Acids 2013, 88, 121–125. [Google Scholar] [CrossRef] [Green Version]
- Wood, J.T.; Williams, J.S.; Pandarinathan, L.; Janero, D.R.; Lammi-Keefe, C.J.; Makriyannis, A. Dietary docosahexaenoic acid supplementation alters select physiological endocannabinoid-system metabolites in brain and plasma. J. Lipid Res. 2010, 51, 1416–1423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yurko-Mauro, K.; McCarthy, D.; Rom, D.; Nelson, E.B.; Ryan, A.S.; Blackwell, A.; Salem, N.; Stedman, M. MIDAS Investigators Beneficial effects of docosahexaenoic acid on cognition in age-related cognitive decline. Alzheimer’s Dement. 2010, 6, 456–464. [Google Scholar] [CrossRef]
- Kharebava, G.; Rashid, M.A.; Lee, J.-W.; Sarkar, S.; Kevala, K.; Kim, H.-Y. N-docosahexaenoylethanolamine regulates Hedgehog signaling and promotes growth of cortical axons. Biol. Open 2015, 4, 1660–1670. [Google Scholar] [CrossRef] [Green Version]
- Tyrtyshnaia, A.A.; Egorova, E.L.; Starinets, A.A.; Ponomarenko, A.I.; Ermolenko, E.V.; Manzhulo, I.V. N-Docosahexaenoylethanolamine Attenuates Neuroinflammation and Improves Hippocampal Neurogenesis in Rats with Sciatic Nerve Chronic Constriction Injury. Mar. Drugs 2020, 18, 516. [Google Scholar] [CrossRef]
- Kawakita, E.; Hashimoto, M.; Shido, O. Docosahexaenoic acid promotes neurogenesis in vitro and in vivo. Neuroscience 2006, 139, 991–997. [Google Scholar] [CrossRef]
- Cao, D.; Kevala, K.; Kim, J.; Moon, H.-S.; Jun, S.B.; Lovinger, D.; Kim, H.-Y. Docosahexaenoic acid promotes hippocampal neuronal development and synaptic function. J. Neurochem. 2009, 111, 510–521. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Calder, P. Omega-3 Fatty Acids and Inflammatory Processes. Nutrients 2010, 2, 355–374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sonti, S.; Duclos, R.I.; Tolia, M.; Gatley, S.J. N-Docosahexaenoylethanolamine (synaptamide): Carbon-14 radiolabeling and metabolic studies. Chem. Phys. Lipids 2018, 210, 90–97. [Google Scholar] [CrossRef] [PubMed]
- Walter, L.; Stella, N. Cannabinoids and neuroinflammation. Br. J. Pharmacol. 2004, 141, 775–785. [Google Scholar] [CrossRef] [Green Version]
- Eminatohara, K.; Eakiyoshi, M.; Okuno, H. Role of Immediate-Early Genes in Synaptic Plasticity and Neuronal Ensembles Underlying the Memory Trace. Front. Mol. Neurosci. 2016, 8, 78. [Google Scholar] [CrossRef] [Green Version]
- Orr, S.K.; Palumbo, S.; Bosetti, F.; Mount, H.T.; Kang, J.X.; Greenwood, C.E.; Ma, D.W.L.; Serhan, C.N.; Bazinet, R.P. Unesterified docosahexaenoic acid is protective in neuroinflammation. J. Neurochem. 2013, 127, 378–393. [Google Scholar] [CrossRef] [Green Version]
- Park, T.; Chen, H.; Kim, H.-Y. GPR110 (ADGRF1) mediates anti-inflammatory effects of N-docosahexaenoylethanolamine. J. Neuroinflamm. 2019, 16, 1–13. [Google Scholar] [CrossRef]
- Raker, V.K.; Ebecker, C.; Esteinbrink, K. The cAMP Pathway as Therapeutic Target in Autoimmune and Inflammatory Diseases. Front. Immunol. 2016, 7, 123. [Google Scholar] [CrossRef] [Green Version]
- Berger, A.; Crozier, G.; Bisogno, T.; Cavaliere, P.; Innis, S.; Di Marzo, V. Anandamide and diet: Inclusion of dietary arachidonate and docosahexaenoate leads to increased brain levels of the corresponding N-acylethanolamines in piglets. Proc. Natl. Acad. Sci. USA 2001, 98, 6402–6406. [Google Scholar] [CrossRef] [Green Version]
