High Dosage Lithium Treatment Induces DNA Damage and p57Kip2 Decrease

 , ,
, , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
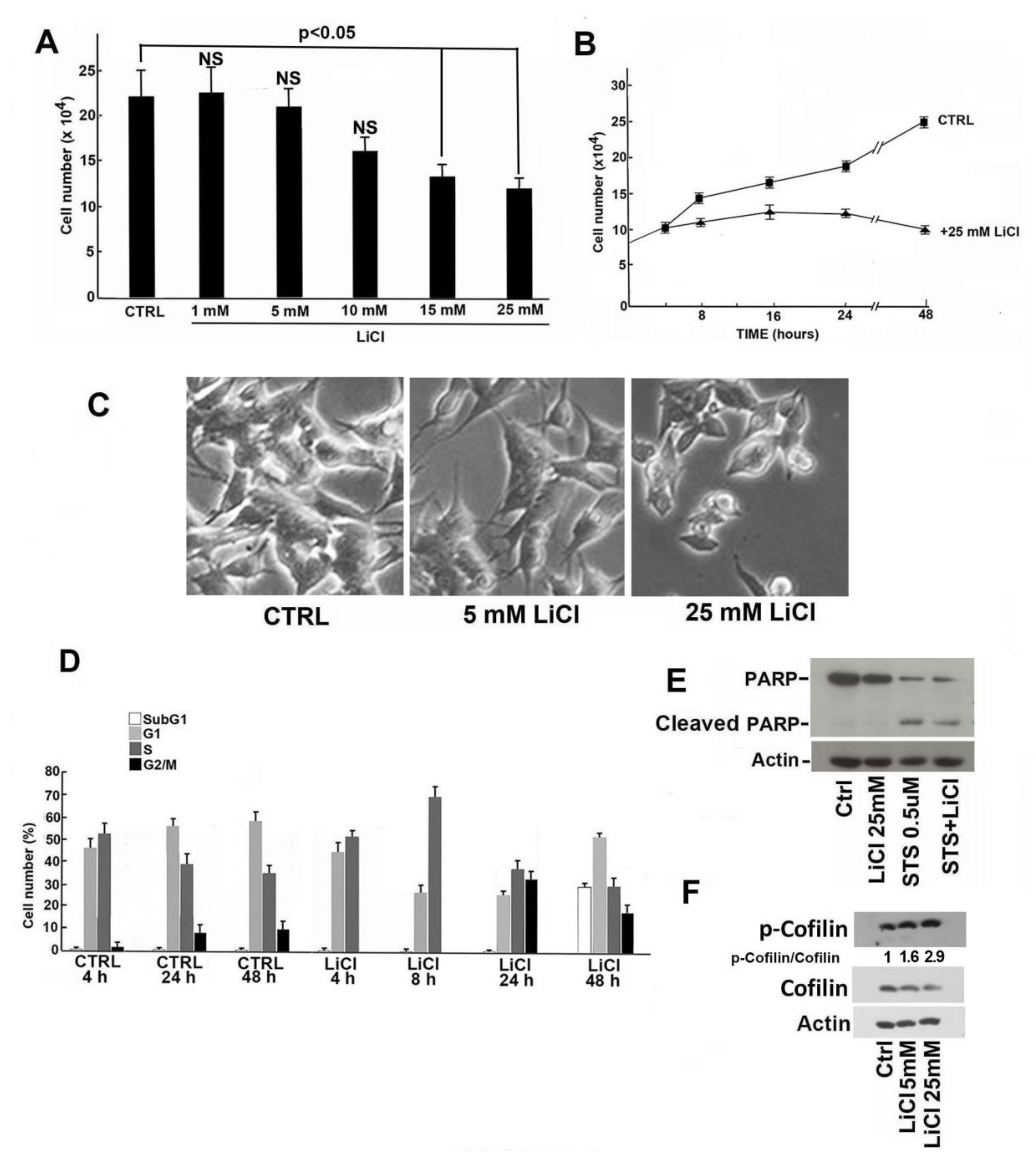
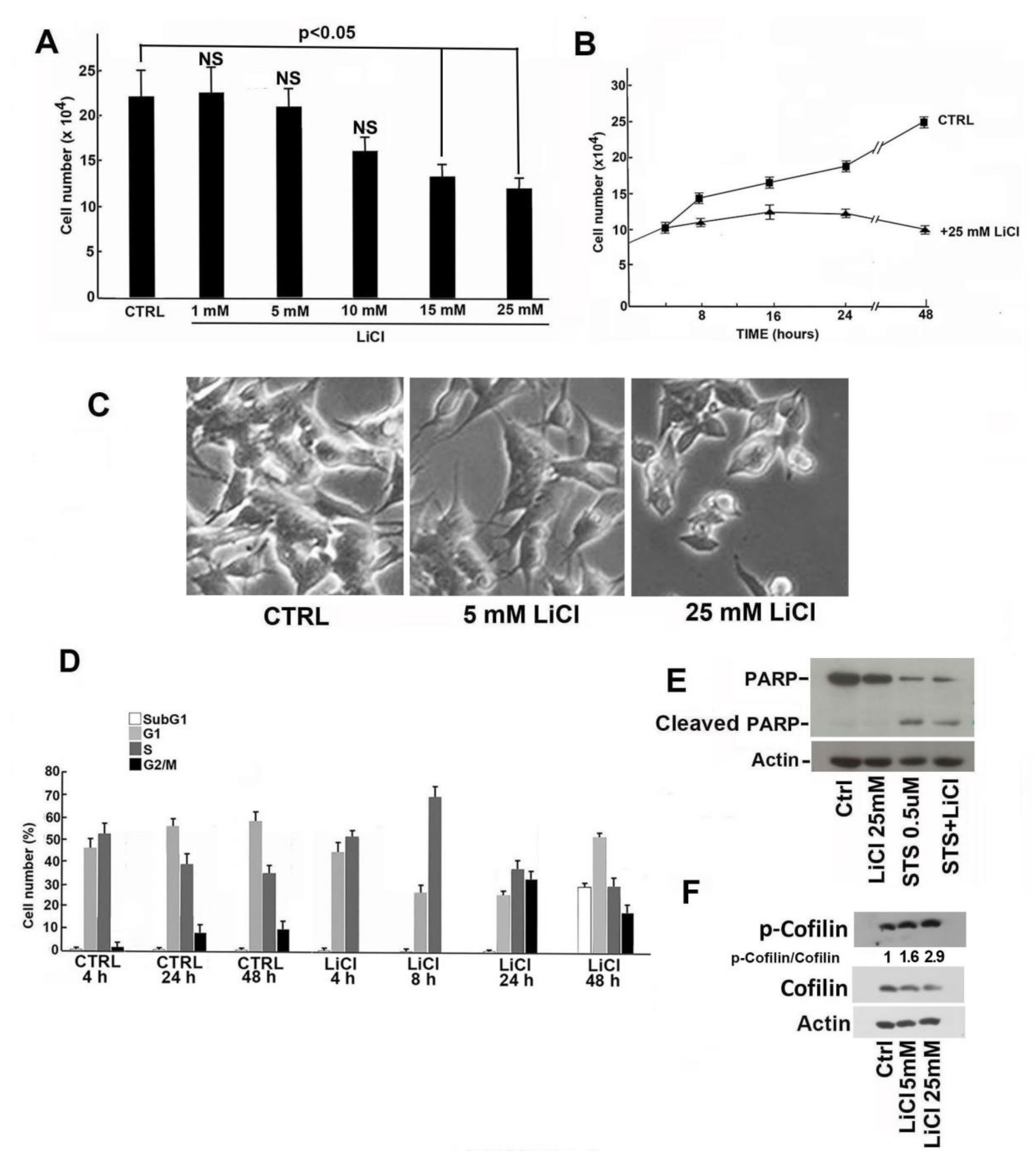
2.1. Proliferation Rate and Viability Reduction of SH-SY5Y Cells Induced by LiCl Treatment
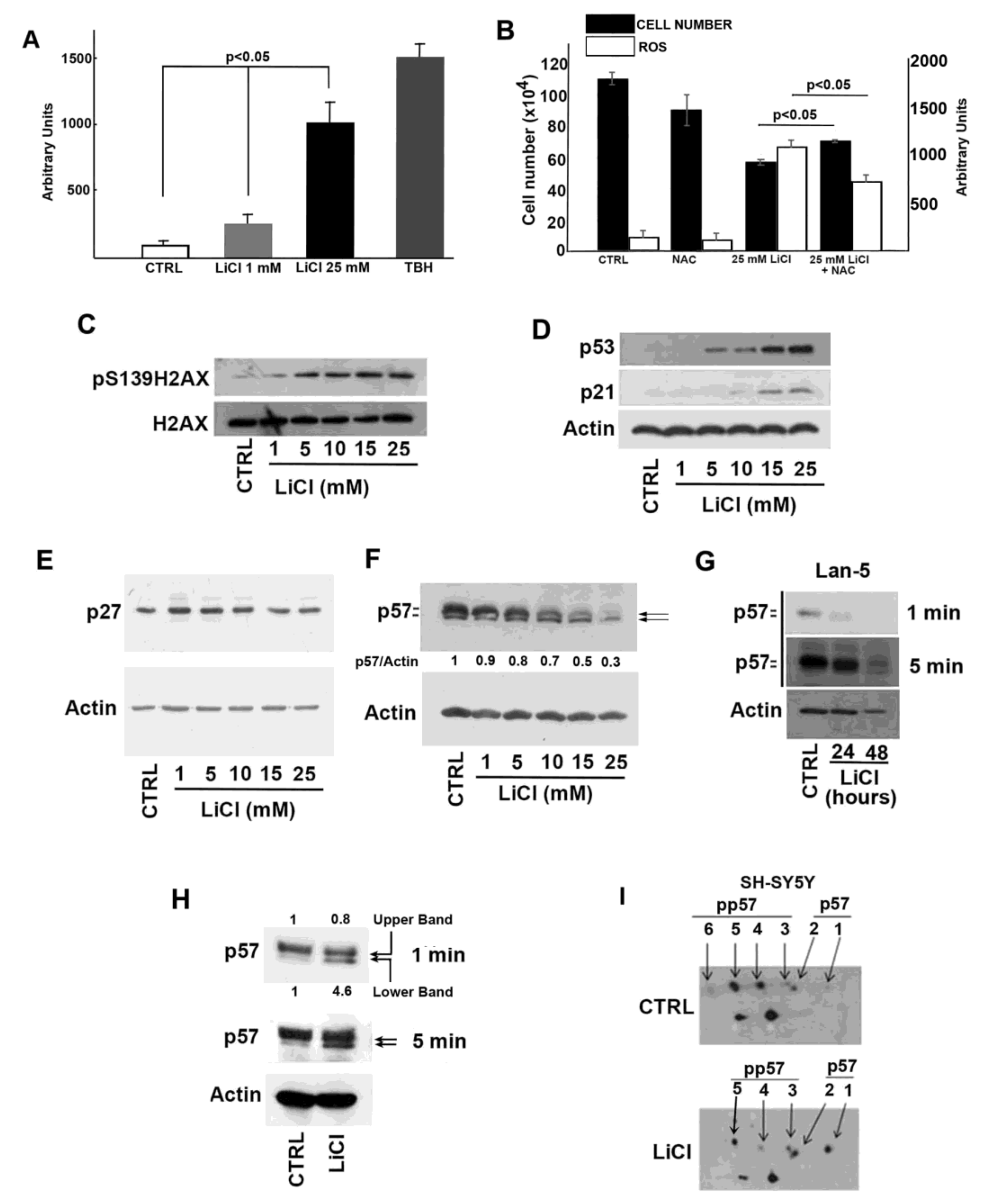
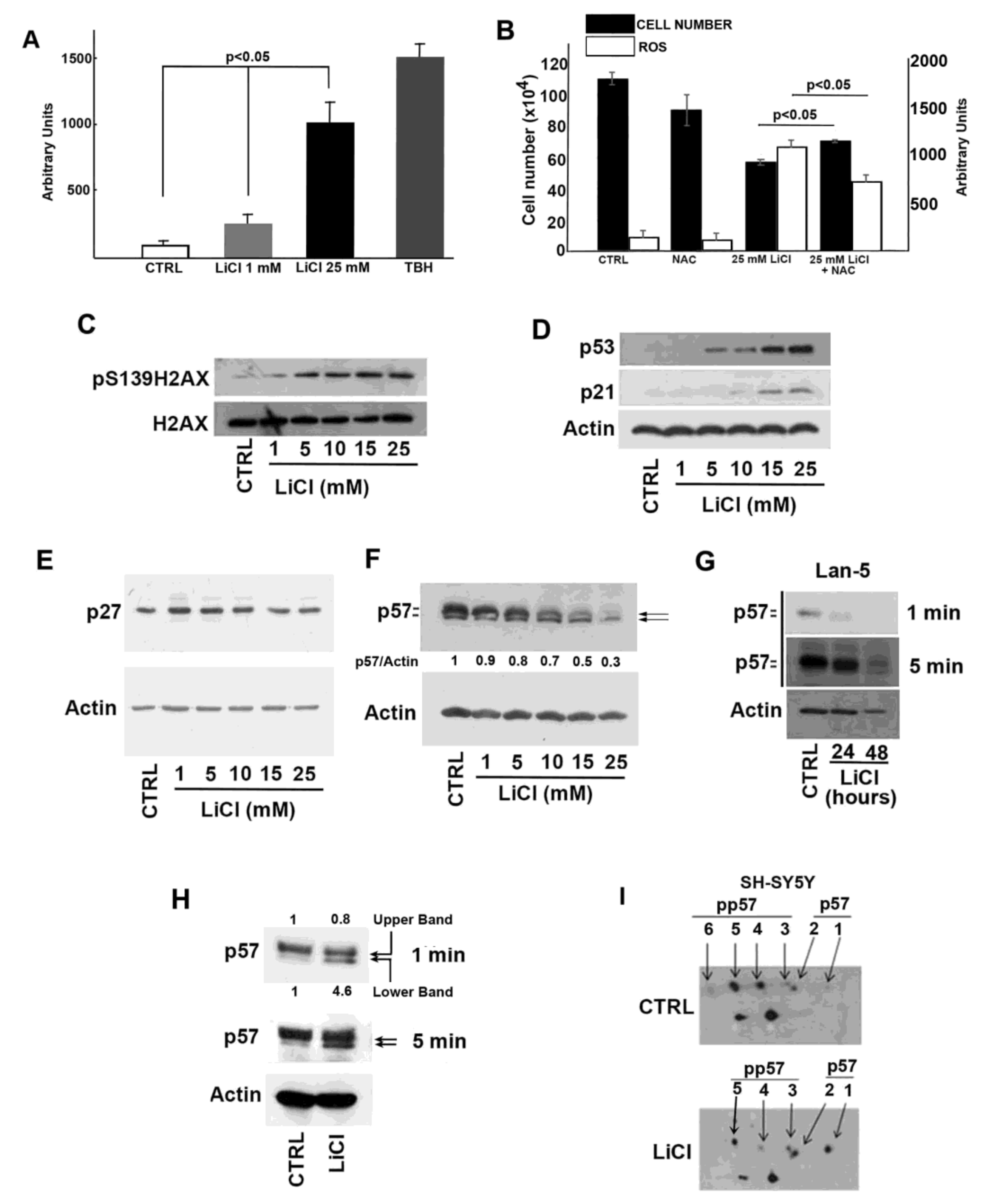
2.2. Oxidative and Genotoxic Stress Determined by LiCl Treatment
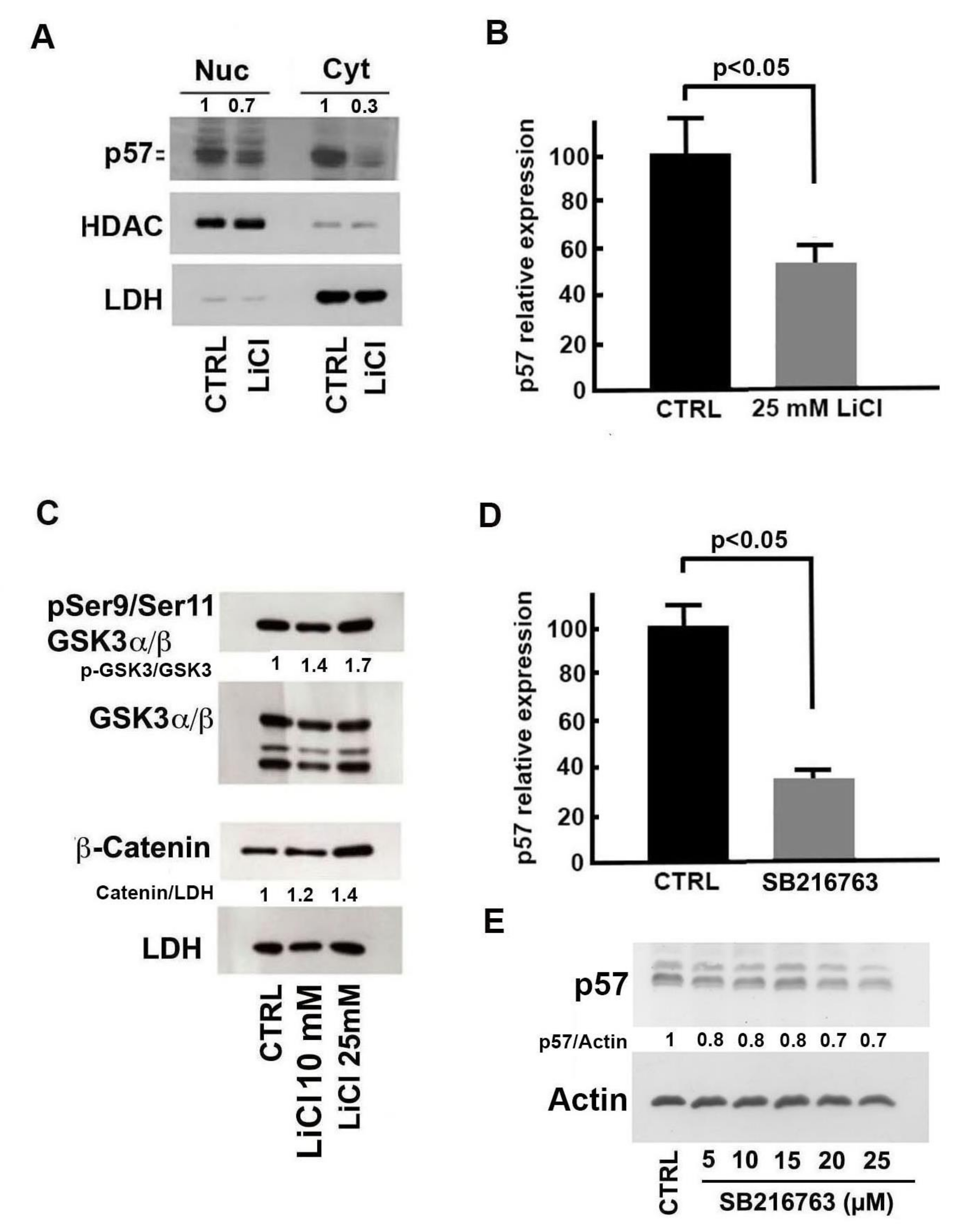
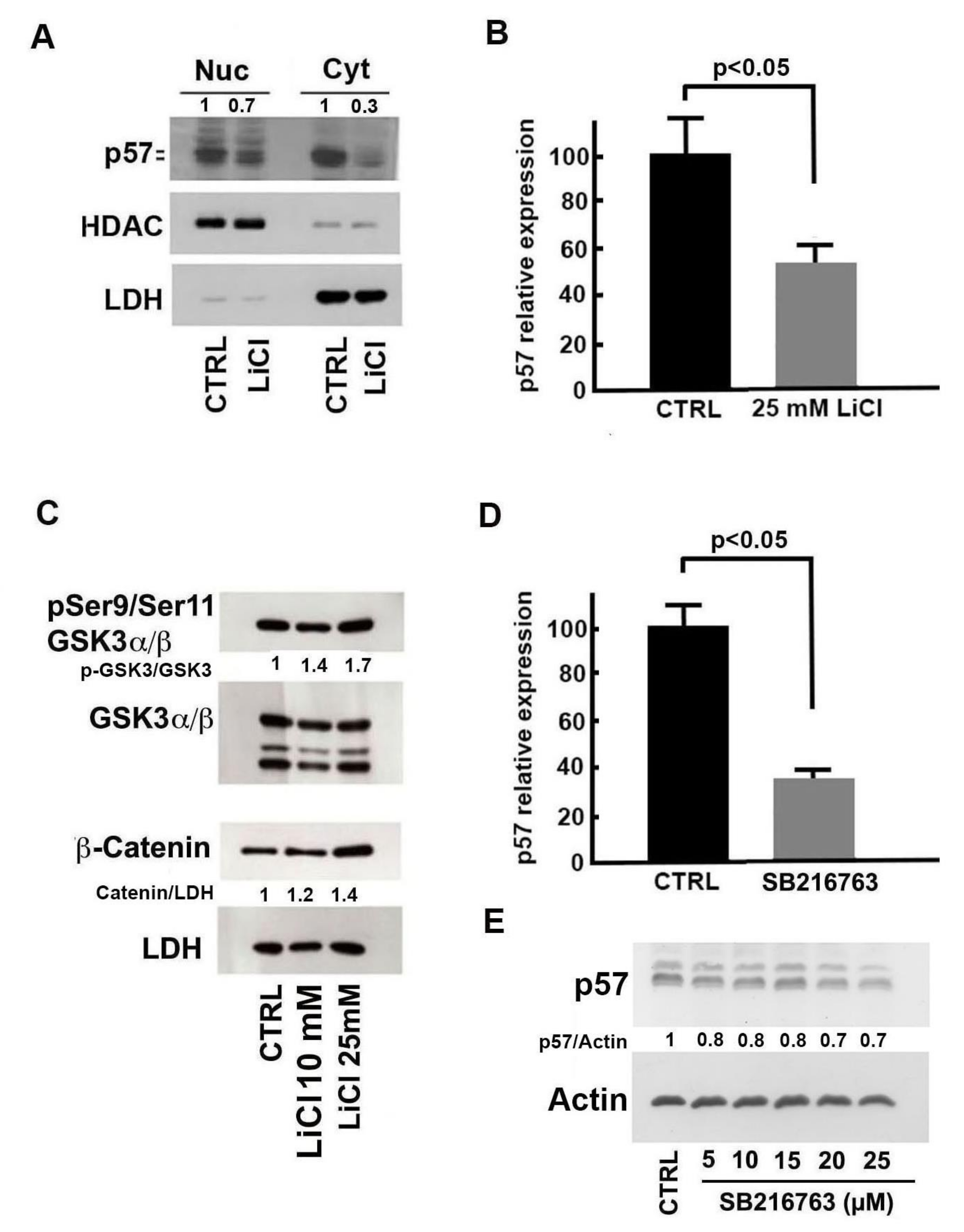
2.3. Molecular Bases of Li-Dependent Effects on p57 Protein
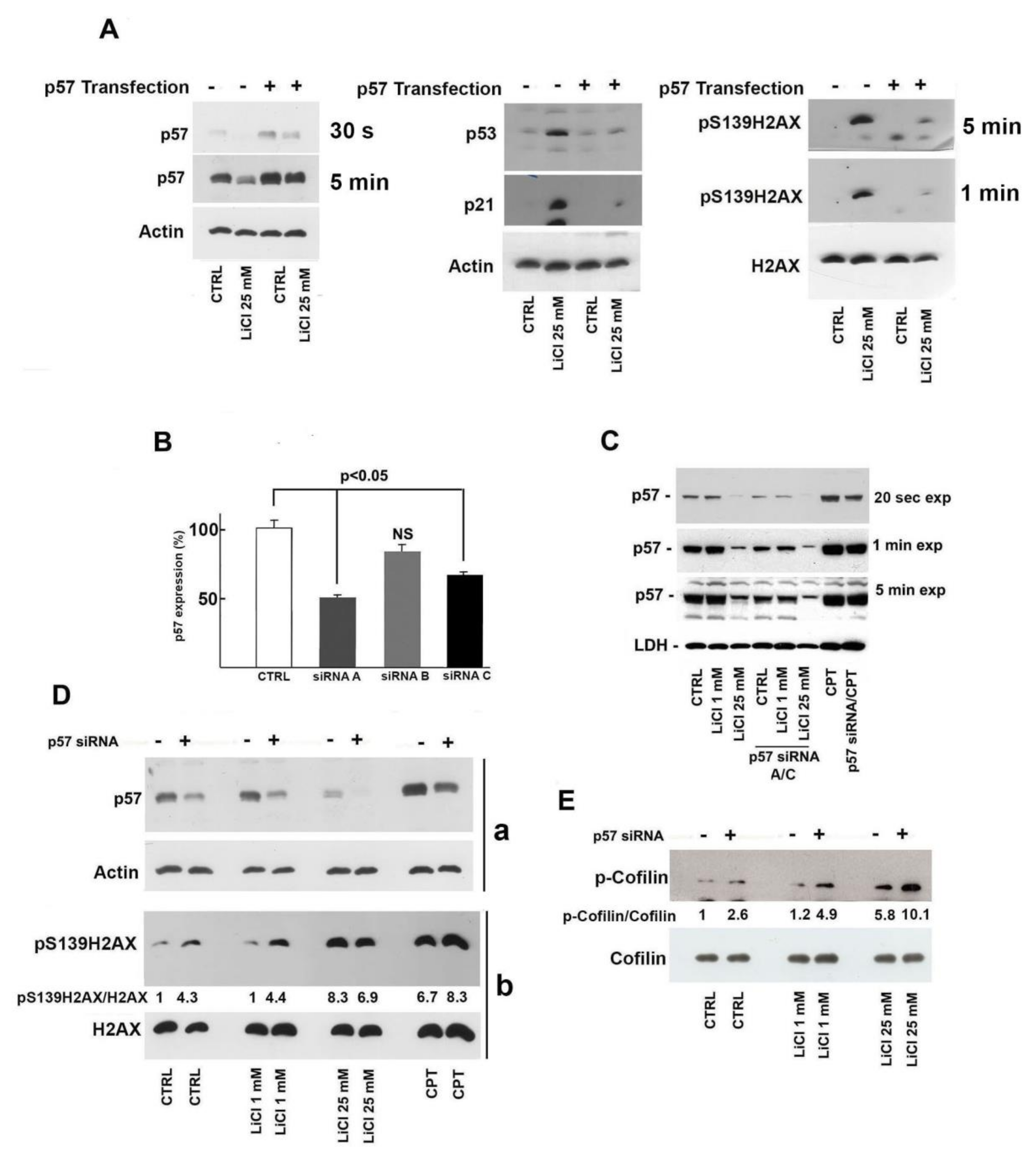
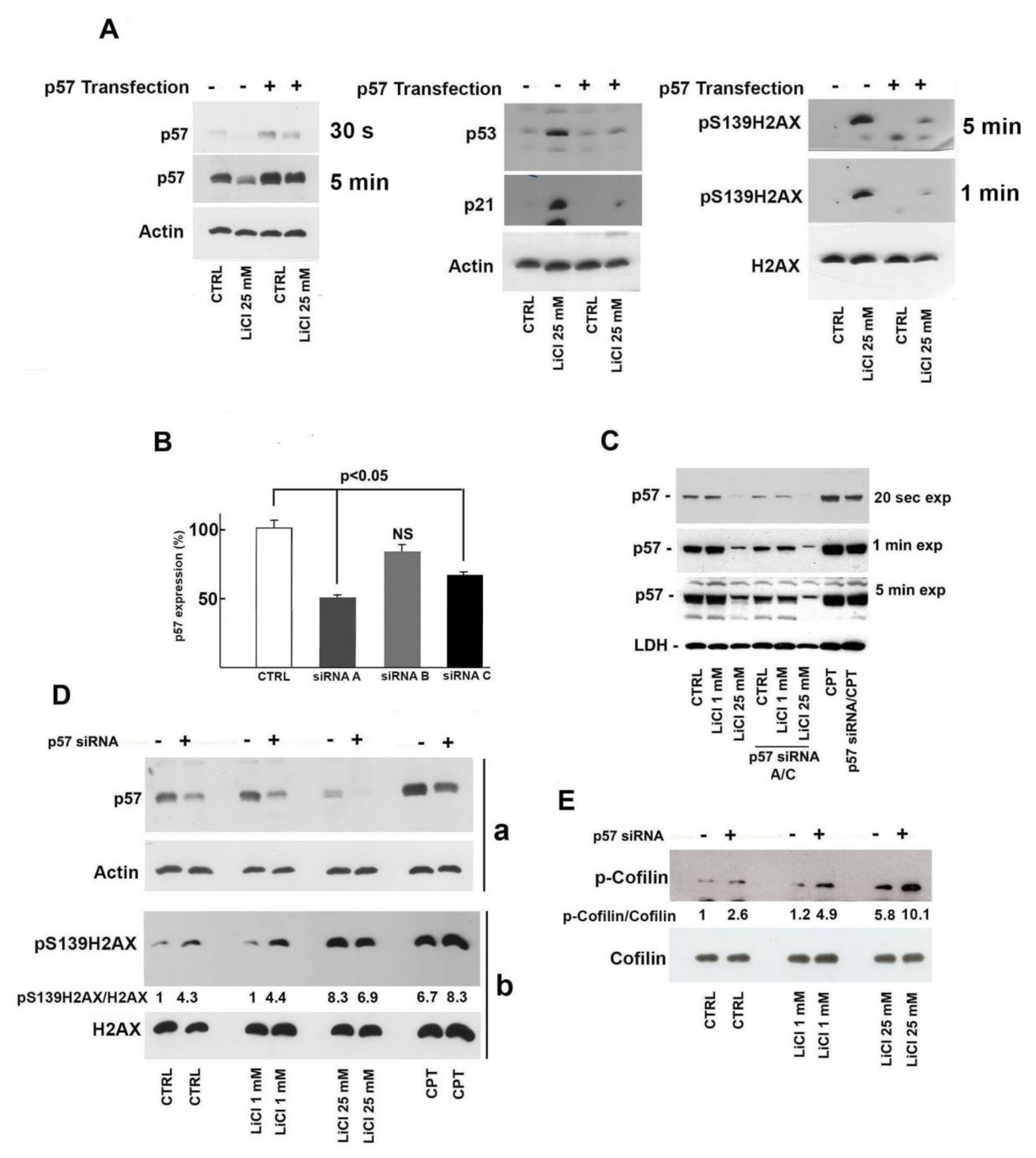
2.4. Role of p57 Decrease in Li-Dependent DNA Damage Response
3. Discussion
4. Critical Assessment on Lithium-Employed Concentrations and Dosages
5. Materials and Methods
5.1. Cell Culture and Treatments
5.2. Analysis of Cell Cycle Distribution
5.3. Real-Time RT-PCR Analyses
5.4. Cell Extracts Preparation and Mono- and Two-Dimensional Western Blotting Analyses
5.5. Plasmid Transfection and Gene Silencing Using Small Interfering RNA (siRNA)
5.6. Evaluation of Intracellular ROS Level and DNA Damage Analysis
5.7. Statistical Analysis
Author Contributions
Funding
Conflicts of Interest
References
- Grof, P.; Müller-Oerlinghausen, B. A Critical Appraisal of Lithium’s Efficacy and Effectiveness: the Last 60 Years. Bipolar Disord. 2009, 11, 10–19. [Google Scholar] [CrossRef] [PubMed]
- Curran, G.; Ravindran, A. Lithium for Bipolar Disorder: a Review of the Recent Literature. Expert Rev. Neurother. 2014, 14, 1079–1098. [Google Scholar] [CrossRef] [PubMed]
- Yatham, L.N.; Kennedy, S.H.; Parikh, S.V.; Schaffer, A.; Beaulieu, S.; Alda, M.; O’Donovan, C.; Macqueen, G.; McIntyre, R.S.; Sharma, V. Canadian Network for Mood and Anxiety Treatments (CANMAT) and International Society for Bipolar Disorders (ISBD) Collaborative Update of CANMAT Guidelines for the Management of Patients with Bipolar Disorder: Update 2013. Bipolar Disord. 