NTRK Fusions in Central Nervous System Tumors: A Rare, but Worthy Target

 ,
,  , ,
, , 
Abstract
:1. Introduction
2. Biology of TRK Signaling
2.1. Characteristics of NTRK Genes and of TRK Signaling
2.2. The Physiological Role of NTRK Signaling and Its Role in Non-Neoplastic Diseases
3. NTRK in Tumor Development
3.1. The Oncogenic Role of NTRK: Fusions Versus Other Alterations
3.2. NTRK Alterations in Non-CNS Tumors
3.3. NTRK Fusions in Pediatric CNS Tumors
3.4. NTRK Fusions in Adult CNS Tumors
4. NTRK as a Novel Therapeutic Target
4.1. NTRK-Fusions Targeting: A Novel, Effective, Histology-Independent Anti-Neoplastic Treatment
4.2. Resistance Mechanisms to First-Generation NTRK Inhibitors
5. Testing for NTRK Fusions. Where Is Waldo?
5.1. Immunohistochemistry
5.2. Fluorescence In-Situ Hybridization
5.3. DNA and RNA Molecular Testing
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Chen, D.S.; Mellman, I. Elements of cancer immunity and the cancer-immune set point. Nature 2017, 541, 321–330. [Google Scholar] [CrossRef] [PubMed]
- Herbst, R.S.; Morgensztern, D.; Boshoff, C. The biology and management of non-small cell lung cancer. Nature 2018, 553, 446–454. [Google Scholar] [CrossRef] [PubMed]
- Ducreux, M.; Chamseddine, A.; Laurent-Puig, P.; Smolenschi, C.; Hollebecque, A.; Dartigues, P.; Samallin, E.; Boige, V.; Malka, D.; Gelli, M. Molecular targeted therapy of BRAF-mutant colorectal cancer. Ther. Adv. Med. Oncol. 2019, 11, 1758835919856494. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fussey, J.M.; Vaidya, B.; Kim, D.; Clark, J.; Ellard, S.; Smith, J.A. The role of molecular genetics in the clinical management of sporadic medullary thyroid carcinoma: A systematic review. Clin. Endocrinol. (Oxf.) 2019, 91, 697–707. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jorgensen, J.T. A paradigm shift in biomarker guided oncology drug development. Ann. Transl. Med. 2019, 7, 148. [Google Scholar] [CrossRef]
- Louis, D.N.; Ohgaki, H.; Wiestler, O.D.; Cavenee, W.K.; Ellison, D.W.; Figarella-Branger, D.; Perry, A.; Reifenberger, G.; von Deimling, A. International Agency for Research on Cancer. In WHO Classification of Tumours of the Central Nervous System, Revised 4th ed.; International Agency for Research on Cancer: Lyon, France, 2016; 408p. [Google Scholar]
- Scheie, D.; Kufaishi, H.H.A.; Broholm, H.; Lund, E.L.; de Stricker, K.; Melchior, L.C.; Grauslund, M. Biomarkers in tumors of the central nervous system—a review. APMIS 2019, 127, 265–287. [Google Scholar] [CrossRef] [Green Version]
- Burford, C.; Laxton, R.; Sidhu, Z.; Aizpurua, M.; King, A.; Bodi, I.; Ashkan, K.; Al-Sarraj, S. ATRX immunohistochemistry can help refine ‘not elsewhere classified’ categorisation for grade II/III gliomas. Br. J. Neurosurg. 2019, 33, 536–540. [Google Scholar] [CrossRef]
- Capper, D.; Jones, D.T.W.; Sill, M.; Hovestadt, V.; Schrimpf, D.; Sturm, D.; Koelsche, C.; Sahm, F.; Chavez, L.; Reuss, D.E.; et al. DNA methylation-based classification of central nervous system tumours. Nature 2018, 555, 469–474. [Google Scholar] [CrossRef]
- Ellison, D.W.; Hawkins, C.; Jones, D.T.W.; Onar-Thomas, A.; Pfister, S.M.; Reifenberger, G.; Louis, D.N. cIMPACT-NOW update 4: Diffuse gliomas characterized by MYB, MYBL1, or FGFR1 alterations or BRAF(V600E) mutation. Acta Neuropathol. 2019, 137, 683–687. [Google Scholar] [CrossRef]
- Ghasemi, D.R.; Sill, M.; Okonechnikov, K.; Korshunov, A.; Yip, S.; Schutz, P.W.; Scheie, D.; Kruse, A.; Harter, P.N.; Kastelan, M.; et al. MYCN amplification drives an aggressive form of spinal ependymoma. Acta Neuropathol. 2019, 138, 1075–1089. [Google Scholar] [CrossRef] [Green Version]
- Wefers, A.K.; Stichel, D.; Schrimpf, D.; Coras, R.; Pages, M.; Tauziede-Espariat, A.; Varlet, P.; Schwarz, D.; Soylemezoglu, F.; Pohl, U.; et al. Isomorphic diffuse glioma is a morphologically and molecularly distinct tumour entity with recurrent gene fusions of MYBL1 or MYB and a benign disease course. Acta Neuropathol. 2019. [Google Scholar] [CrossRef] [PubMed]
- Zwick, E.; Bange, J.; Ullrich, A. Receptor tyrosine kinase signalling as a target for cancer intervention strategies. Endocr. Relat. Cancer 2001, 8, 161–173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lemmon, M.A.; Schlessinger, J. Cell signaling by receptor tyrosine kinases. Cell 2010, 141, 1117–1134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schlessinger, J. Receptor tyrosine kinases: Legacy of the first two decades. Cold Spring Harb. Perspect. Biol. 2014, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Du, Z.; Lovly, C.M. Mechanisms of receptor tyrosine kinase activation in cancer. Mol. Cancer 2018, 17, 58. [Google Scholar] [CrossRef]
- Miles, J.; White, Y. Neratinib for the Treatment of Early-Stage HER2-Positive Breast Cancer. J. Adv. Pract. Oncol. 2018, 9, 750–754. [Google Scholar]
- Russo, A.; Franchina, T.; Ricciardi, G.; Battaglia, A.; Picciotto, M.; Adamo, V. Heterogeneous Responses to Epidermal Growth Factor Receptor (EGFR) Tyrosine Kinase Inhibitors (TKIs) in Patients with Uncommon EGFR Mutations: New Insights and Future Perspectives in this Complex Clinical Scenario. Int. J. Mol. Sci. 2019, 20, 1431. [Google Scholar] [CrossRef] [Green Version]
- Teishima, J.; Hayashi, T.; Nagamatsu, H.; Shoji, K.; Shikuma, H.; Yamanaka, R.; Sekino, Y.; Goto, K.; Inoue, S.; Matsubara, A. Fibroblast Growth Factor Family in the Progression of Prostate Cancer. J. Clin. Med. 2019, 8, 183. [Google Scholar] [CrossRef] [Green Version]
- Carlisle, J.W.; Ramalingam, S.S. Role of osimertinib in the treatment of EGFR-mutation positive non-small-cell lung cancer. Future Oncol. 2019, 15, 805–816. [Google Scholar] [CrossRef]
- Roskoski, R., Jr. Small molecule inhibitors targeting the EGFR/ErbB family of protein-tyrosine kinases in human cancers. Pharmacol. Res. 2019, 139, 395–411. [Google Scholar] [CrossRef]
- Goetze, T.O.; Al-Batran, S.E.; Berlth, F.; Hoelscher, A.H. Multimodal Treatment Strategies in Esophagogastric Junction Cancer: A Western Perspective. J. Gastric Cancer 2019, 19, 148–156. [Google Scholar] [CrossRef] [PubMed]
