Revealing and Harnessing Tumour-Associated Microglia/Macrophage Heterogeneity in Glioblastoma


Abstract
:1. Introduction
2. Tumour-Associated Microglia/Macrophages in Glioblastoma
2.1. M1 and M2 Polarization States: The Basic School of Thought
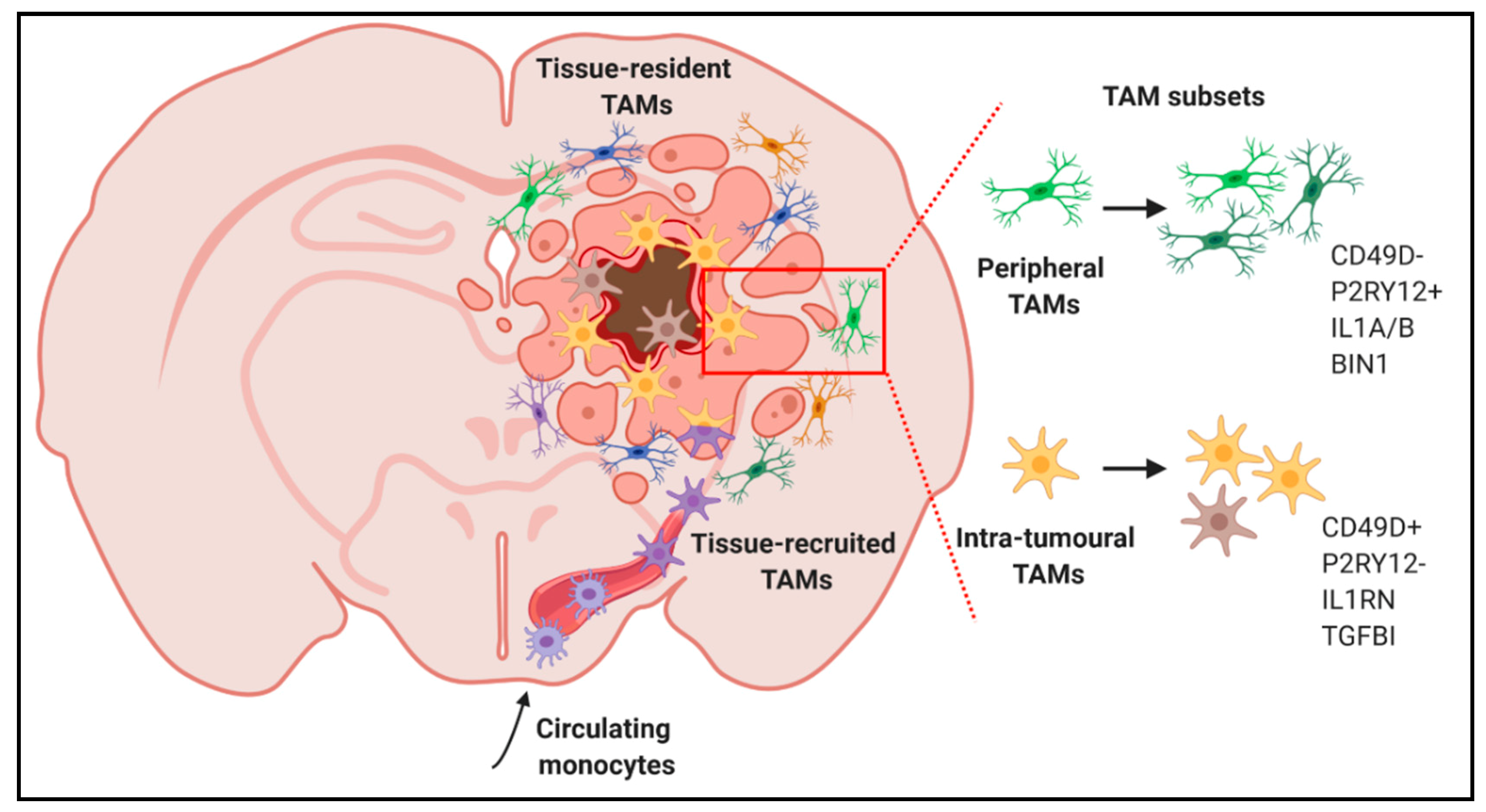
2.2. Impact of Ontogeny on Tumour-Associated Microglia/Macrophage Functionality
3. Tumour-Associated Microglia/Macrophages as Therapeutic Targets in Glioblastoma
3.1. Effect of Chemotherapy and Radiotherapy on Tumour-Associated Microglia/Macrophages
