EHMT2 Inhibition Induces Cell Death in Human Non-Small Cell Lung Cancer by Altering the Cholesterol Biosynthesis Pathway


Abstract
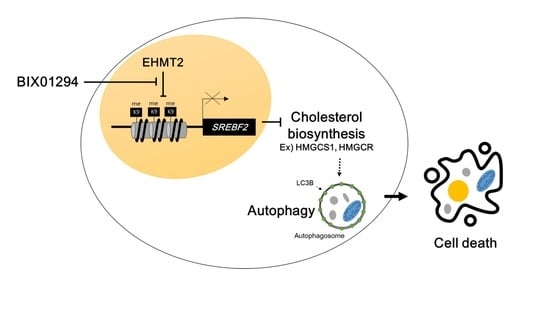
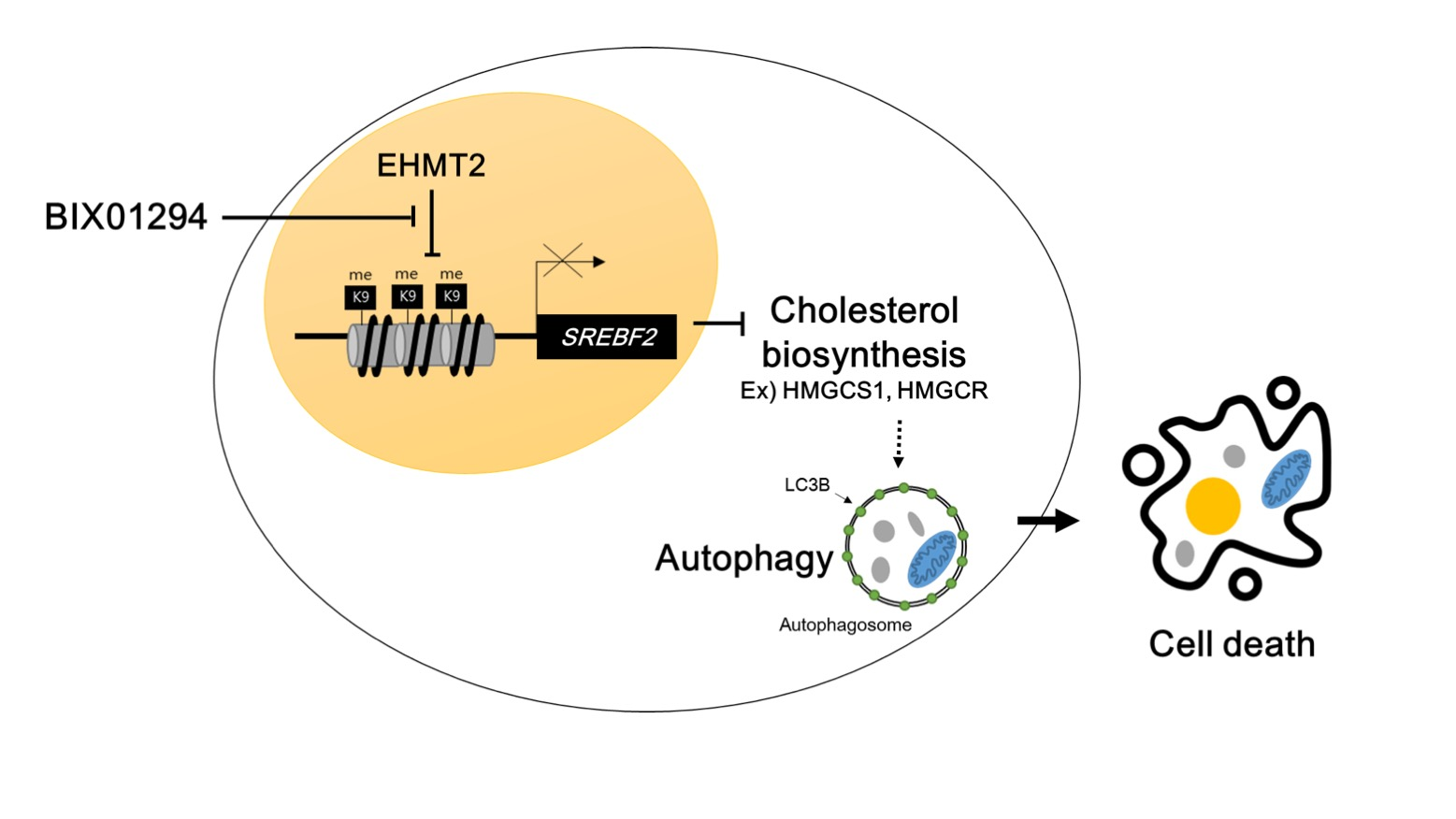
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
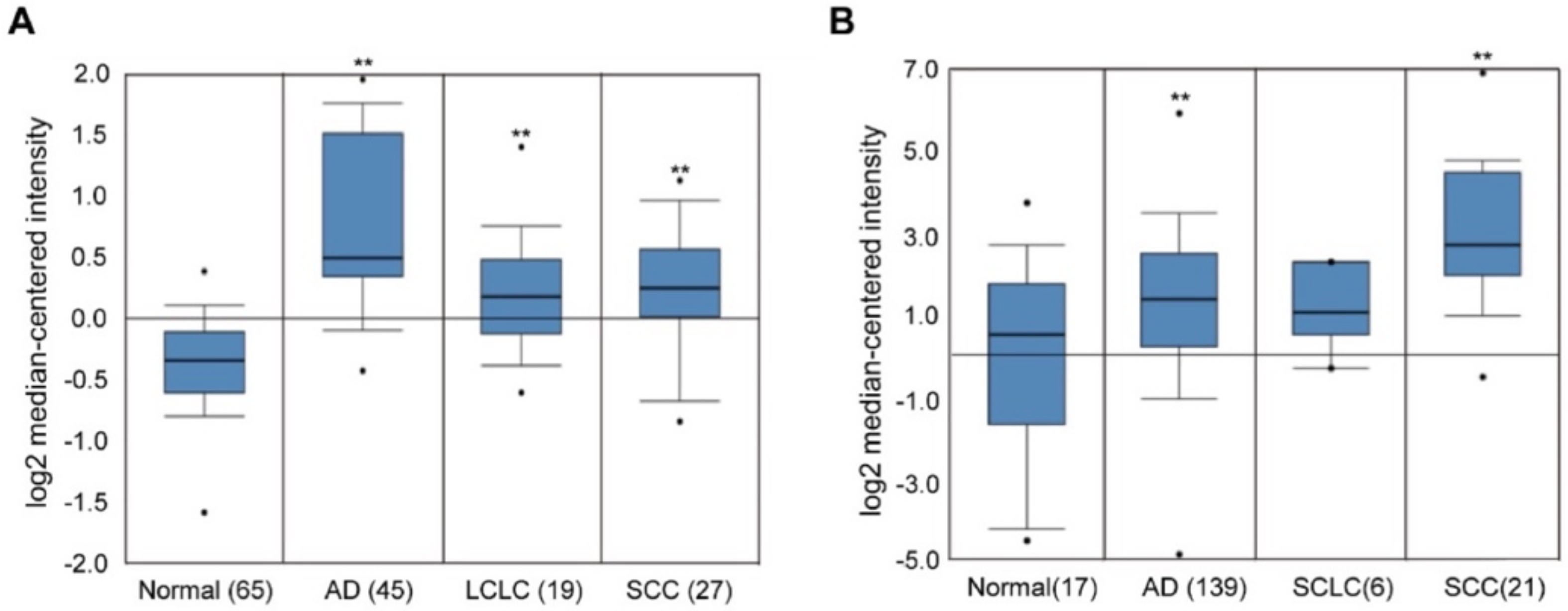
2.1. Overexpression of EHMT2 in NSCLC
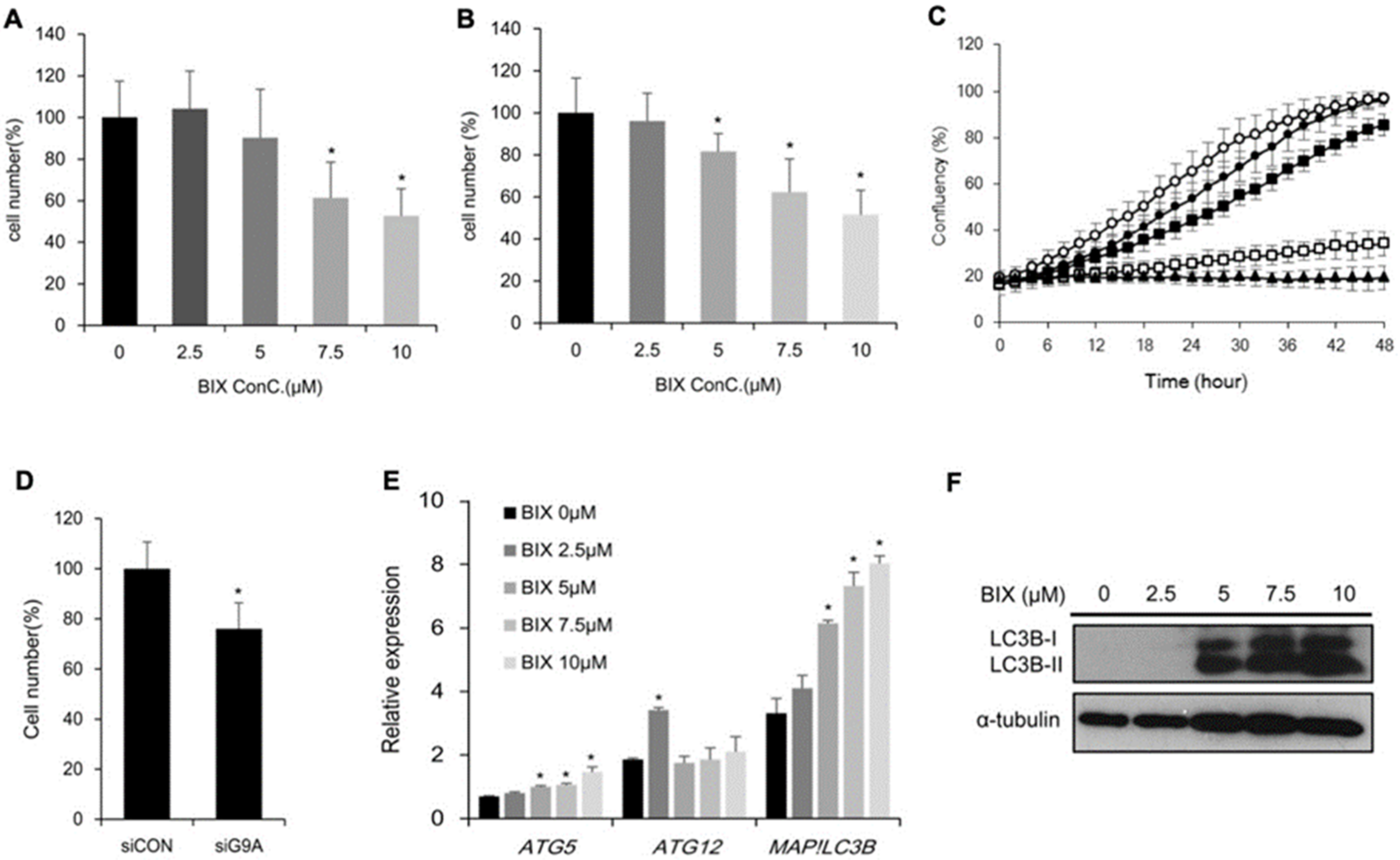
2.2. Effects of EHMT2 Regulation on Cell Viability
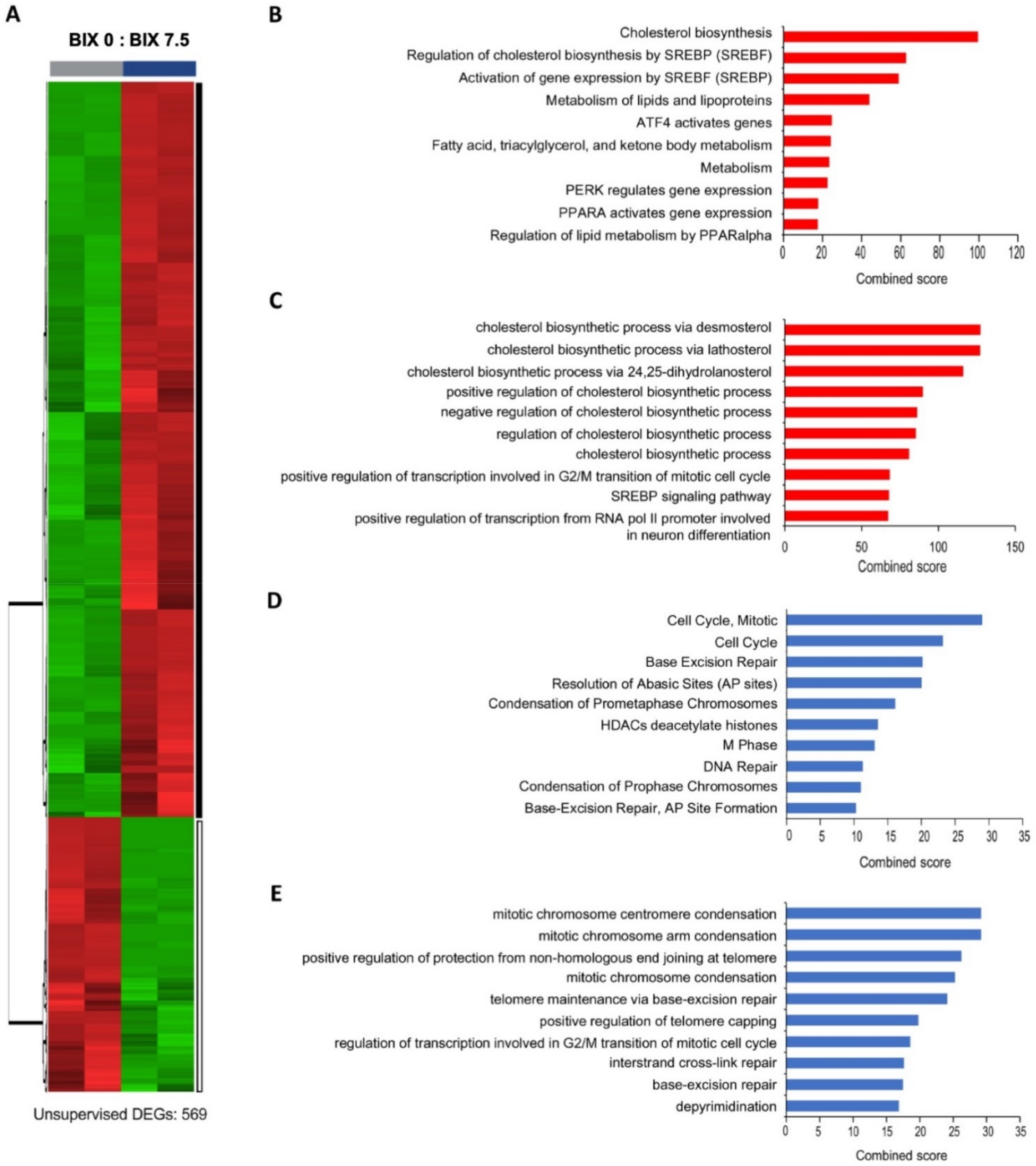
2.3. Distinct Gene Expression Profiles with EHMT2 Inhibition in H1299 Cells
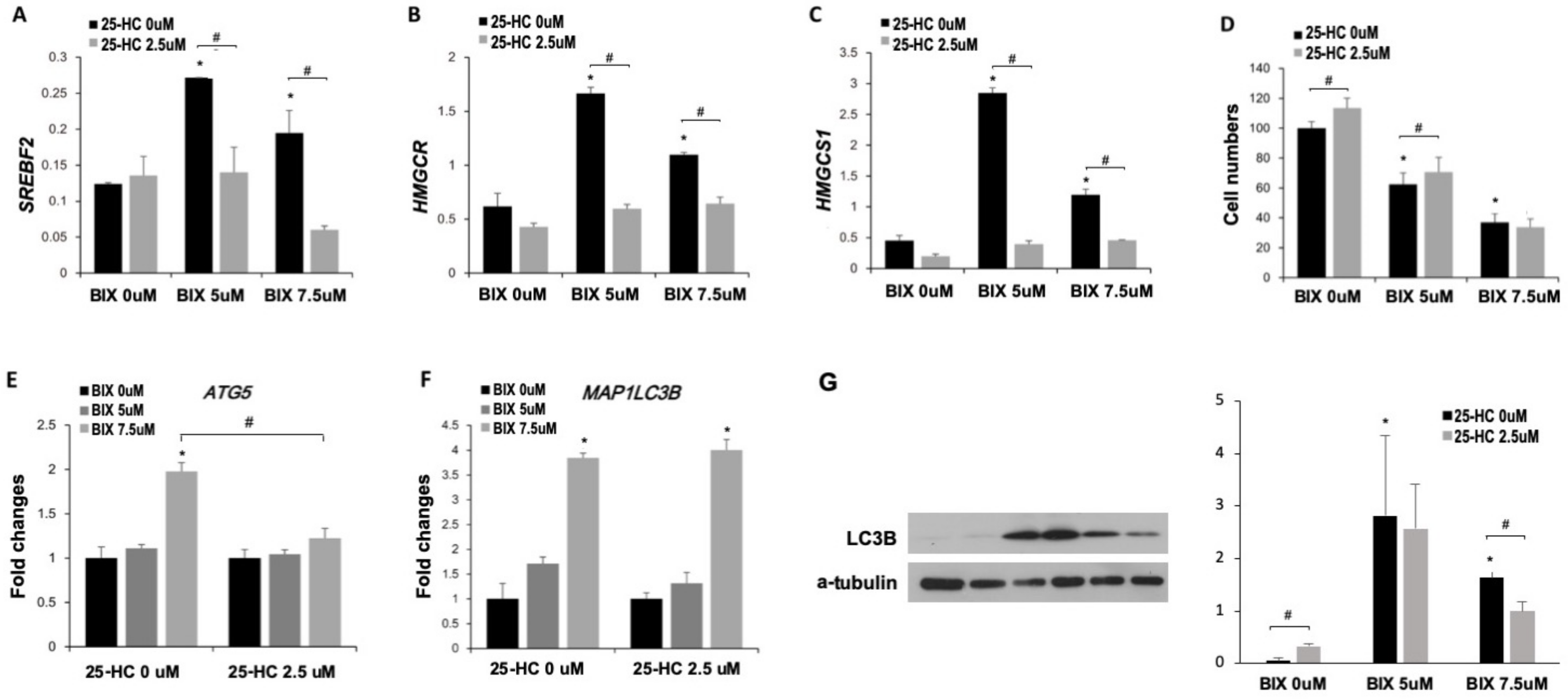
2.4. Induction of Cholesterol Biosynthesis Pathway by BIX Treatment
2.5. Involvement of Cholesterol Biosynthesis in BIX-Induced Cell Death
3. Discussion
4. Materials and Methods
4.1. Cell Culture and Treatments
4.2. Small Interfering RNA Transfection
4.3. MTT Assay for Cell Viability
4.4. IncuCyte Zoom Assays
4.5. RNA Isolation and Quantitative Reverse Transcription RT-PCR
4.6. Chromatin Immunoprecipitation (ChIP) Analysis
4.7. Western Blot Analysis
4.8. RNA-Sequencing Analysis
4.9. Statistics
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| 25-HC | 25-hydroxycholesterol |
| AD | Adenocarcinoma |
| ATF4 | Activating transcription factor 4 |
| ATG | Autophagy-related gene |
| BIX | BIX01294 |
| BP | Biological process |
| ChIP | Chromatin immunoprecipitation |
| DEG | Differentially expressed gene |
| DMSO | Dimethyl sulforxide |
| EHMT2 | Euchromatic histone-lysine N-methyltransferase 2 |
| FACS | Flow cytometry analysis |
| GO | Gene Ontology |
| H3K9 | Histone H3 lysine 9 |
| H3K9me1 | Histone H3 lysine 9 mono-methylation |
| H3K9me2 | Histone H3 lysine 9 di-methylation |
| HMGCR | HMG-CoA synthase 1 |
| HMGCS1 | HMG-CoA reductase |