- Katoh, M. Fibroblast growth factor receptors as treatment targets in clinical oncology. Nat. Rev. Clin. Oncol. 2019, 16, 105–122. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Gutierrez, V.; Hernandez-Boluda, J.C. Tyrosine Kinase Inhibitors Available for Chronic Myeloid Leukemia: Efficacy and Safety. Front. Oncol. 2019, 9, 603. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pathak, M.; Dwivedi, S.N.; Deo, S.V.S.; Thakur, B.; Sreenivas, V.; Rath, G.K. Effectiveness of Added Targeted Therapies to Neoadjuvant Chemotherapy for Breast Cancer: A Systematic Review and Meta-analysis. Clin. Breast Cancer 2019. [Google Scholar] [CrossRef] [PubMed]
- Valent, A.; Danglot, G.; Bernheim, A. Mapping of the tyrosine kinase receptors trkA (NTRK1), trkB (NTRK2) and trkC(NTRK3) to human chromosomes 1q22, 9q22 and 15q25 by fluorescence in situ hybridization. Eur. J. Hum. Genet. 1997, 5, 102–104. [Google Scholar] [CrossRef] [PubMed]
- Pulciani, S.; Santos, E.; Lauver, A.V.; Long, L.K.; Aaronson, S.A.; Barbacid, M. Oncogenes in solid human tumours. Nature 1982, 300, 539–542. [Google Scholar] [CrossRef] [PubMed]
- Martin-Zanca, D.; Oskam, R.; Mitra, G.; Copeland, T.; Barbacid, M. Molecular and biochemical characterization of the human trk proto-oncogene. Mol. Cell Biol. 1989, 9, 24–33. [Google Scholar] [CrossRef] [Green Version]
- Klein, R.; Jing, S.Q.; Nanduri, V.; O’Rourke, E.; Barbacid, M. The trk proto-oncogene encodes a receptor for nerve growth factor. Cell 1991, 65, 189–197. [Google Scholar] [CrossRef]
- Soppet, D.; Escandon, E.; Maragos, J.; Middlemas, D.S.; Reid, S.W.; Blair, J.; Burton, L.E.; Stanton, B.R.; Kaplan, D.R.; Hunter, T.; et al. The neurotrophic factors brain-derived neurotrophic factor and neurotrophin-3 are ligands for the trkB tyrosine kinase receptor. Cell 1991, 65, 895–903. [Google Scholar] [CrossRef]
- Davies, A.M.; Horton, A.; Burton, L.E.; Schmelzer, C.; Vandlen, R.; Rosenthal, A. Neurotrophin-4/5 is a mammalian-specific survival factor for distinct populations of sensory neurons. J. Neurosci. 1993, 13, 4961–4967. [Google Scholar] [CrossRef]
- Dechant, G.; Biffo, S.; Okazawa, H.; Kolbeck, R.; Pottgiesser, J.; Barde, Y.A. Expression and binding characteristics of the BDNF receptor chick trkB. Development 1993, 119, 545–558. [Google Scholar] [PubMed]
- Chao, M.V. Neurotrophins and their receptors: A convergence point for many signalling pathways. Nat. Rev. Neurosci. 2003, 4, 299–309. [Google Scholar] [CrossRef] [PubMed]
- Deinhardt, K.; Chao, M.V. Trk receptors. Handb. Exp. Pharmacol. 2014, 220, 103–119. [Google Scholar] [CrossRef] [PubMed]
- Marchetti, L.; Bonsignore, F.; Gobbo, F.; Amodeo, R.; Calvello, M.; Jacob, A.; Signore, G.; Schirripa Spagnolo, C.; Porciani, D.; Mainardi, M.; et al. Fast-diffusing p75(NTR) monomers support apoptosis and growth cone collapse by neurotrophin ligands. Proc. Natl. Acad. Sci. USA 2019, 116, 21563–21572. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mahadeo, D.; Kaplan, L.; Chao, M.V.; Hempstead, B.L. High affinity nerve growth factor binding displays a faster rate of association than p140trk binding. Implications for multi-subunit polypeptide receptors. J. Biol. Chem. 1994, 269, 6884–6891. [Google Scholar]
- Saadipour, K.; MacLean, M.; Pirkle, S.; Ali, S.; Lopez-Redondo, M.L.; Stokes, D.L.; Chao, M.V. The transmembrane domain of the p75 neurotrophin receptor stimulates phosphorylation of the TrkB tyrosine kinase receptor. J. Biol. Chem. 2017, 292, 16594–16604. [Google Scholar] [CrossRef] [Green Version]
- Vilar, M. Structural Characterization of the p75 Neurotrophin Receptor: A Stranger in the TNFR Superfamily. Vitam. Horm. 2017, 104, 57–87. [Google Scholar] [CrossRef]
- Alshehri, M.M.; Robbins, S.M.; Senger, D.L. The Role of Neurotrophin Signaling in Gliomagenesis: A Focus on the p75 Neurotrophin Receptor (p75(NTR)/CD271). Vitam. Horm. 2017, 104, 367–404. [Google Scholar] [CrossRef]
- Vaishnavi, A.; Le, A.T.; Doebele, R.C. TRKing down an old oncogene in a new era of targeted therapy. Cancer Discov. 2015, 5, 25–34. [Google Scholar] [CrossRef] [Green Version]
- Cocco, E.; Scaltriti, M.; Drilon, A. NTRK fusion-positive cancers and TRK inhibitor therapy. Nat. Rev. Clin. Oncol. 2018, 15, 731–747. [Google Scholar] [CrossRef]
- Yamashita, N.; Kuruvilla, R. Neurotrophin signaling endosomes: Biogenesis, regulation, and functions. Curr. Opin. Neurobiol. 2016, 39, 139–145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barford, K.; Deppmann, C.; Winckler, B. The neurotrophin receptor signaling endosome: Where trafficking meets signaling. Dev. Neurobiol. 2017, 77, 405–418. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clary, D.O.; Reichardt, L.F. An alternatively spliced form of the nerve growth factor receptor TrkA confers an enhanced response to neurotrophin 3. Proc. Natl. Acad. Sci. USA 1994, 91, 11133–11137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, E.J.; Reichardt, L.F. Trk receptors: Roles in neuronal signal transduction. Annu. Rev. Biochem. 2003, 72, 609–642. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luberg, K.; Wong, J.; Weickert, C.S.; Timmusk, T. Human TrkB gene: Novel alternative transcripts, protein isoforms and expression pattern in the prefrontal cerebral cortex during postnatal development. J. Neurochem. 2010, 113, 952–964. [Google Scholar] [CrossRef] [PubMed]
- Stoilov, P.; Castren, E.; Stamm, S. Analysis of the human TrkB gene genomic organization reveals novel TrkB isoforms, unusual gene length, and splicing mechanism. Biochem. Biophys. Res. Commun. 2002, 290, 1054–1065. [Google Scholar] [CrossRef] [Green Version]
- Brodeur, G.M.; Minturn, J.E.; Ho, R.; Simpson, A.M.; Iyer, R.; Varela, C.R.; Light, J.E.; Kolla, V.; Evans, A.E. Trk receptor expression and inhibition in neuroblastomas. Clin. Cancer Res. 2009, 15, 3244–3250. [Google Scholar] [CrossRef] [Green Version]
- Tacconelli, A.; Farina, A.R.; Cappabianca, L.; Desantis, G.; Tessitore, A.; Vetuschi, A.; Sferra, R.; Rucci, N.; Argenti, B.; Screpanti, I.; et al. TrkA alternative splicing: A regulated tumor-promoting switch in human neuroblastoma. Cancer Cell 2004, 6, 347–360. [Google Scholar] [CrossRef] [Green Version]