2013, 15, 1–44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vadnal, R.; Parthasarathy, R. Myo-inositol Monophosphatase: Diverse Effects of Lithium, Carbamazepine, and Valproate. Neuropsychopharmacology 1995, 12, 277–285. [Google Scholar] [CrossRef]
- Risby, E.D.; Hsiao, J.K.; Manji, H.K.; Bitran, J.; Moses, F.; Zhou, D.F.; Potter, W.Z. The Mechanisms of Action of Lithium. II. Effects on Adenylate Cyclase Activity and Beta-Adrenergic Receptor Binding in Normal Subjects. Arch Gen. Psychiatry 1991, 48, 513–524. [Google Scholar] [CrossRef] [PubMed]
- Manji, H.K.; Bersudsky, Y.; Chen, G.; Belmaker, R.H.; Potter, W.Z. Modulation of Protein Kinase C Isozymes and Substrates by Lithium: the Role of Myo-Inositol. Neuropsychopharmacology 1996, 15, 370–381. [Google Scholar] [CrossRef] [Green Version]
- Chalecka-Franaszek, E.; Chuang, D.M. Lithium Activates the Serine/Threonine Kinase Akt-1 and Suppresses Glutamate-induced Inhibition of Akt-1 Activity in Neurons. Proc. Natl. Acad. Sci. USA 1999, 96, 8745–8750. [Google Scholar] [CrossRef] [Green Version]
- Meffre, D.; Grenier, J.; Bernard, S.; Courtin, F.; Dudev, T.; Shackleford, G.; Jafarian-Tehrani, M.; Massaad, C. Wnt and Lithium: A Common Destiny in the Therapy of Nervous System Pathologies? Cell. Mol. Life Sci. 2014, 71, 1123–1148. [Google Scholar] [CrossRef]
- Jope, R.S. Lithium and GSK-3: One Inhibitor, Two Inhibitory Actions, Multiple Outcomes. Trends Pharmacol. Sci. 2003, 24, 441–443. [Google Scholar] [CrossRef]
- Beurel, E.; Grieco, S.F.; Jope, R.S. Glycogen Synthase Kinase-3 (GSK3): Regulation, Actions, and Diseases. Pharmacol. Ther. 2015, 148, 114–131. [Google Scholar] [CrossRef] [Green Version]
- Qing, Z.; Yang, J.; Han, S.; Liu, J.; Holzbeierlein, J.; Thrasher, J.B.; Li, B. Suppression of Glycogen Synthase Kinase 3 Activity Reduces Tumor Growth of Prostate Cancer in Vivo. Prostate 2011, 71, 835–845. [Google Scholar] [CrossRef]