- Hou, Y.; Xie, G.; Liu, X.; Li, G.; Jia, C.; Xu, J.; Wang, B. Minocycline protects against lipopolysaccharide-induced cognitive impairment in mice. Psychopharmacol. 2015, 233, 905–916. [Google Scholar] [CrossRef]
- Song, J.; Kang, S.M.; Lee, K.M.; Lee, J.E. The Protective Effect of Melatonin on Neural Stem Cell against LPS-Induced Inflammation. BioMed Res. Int. 2015, 2015, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Magó, Á.; Weber, J.P.; Ujfalussy, B.B.; Makara, J.K. Synaptic Plasticity Depends on the Fine-Scale Input Pattern in Thin Dendrites of CA1 Pyramidal Neurons. J. Neurosci. 2020, 40, 2593–2605. [Google Scholar] [CrossRef]
- Neumann, H.; Schweigreiter, R.; Yamashita, T.; Rosenkranz, K.; Wekerle, H.; Barde, Y.-A. Tumor Necrosis Factor Inhibits Neurite Outgrowth and Branching of Hippocampal Neurons by a Rho-Dependent Mechanism. J. Neurosci. 2002, 22, 854–862. [Google Scholar] [CrossRef] [PubMed]
- Clarke, D.J.; Branton, R.L. IL-1β is released from the host brain following transplantation but does not compromise embryonic dopaminergic neuron survival. Brain Res. 2002, 952, 78–85. [Google Scholar] [CrossRef]
- Li, Y.; Liu, L.; Barger, S.W.; Griffin, W.S.T. Interleukin-1 Mediates Pathological Effects of Microglia on Tau Phosphorylation and on Synaptophysin Synthesis in Cortical Neurons through a p38-MAPK Pathway. J. Neurosci. 2003, 23, 1605–1611. [Google Scholar] [CrossRef] [Green Version]
- Sheppard, O.; Coleman, M.P.; Durrant, C.S. Lipopolysaccharide-induced neuroinflammation induces presynaptic disruption through a direct action on brain tissue involving microglia-derived interleukin 1 beta. J. Neuroinflamm. 2019, 16, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Wang, K.; White, T.D. The Bacterial Endotoxin Lipopolysaccharide Causes Rapid Inappropriate Excitation in Rat Cortex. J. Neurochem. 1999, 72, 652–660. [Google Scholar] [CrossRef]
- Jo, J.-H.; Park, E.-J.; Lee, J.-K.; Jung, M.-W.; Lee, C.-J. Lipopolysaccharide inhibits induction of long-term potentiation and depression in the rat hippocampal CA1 area. Eur. J. Pharmacol. 2001, 422, 69–76. [Google Scholar] [CrossRef]
- Hoshino, K.; Hasegawa, K.; Kamiya, H.; Morimoto, Y. Synapse-specific effects of IL-1β on long-term potentiation in the mouse hippocampus. Biomed. Res. 2017, 38, 183–188. [Google Scholar] [CrossRef] [Green Version]
- Bellinger, F.; Madamba, S.; Campbell, I.; Siggins, G. Reduced long-term potentiation in the dentate gyrus of transgenic mice with cerebral overexpression of interleukin-6. Neurosci. Lett. 1995, 198, 95–98. [Google Scholar] [CrossRef]
- Nelson, T.E.; Engberink, A.O.; Hernandez, R.; Puro, A.; Huitron-Resendiz, S.; Hao, C.; De Graan, P.; Gruol, D.L. Altered synaptic transmission in the hippocampus of transgenic mice with enhanced central nervous systems expression of interleukin-6. Brain Behav. Immun. 2012, 26, 959–971. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosi, S.; Ramirez-Amaya, V.; Hauss-Wegrzyniak, B.; Wenk, G.L. Chronic brain inflammation leads to a decline in hippocampal NMDA-R1 receptors. J. Neuroinflamm. 2004, 1, 12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zou, J.; Crews, F. Glutamate/NMDA excitotoxicity and HMGB1/TLR4 neuroimmune toxicity converge as components of neurodegeneration. AIMS Mol. Sci. 2015, 2, 77–100. [Google Scholar] [CrossRef]