- Tacconelli, A.; Farina, A.R.; Cappabianca, L.; Gulino, A.; Mackay, A.R. Alternative TrkAIII splicing: A potential regulated tumor-promoting switch and therapeutic target in neuroblastoma. Future Oncol. 2005, 1, 689–698. [Google Scholar] [CrossRef]
- Snider, W.D. Functions of the neurotrophins during nervous system development: What the knockouts are teaching us. Cell 1994, 77, 627–638. [Google Scholar] [CrossRef]
- Bibel, M.; Barde, Y.A. Neurotrophins: Key regulators of cell fate and cell shape in the vertebrate nervous system. Genes Dev. 2000, 14, 2919–2937. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Minichiello, L.; Korte, M.; Wolfer, D.; Kuhn, R.; Unsicker, K.; Cestari, V.; Rossi-Arnaud, C.; Lipp, H.P.; Bonhoeffer, T.; Klein, R. Essential role for TrkB receptors in hippocampus-mediated learning. Neuron 1999, 24, 401–414. [Google Scholar] [CrossRef] [Green Version]
- Schropel, A.; von Schack, D.; Dechant, G.; Barde, Y.A. Early expression of the nerve growth factor receptor ctrkA in chick sympathetic and sensory ganglia. Mol. Cell Neurosci. 1995, 6, 544–566. [Google Scholar] [CrossRef] [PubMed]
- Minichiello, L.; Klein, R. TrkB and TrkC neurotrophin receptors cooperate in promoting survival of hippocampal and cerebellar granule neurons. Genes Dev. 1996, 10, 2849–2858. [Google Scholar] [CrossRef] [Green Version]
- Tchetchelnitski, V.; van den Eijnden, M.; Schmidt, F.; Stoker, A.W. Developmental co-expression and functional redundancy of tyrosine phosphatases with neurotrophin receptors in developing sensory neurons. Int. J. Dev. Neurosci. 2014, 34, 48–59. [Google Scholar] [CrossRef]
- Ito, A.; Nosrat, C.A. Gustatory papillae and taste bud development and maintenance in the absence of TrkB ligands BDNF and NT-4. Cell Tissue Res. 2009, 337, 349–359. [Google Scholar] [CrossRef]
- Nittoli, V.; Sepe, R.M.; Coppola, U.; D’Agostino, Y.; De Felice, E.; Palladino, A.; Vassalli, Q.A.; Locascio, A.; Ristoratore, F.; Spagnuolo, A.; et al. A comprehensive analysis of neurotrophins and neurotrophin tyrosine kinase receptors expression during development of zebrafish. J. Comp. Neurol. 2018, 526, 1057–1072. [Google Scholar] [CrossRef]
- Minichiello, L.; Calella, A.M.; Medina, D.L.; Bonhoeffer, T.; Klein, R.; Korte, M. Mechanism of TrkB-mediated hippocampal long-term potentiation. Neuron 2002, 36, 121–137. [Google Scholar] [CrossRef] [Green Version]
- Gorski, J.A.; Zeiler, S.R.; Tamowski, S.; Jones, K.R. Brain-derived neurotrophic factor is required for the maintenance of cortical dendrites. J. Neurosci. 2003, 23, 6856–6865. [Google Scholar] [CrossRef]
- Medina, D.L.; Sciarretta, C.; Calella, A.M.; Von Bohlen Und Halbach, O.; Unsicker, K.; Minichiello, L. TrkB regulates neocortex formation through the Shc/PLCgamma-mediated control of neuronal migration. EMBO J. 2004, 23, 3803–3814. [Google Scholar] [CrossRef] [Green Version]
- Calella, A.M.; Nerlov, C.; Lopez, R.G.; Sciarretta, C.; von Bohlen und Halbach, O.; Bereshchenko, O.; Minichiello, L. Neurotrophin/Trk receptor signaling mediates C/EBPalpha, -beta and NeuroD recruitment to immediate-early gene promoters in neuronal cells and requires C/EBPs to induce immediate-early gene transcription. Neural Dev. 2007, 2, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farhang, S.; Barar, J.; Fakhari, A.; Mesgariabbasi, M.; Khani, S.; Omidi, Y.; Farnam, A. Asymmetrical expression of BDNF and NTRK3 genes in frontoparietal cortex of stress-resilient rats in an animal model of depression. Synapse 2014, 68, 387–393. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Simmons, M.S.; Perry, R.T.; Wiener, H.W.; Harrell, L.E.; Go, R.C. Genetic association of neurotrophic tyrosine kinase receptor type 2 (NTRK2) With Alzheimer’s disease. Am. J. Med. Genet. B Neuropsychiatr. Genet. 2008, 147, 363–369. [Google Scholar] [CrossRef] [PubMed]
- Weickert, C.S.; Ligons, D.L.; Romanczyk, T.; Ungaro, G.; Hyde, T.M.; Herman, M.M.; Weinberger, D.R.; Kleinman, J.E. Reductions in neurotrophin receptor mRNAs in the prefrontal cortex of patients with schizophrenia. Mol. Psychiatry 2005, 10, 637–650. [Google Scholar] [CrossRef]
- Otnaess, M.K.; Djurovic, S.; Rimol, L.M.; Kulle, B.; Kahler, A.K.; Jonsson, E.G.; Agartz, I.; Sundet, K.; Hall, H.; Timm, S.; et al. Evidence for a possible association of neurotrophin receptor (NTRK-3) gene polymorphisms with hippocampal function and schizophrenia. Neurobiol. Dis. 2009, 34, 518–524. [Google Scholar] [CrossRef]
- Autry, A.E.; Monteggia, L.M. Brain-derived neurotrophic factor and neuropsychiatric disorders. Pharmacol. Rev. 2012, 64, 238–258. [Google Scholar] [CrossRef] [Green Version]
- Boulle, F.; Kenis, G.; Cazorla, M.; Hamon, M.; Steinbusch, H.W.; Lanfumey, L.; van den Hove, D.L. TrkB inhibition as a therapeutic target for CNS-related disorders. Prog. Neurobiol. 2012, 98, 197–206. [Google Scholar] [CrossRef]
- Drilon, A.; Siena, S.; Ou, S.I.; Patel, M.; Ahn, M.J.; Lee, J.; Bauer, T.M.; Farago, A.F.; Wheler, J.J.; Liu, S.V.; et al. Safety and Antitumor Activity of the Multitargeted Pan-TRK, ROS1, and ALK Inhibitor Entrectinib: Combined Results from Two Phase I Trials (ALKA-372-001 and STARTRK-1). Cancer Discov. 2017, 7, 400–409. [Google Scholar] [CrossRef] [Green Version]
- Okamura, R.; Boichard, A.; Kato, S.; Sicklick, J.K.; Bazhenova, L.; Kurzrock, R. Analysis of NTRK Alterations in Pan-Cancer Adult and Pediatric Malignancies: Implications for NTRK-Targeted Therapeutics. JCO Precis. Oncol. 2018, 2018. [Google Scholar] [CrossRef]
- Shaw, A.T.; Hsu, P.P.; Awad, M.M.; Engelman, J.A. Tyrosine kinase gene rearrangements in epithelial malignancies. Nat. Rev. Cancer 2013, 13, 772–787. [Google Scholar] [CrossRef]
- Stransky, N.; Cerami, E.; Schalm, S.; Kim, J.L.; Lengauer, C. The landscape of kinase fusions in cancer. Nat. Commun. 2014, 5, 4846. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gross, S.; Rahal, R.; Stransky, N.; Lengauer, C.; Hoeflich, K.P. Targeting cancer with kinase inhibitors. J. Clin. Invest. 2015, 125, 1780–1789. [Google Scholar] [CrossRef] [PubMed]