3.2. Depletion of Tumour-Associated Microglia/Macrophages in Glioblastoma
3.3. Immune Checkpoint Inhibitors and Reprogramming of Tumour-Associated Microglia/Macrophages in Glioblastoma
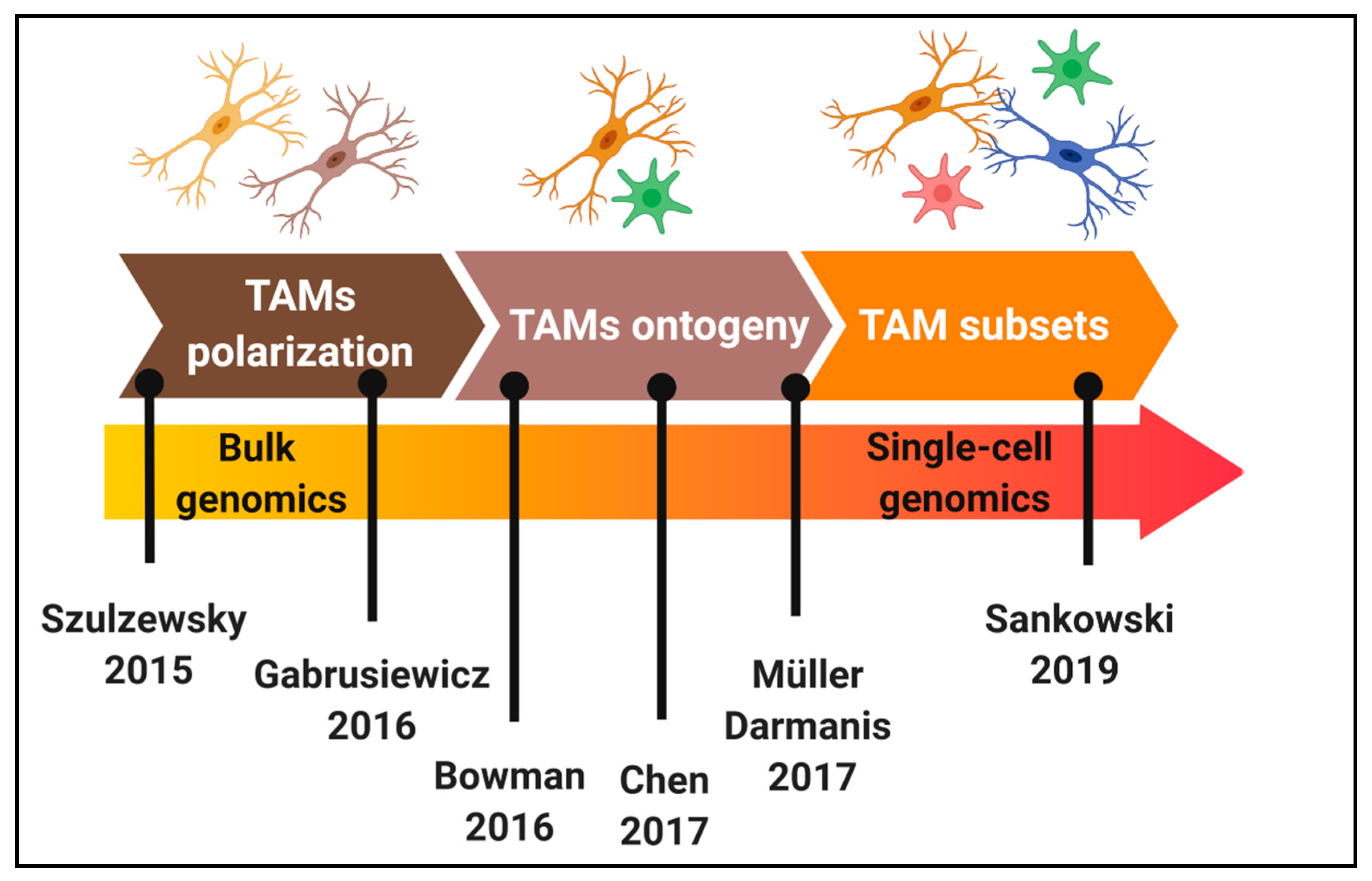
4. Dissecting Tumour-Associated Microglia/Macrophages Diversity at Single-Cell Resolution
4.1. Glioblastoma Subtyping and Single-Cell Analyses
4.2. Single-Cell Analyses of Microglia and Macrophages in Glioblastoma: Heterogeneity beyond Polarization States and Ontogeny?