- Flores-Soto, M.E.; Chaparro-Huerta, V.; Escoto-Delgadillo, M.; Vázquez-Valls, E.; Gonzalez-Castañeda, R.; Beas-Zárate, C. Estructura y función de las subunidades del receptor a glutamato tipo NMDA. Neurología 2012, 27, 301–310. [Google Scholar] [CrossRef] [PubMed]
- Bal-Price, A.; Brown, G.C. Inflammatory Neurodegeneration Mediated by Nitric Oxide from Activated Glia-Inhibiting Neuronal Respiration, Causing Glutamate Release and Excitotoxicity. J. Neurosci. 2001, 21, 6480–6491. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Blázquez, P.; Rodríguez-Muñoz, M.; Garzón-Niño, J. The cannabinoid receptor 1 associates with NMDA receptors to produce glutamatergic hypofunction: Implications in psychosis and schizophrenia. Front. Pharmacol. 2014, 4, 169. [Google Scholar] [CrossRef] [Green Version]
- Hu, H.; Ho, W.; Mackie, K.; Pittman, Q.J.; Sharkey, K.A. Brain CB1 receptor expression following lipopolysaccharide-induced inflammation. Neuroscience 2012, 227, 211–222. [Google Scholar] [CrossRef] [Green Version]
- Redondo, R.L.; Morris, R.G.M. Making memories last: The synaptic tagging and capture hypothesis. Nat. Rev. Neurosci. 2011, 12, 17–30. [Google Scholar] [CrossRef]
- Liu, X.; Ramirez, S.; Pang, P.T.; Puryear, C.B.; Govindarajan, A.; Deisseroth, K.; Tonegawa, S. Optogenetic stimulation of a hippocampal engram activates fear memory recall. Nat. Cell Biol. 2012, 484, 381–385. [Google Scholar] [CrossRef] [Green Version]
- Denny, C.A.; Kheirbek, M.A.; Alba, E.L.; Tanaka, K.F.; Brachman, R.A.; Laughman, K.B.; Tomm, N.K.; Turi, G.F.; Losonczy, A.; Hen, R. Hippocampal Memory Traces Are Differentially Modulated by Experience, Time, and Adult Neurogenesis. Neuron 2014, 83, 189–201. [Google Scholar] [CrossRef] [Green Version]
- Tanaka, K.Z.; Pevzner, A.; Hamidi, A.B.; Nakazawa, Y.; Graham, J.; Wiltgen, B.J. Cortical Representations Are Reinstated by the Hippocampus during Memory Retrieval. Neuron 2014, 84, 347–354. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Messaoudi, E.; Kanhema, T.; Soulé, J.; Tiron, A.; Dagyte, G.; Da Silva, B.; Bramham, C.R. Sustained Arc/Arg3.1 Synthesis Controls Long-Term Potentiation Consolidation through Regulation of Local Actin Polymerization in the Dentate Gyrus In Vivo. J. Neurosci. 2007, 27, 10445–10455. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bruel-Jungerman, E.; Laroche, S.; Rampon, C. New neurons in the dentate gyrus are involved in the expression of enhanced long-term memory following environmental enrichment. Eur. J. Neurosci. 2005, 21, 513–521. [Google Scholar] [CrossRef] [PubMed]
- Jessberger, S.; Clark, R.E.; Broadbent, N.J.; Clemenson, J.G.D.; Consiglio, A.; Lie, D.C.; Squire, L.R.; Gage, F.H. Dentate gyrus-specific knockdown of adult neurogenesis impairs spatial and object recognition memory in adult rats. Learn. Mem. 2009, 16, 147–154. [Google Scholar] [CrossRef] [Green Version]