- Khotskaya, Y.B.; Holla, V.R.; Farago, A.F.; Mills Shaw, K.R.; Meric-Bernstam, F.; Hong, D.S. Targeting TRK family proteins in cancer. Pharmacol. Ther. 2017, 173, 58–66. [Google Scholar] [CrossRef] [PubMed]
- Farago, A.F.; Azzoli, C.G. Beyond ALK and ROS1: RET, NTRK, EGFR and BRAF gene rearrangements in non-small cell lung cancer. Transl. Lung Cancer Res. 2017, 6, 550–559. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tognon, C.; Knezevich, S.R.; Huntsman, D.; Roskelley, C.D.; Melnyk, N.; Mathers, J.A.; Becker, L.; Carneiro, F.; MacPherson, N.; Horsman, D.; et al. Expression of the ETV6-NTRK3 gene fusion as a primary event in human secretory breast carcinoma. Cancer Cell 2002, 2, 367–376. [Google Scholar] [CrossRef] [Green Version]
- Hung, Y.P.; Fletcher, C.D.M.; Hornick, J.L. Evaluation of pan-TRK immunohistochemistry in infantile fibrosarcoma, lipofibromatosis-like neural tumour and histological mimics. Histopathology 2018, 73, 634–644. [Google Scholar] [CrossRef] [PubMed]
- Hung, Y.P.; Jo, V.Y.; Hornick, J.L. Immunohistochemistry with a pan-TRK antibody distinguishes secretory carcinoma of the salivary gland from acinic cell carcinoma. Histopathology 2019, 75, 54–62. [Google Scholar] [CrossRef] [PubMed]
- Xu, B.; Haroon Al Rasheed, M.R.; Antonescu, C.R.; Alex, D.; Frosina, D.; Ghossein, R.; Jungbluth, A.A.; Katabi, N. Pan-Trk immunohistochemistry is a sensitive and specific ancillary tool in diagnosing secretory carcinoma of salivary gland and detecting ETV6-NTRK3 fusion. Histopathology 2019. [Google Scholar] [CrossRef]
- Harrison, B.T.; Fowler, E.; Krings, G.; Chen, Y.Y.; Bean, G.R.; Vincent-Salomon, A.; Fuhrmann, L.; Barnick, S.E.; Chen, B.; Hosfield, E.M.; et al. Pan-TRK Immunohistochemistry: A Useful Diagnostic Adjunct for Secretory Carcinoma of the Breast. Am. J. Surg. Pathol. 2019. [Google Scholar] [CrossRef]
- Gao, Q.; Liang, W.W.; Foltz, S.M.; Mutharasu, G.; Jayasinghe, R.G.; Cao, S.; Liao, W.W.; Reynolds, S.M.; Wyczalkowski, M.A.; Yao, L.; et al. Driver Fusions and Their Implications in the Development and Treatment of Human Cancers. Cell Rep. 2018, 23, 227–238 e3. [Google Scholar] [CrossRef] [Green Version]
- Reuther, G.W.; Lambert, Q.T.; Caligiuri, M.A.; Der, C.J. Identification and characterization of an activating TrkA deletion mutation in acute myeloid leukemia. Mol. Cell. Biol. 2000, 20, 8655–8666. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thiele, C.J.; Li, Z.; McKee, A.E. On Trk--the TrkB signal transduction pathway is an increasingly important target in cancer biology. Clin. Cancer Res. 2009, 15, 5962–5967. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Light, J.E.; Koyama, H.; Minturn, J.E.; Ho, R.; Simpson, A.M.; Iyer, R.; Mangino, J.L.; Kolla, V.; London, W.B.; Brodeur, G.M. Clinical significance of NTRK family gene expression in neuroblastomas. Pediatr. Blood Cancer 2012, 59, 226–232. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.J.; Kim, N.K.D.; Lee, S.-H.; Kim, S.T.; Park, S.H.; Park, J.O.; Park, Y.S.; Lim, H.Y.; Kang, W.K.; Park, W.Y.; et al. NTRK gene amplification in patients with metastatic cancer. Precis Future Med. 2017, 1, 129–137. [Google Scholar] [CrossRef] [Green Version]
- Farina, A.R.; Cappabianca, L.; Ruggeri, P.; Gneo, L.; Pellegrini, C.; Fargnoli, M.C.; Mackay, A.R. The oncogenic neurotrophin receptor tropomyosin-related kinase variant, TrkAIII. J. Exp. Clin. Cancer Res. 2018, 37, 119. [Google Scholar] [CrossRef] [PubMed]
- Fuse, M.J.; Okada, K.; Oh-Hara, T.; Ogura, H.; Fujita, N.; Katayama, R. Mechanisms of Resistance to NTRK Inhibitors and Therapeutic Strategies in NTRK1-Rearranged Cancers. Mol. Cancer Ther. 2017, 16, 2130–2143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martin-Zanca, D.; Hughes, S.H.; Barbacid, M. A human oncogene formed by the fusion of truncated tropomyosin and protein tyrosine kinase sequences. Nature 1986, 319, 743–748. [Google Scholar] [CrossRef]
- Ardini, E.; Bosotti, R.; Borgia, A.L.; De Ponti, C.; Somaschini, A.; Cammarota, R.; Amboldi, N.; Raddrizzani, L.; Milani, A.; Magnaghi, P.; et al. The TPM3-NTRK1 rearrangement is a recurring event in colorectal carcinoma and is associated with tumor sensitivity to TRKA kinase inhibition. Mol. Oncol. 2014, 8, 1495–1507. [Google Scholar] [CrossRef]
- Creancier, L.; Vandenberghe, I.; Gomes, B.; Dejean, C.; Blanchet, J.C.; Meilleroux, J.; Guimbaud, R.; Selves, J.; Kruczynski, A. Chromosomal rearrangements involving the NTRK1 gene in colorectal carcinoma. Cancer Lett. 2015, 365, 107–111. [Google Scholar] [CrossRef]
- Lee, S.J.; Li, G.G.; Kim, S.T.; Hong, M.E.; Jang, J.; Yoon, N.; Ahn, S.M.; Murphy, D.; Christiansen, J.; Wei, G.; et al. NTRK1 rearrangement in colorectal cancer patients: Evidence for actionable target using patient-derived tumor cell line. Oncotarget 2015, 6, 39028–39035. [Google Scholar] [CrossRef] [Green Version]
- Sartore-Bianchi, A.; Ardini, E.; Bosotti, R.; Amatu, A.; Valtorta, E.; Somaschini, A.; Raddrizzani, L.; Palmeri, L.; Banfi, P.; Bonazzina, E.; et al. Sensitivity to Entrectinib Associated With a Novel LMNA-NTRK1 Gene Fusion in Metastatic Colorectal Cancer. J. Natl. Cancer Inst. 2016, 108. [Google Scholar] [CrossRef]
- Hechtman, J.F.; Zehir, A.; Yaeger, R.; Wang, L.; Middha, S.; Zheng, T.; Hyman, D.M.; Solit, D.; Arcila, M.E.; Borsu, L.; et al. Identification of Targetable Kinase Alterations in Patients with Colorectal Carcinoma That are Preferentially Associated with Wild-Type RAS/RAF. Mol. Cancer Res. 2016, 14, 296–301. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pietrantonio, F.; Di Nicolantonio, F.; Schrock, A.B.; Lee, J.; Tejpar, S.; Sartore-Bianchi, A.; Hechtman, J.F.; Christiansen, J.; Novara, L.; Tebbutt, N.; et al. ALK, ROS1, and NTRK Rearrangements in Metastatic Colorectal Cancer. J. Natl. Cancer Inst. 2017, 109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vaishnavi, A.; Capelletti, M.; Le, A.T.; Kako, S.; Butaney, M.; Ercan, D.; Mahale, S.; Davies, K.D.; Aisner, D.L.; Pilling, A.B.; et al. Oncogenic and drug-sensitive NTRK1 rearrangements in lung cancer. Nat. Med. 2013, 19, 1469–1472. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bongarzone, I.; Pierotti, M.A.; Monzini, N.; Mondellini, P.; Manenti, G.; Donghi, R.; Pilotti, S.; Grieco, M.; Santoro, M.; Fusco, A.; et al. High frequency of activation of tyrosine kinase oncogenes in human papillary thyroid carcinoma. Oncogene 1989, 4, 1457–1462. [Google Scholar] [PubMed]