5. Conclusions and Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- Goodenberger, M.L.; Jenkins, R.B. Genetics of adult glioma. Cancer Genet. 2012, 205, 613–621. [Google Scholar] [CrossRef]
- Stupp, R.; Mason, W.P.; van den Bent, M.J.; Weller, M.; Fisher, B.; Taphoorn, M.J.; Belanger, K.; Brandes, A.A.; Marosi, C.; Bogdahn, U.; et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N. Engl. J. Med. 2005, 352, 987–996. [Google Scholar] [CrossRef]
- Quail, D.F.; Joyce, J.A. The Microenvironmental Landscape of Brain Tumors. Cancer Cell 2017, 31, 326–341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Glass, R.; Synowitz, M. CNS macrophages and peripheral myeloid cells in brain tumours. Acta Neuropathol. 2014, 128, 347–362. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lim, M.; Xia, Y.; Bettegowda, C.; Weller, M. Current state of immunotherapy for glioblastoma. Nat. Rev. Clin. Oncol. 2018, 15, 422–442. [Google Scholar] [CrossRef] [PubMed]
- Thorsson, V.; Gibbs, D.L.; Brown, S.D.; Wolf, D.; Bortone, D.S.; Ou Yang, T.H.; Porta-Pardo, E.; Gao, G.F.; Plaisier, C.L.; Eddy, J.A.; et al. The Immune Landscape of Cancer. Immunity 2018, 48, 812–830.e14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coniglio, S.J.; Eugenin, E.; Dobrenis, K.; Stanley, E.R.; West, B.L.; Symons, M.H.; Segall, J.E. Microglial stimulation of glioblastoma invasion involves epidermal growth factor receptor (EGFR) and colony stimulating factor 1 receptor (CSF-1R) signaling. Mol. Med. 2012, 18, 519–527. [Google Scholar] [CrossRef] [PubMed]
- Patel, S.; Player, M.R. Colony-stimulating factor-1 receptor inhibitors for the treatment of cancer and inflammatory disease. Curr. Top. Med. Chem. 2009, 9, 599–610. [Google Scholar] [CrossRef] [PubMed]
- Pyonteck, S.M.; Akkari, L.; Schuhmacher, A.J.; Bowman, R.L.; Sevenich, L.; Quail, D.F.; Olson, O.C.; Quick, M.L.; Huse, J.T.; Teijeiro, V.; et al. CSF-1R inhibition alters macrophage polarization and blocks glioma progression. Nat. Med. 2013, 19, 1264–1272. [Google Scholar] [CrossRef] [Green Version]
- Quail, D.F.; Joyce, J.A. Molecular Pathways: Deciphering Mechanisms of Resistance to Macrophage-Targeted Therapies. Clin. Cancer Res. 2017, 23, 876–884. [Google Scholar] [CrossRef] [Green Version]
- Mills, C.D.; Kincaid, K.; Alt, J.M.; Heilman, M.J.; Hill, A.M. Pillars Article: M-1/M-2 Macrophages and the Th1/Th2 Paradigm. J. Immunol. 2017, 164, 6166–6173, Erratum in 2017, 199, 2194–2201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Michelucci, A.; Heurtaux, T.; Grandbarbe, L.; Morga, E.; Heuschling, P. Characterization of the microglial phenotype under specific pro-inflammatory and anti-inflammatory conditions: Effects of oligomeric and fibrillar amyloid-beta. J. Neuroimmunol. 2009, 210, 3–12. [Google Scholar] [CrossRef]
- Mantovani, A.; Sozzani, S.; Locati, M.; Allavena, P.; Sica, A. Macrophage polarization: Tumor-associated macrophages as a paradigm for polarized M2 mononuclear phagocytes. Trends Immunol. 2002, 23, 549–555. [Google Scholar] [CrossRef]
- Roesch, S.; Rapp, C.; Dettling, S.; Herold-Mende, C. When Immune Cells Turn Bad-Tumor-Associated Microglia/Macrophages in Glioma. Int. J. Mol. Sci. 2018, 19, 436. [Google Scholar] [CrossRef] [Green Version]
- Mantovani, A.; Marchesi, F.; Malesci, A.; Laghi, L.; Allavena, P. Tumour-associated macrophages as treatment targets in oncology. Nat. Rev. Clin. Oncol. 2017, 14, 399–416. [Google Scholar] [CrossRef] [PubMed]