- Duffy, D.J.; Krstic, A.; Schwarzl, D.; Higgins, D.G.; Kolch, W. GSK3 Inhibitors Regulate MYCN mRNA Levels and Reduce Neuroblastoma Cell Viability through Multiple Mechanisms, Including p53 and Wnt Signaling. Mol. Cancer Ther. 2014, 13, 454–467. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Huang, K.; Liu, X.; Liu, J.; Lu, X.; Tao, K.; Wang, G.; Wang, J. Lithium Chloride Suppresses Colorectal Cancer Cell Survival and Proliferation through ROS/GSK-3β/NF-κB Signaling Pathway. Oxid. Med. Cell. Longev. 2014, 2014, 241864. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Forlenza, O.V.; De-Paula, V.J.R.; Diniz, B.S.O. Neuroprotective Effects of Lithium: Implications for the Treatment of Alzheimer’s Disease and Related Neurodegenerative Disorders. ACS Chem. Neurosci. 2014, 5, 443–450. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guidotti, S.; Minguzzi, M.; Platano, D.; Cattini, L.; Trisolino, G.; Mariani, E.; Borzì, R.M. Lithium Chloride Dependent Glycogen Synthase Kinase 3 Inactivation Links Oxidative DNA Damage, Hypertrophy and Senescence in Human Articular Chondrocytes and Reproduces Chondrocyte Phenotype of Obese Osteoarthritis Patients. PLoS ONE 2015, 10, e0143865. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, E.S.; Wang, H.; Jiang, G.; Nowsheen, S.; Fu, A.; Hallahan, D.E.; Xia, F. Lithium-mediated Protection of Hippocampal Cells Involves Enhancement of DNA-PK-Dependent Repair in Mice. J. Clin. Invest. 2009, 119, 1124–1135. [Google Scholar] [CrossRef]
- McKinnon, P.J. Maintaining Genome Stability in the Nervous System. Nat. Neurosci. 2013, 16, 1523–1529. [Google Scholar] [CrossRef] [Green Version]
- Jacoby, A.S.; Vinberg, M.; Poulsen, H.E.; Kessing, L.V.; Munkholm, K. Increased DNA and RNA Damage by Oxidation in Patients with Bipolar I Disorder. Transl. Psychiatry 2016, 6, e867. [Google Scholar] [CrossRef] [Green Version]