- Butti, M.G.; Bongarzone, I.; Ferraresi, G.; Mondellini, P.; Borrello, M.G.; Pierotti, M.A. A sequence analysis of the genomic regions involved in the rearrangements between TPM3 and NTRK1 genes producing TRK oncogenes in papillary thyroid carcinomas. Genomics 1995, 28, 15–24. [Google Scholar] [CrossRef] [PubMed]
- Leeman-Neill, R.J.; Kelly, L.M.; Liu, P.; Brenner, A.V.; Little, M.P.; Bogdanova, T.I.; Evdokimova, V.N.; Hatch, M.; Zurnadzy, L.Y.; Nikiforova, M.N.; et al. ETV6-NTRK3 is a common chromosomal rearrangement in radiation-associated thyroid cancer. Cancer 2014, 120, 799–807. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Knezevich, S.R.; Garnett, M.J.; Pysher, T.J.; Beckwith, J.B.; Grundy, P.E.; Sorensen, P.H. ETV6-NTRK3 gene fusions and trisomy 11 establish a histogenetic link between mesoblastic nephroma and congenital fibrosarcoma. Cancer Res. 1998, 58, 5046–5048. [Google Scholar]
- Knezevich, S.R.; McFadden, D.E.; Tao, W.; Lim, J.F.; Sorensen, P.H. A novel ETV6-NTRK3 gene fusion in congenital fibrosarcoma. Nat. Genet. 1998, 18, 184–187. [Google Scholar] [CrossRef]
- Skalova, A.; Vanecek, T.; Sima, R.; Laco, J.; Weinreb, I.; Perez-Ordonez, B.; Starek, I.; Geierova, M.; Simpson, R.H.; Passador-Santos, F.; et al. Mammary analogue secretory carcinoma of salivary glands, containing the ETV6-NTRK3 fusion gene: A hitherto undescribed salivary gland tumor entity. Am. J. Surg. Pathol. 2010, 34, 599–608. [Google Scholar] [CrossRef]
- Lannon, C.L.; Sorensen, P.H. ETV6-NTRK3: A chimeric protein tyrosine kinase with transformation activity in multiple cell lineages. Semin. Cancer Biol. 2005, 15, 215–223. [Google Scholar] [CrossRef] [PubMed]
- Yeh, I.; Tee, M.K.; Botton, T.; Shain, A.H.; Sparatta, A.J.; Gagnon, A.; Vemula, S.S.; Garrido, M.C.; Nakamaru, K.; Isoyama, T.; et al. NTRK3 kinase fusions in Spitz tumours. J. Pathol. 2016, 240, 282–290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnson, M.D.; Stone, B.; Thibodeau, B.J.; Baschnagel, A.M.; Galoforo, S.; Fortier, L.E.; Ketelsen, B.; Ahmed, S.; Kelley, Z.; Hana, A.; et al. The significance of Trk receptors in pancreatic cancer. Tumour. Biol. 2017, 39, 1010428317692256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sigal, D.; Tartar, M.; Xavier, M.; Bao, F.; Foley, P.; Luo, D.; Christiansen, J.; Hornby, Z.; Maneval, E.C.; Multani, P. Activity of Entrectinib in a Patient With the First Reported NTRK Fusion in Neuroendocrine Cancer. J. Natl. Compr. Canc. Netw. 2017, 15, 1317–1322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rudzinski, E.R.; Lockwood, C.M.; Stohr, B.A.; Vargas, S.O.; Sheridan, R.; Black, J.O.; Rajaram, V.; Laetsch, T.W.; Davis, J.L. Pan-Trk Immunohistochemistry Identifies NTRK Rearrangements in Pediatric Mesenchymal Tumors. Am. J. Surg. Pathol. 2018, 42, 927–935. [Google Scholar] [CrossRef] [PubMed]
- Sigal, D.S.; Bhangoo, M.S.; Hermel, J.A.; Pavlick, D.C.; Frampton, G.; Miller, V.A.; Ross, J.S.; Ali, S.M. Comprehensive genomic profiling identifies novel NTRK fusions in neuroendocrine tumors. Oncotarget 2018, 9, 35809–35812. [Google Scholar] [CrossRef] [Green Version]
- Lezcano, C.; Shoushtari, A.N.; Ariyan, C.; Hollmann, T.J.; Busam, K.J. Primary and Metastatic Melanoma With NTRK Fusions. Am. J. Surg. Pathol. 2018, 42, 1052–1058. [Google Scholar] [CrossRef]
- Miettinen, M.; Felisiak-Golabek, A.; Luina Contreras, A.; Glod, J.; Kaplan, R.N.; Killian, J.K.; Lasota, J. New fusion sarcomas: Histopathology and clinical significance of selected entities. Hum. Pathol. 2019, 86, 57–65. [Google Scholar] [CrossRef]
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [Green Version]
- Ostrom, Q.T.; Cioffi, G.; Gittleman, H.; Patil, N.; Waite, K.; Kruchko, C.; Barnholtz-Sloan, J.S. CBTRUS Statistical Report: Primary Brain and Other Central Nervous System Tumors Diagnosed in the United States in 2012-2016. Neuro. Oncol. 2019, 21, v1–v100. [Google Scholar] [CrossRef]
- Zhang, J.; Wu, G.; Miller, C.P.; Tatevossian, R.G.; Dalton, J.D.; Tang, B.; Orisme, W.; Punchihewa, C.; Parker, M.; Qaddoumi, I.; et al. Whole-genome sequencing identifies genetic alterations in pediatric low-grade gliomas. Nat. Genet. 2013, 45, 602–612. [Google Scholar] [CrossRef] [PubMed]
- Lake, J.A.; Donson, A.M.; Prince, E.; Davies, K.D.; Nellan, A.; Green, A.L.; Mulcahy Levy, J.; Dorris, K.; Vibhakar, R.; Hankinson, T.C.; et al. Targeted fusion analysis can aid in the classification and treatment of pediatric glioma, ependymoma, and glioneuronal tumors. Pediatr. Blood Cancer 2020, 67, e28028. [Google Scholar] [CrossRef] [PubMed]
- Jones, D.T.; Hutter, B.; Jager, N.; Korshunov, A.; Kool, M.; Warnatz, H.J.; Zichner, T.; Lambert, S.R.; Ryzhova, M.; Quang, D.A.; et al. Recurrent somatic alterations of FGFR1 and NTRK2 in pilocytic astrocytoma. Nat. Genet. 2013, 45, 927–932. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.; Han, S.; Meng, L.; Li, Z.; Zhang, X.; Wu, A. TERT promoter mutations lead to high transcriptional activity under hypoxia and temozolomide treatment and predict poor prognosis in gliomas. PLoS One 2014, 9, e100297. [Google Scholar] [CrossRef] [Green Version]
- Lassaletta, A.; Zapotocky, M.; Bouffet, E.; Hawkins, C.; Tabori, U. An integrative molecular and genomic analysis of pediatric hemispheric low-grade gliomas: An update. Childs Nerv. Syst. 2016, 32, 1789–1797. [Google Scholar] [CrossRef] [PubMed]
- Vanan, M.I.; Underhill, D.A.; Eisenstat, D.D. Targeting Epigenetic Pathways in the Treatment of Pediatric Diffuse (High Grade) Gliomas. Neurotherapeutics 2017, 14, 274–283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bornhorst, M.; Hwang, E.I. Molecularly Targeted Agents in the Therapy of Pediatric Brain Tumors. Paediatr. Drugs 2019. [Google Scholar] [CrossRef]
- Collins, V.P.; Jones, D.T.; Giannini, C. Pilocytic astrocytoma: Pathology, molecular mechanisms and markers. Acta Neuropathol. 2015, 129, 775–788. [Google Scholar] [CrossRef] [Green Version]