- Szulzewsky, F.; Pelz, A.; Feng, X.; Synowitz, M.; Markovic, D.; Langmann, T.; Holtman, I.R.; Wang, X.; Eggen, B.J.; Boddeke, H.W.; et al. Glioma-associated microglia/macrophages display an expression profile different from M1 and M2 polarization and highly express Gpnmb and Spp1. PLoS ONE 2015, 10, e0116644. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gabrusiewicz, K.; Rodriguez, B.; Wei, J.; Hashimoto, Y.; Healy, L.M.; Maiti, S.N.; Thomas, G.; Zhou, S.; Wang, Q.; Elakkad, A.; et al. Glioblastoma-infiltrated innate immune cells resemble M0 macrophage phenotype. JCI Insight 2016, 1, e85841. [Google Scholar] [CrossRef]
- Goldmann, T.; Wieghofer, P.; Jordao, M.J.; Prutek, F.; Hagemeyer, N.; Frenzel, K.; Amann, L.; Staszewski, O.; Kierdorf, K.; Krueger, M.; et al. Origin, fate and dynamics of macrophages at central nervous system interfaces. Nat. Immunol. 2016, 17, 797–805. [Google Scholar] [CrossRef]
- Ginhoux, F.; Greter, M.; Leboeuf, M.; Nandi, S.; See, P.; Gokhan, S.; Mehler, M.F.; Conway, S.J.; Ng, L.G.; Stanley, E.R.; et al. Fate mapping analysis reveals that adult microglia derive from primitive macrophages. Science 2010, 330, 841–845. [Google Scholar] [CrossRef] [Green Version]
- Colonna, M.; Butovsky, O. Microglia Function in the Central Nervous System During Health and Neurodegeneration. Annu. Rev. Immunol. 2017, 35, 441–468. [Google Scholar] [CrossRef]
- Salter, M.W.; Stevens, B. Microglia emerge as central players in brain disease. Nat. Med. 2017, 23, 1018–1027. [Google Scholar] [CrossRef] [PubMed]
- Crotti, A.; Ransohoff, R.M. Microglial Physiology and Pathophysiology: Insights from Genome-wide Transcriptional Profiling. Immunity 2016, 44, 505–515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sousa, C.; Biber, K.; Michelucci, A. Cellular and Molecular Characterization of Microglia: A Unique Immune Cell Population. Front. Immunol. 2017, 8, 198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hambardzumyan, D.; Gutmann, D.H.; Kettenmann, H. The role of microglia and macrophages in glioma maintenance and progression. Nat. Neurosci. 2016, 19, 20–27. [Google Scholar] [CrossRef] [Green Version]
- Ricard, C.; Tchoghandjian, A.; Luche, H.; Grenot, P.; Figarella-Branger, D.; Rougon, G.; Malissen, M.; Debarbieux, F. Phenotypic dynamics of microglial and monocyte-derived cells in glioblastoma-bearing mice. Sci. Rep. 2016, 6, 26381. [Google Scholar] [CrossRef] [Green Version]
- Cassetta, L.; Pollard, J.W. Targeting macrophages: Therapeutic approaches in cancer. Nat. Rev. Drug Discov. 2018, 17, 887–904. [Google Scholar] [CrossRef]
- Li, W.; Graeber, M.B. The molecular profile of microglia under the influence of glioma. Neuro-Oncol. 2012, 14, 958–978. [Google Scholar] [CrossRef] [Green Version]
- Wei, J.; Marisetty, A.; Schrand, B.; Gabrusiewicz, K.; Hashimoto, Y.; Ott, M.; Grami, Z.; Kong, L.Y.; Ling, X.; Caruso, H.; et al. Osteopontin mediates glioblastoma-associated macrophage infiltration and is a potential therapeutic target. J. Clin. Investig. 2019, 129, 137–149. [Google Scholar] [CrossRef]
- Zhang, J.; Sarkar, S.; Cua, R.; Zhou, Y.; Hader, W.; Yong, V.W. A dialog between glioma and microglia that promotes tumor invasiveness through the CCL2/CCR2/interleukin-6 axis. Carcinogenesis 2012, 33, 312–319. [Google Scholar] [CrossRef]
- Graeber, M.B.; Scheithauer, B.W.; Kreutzberg, G.W. Microglia in brain tumors. Glia 2002, 40, 252–259. [Google Scholar] [CrossRef]
- London, A.; Cohen, M.; Schwartz, M. Microglia and monocyte-derived macrophages: Functionally distinct populations that act in concert in CNS plasticity and repair. Front. Cell. Neurosci. 2013, 7, 34. [Google Scholar] [CrossRef] [Green Version]
- Pinton, L.; Masetto, E.; Vettore, M.; Solito, S.; Magri, S.; D’Andolfi, M.; Del Bianco, P.; Lollo, G.; Benoit, J.P.; Okada, H.; et al. The immune suppressive microenvironment of human gliomas depends on the accumulation of bone marrow-derived macrophages in the center of the lesion. J. Immunother. Cancer 2019, 7, 58. [Google Scholar] [CrossRef] [PubMed]