| LCLC | Large cell carcinoma |
| MAP1LC3B | Microtubule-associated proteins 1A/1B light chain 3B |
| MTT | Thiazolyl blue tetrazolium bromide |
| NSCLC | Non-small cell lung cancer |
| qRT-PCR | Quantitative real-time PCR |
| SCC | Squamous cell cancer |
| SD | Standard deviation |
| siATF4 | Small interfering RNA targeting ATF4 |
| siCON | Non-targeting small interfering RNA |
| siEHMT2 | Small interfering RNA targeting EHMT2 |
| SREBF | Sterol regulatory element-binding transcription factor |
References
- World Health Organization; International Agency for Research on Cancer. World Cancer Report 2014; WHO Press: Geneva, Switzerland, 2015; pp. 17–19. [Google Scholar]
- Duma, N.; Santana-Davila, R.; Molina, J.R. Non-Small Cell Lung Cancer: Epidemiology, Screening, Diagnosis and Treatment. Mayo Clin. Proc. 2019, 94, 1623–1640. [Google Scholar]
- Zochbauer-Muller, S.; Fong, K.M.; Virmani, A.K.; Geradts, J.; Gazdar, A.F.; Minna, J.D. Aberrant promoter methylation of multiple genes in non-small cell lung cancers. Cancer Res. 2001, 61, 249–255. [Google Scholar]
- Sasaki, H.; Moriyama, S.; Nakashima, Y.; Kobayashi, Y.; Kiriyama, M.; Fukai, I.; Yamakawa, Y.; Fujii, Y. Histone deacetylase 1 mRNA expression in lung cancer. Lung Cancer 2004, 46, 171–178. [Google Scholar] [CrossRef]
- Flavahan, W.A.; Gaskell, E.; Bernstein, B.E. Epigenetic plasticity and the hallmarks of cancer. Science 2017, 357. [Google Scholar] [CrossRef] [Green Version]
- Lindroth, A.M.; Park, Y.J.; Plass, C. Epigenetic Reprogramming in Cancer. In Epigenetic Mechanisms in Cellular Reprogramming; Meissner, A., Walter, J., Eds.; Springer: Berlin/Heidelberg, Germany, 2015; pp. 193–223. [Google Scholar] [CrossRef]