- Nobusawa, S.; Hirato, J.; Yokoo, H. Molecular genetics of ependymomas and pediatric diffuse gliomas: A short review. Brain Tumor Pathol. 2014, 31, 229–233. [Google Scholar] [CrossRef]
- Wu, G.; Diaz, A.K.; Paugh, B.S.; Rankin, S.L.; Ju, B.; Li, Y.; Zhu, X.; Qu, C.; Chen, X.; Zhang, J.; et al. The genomic landscape of diffuse intrinsic pontine glioma and pediatric non-brainstem high-grade glioma. Nat. Genet. 2014, 46, 444–450. [Google Scholar] [CrossRef]
- Chamdine, O.; Gajjar, A. Molecular characteristics of pediatric high-grade gliomas. CNS Oncol. 2014, 3, 433–443. [Google Scholar] [CrossRef] [PubMed]
- Albert, C.M.; Davis, J.L.; Federman, N.; Casanova, M.; Laetsch, T.W. TRK Fusion Cancers in Children: A Clinical Review and Recommendations for Screening. J. Clin. Oncol. 2019, 37, 513–524. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Z.; Liebers, M.; Zhelyazkova, B.; Cao, Y.; Panditi, D.; Lynch, K.D.; Chen, J.; Robinson, H.E.; Shim, H.S.; Chmielecki, J.; et al. Anchored multiplex PCR for targeted next-generation sequencing. Nat. Med. 2014, 20, 1479–1484. [Google Scholar] [CrossRef] [PubMed]
- Deng, M.Y.; Sill, M.; Chiang, J.; Schittenhelm, J.; Ebinger, M.; Schuhmann, M.U.; Monoranu, C.M.; Milde, T.; Wittmann, A.; Hartmann, C.; et al. Molecularly defined diffuse leptomeningeal glioneuronal tumor (DLGNT) comprises two subgroups with distinct clinical and genetic features. Acta Neuropathol. 2018, 136, 239–253. [Google Scholar] [CrossRef] [Green Version]
- Prabhakaran, N.; Guzman, M.A.; Navalkele, P.; Chow-Maneval, E.; Batanian, J.R. Novel TLE4-NTRK2 fusion in a ganglioglioma identified by array-CGH and confirmed by NGS: Potential for a gene targeted therapy. Neuropathology 2018. [Google Scholar] [CrossRef]
- Kurozumi, K.; Nakano, Y.; Ishida, J.; Tanaka, T.; Doi, M.; Hirato, J.; Yoshida, A.; Washio, K.; Shimada, A.; Kohno, T.; et al. High-grade glioneuronal tumor with an ARHGEF2-NTRK1 fusion gene. Brain Tumor Pathol. 2019, 36, 121–128. [Google Scholar] [CrossRef]
- Segal, R.A.; Goumnerova, L.C.; Kwon, Y.K.; Stiles, C.D.; Pomeroy, S.L. Expression of the neurotrophin receptor TrkC is linked to a favorable outcome in medulloblastoma. Proc. Natl. Acad. Sci. USA 1994, 91, 12867–12871. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.Y.; Sutton, M.E.; Lu, D.J.; Cho, T.A.; Goumnerova, L.C.; Goritchenko, L.; Kaufman, J.R.; Lam, K.K.; Billet, A.L.; Tarbell, N.J.; et al. Activation of neurotrophin-3 receptor TrkC induces apoptosis in medulloblastomas. Cancer Res. 1999, 59, 711–719. [Google Scholar]
- Grotzer, M.A.; Janss, A.J.; Fung, K.; Biegel, J.A.; Sutton, L.N.; Rorke, L.B.; Zhao, H.; Cnaan, A.; Phillips, P.C.; Lee, V.M.; et al. TrkC expression predicts good clinical outcome in primitive neuroectodermal brain tumors. J. Clin. Oncol. 2000, 18, 1027–1035. [Google Scholar] [CrossRef]
- Brandes, A.A.; Franceschi, E. Shedding light on adult medulloblastoma: Current management and opportunities for advances. Am. Soc. Clin. Oncol. Educ. Book 2014, e82–e87. [Google Scholar] [CrossRef]
- Amatu, A.; Sartore-Bianchi, A.; Siena, S. NTRK gene fusions as novel targets of cancer therapy across multiple tumour types. ESMO Open 2016, 1, e000023. [Google Scholar] [CrossRef] [PubMed]
- Ferguson, S.D.; Zhou, S.; Huse, J.T.; de Groot, J.F.; Xiu, J.; Subramaniam, D.S.; Mehta, S.; Gatalica, Z.; Swensen, J.; Sanai, N.; et al. Targetable Gene Fusions Associate With the IDH Wild-Type Astrocytic Lineage in Adult Gliomas. J. Neuropathol. Exp. Neurol. 2018, 77, 437–442. [Google Scholar] [CrossRef] [Green Version]
- Frattini, V.; Trifonov, V.; Chan, J.M.; Castano, A.; Lia, M.; Abate, F.; Keir, S.T.; Ji, A.X.; Zoppoli, P.; Niola, F.; et al. The integrated landscape of driver genomic alterations in glioblastoma. Nat. Genet. 2013, 45, 1141–1149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shah, N.; Lankerovich, M.; Lee, H.; Yoon, J.G.; Schroeder, B.; Foltz, G. Exploration of the gene fusion landscape of glioblastoma using transcriptome sequencing and copy number data. BMC Genomics 2013, 14, 818. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.; Lee, Y.; Cho, H.J.; Lee, Y.E.; An, J.; Cho, G.H.; Ko, Y.H.; Joo, K.M.; Nam, D.H. NTRK1 fusion in glioblastoma multiforme. PLoS One 2014, 9, e91940. [Google Scholar] [CrossRef] [Green Version]
- Cook, P.J.; Thomas, R.; Kannan, R.; de Leon, E.S.; Drilon, A.; Rosenblum, M.K.; Scaltriti, M.; Benezra, R.; Ventura, A. Somatic chromosomal engineering identifies BCAN-NTRK1 as a potent glioma driver and therapeutic target. Nat. Commun. 2017, 8, 15987. [Google Scholar] [CrossRef] [PubMed]
- Assimakopoulou, M.; Kondyli, M.; Gatzounis, G.; Maraziotis, T.; Varakis, J. Neurotrophin receptors expression and JNK pathway activation in human astrocytomas. BMC Cancer 2007, 7, 202. [Google Scholar] [CrossRef] [Green Version]
- Palani, M.; Arunkumar, R.; Vanisree, A.J. Methylation and expression patterns of tropomyosin-related kinase genes in different grades of glioma. Neuromolecular Med. 2014, 16, 529–539. [Google Scholar] [CrossRef]
- Pajtler, K.W.; Rebmann, V.; Lindemann, M.; Schulte, J.H.; Schulte, S.; Stauder, M.; Leuschner, I.; Schmid, K.W.; Kohl, U.; Schramm, A.; et al. Expression of NTRK1/TrkA affects immunogenicity of neuroblastoma cells. Int. J. Cancer 2013, 133, 908–919. [Google Scholar] [CrossRef]
- Offin, M.; Liu, D.; Drilon, A. Tumor-Agnostic Drug Development. Am. Soc. Clin. Oncol. Educ. Book 2018, 38, 184–187. [Google Scholar] [CrossRef]
- Liu, D.; Offin, M.; Harnicar, S.; Li, B.T.; Drilon, A. Entrectinib: An orally available, selective tyrosine kinase inhibitor for the treatment of NTRK, ROS1, and ALK fusion-positive solid tumors. Ther. Clin. Risk Manag. 2018, 14, 1247–1252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ardini, E.; Menichincheri, M.; Banfi, P.; Bosotti, R.; De Ponti, C.; Pulci, R.; Ballinari, D.; Ciomei, M.; Texido, G.; Degrassi, A.; et al. Entrectinib, a Pan-TRK, ROS1, and ALK Inhibitor with Activity in Multiple Molecularly Defined Cancer Indications. Mol. Cancer Ther. 2016, 15, 628–639. [Google Scholar] [CrossRef] [Green Version]