- Sedgwick, J.D.; Schwender, S.; Imrich, H.; Dorries, R.; Butcher, G.W.; ter Meulen, V. Isolation and direct characterization of resident microglial cells from the normal and inflamed central nervous system. Proc. Natl. Acad. Sci. USA 1991, 88, 7438–7442. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muller, A.; Brandenburg, S.; Turkowski, K.; Muller, S.; Vajkoczy, P. Resident microglia, and not peripheral macrophages, are the main source of brain tumor mononuclear cells. Int. J. Cancer 2015, 137, 278–288. [Google Scholar] [CrossRef]
- Bowman, R.L.; Klemm, F.; Akkari, L.; Pyonteck, S.M.; Sevenich, L.; Quail, D.F.; Dhara, S.; Simpson, K.; Gardner, E.E.; Iacobuzio-Donahue, C.A.; et al. Macrophage Ontogeny Underlies Differences in Tumor-Specific Education in Brain Malignancies. Cell Rep. 2016, 17, 2445–2459. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haage, V.; Semtner, M.; Vidal, R.O.; Hernandez, D.P.; Pong, W.W.; Chen, Z.; Hambardzumyan, D.; Magrini, V.; Ly, A.; Walker, J.; et al. Comprehensive gene expression meta-analysis identifies signature genes that distinguish microglia from peripheral monocytes/macrophages in health and glioma. Acta Neuropathol. Commun. 2019, 7, 20. [Google Scholar] [CrossRef] [Green Version]
- Greenhalgh, A.D.; Zarruk, J.G.; Healy, L.M.; Baskar Jesudasan, S.J.; Jhelum, P.; Salmon, C.K.; Formanek, A.; Russo, M.V.; Antel, J.P.; McGavern, D.B.; et al. Peripherally derived macrophages modulate microglial function to reduce inflammation after CNS injury. PLoS Biol. 2018, 16, e2005264. [Google Scholar] [CrossRef] [Green Version]
- Hagemann, T.; Lawrence, T.; McNeish, I.; Charles, K.A.; Kulbe, H.; Thompson, R.G.; Robinson, S.C.; Balkwill, F.R. “Re-educating” tumor-associated macrophages by targeting NF-kappaB. J. Exp. Med. 2008, 205, 1261–1268. [Google Scholar] [CrossRef]
- Nesseler, J.P.; Schaue, D.; McBride, W.H.; Lee, M.H.; Kaprealian, T.; Niclou, S.P.; Nickers, P. Irradiation to Improve the Response to Immunotherapeutic Agents in Glioblastomas. Adv. Radiat. Oncol. 2019, 4, 268–282. [Google Scholar] [CrossRef] [Green Version]
- Brown, N.F.; Carter, T.J.; Ottaviani, D.; Mulholland, P. Harnessing the immune system in glioblastoma. Br. J. Cancer 2018, 119, 1171–1181. [Google Scholar] [CrossRef] [Green Version]
- Olnes, M.J.; Kotliarov, Y.; Biancotto, A.; Cheung, F.; Chen, J.; Shi, R.; Zhou, H.; Wang, E.; Tsang, J.S.; Nussenblatt, R.; et al. Effects of Systemically Administered Hydrocortisone on the Human Immunome. Sci. Rep. 2016, 6, 23002. [Google Scholar] [CrossRef] [Green Version]
- Kalbasi, A.; June, C.H.; Haas, N.; Vapiwala, N. Radiation and immunotherapy: A synergistic combination. J. Clin. Investig. 2013, 123, 2756–2763. [Google Scholar] [CrossRef]
- Deng, L.; Liang, H.; Xu, M.; Yang, X.; Burnette, B.; Arina, A.; Li, X.D.; Mauceri, H.; Beckett, M.; Darga, T.; et al. STING-Dependent Cytosolic DNA Sensing Promotes Radiation-Induced Type I Interferon-Dependent Antitumor Immunity in Immunogenic Tumors. Immunity 2014, 41, 843–852. [Google Scholar] [CrossRef] [Green Version]
- Markovic, D.S.; Glass, R.; Synowitz, M.; Rooijen, N.; Kettenmann, H. Microglia stimulate the invasiveness of glioma cells by increasing the activity of metalloprotease-2. J. Neuropathol. Exp. Neurol. 2005, 64, 754–762. [Google Scholar] [CrossRef] [Green Version]
- Han, X.; Li, Q.; Lan, X.; El-Mufti, L.; Ren, H.; Wang, J. Microglial Depletion with Clodronate Liposomes Increases Proinflammatory Cytokine Levels, Induces Astrocyte Activation, and Damages Blood Vessel Integrity. Mol. Neurobiol. 2019, 56, 6184–6196. [Google Scholar] [CrossRef]
- Chen, Z.; Feng, X.; Herting, C.J.; Garcia, V.A.; Nie, K.; Pong, W.W.; Rasmussen, R.; Dwivedi, B.; Seby, S.; Wolf, S.A.; et al. Cellular and Molecular Identity of Tumor-Associated Macrophages in Glioblastoma. Cancer Res. 2017, 77, 2266–2278. [Google Scholar] [CrossRef] [Green Version]