- Tachibana, M.; Sugimoto, K.; Fukushima, T.; Shinkai, Y. Set domain-containing protein, G9a, is a novel lysine-preferring mammalian histone methyltransferase with hyperactivity and specific selectivity to lysines 9 and 27 of histone H3. J. Biol. Chem. 2001, 276, 25309–25317. [Google Scholar] [CrossRef] [Green Version]
- Chen, M.W.; Hua, K.T.; Kao, H.J.; Chi, C.C.; Wei, L.H.; Johansson, G.; Shiah, S.G.; Chen, P.S.; Jeng, Y.M.; Cheng, T.Y.; et al. H3K9 histone methyltransferase G9a promotes lung cancer invasion and metastasis by silencing the cell adhesion molecule Ep-CAM. Cancer Res. 2010, 70, 7830–7840. [Google Scholar] [CrossRef] [Green Version]
- Hua, K.T.; Wang, M.Y.; Chen, M.W.; Wei, L.H.; Chen, C.K.; Ko, C.H.; Jeng, Y.M.; Sung, P.L.; Jan, Y.H.; Hsiao, M.; et al. The H3K9 methyltransferase G9a is a marker of aggressive ovarian cancer that promotes peritoneal metastasis. Mol. Cancer 2014, 13, 189. [Google Scholar] [CrossRef] [Green Version]
- Huang, T.; Zhang, P.; Li, W.; Zhao, T.; Zhang, Z.; Chen, S.; Yang, Y.; Feng, Y.; Li, F.; Shirley Liu, X.; et al. G9A promotes tumor cell growth and invasion by silencing CASP1 in non-small-cell lung cancer cells. Cell Death Dis. 2017, 8, e2726. [Google Scholar] [CrossRef] [Green Version]
- Cho, H.S.; Kelly, J.D.; Hayami, S.; Toyokawa, G.; Takawa, M.; Yoshimatsu, M.; Tsunoda, T.; Field, H.I.; Neal, D.E.; Ponder, B.A.; et al. Enhanced expression of EHMT2 is involved in the proliferation of cancer cells through negative regulation of SIAH1. Neoplasia 2011, 13, 676–684. [Google Scholar] [CrossRef] [Green Version]
- Ding, J.; Li, T.; Wang, X.; Zhao, E.; Choi, J.H.; Yang, L.; Zha, Y.; Dong, Z.; Huang, S.; Asara, J.M.; et al. The histone H3 methyltransferase G9A epigenetically activates the serine-glycine synthesis pathway to sustain cancer cell survival and proliferation. Cell Metab. 2013, 18, 896–907. [Google Scholar] [CrossRef] [Green Version]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [Green Version]