- Drilon, A.; Laetsch, T.W.; Kummar, S.; DuBois, S.G.; Lassen, U.N.; Demetri, G.D.; Nathenson, M.; Doebele, R.C.; Farago, A.F.; Pappo, A.S.; et al. Efficacy of Larotrectinib in TRK Fusion-Positive Cancers in Adults and Children. N. Engl. J. Med. 2018, 378, 731–739. [Google Scholar] [CrossRef] [PubMed]
- Laetsch, T.W.; DuBois, S.G.; Mascarenhas, L.; Turpin, B.; Federman, N.; Albert, C.M.; Nagasubramanian, R.; Davis, J.L.; Rudzinski, E.; Feraco, A.M.; et al. Larotrectinib for paediatric solid tumours harbouring NTRK gene fusions: Phase 1 results from a multicentre, open-label, phase 1/2 study. Lancet. Oncol. 2018, 19, 705–714. [Google Scholar] [CrossRef]
- Drilon, A.E.; DuBois, S.G.; Farago, A.F.; Geoerger, B.; Grilley-Olson, J.E.; Hong, D.S.; Sohal, D.; van Tilburg, C.M.; Ziegler, D.S.; Ku, N.; et al. Activity of larotrectinib in TRK fusion cancer patients with brain metastases or primary central nervous system tumors. J. Clin. Oncol. 2019, 37, 2006. [Google Scholar] [CrossRef]
- Drilon, A.; Nagasubramanian, R.; Blake, J.F.; Ku, N.; Tuch, B.B.; Ebata, K.; Smith, S.; Lauriault, V.; Kolakowski, G.R.; Brandhuber, B.J.; et al. A Next-Generation TRK Kinase Inhibitor Overcomes Acquired Resistance to Prior TRK Kinase Inhibition in Patients with TRK Fusion-Positive Solid Tumors. Cancer Discov. 2017, 7, 963–972. [Google Scholar] [CrossRef] [Green Version]
- Drilon, A.; Ou, S.I.; Cho, B.C.; Kim, D.W.; Lee, J.; Lin, J.J.; Zhu, V.W.; Ahn, M.J.; Camidge, D.R.; Nguyen, J.; et al. Repotrectinib (TPX-0005) Is a Next-Generation ROS1/TRK/ALK Inhibitor That Potently Inhibits ROS1/TRK/ALK Solvent- Front Mutations. Cancer Discov. 2018, 8, 1227–1236. [Google Scholar] [CrossRef] [Green Version]
- Ercan, D.; Choi, H.G.; Yun, C.H.; Capelletti, M.; Xie, T.; Eck, M.J.; Gray, N.S.; Janne, P.A. EGFR Mutations and Resistance to Irreversible Pyrimidine-Based EGFR Inhibitors. Clin. Cancer Res. 2015, 21, 3913–3923. [Google Scholar] [CrossRef] [Green Version]
- Gainor, J.F.; Dardaei, L.; Yoda, S.; Friboulet, L.; Leshchiner, I.; Katayama, R.; Dagogo-Jack, I.; Gadgeel, S.; Schultz, K.; Singh, M.; et al. Molecular Mechanisms of Resistance to First- and Second-Generation ALK Inhibitors in ALK-Rearranged Lung Cancer. Cancer Discov. 2016, 6, 1118–1133. [Google Scholar] [CrossRef] [Green Version]
- Bennouna, J.; Girard, N.; Audigier-Valette, C.; le Thuaut, A.; Gervais, R.; Masson, P.; Marcq, M.; Molinier, O.; Cortot, A.; Debieuvre, D.; et al. Phase II Study Evaluating the Mechanisms of Resistance on Tumor Tissue and Liquid Biopsy in Patients With EGFR-mutated Non-pretreated Advanced Lung Cancer Receiving Osimertinib Until and Beyond Radiologic Progression: The MELROSE Trial. Clin. Lung Cancer 2019. [Google Scholar] [CrossRef] [Green Version]
- Recondo, G.; Mezquita, L.; Facchinetti, F.; Planchard, D.; Gazzah, A.; Bigot, L.; Rizvi, A.Z.; Frias, R.L.; Thiery, J.P.; Scoazec, J.Y.; et al. Diverse resistance mechanisms to the third-generation ALK inhibitor lorlatinib in ALK-rearranged lung cancer. Clin. Cancer Res. 2019. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morris, T.A.; Khoo, C.; Solomon, B.J. Targeting ROS1 Rearrangements in Non-small Cell Lung Cancer: Crizotinib and Newer Generation Tyrosine Kinase Inhibitors. Drugs 2019, 79, 1277–1286. [Google Scholar] [CrossRef]
- Roys, A.; Chang, X.; Liu, Y.; Xu, X.; Wu, Y.; Zuo, D. Resistance mechanisms and potent-targeted therapies of ROS1-positive lung cancer. Cancer Chemother Pharmacol. 2019, 84, 679–688. [Google Scholar] [CrossRef]
- Ricciuti, B.; Genova, C.; Crino, L.; Libra, M.; Leonardi, G.C. Antitumor activity of larotrectinib in tumors harboring NTRK gene fusions: A short review on the current evidence. Onco. Targets Ther. 2019, 12, 3171–3179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cocco, E.; Schram, A.M.; Kulick, A.; Misale, S.; Won, H.H.; Yaeger, R.; Razavi, P.; Ptashkin, R.; Hechtman, J.F.; Toska, E.; et al. Resistance to TRK inhibition mediated by convergent MAPK pathway activation. Nat. Med. 2019, 25, 1422–1427. [Google Scholar] [CrossRef] [PubMed]
- Kummar, S.; Lassen, U.N. TRK Inhibition: A New Tumor-Agnostic Treatment Strategy. Target. Oncol. 2018, 13, 545–556. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hechtman, J.F.; Benayed, R.; Hyman, D.M.; Drilon, A.; Zehir, A.; Frosina, D.; Arcila, M.E.; Dogan, S.; Klimstra, D.S.; Ladanyi, M.; et al. Pan-Trk Immunohistochemistry Is an Efficient and Reliable Screen for the Detection of NTRK Fusions. Am. J. Surg. Pathol. 2017, 41, 1547–1551. [Google Scholar] [CrossRef]
- Bourhis, A.; Redoulez, G.; Quintin-Roue, I.; Marcorelles, P.; Uguen, A. Screening for NTRK-rearranged Tumors Using Immunohistochemistry: Comparison of 2 Different pan-TRK Clones in Melanoma Samples. Appl. Immunohistochem. Mol. Morphol. 2019. [Google Scholar] [CrossRef]
- Solomon, J.P.; Linkov, I.; Rosado, A.; Mullaney, K.; Rosen, E.Y.; Frosina, D.; Jungbluth, A.A.; Zehir, A.; Benayed, R.; Drilon, A.; et al. NTRK fusion detection across multiple assays and 33,997 cases: Diagnostic implications and pitfalls. Mod. Pathol. 2019. [Google Scholar] [CrossRef]
- Solomon, J.P.; Hechtman, J.F. Detection of NTRK Fusions: Merits and Limitations of Current Diagnostic Platforms. Cancer Res. 2019, 79, 3163–3168. [Google Scholar] [CrossRef]
- Gatalica, Z.; Xiu, J.; Swensen, J.; Vranic, S. Molecular characterization of cancers with NTRK gene fusions. Mod. Pathol. 2019, 32, 147–153. [Google Scholar] [CrossRef] [PubMed]
- Murphy, D.A.; Ely, H.A.; Shoemaker, R.; Boomer, A.; Culver, B.P.; Hoskins, I.; Haimes, J.D.; Walters, R.D.; Fernandez, D.; Stahl, J.A.; et al. Detecting Gene Rearrangements in Patient Populations Through a 2-Step Diagnostic Test Comprised of Rapid IHC Enrichment Followed by Sensitive Next-Generation Sequencing. Appl. Immunohistochem. Mol. Morphol. 2017, 25, 513–523. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marchio, C.; Scaltriti, M.; Ladanyi, M.; Iafrate, A.J.; Bibeau, F.; Dietel, M.; Hechtman, J.F.; Troiani, T.; Lopez-Rios, F.; Douillard, J.Y.; et al. ESMO recommendations on the standard methods to detect NTRK fusions in daily practice and clinical research. Ann. Oncol. 2019, 30, 1417–1427. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Penault-Llorca, F.; Rudzinski, E.R.; Sepulveda, A.R. Testing algorithm for identification of patients with TRK fusion cancer. J. Clin. Pathol. 2019, 72, 460–467. [Google Scholar] [CrossRef] [PubMed]