- Galarneau, H.; Villeneuve, J.; Gowing, G.; Julien, J.P.; Vallieres, L. Increased glioma growth in mice depleted of macrophages. Cancer Res. 2007, 67, 8874–8881. [Google Scholar] [CrossRef] [Green Version]
- Alban, T.J.; Alvarado, A.G.; Sorensen, M.D.; Bayik, D.; Volovetz, J.; Serbinowski, E.; Mulkearns-Hubert, E.E.; Sinyuk, M.; Hale, J.S.; Onzi, G.R.; et al. Global immune fingerprinting in glioblastoma patient peripheral blood reveals immune-suppression signatures associated with prognosis. JCI Insight 2018, 3, e122264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, Y.; Lu, Y.; Chen, W.; Fu, J.; Fan, R. In silico experimentation of glioma microenvironment development and anti-tumor therapy. PLoS Comput. Biol. 2012, 8, e1002355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boussiotis, V.A.; Charest, A. Immunotherapies for malignant glioma. Oncogene 2018, 37, 1121–1141. [Google Scholar] [CrossRef] [PubMed]
- Tomaszewski, W.; Sanchez-Perez, L.; Gajewski, T.F.; Sampson, J.H. Brain Tumor Microenvironment and Host State: Implications for Immunotherapy. Clin. Cancer Res. 2019, 25, 4202–4210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reardon, D.A.; Gokhale, P.C.; Klein, S.R.; Ligon, K.L.; Rodig, S.J.; Ramkissoon, S.H.; Jones, K.L.; Conway, A.S.; Liao, X.; Zhou, J.; et al. Glioblastoma Eradication Following Immune Checkpoint Blockade in an Orthotopic, Immunocompetent Model. Cancer Immunol. Res. 2016, 4, 124–135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yarchoan, M.; Hopkins, A.; Jaffee, E.M. Tumor Mutational Burden and Response Rate to PD-1 Inhibition. N. Engl. J. Med. 2017, 377, 2500–2501. [Google Scholar] [CrossRef] [PubMed]
- Gholamin, S.; Mitra, S.S.; Feroze, A.H.; Liu, J.; Kahn, S.A.; Zhang, M.; Esparza, R.; Richard, C.; Ramaswamy, V.; Remke, M.; et al. Disrupting the CD47-SIRPalpha anti-phagocytic axis by a humanized anti-CD47 antibody is an efficacious treatment for malignant pediatric brain tumors. Sci. Transl. Med. 2017, 9, eaaf2968. [Google Scholar] [CrossRef] [Green Version]
- Hutter, G.; Theruvath, J.; Graef, C.M.; Zhang, M.; Schoen, M.K.; Manz, E.M.; Bennett, M.L.; Olson, A.; Azad, T.D.; Sinha, R.; et al. Microglia are effector cells of CD47-SIRPalpha antiphagocytic axis disruption against glioblastoma. Proc. Natl. Acad. Sci. USA 2019, 116, 997–1006. [Google Scholar] [CrossRef] [Green Version]
- Castro, B.A.; Flanigan, P.; Jahangiri, A.; Hoffman, D.; Chen, W.; Kuang, R.; De Lay, M.; Yagnik, G.; Wagner, J.R.; Mascharak, S.; et al. Macrophage migration inhibitory factor downregulation: A novel mechanism of resistance to anti-angiogenic therapy. Oncogene 2017, 36, 3749–3759. [Google Scholar] [CrossRef] [Green Version]
- Wiehagen, K.R.; Girgis, N.M.; Yamada, D.H.; Smith, A.A.; Chan, S.R.; Grewal, I.S.; Quigley, M.; Verona, R.I. Combination of CD40 Agonism and CSF-1R Blockade Reconditions Tumor-Associated Macrophages and Drives Potent Antitumor Immunity. Cancer Immunol. Res. 2017, 5, 1109–1121. [Google Scholar] [CrossRef] [Green Version]
- Saha, D.; Martuza, R.L.; Rabkin, S.D. Macrophage Polarization Contributes to Glioblastoma Eradication by Combination Immunovirotherapy and Immune Checkpoint Blockade. Cancer Cell 2017, 32, 253–267.e5. [Google Scholar] [CrossRef] [Green Version]
- Phillips, H.S.; Kharbanda, S.; Chen, R.; Forrest, W.F.; Soriano, R.H.; Wu, T.D.; Misra, A.; Nigro, J.M.; Colman, H.; Soroceanu, L.; et al. Molecular subclasses of high-grade glioma predict prognosis, delineate a pattern of disease progression, and resemble stages in neurogenesis. Cancer Cell 2006, 9, 157–173. [Google Scholar] [CrossRef] [Green Version]