- McKinnon, P.J. Genome Integrity and Disease Prevention in the Nervous System. Genes Dev. 2017, 31, 1180–1194. [Google Scholar] [CrossRef] [Green Version]
- Lovejoy, C.A.; Cortez, D. Common Mechanisms of PIKK Regulation. DNA Repair 2009, 8, 1004–1008. [Google Scholar] [CrossRef] [Green Version]
- Paull, T.T.; Rogakou, E.P.; Yamazaki, V.; Kirchgessner, C.U.; Gellert, M.; Bonner, W.M. A Critical Role for Histone H2AX in Recruitment of Repair Factors to Nuclear Foci After DNA Damage. Curr. Biol. 2000, 10, 886–895. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.; Yu, Y.; Hamrick, H.E.; Duerksen-Hughes, P.J. ATM, ATR and DNA-PK: Initiators of the Cellular Genotoxic Stress Responses. Carcinogenesis 2003, 24, 1571–1580. [Google Scholar] [CrossRef] [Green Version]
- Macleod, K.F.; Sherry, N.; Hannon, G.; Beach, D.; Tokino, T.; Kinzler, K.; Vogelstein, B.; Jacks, T. p53-dependent and Independent Expression of p21 During Cell Growth, Differentiation and DNA Damage. Gen. Dev. 1995, 9, 935–944. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Besson, A.; Dowdy, S.F.; Roberts, J.M. CDK Inhibitors: Cell Cycle Regulators and Beyond. Dev. Cell 2008, 14, 159–169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, Y.; Frisén, J.; Lee, M.H.; Massagué, J.; Barbacid, M. Ablation of the CDK Inhibitor p57Kip2 Results in Increased Apoptosis and Delayed Differentiation During Mouse Development. Genes Dev. 1997, 11, 973–983. [Google Scholar] [CrossRef] [Green Version]
- Joaquin, M.; Gubern, A.; González-Nuñez, D.; Josué Ruiz, E.; Ferreiro, I.; de Nadal, E.; Nebreda, A.R.; Posas, F. The p57 CDKi Integrates Stress Signals Into Cell-Cycle Progression to Promote Cell Survival Upon Stress. EMBO J. 2012, 31, 2952–2964. [Google Scholar] [CrossRef]
- Chang, T.S.; Kim, M.J.; Ryoo, K.; Park, J.; Eom, S.J.; Shim, J.; Nakayama, K.I.; Nakayama, K.; Tomita, M.; Takahashi, K.; et al. p57KIP2 Modulates Stress-Activated Signaling by Inhibiting c-Jun NH2-Terminal Kinase/Stress-Activated Protein Kinase. J. Biol. Chem. 2003, 28, 278–48092. [Google Scholar] [CrossRef] [Green Version]