- Carmeliet, P.; Dor, Y.; Herbert, J.M.; Fukumura, D.; Brusselmans, K.; Dewerchin, M.; Neeman, M.; Bono, F.; Abramovitch, R.; Maxwell, P.; et al. Role of HIF-1alpha in hypoxia-mediated apoptosis, cell proliferation and tumour angiogenesis. Nature 1998, 394, 485–490. [Google Scholar] [CrossRef]
- Izuishi, K.; Kato, K.; Ogura, T.; Kinoshita, T.; Esumi, H. Remarkable Tolerance of Tumor Cells to Nutrient Deprivation: Possible New Biochemical Target for Cancer Therapy. Cancer Res. 2000, 60, 6201. [Google Scholar]
- Boroughs, L.K.; DeBerardinis, R.J. Metabolic pathways promoting cancer cell survival and growth. Nat. Cell Biol. 2015, 17, 351–359. [Google Scholar] [CrossRef] [Green Version]
- Furuta, E.; Okuda, H.; Kobayashi, A.; Watabe, K. Metabolic genes in cancer: Their roles in tumor progression and clinical implications. Biochim. Biophys. Acta 2010, 1805, 141–152. [Google Scholar] [CrossRef] [Green Version]
- Santos, C.R.; Schulze, A. Lipid metabolism in cancer. FEBS J. 2012, 279, 2610–2623. [Google Scholar] [CrossRef]
- Maxfield, F.R.; van Meer, G. Cholesterol, the central lipid of mammalian cells. Curr. Opin. Cell Biol. 2010, 22, 422–429. [Google Scholar] [CrossRef] [Green Version]
- Larsson, O. HMG-CoA reductase inhibitors: Role in normal and malignant cells. Crit. Rev. Oncol. Hematol. 1996, 22, 197–212. [Google Scholar]
- Kuzu, O.F.; Noory, M.A.; Robertson, G.P. The Role of Cholesterol in Cancer. Cancer Res. 2016, 76, 2063. [Google Scholar]
- Horton, J.D. Sterol regulatory element-binding proteins: Transcriptional activators of lipid synthesis. Biochem. Soc. Trans. 2002, 30, 1091–1095. [Google Scholar] [CrossRef] [Green Version]