- Verhaak, R.G.; Hoadley, K.A.; Purdom, E.; Wang, V.; Qi, Y.; Wilkerson, M.D.; Miller, C.R.; Ding, L.; Golub, T.; Mesirov, J.P.; et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell 2010, 17, 98–110. [Google Scholar] [CrossRef] [Green Version]
- Ceccarelli, M.; Barthel, F.P.; Malta, T.M.; Sabedot, T.S.; Salama, S.R.; Murray, B.A.; Morozova, O.; Newton, Y.; Radenbaugh, A.; Pagnotta, S.M.; et al. Molecular Profiling Reveals Biologically Discrete Subsets and Pathways of Progression in Diffuse Glioma. Cell 2016, 164, 550–563. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Hu, B.; Hu, X.; Kim, H.; Squatrito, M.; Scarpace, L.; de Carvalho, A.C.; Lyu, S.; Li, P.; Li, Y.; et al. Tumor Evolution of Glioma-Intrinsic Gene Expression Subtypes Associates with Immunological Changes in the Microenvironment. Cancer Cell 2017, 32, 42–56.e6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sottoriva, A.; Spiteri, I.; Piccirillo, S.G.; Touloumis, A.; Collins, V.P.; Marioni, J.C.; Curtis, C.; Watts, C.; Tavare, S. Intratumor heterogeneity in human glioblastoma reflects cancer evolutionary dynamics. Proc. Natl. Acad. Sci. USA 2013, 110, 4009–4014. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neftel, C.; Laffy, J.; Filbin, M.G.; Hara, T.; Shore, M.E.; Rahme, G.J.; Richman, A.R.; Silverbush, D.; Shaw, M.L.; Hebert, C.M.; et al. An Integrative Model of Cellular States, Plasticity, and Genetics for Glioblastoma. Cell 2019, 178, 835–849.e21. [Google Scholar] [CrossRef] [PubMed]
- Patel, A.P.; Tirosh, I.; Trombetta, J.J.; Shalek, A.K.; Gillespie, S.M.; Wakimoto, H.; Cahill, D.P.; Nahed, B.V.; Curry, W.T.; Martuza, R.L.; et al. Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma. Science 2014, 344, 1396–1401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Capper, D.; Jones, D.T.W.; Sill, M.; Hovestadt, V.; Schrimpf, D.; Sturm, D.; Koelsche, C.; Sahm, F.; Chavez, L.; Reuss, D.E.; et al. DNA methylation-based classification of central nervous system tumours. Nature 2018, 555, 469–474. [Google Scholar] [CrossRef]
- Van Hove, H.; Martens, L.; Scheyltjens, I.; De Vlaminck, K.; Pombo Antunes, A.R.; De Prijck, S.; Vandamme, N.; De Schepper, S.; Van Isterdael, G.; Scott, C.L.; et al. A single-cell atlas of mouse brain macrophages reveals unique transcriptional identities shaped by ontogeny and tissue environment. Nat. Neurosci. 2019, 22, 1021–1035. [Google Scholar] [CrossRef]
- Mrdjen, D.; Pavlovic, A.; Hartmann, F.J.; Schreiner, B.; Utz, S.G.; Leung, B.P.; Lelios, I.; Heppner, F.L.; Kipnis, J.; Merkler, D.; et al. High-Dimensional Single-Cell Mapping of Central Nervous System Immune Cells Reveals Distinct Myeloid Subsets in Health, Aging, and Disease. Immunity 2018, 48, 380–395.e6. [Google Scholar] [CrossRef] [Green Version]
- Keren-Shaul, H.; Spinrad, A.; Weiner, A.; Matcovitch-Natan, O.; Dvir-Szternfeld, R.; Ulland, T.K.; David, E.; Baruch, K.; Lara-Astaiso, D.; Toth, B.; et al. A Unique Microglia Type Associated with Restricting Development of Alzheimer’s Disease. Cell 2017, 169, 1276–1290.e17. [Google Scholar] [CrossRef]
- Sousa, C.; Golebiewska, A.; Poovathingal, S.K.; Kaoma, T.; Pires-Afonso, Y.; Martina, S.; Coowar, D.; Azuaje, F.; Skupin, A.; Balling, R.; et al. Single-cell transcriptomics reveals distinct inflammation-induced microglia signatures. EMBO Rep. 2018, 19, e46171. [Google Scholar] [CrossRef]
- Jordao, M.J.C.; Sankowski, R.; Brendecke, S.M.; Locatelli, G.; Tai, Y.H.; Tay, T.L.; Schramm, E.; Armbruster, S.; Hagemeyer, N.; Gross, O.; et al. Single-cell profiling identifies myeloid cell subsets with distinct fates during neuroinflammation. Science 2019, 363, eaat7554. [Google Scholar] [CrossRef] [PubMed]