- Rossi, M.N.; Antonangeli, F. Cellular Response upon Stress: p57 Contribution to the Final Outcome. Mediators Inflamm. 2015, 259325. [Google Scholar] [CrossRef] [Green Version]
- Jia, H.; Cong, Q.; Chua, J.F.; Liu, H.; Xia, X.; Zhang, X.; Lin, J.; Habib, S.L.; Ao, J.; Zuo, Q.; et al. p57Kip2 is an Unrecognized DNA Damage Response Effector Molecule that Functions in Tumor Suppression and Chemoresistance. Oncogene 2015, 34, 3568–3581. [Google Scholar] [CrossRef]
- Cassimere, E.K.; Mauvais, C.; Denicourt, C. p27Kip1 Is Required to Mediate a G1 Cell Cycle Arrest Downstream of ATM following Genotoxic Stress. PLoS ONE 2016, 11, e0162806. [Google Scholar] [CrossRef]
- Vlachos, P.; Nyman, U.; Hajji, N.; Joseph, B. The Cell Cycle Inhibitor p57(Kip2) Promotes Cell Death via the Mitochondrial Apoptotic Pathway. Cell Death Differ. 2007, 14, 1497–1507. [Google Scholar] [CrossRef]
- Stampone, E.; Caldarelli, I.; Zullo, A.; Bencivenga, D.; Mancini, F.P.; Della Ragione, F.; Borriello, A. Genetic and Epigenetic Control of CDKN1C Expression: Importance in Cell Commitment and Differentiation, Tissue Homeostasis and Human Diseases. Int. J. Mol. Sci. 2018, 19, 1055. [Google Scholar] [CrossRef] [Green Version]
- Guo, H.; Tian, T.; Nan, K.; Wang, W. p57: A Multifunctional Protein in Cancer. Int. J. Oncol. 2010, 36, 1321–1329. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, F.; Phiel, C.J.; Spece, L.; Gurvich, N.; Klein, P.S. Inhibitory Phosphorylation of Glycogen Synthase Kinase-3 (GSK-3) in Response to Lithium. Evidence for autoregulation of GSK-3. J. Biol. Chem. 2003, 278, 33067–33077. [Google Scholar] [CrossRef] [Green Version]
- Castelo-Branco, G.; Wagner, J.; Rodriguez, F.J.; Kele, J.; Sousa, K.; Rawal, N.; Pasolli, H.A.; Fuchs, E.; Kitajewski, J.; Arenas, E. Differential Regulation of Midbrain Dopaminergic Neuron Development by Wnt-1, Wnt-3a, and Wnt-5a. Proc. Natl. Acad. Sci. USA 2003, 100, 12747–12752. [Google Scholar] [CrossRef] [Green Version]