- Lewis, C.A.; Brault, C.; Peck, B.; Bensaad, K.; Griffiths, B.; Mitter, R.; Chakravarty, P.; East, P.; Dankworth, B.; Alibhai, D.; et al. SREBP maintains lipid biosynthesis and viability of cancer cells under lipid- and oxygen-deprived conditions and defines a gene signature associated with poor survival in glioblastoma multiforme. Oncogene 2015, 34, 5128–5140. [Google Scholar] [CrossRef]
- Rhodes, D.R.; Yu, J.; Shanker, K.; Deshpande, N.; Varambally, R.; Ghosh, D.; Barrette, T.; Pandey, A.; Chinnaiyan, A.M. ONCOMINE: A cancer microarray database and integrated data-mining platform. Neoplasia 2004, 6, 1–6. [Google Scholar]
- Hou, J.; Aerts, J.; den Hamer, B.; van Ijcken, W.; den Bakker, M.; Riegman, P.; van der Leest, C.; van der Spek, P.; Foekens, J.A.; Hoogsteden, H.C.; et al. Gene expression-based classification of non-small cell lung carcinomas and survival prediction. PLoS ONE 2010, 5, e10312. [Google Scholar] [CrossRef]
- Bhattacharjee, A.; Richards, W.G.; Staunton, J.; Li, C.; Monti, S.; Vasa, P.; Ladd, C.; Beheshti, J.; Bueno, R.; Gillette, M.; et al. Classification of human lung carcinomas by mRNA expression profiling reveals distinct adenocarcinoma subclasses. Proc. Natl. Acad. Sci. USA 2001, 98, 13790–13795. [Google Scholar] [CrossRef] [Green Version]
- Faust, P.L.; Kovacs, W.J. Cholesterol biosynthesis and ER stress in peroxisome deficiency. Biochimie 2014, 98, 75–85. [Google Scholar] [CrossRef]
- Adams, C.M.; Reitz, J.; De Brabander, J.K.; Feramisco, J.D.; Li, L.; Brown, M.S.; Goldstein, J.L. Cholesterol and 25-hydroxycholesterol inhibit activation of SREBPs by different mechanisms, both involving SCAP and Insigs. J. Biol. Chem. 2004, 279, 52772–52780. [Google Scholar] [CrossRef] [Green Version]