- Muller, S.; Kohanbash, G.; Liu, S.J.; Alvarado, B.; Carrera, D.; Bhaduri, A.; Watchmaker, P.B.; Yagnik, G.; Di Lullo, E.; Malatesta, M.; et al. Single-cell profiling of human gliomas reveals macrophage ontogeny as a basis for regional differences in macrophage activation in the tumor microenvironment. Genome Biol. 2017, 18, 234. [Google Scholar] [CrossRef] [PubMed]
- Bennett, M.L.; Bennett, F.C.; Liddelow, S.A.; Ajami, B.; Zamanian, J.L.; Fernhoff, N.B.; Mulinyawe, S.B.; Bohlen, C.J.; Adil, A.; Tucker, A.; et al. New tools for studying microglia in the mouse and human CNS. Proc. Natl. Acad. Sci. USA 2016, 113, E1738–E1746. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Darmanis, S.; Sloan, S.A.; Croote, D.; Mignardi, M.; Chernikova, S.; Samghababi, P.; Zhang, Y.; Neff, N.; Kowarsky, M.; Caneda, C.; et al. Single-Cell RNA-Seq Analysis of Infiltrating Neoplastic Cells at the Migrating Front of Human Glioblastoma. Cell Rep. 2017, 21, 1399–1410. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lucas, C.G.; Solomon, D.A.; Perry, A. A review of recently described genetic alterations in central nervous system tumors. Hum. Pathol. 2019. [Google Scholar] [CrossRef]
- Venteicher, A.S.; Tirosh, I.; Hebert, C.; Yizhak, K.; Neftel, C.; Filbin, M.G.; Hovestadt, V.; Escalante, L.E.; Shaw, M.L.; Rodman, C.; et al. Decoupling genetics, lineages, and microenvironment in IDH-mutant gliomas by single-cell RNA-seq. Science 2017, 355, eaai8478. [Google Scholar] [CrossRef] [Green Version]
- Munoz-Fontela, C.; Mandinova, A.; Aaronson, S.A.; Lee, S.W. Emerging roles of p53 and other tumour-suppressor genes in immune regulation. Nat. Rev. Immunol. 2016, 16, 741–750. [Google Scholar] [CrossRef] [Green Version]
- Sankowski, R.; Bottcher, C.; Masuda, T.; Geirsdottir, L.; Sagar; Sindram, E.; Seredenina, T.; Muhs, A.; Scheiwe, C.; Shah, M.J.; et al. Mapping microglia states in the human brain through the integration of high-dimensional techniques. Nat. Neurosci. 2019, 22, 2098–2110. [Google Scholar] [CrossRef]
- Kumar, M.P.; Du, J.; Lagoudas, G.; Jiao, Y.; Sawyer, A.; Drummond, D.C.; Lauffenburger, D.A.; Raue, A. Analysis of Single-Cell RNA-Seq Identifies Cell-Cell Communication Associated with Tumor Characteristics. Cell Rep. 2018, 25, 1458–1468.e4. [Google Scholar] [CrossRef] [Green Version]



{kind=link}
{kind=link}
| Myeloid Target | Drug Name | Additional Treatment | Study Phase | Tumour Type | Study Identifier |
|---|---|---|---|---|---|
| CSF-1R inhibitor | Cabiralizumab | Nivolumab (anti-PD-1) | I | GBM | NCT02526017 |
| CSF-1R inhibitor | BLZ945 | PDR001 (anti-PD-1) | I/II | GBM/rGBM | NCT02829723 |
| CSF-1R inhibitor | Pexidartinib | RT + TMZ | I/II | GBM | NCT01790503 |
| CXCR4 inhibitor | USL311 | Lomustine | II | rGBM | NCT02765165 |
| PD-L1 inhibitor | Avelumab | MRI-guided LITT therapy | I | rGBM | NCT03341806 |
| STAT3 inhibitor | WP1066 | - | I | rGBM | NCT01904123 |
| GM-CSF | VBI-1901 | - | I/II | rGBM | NCT03382977 |
| MIF inhibitor | Ibudilast | TMZ | I/II | GBM/rGBM | NCT03782415 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pires-Afonso, Y.; Niclou, S.P.; Michelucci, A. Revealing and Harnessing Tumour-Associated Microglia/Macrophage Heterogeneity in Glioblastoma. Int. J. Mol. Sci. 2020, 21, 689. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21030689
Pires-Afonso Y, Niclou SP, Michelucci A. Revealing and Harnessing Tumour-Associated Microglia/Macrophage Heterogeneity in Glioblastoma. International Journal of Molecular Sciences. 2020; 21(3):689. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21030689
Chicago/Turabian StylePires-Afonso, Yolanda, Simone P. Niclou, and Alessandro Michelucci. 2020. "Revealing and Harnessing Tumour-Associated Microglia/Macrophage Heterogeneity in Glioblastoma" International Journal of Molecular Sciences 21, no. 3: 689. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21030689