- Ferrara, L.; Kmiec, E.B. Camptothecin Enhances the Frequency of Oligonucleotide-Directed Gene Repair in Mammalian Cells by Inducing DNA Damage and Activating Homologous Recombination. Nucl. Acids Res. 2004, 32, 5239–5248. [Google Scholar] [CrossRef] [Green Version]
- Duquesnes, N.; Callot, C.; Jeannot, P.; Daburon, V.; Nakayama, K.I.; Manenti, S.; Davy, A.; Besson, A. p57(Kip2) Knock-In Mouse Reveals CDK-Independent Contribution in the Development of Beckwith-Wiedemann Syndrome. J. Pathol. 2016, 239, 250–261. [Google Scholar] [CrossRef]
- Borriello, A.; Caldarelli, I.; Bencivenga, D.; Criscuolo, M.; Cucciolla, V.; Tramontano, A.; Oliva, A.; Perrotta, S.; Della Ragione, F. p57(Kip2) and Cancer: Time for a Critical Appraisal. Mol. Cancer Res. 2011, 9, 1269–1284. [Google Scholar] [CrossRef] [Green Version]
- Kim, W.Y.; Wang, X.; Wu, Y.; Doble, B.W.; Patel, S.; Woodgett, J.R.; Snider, W.D. GSK-3 is a Master Regulator of Neural Progenitor Homeostasis. Nat. Neurosci. 2009, 12, 1390–1397. [Google Scholar] [CrossRef] [Green Version]
- Wu, D.; Pan, W. GSK3: A Multifaceted Kinase in Wnt Signaling. Trends Biochem. Sci. 2010, 35, 161–168. [Google Scholar] [CrossRef] [Green Version]
- Liu, C.; Li, Y.; Semenov, M.; Han, C.; Baeg, G.H.; Tan, Y.; Zhang, Z.; Lin, X.; He, X. Control of Beta-Catenin Phosphorylation/Degradation by a Dual-Kinase Mechanism. Cell 2002, 108, 837–847. [Google Scholar] [CrossRef] [Green Version]
- Gilardini Montani, M.S.; Granato, M.; Cuomo, L.; Valia, S.; Di Renzo, L.; D’Orazi, G.; Faggioni, A.; Cirone, M. High glucose and hyperglycemic sera from type 2 diabetic patients impair DC differentiation by inducing ROS and activating Wnt/β-catenin and p38 MAPK. Biochim. Biophys. Acta 2016, 1862, 805–813. [Google Scholar] [CrossRef] [PubMed]