- Cui, J.; Sun, W.; Hao, X.; Wei, M.; Su, X.; Zhang, Y.; Su, L.; Liu, X. EHMT2 inhibitor BIX-01294 induces apoptosis through PMAIP1-USP9X-MCL1 axis in human bladder cancer cells. Cancer Cell Int. 2015, 15, 4. [Google Scholar] [CrossRef] [Green Version]
- Kim, Y.; Kim, Y.S.; Kim, D.E.; Lee, J.S.; Song, J.H.; Kim, H.G.; Cho, D.H.; Jeong, S.Y.; Jin, D.H.; Jang, S.J.; et al. BIX-01294 induces autophagy-associated cell death via EHMT2/G9a dysfunction and intracellular reactive oxygen species production. Autophagy 2013, 9, 2126–2139. [Google Scholar] [CrossRef]
- Shimizu, S.; Kanaseki, T.; Mizushima, N.; Mizuta, T.; Arakawa-Kobayashi, S.; Thompson, C.B.; Tsujimoto, Y. Role of Bcl-2 family proteins in a non-apoptotic programmed cell death dependent on autophagy genes. Nat. Cell Biol. 2004, 6, 1221–1228. [Google Scholar] [CrossRef]
- Wesselborg, S.; Stork, B. Autophagy signal transduction by ATG proteins: From hierarchies to networks. Cell. Mol. Life Sci. 2015, 72, 4721–4757. [Google Scholar] [CrossRef] [Green Version]
- Otomo, C.; Metlagel, Z.; Takaesu, G.; Otomo, T. Structure of the human ATG12~ATG5 conjugate required for LC3 lipidation in autophagy. Nat. Struct. Mol. Biol. 2013, 20, 59–66. [Google Scholar] [CrossRef] [Green Version]
- An, P.N.T.; Shimaji, K.; Tanaka, R.; Yoshida, H.; Kimura, H.; Fukusaki, E.; Yamaguchi, M. Epigenetic regulation of starvation-induced autophagy in Drosophila by histone methyltransferase G9a. Sci. Rep. 2017, 7, 7343. [Google Scholar] [CrossRef] [Green Version]
- Fan, J.D.; Lei, P.J.; Zheng, J.Y.; Wang, X.; Li, S.; Liu, H.; He, Y.L.; Wang, Z.N.; Wei, G.; Zhang, X.; et al. The selective activation of p53 target genes regulated by SMYD2 in BIX-01294 induced autophagy-related cell death. PLoS ONE 2015, 10, e0116782. [Google Scholar] [CrossRef] [Green Version]