- Vallières, L.; Campbell, I.L.; Gage, F.H.; Sawchenko, P.E. Reduced Hippocampal Neurogenesis in Adult Transgenic Mice with Chronic Astrocytic Production of Interleukin-6. J. Neurosci. 2002, 22, 486–492. [Google Scholar] [CrossRef] [Green Version]
- Perez-Asensio, F.J.; Perpiñá, U.; Planas, A.M.; Pozas, E. Interleukin-10 regulates progenitor differentiation and modulates neurogenesis in adult brain. J. Cell Sci. 2013, 126, 4208–4219. [Google Scholar] [CrossRef] [Green Version]
- Pereira, B.C.; Da Rocha, A.; Pauli, J.R.; Ropelle, E.R.; De Souza, C.; E Cintra, D.; Sant’Ana, M.; Da Silva, A.S. Excessive eccentric exercise leads to transitory hypothalamic inflammation, which may contribute to the low body weight gain and food intake in overtrained mice. Neuroscience 2015, 311, 231–242. [Google Scholar] [CrossRef]
- Chen, S.; Hillman, D.E. Transient c-fos expression and dendritic spine plasticity in hippocampal granule cells. Brain Res. 1992, 577, 169–174. [Google Scholar] [CrossRef]
- Ermolenko, E.V.; Latyshev, N.; Sultanov, R.; Kasyanov, S. Technological approach of 1-O-alkyl-sn-glycerols separation from Berryteuthis magister squid liver oil. J. Food Sci. Technol. 2016, 53, 1722–1726. [Google Scholar] [CrossRef] [Green Version]
- Ishiyama, T.; Tokuda, K.; Ishibashi, T.; Ito, A.; Toma, S.; Ohno, Y. Lurasidone (SM-13496), a novel atypical antipsychotic drug, reverses MK-801-induced impairment of learning and memory in the rat passive-avoidance test. Eur. J. Pharmacol. 2007, 572, 160–170. [Google Scholar] [CrossRef]
- Bevins, R.A.; Besheer, J. Object recognition in rats and mice: A one-trial non-matching-to-sample learning task to study ’recognition memory’. Nat. Protoc. 2006, 1, 1306–1311. [Google Scholar] [CrossRef] [PubMed]
- Sholl, D.A. Dendritic organization in the neurons of the visual and motor cortices of the cat. J. Anat. 1953, 87, 387–406. [Google Scholar] [PubMed]
- Bastian, T.W.; Duck, K.A.; Michalopoulos, G.C.; Chen, M.J.; Liu, Z.-J.; Connor, J.R.; Lanier, L.M.; Sola-Visner, M.C.; Georgieff, M.K. Eltrombopag, a thrombopoietin mimetic, crosses the blood-brain barrier and impairs iron-dependent hippocampal neuron dendrite development. J. Thromb. Haemost. 2017, 15, 565–574. [Google Scholar] [CrossRef] [PubMed] [Green Version]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tyrtyshnaia, A.; Bondar, A.; Konovalova, S.; Sultanov, R.; Manzhulo, I. N-Docosahexanoylethanolamine Reduces Microglial Activation and Improves Hippocampal Plasticity in a Murine Model of Neuroinflammation. Int. J. Mol. Sci. 2020, 21, 9703. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21249703
Tyrtyshnaia A, Bondar A, Konovalova S, Sultanov R, Manzhulo I. N-Docosahexanoylethanolamine Reduces Microglial Activation and Improves Hippocampal Plasticity in a Murine Model of Neuroinflammation. International Journal of Molecular Sciences. 2020; 21(24):9703. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21249703
Chicago/Turabian StyleTyrtyshnaia, Anna, Anatoly Bondar, Sophia Konovalova, Ruslan Sultanov, and Igor Manzhulo. 2020. "N-Docosahexanoylethanolamine Reduces Microglial Activation and Improves Hippocampal Plasticity in a Murine Model of Neuroinflammation" International Journal of Molecular Sciences 21, no. 24: 9703. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21249703