- Nolen, W.A.; Licht, R.W.; Young, A.H.; Malhi, G.S.; Tohen, M.; Vieta, E.; Kupka, R.W.; Zarate, C.; Nielsen, R.E.; Baldessarini, R.J.; et al. What is the Optimal Serum Level for Lithium in the Maintenance Treatment of Bipolar Disorder? A Systematic Review and Recommendations from the ISBD/IGSLI Task Force on Treatment with Lithium. Bipolar Disord. 2019, 21, 394–409. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nowicki, M.O.; Dmitrieva, N.; Stein, A.M.; Cutter, J.L.; Godlewski, J.; Saeki, Y.; Nita, M.; Berens, M.E.; Sander, L.M.; Newton, H.B.; et al. Lithium Inhibits Invasion of Glioma Cells; Possible Involvement of Glycogen Synthase Kinase-3. Neuro Oncol. 2008, 10, 690–699. [Google Scholar] [CrossRef] [Green Version]
- Vidal, F.; de Araujo, W.M.; Cruz, A.L.; Tanaka, M.N.; Viola, J.P.; Morgado-Díaz, J.A. Lithium Reduces Tumorigenic Potential in Response to EGF Signaling in Human Colorectal Cancer Cells. Int. J. Oncol. 2011, 38, 1365–1373. [Google Scholar] [CrossRef]
- Ronchi, A.; Salaroli, R.; Rivetti, S.; Della Bella, E.; Di Tomaso, T.; Voltattorni, M.; Cammelli, S.; Ceccarelli, C.; Giangaspero, F.; Barbieri, E.; et al. Lithium Induces Mortality in Medulloblastoma Cell Lines. Int. J. Oncol. 2010, 37, 745–752. [Google Scholar] [CrossRef]
- Erdal, E.; Ozturk, N.; Cagatay, T.; Eksioglu-Demiralp, E.; Ozturk, M. Lithium-Mediated Downregulation of PKB/Akt and Cyclin E with Growth Inhibition in Hepatocellular Carcinoma Cells. Int. J. Cancer 2005, 115, 903–910. [Google Scholar] [CrossRef]
- Beyaert, R.; Vanhaesebroeck, B.; Suffys, P.; Van Roy, F.; Fiers, W. Lithium Chloride Potentiates Tumor Necrosis Factor-Mediated Cytotoxicity in Vitro and in Vivo. Proc. Natl. Acad. Sci. USA 1989, 86, 9494–9498. [Google Scholar] [CrossRef] [Green Version]
- Borriello, A.; Caldarelli, I.; Speranza, M.C.; Scianguetta, S.; Tramontano, A.; Bencivenga, D.; Stampone, E.; Negri, A.; Nobili, B.; Locatelli, F.; et al. Iron Overload Enhances Human Mesenchymal Stromal Cell Growth and Hampers Matrix Calcification. Biochim. Biophys. Acta 2016, 1860, 1211–1223. [Google Scholar] [CrossRef]