- Cao, W.; Ribeiro Rde, O.; Liu, D.; Saintigny, P.; Xia, R.; Xue, Y.; Lin, R.; Mao, L.; Ren, H. EZH2 promotes malignant behaviors via cell cycle dysregulation and its mRNA level associates with prognosis of patient with non-small cell lung cancer. PLoS ONE 2012, 7, e52984. [Google Scholar] [CrossRef] [Green Version]
- Seo, Y.K.; Jeon, T.-I.; Chong, H.K.; Beisinger, J.; Xie, X.; Osborne, T.F. Genome-wide Localization of SREBP-2 in Hepatic Chromatin Predicts a Role in Autophagy. Cell Metab. 2011, 13, 367–375. [Google Scholar] [CrossRef] [Green Version]
- Lim, S.C.; Parajuli, K.R.; Duong, H.Q.; Choi, J.E.; Han, S.I. Cholesterol induces autophagic and apoptotic death in gastric carcinoma cells. Int. J. Oncol. 2014, 44, 805–811. [Google Scholar] [CrossRef] [Green Version]
- Cheng, J.; Ohsaki, Y.; Tauchi-Sato, K.; Fujita, A.; Fujimoto, T. Cholesterol depletion induces autophagy. Biochem. Biophys. Res. Commun. 2006, 351, 246–252. [Google Scholar] [CrossRef]
- Cruz, P.M.R.; Mo, H.; McConathy, W.J.; Sabnis, N.; Lacko, A.G. The role of cholesterol metabolism and cholesterol transport in carcinogenesis: A review of scientific findings, relevant to future cancer therapeutics. Front. Pharmacol. 2013, 4, 119. [Google Scholar] [CrossRef] [Green Version]
- Sloan, C.A.; Chan, E.T.; Davidson, J.M.; Malladi, V.S.; Strattan, J.S.; Hitz, B.C.; Gabdank, I.; Narayanan, A.K.; Ho, M.; Lee, B.T.; et al. ENCODE data at the ENCODE portal. Nucleic Acids Res. 2016, 44, D726–D732. [Google Scholar] [CrossRef] [Green Version]
- Dunham, I.; Kundaje, A.; Aldred, S. An integrated encyclopedia of DNA elements in the human genome. Nature 2012, 489, 57–74. [Google Scholar] [CrossRef]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T.; et al. Gene ontology: Tool for the unification of biology. The Gene Ontology Consortium. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef] [Green Version]
- Croft, D.; O’Kelly, G.; Wu, G.; Haw, R.; Gillespie, M.; Matthews, L.; Caudy, M.; Garapati, P.; Gopinath, G.; Jassal, B.; et al. Reactome: A database of reactions, pathways and biological processes. Nucleic Acids Res. 2011, 39, D691–D697. [Google Scholar] [CrossRef]
- Kuleshov, M.V.; Jones, M.R.; Rouillard, A.D.; Fernandez, N.F.; Duan, Q.; Wang, Z.; Koplev, S.; Jenkins, S.L.; Jagodnik, K.M.; Lachmann, A.; et al. Enrichr: A comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res. 2016, 44, W90–W97. [Google Scholar] [CrossRef] [Green Version]





© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, H.; Choi, S.Y.; Lim, J.; Lindroth, A.M.; Park, Y.J. EHMT2 Inhibition Induces Cell Death in Human Non-Small Cell Lung Cancer by Altering the Cholesterol Biosynthesis Pathway. Int. J. Mol. Sci. 2020, 21, 1002. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21031002
Kim H, Choi SY, Lim J, Lindroth AM, Park YJ. EHMT2 Inhibition Induces Cell Death in Human Non-Small Cell Lung Cancer by Altering the Cholesterol Biosynthesis Pathway. International Journal of Molecular Sciences. 2020; 21(3):1002. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21031002
Chicago/Turabian StyleKim, Haeun, Seo Yoon Choi, Jinyeong Lim, Anders M. Lindroth, and Yoon Jung Park. 2020. "EHMT2 Inhibition Induces Cell Death in Human Non-Small Cell Lung Cancer by Altering the Cholesterol Biosynthesis Pathway" International Journal of Molecular Sciences 21, no. 3: 1002. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21031002