- Iolascon, A.; Giordani, L.; Moretti, A.; Tonini, G.P.; Lo Cunsolo, C.; Mastropietro, S.; Borriello, A.; Della Ragione, F. Structural and functional analysis of cyclin-dependent kinase inhibitor genes (CDKN2A, CDKN2B, and CDKN2C) in neuroblastoma. Pediatr. Res. 1998, 43, 139–144. [Google Scholar] [CrossRef] [Green Version]
- Iolascon, A.; Giordani, L.; Moretti, A.; Basso, A.; Borriello, A.; Della Ragione, F. Analysis of CDKN2A, CDKN2B, CDKN2C and cyclin D gene status in hepatoblastoma. Hepatology 1998, 27, 989–995. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of Relative Gene Expression Data Using Real-Time Quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Borriello, A.; Caldarelli, I.; Bencivenga, D.; Cucciolla, V.; Oliva, A.; Usala, E.; Danise, P.; Ronzoni, L.; Perrotta, S.; Della Ragione, F. p57Kip2 is a Downstream Effector of BCR–ABL Kinase Inhibitors in Chronic Myelogenous Leukemia Cells. Carcinogenesis 2011, 32, 10–18. [Google Scholar] [CrossRef] [PubMed]
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Stampone, E.; Bencivenga, D.; Barone, C.; Aulitto, A.; Verace, F.; Della Ragione, F.; Borriello, A. High Dosage Lithium Treatment Induces DNA Damage and p57Kip2 Decrease. Int. J. Mol. Sci. 2020, 21, 1169. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21031169
Stampone E, Bencivenga D, Barone C, Aulitto A, Verace F, Della Ragione F, Borriello A. High Dosage Lithium Treatment Induces DNA Damage and p57Kip2 Decrease. International Journal of Molecular Sciences. 2020; 21(3):1169. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21031169
Chicago/Turabian StyleStampone, Emanuela, Debora Bencivenga, Clementina Barone, Arianna Aulitto, Federica Verace, Fulvio Della Ragione, and Adriana Borriello. 2020. "High Dosage Lithium Treatment Induces DNA Damage and p57Kip2 Decrease" International Journal of Molecular Sciences 21, no. 3: 1169. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21031169