- Hsiao, S.J.; Zehir, A.; Sireci, A.N.; Aisner, D.L. Detection of Tumor NTRK Gene Fusions to Identify Patients Who May Benefit from Tyrosine Kinase (TRK) Inhibitor Therapy. J. Mol. Diagn. 2019, 21, 553–571. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cocco, E.; Benhamida, J.; Middha, S.; Zehir, A.; Mullaney, K.; Shia, J.; Yaeger, R.; Zhang, L.; Wong, D.; Villafania, L.; et al. Colorectal Carcinomas Containing Hypermethylated MLH1 Promoter and Wild-Type BRAF/KRAS Are Enriched for Targetable Kinase Fusions. Cancer Res. 2019, 79, 1047–1053. [Google Scholar] [CrossRef] [Green Version]



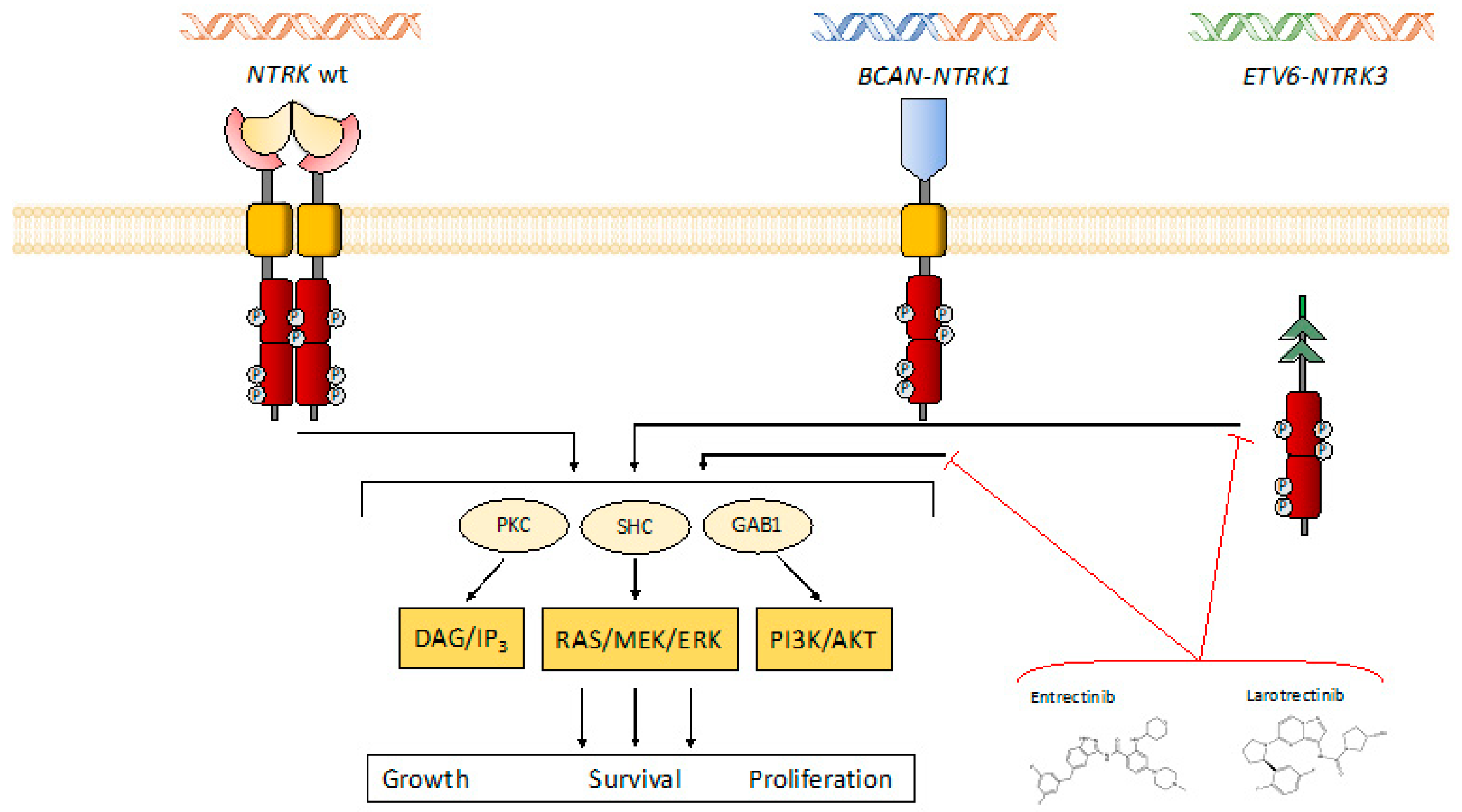{kind=link}
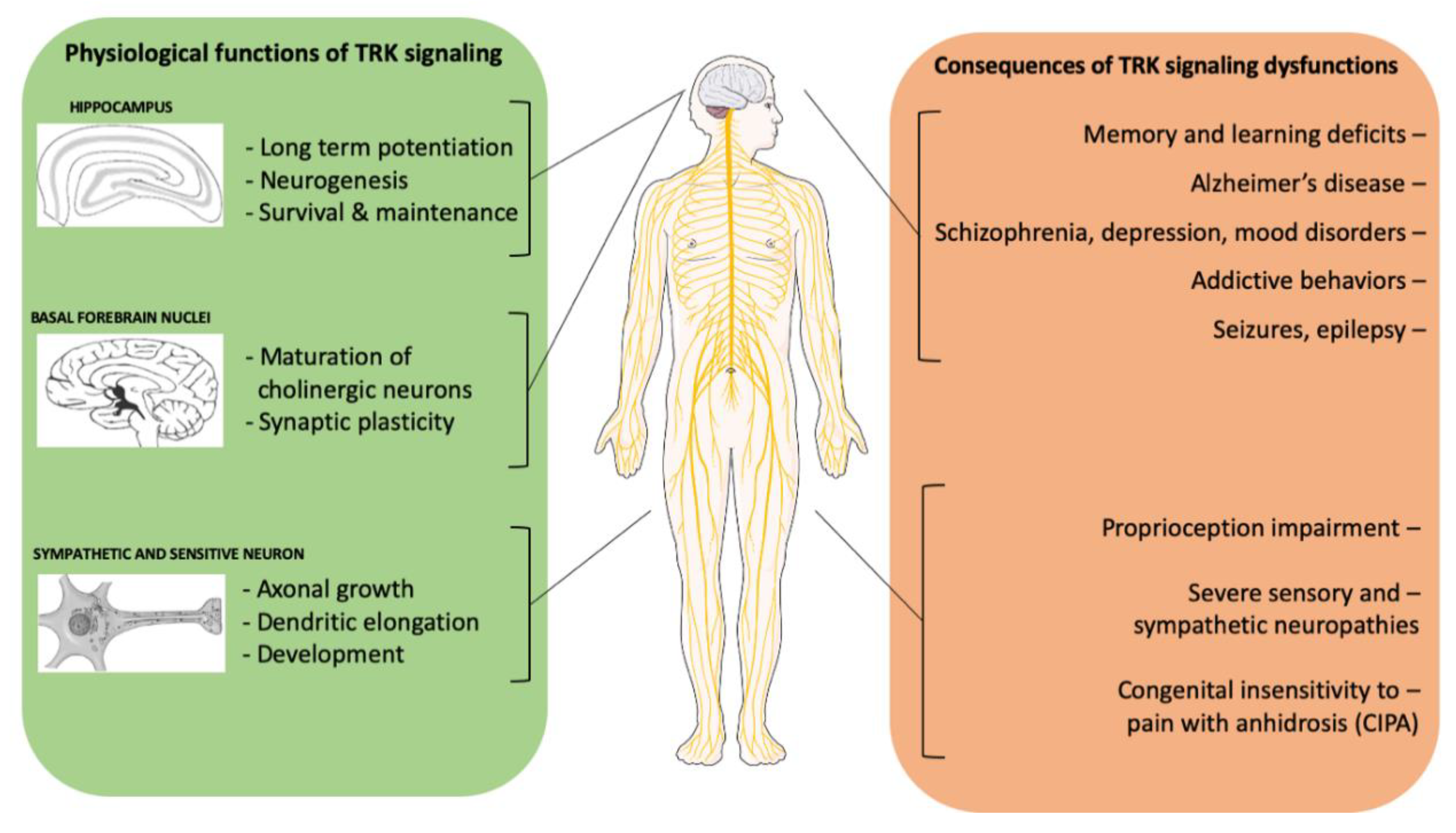{kind=link}
| Tumor Entity | NTRK Fusions Frequency | Most Frequently Reported NTRK Fusions |
|---|---|---|
| Glioblastoma | 1.1% (Frattini et al.) [134] 1.1% (Shah et al.) [135] 2.6% (Zheng et al.) [124] 1.2% (Kim et al.) [136] 1.7% (Ferguson et al.) [133] | BCAN-NTRK1 NFASC-NTRK1 ARHGEF2-NTRK1 CHTOP-NTRK1 GKAP-NTRK2 KCTD8-NTRK2 TBC1D2-NTRK2 EML4-NTRK3 |
| Non-brainstem high-grade glioma | 10%–40% (Wu et al.)# [121] | ETV6-NTRK3 TPM3-NTRK1 BTBD1-NTRK3 VCL-NTRK2 AGBL4-NTRK2 |
| DIPG° | 4% (Wu et al.) [121] | |
| Pilocytic astrocytoma | 16.6% (Ferguson et al.) [133] 3.1% (Jones et al.) [114] | BCAN-NTRK1 NACC2-NTRK2 QKI-NTRK2 |
| Anaplastic astrocytoma | 2.3% (Ferguson et al.) [133] | NOS1AP-NTRK2 |
| Glioma NOS | 4.1% (Ferguson et al.) [133] | SQSTM1-NTRK2 |
| Low-grade glioma | 0.7% (Zhang et al.) [112] 0.43% (Stransky et al.) [72] 4.3% (Ferguson et al.) [133] | ETV6-NTRK3 AFAP1-NTRK2 VCAN-NTRK2 |
| High-grade glioneuronal tumor | Case report (Kurozumi et al.) [127] | ARHGEF2-NTRK1 |
| Ganglioglioma | Case report (Prabhakaran et al.) [124] | TLE4-NTRK2 |
| Molecule | Population and Enrollment | Allocation and Intervention Model | Phase | Primary Outcomes | Start Date and Current Status | Identifier |
|---|---|---|---|---|---|---|
| Entrectinib (RXDX-101) | Adult (minimum age: 18 Years)—84 participants | Non-Randomized—Single Group Assignment | I | Dose limiting toxicity Maximum tolerated dose Recommended Phase II dose Overall response rate | 2014—Active, Not Recruiting | NCT02097810 RXDX-101-01 (STARTRK-1) |
| Entrectinib (RXDX-101) | Adult (minimum age: 18 Years)—300 participants (estimated) | Non-Randomized—Parallel Assignment | II | Objective response rate | 2015—Recruiting | NCT02568267 RXDX-101-02 (STARTRK-2) |
| Entrectinib (RXDX-101) | Pediatric and Adult (maximum age: 22 Years)—65 participants | Non-Randomized—Single Group Assignment | I | Maximum tolerated dose Recommended Phase II dose Objective response rate | 2016—Recruiting | NCT02650401 RXDX-101-03 (STARTRK-NG) |
| Larotrectinib (LOXO-101) | Pediatric and Adult (minimum age: 18 Years)—6452 participants | Non-Randomized—Parallel Assignment | II | Proportion of patients with objective response | 2015—Recruiting | NCT02465060 EAY131 NCI-2015-00054 |
| Larotrectinib (LOXO-101) | Pediatric and Adult (minimum age: 12 Years)—320 participants | Non-Randomized—Parallel Assignment | II | Best overall response rate | 2015—Recruiting | NCT02576431 LOXO-TRK-15002 (NAVIGATE) |
| Larotrectinib (LOXO-101) | Pediatric and Adult (maximum age: 21 Years) —174 participants | Non-Randomized—Parallel Assignment | I/II | Number and severity of adverse events (Phase I) Overall response rate (Phase II) | 2015—Recruiting | NCT02637687 LOXO-TRK-15003 (SCOUT) |
| Larotrectinib (LOXO-101) | Pediatric and Adult (from 12 Months to 21 Years)—1000 participants (estimated) | Non-Randomized—Parallel Assignment | II | Objective response rate | 2017—Recruiting | NCT03155620 APEC1621SC NCI-2017-01251 |
| Larotrectinib (LOXO-101) | Pediatric and Adult (from 12 Months to 21 Years)—49 participants | Non-Randomized—Single Group Assignment | II | Objective response rate | 2017—Recruiting | NCT03213704 APEC1621A NCI-2017-01264 |
| Larotrectinib (LOXO-101) | Pediatric and Adult (maximum age: 30 Years)—70 participants | Non-Randomized—Single Group Assignment | II | Objective response rate | 2019—Recruiting | NCT03834961 ADVL1823 NCI-2019-00015 |
| Repotrectinib (TPX-0005) | Pediatric and Adult (12 Years and older)—450 (estimated) | Non-Randomized—Single Group Assignment | I/II | Dose limiting toxicities Recommended Phase II dose Overall response rate | 2019—Recruiting | NCT03093116 TPX-0005-01 (TRIDENT-1) |
| Repotrectinib (TPX-0005) | Pediatric (4 Years to 12 Years)—12 participants | Non-Randomized—Single Group Assignment | I | Dose limiting toxicities Pediatric recommended Phase II dose | 2019—Recruiting | NCT04094610 TPX-0005-07 |
| Selitrectinib (LOXO-195) | Pediatric and Adult (minimum age: 1 Month) | Expanded Access (Individual Patients) | NA | NA | 2017—Available (Expanded Access) | NCT03206931 |
| Selitrectinib (LOXO-195) | Pediatric and Adult (minimum age: 1 Month)—93 participants | Non-Randomized—Sequential Assignment | I/II | Maximum tolerated dose Recommended dose Overall response rate | 2017—Recruiting | NCT03215511 LOXO-EXT-17005 |
| Assay Type | Advantages | Limitations | Turnaround Time | Main Role in Potential Diagnostic Algorithms |
|---|---|---|---|---|
| IHC |
|
| 1–2 days | Screening |
| FISH |
|
| 3–5 days | Confirmatory |
| RT-PCR |
|
| 5–7 days | Confirmatory |
| Real time-PCR |
|
| 5–7 days | Screening/Confirmatory* |
| RNA-NGS |
|
| 1–3 weeks | Screening/Confirmatory* |
| DNA-NGS |
|
| 1–3 weeks | Screening/Confirmatory* |
| DNA/RNA-NGS |
|
| 1–3 weeks | Screening/Confirmatory* |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gambella, A.; Senetta, R.; Collemi, G.; Vallero, S.G.; Monticelli, M.; Cofano, F.; Zeppa, P.; Garbossa, D.; Pellerino, A.; Rudà, R.; et al. NTRK Fusions in Central Nervous System Tumors: A Rare, but Worthy Target. Int. J. Mol. Sci. 2020, 21, 753. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21030753
Gambella A, Senetta R, Collemi G, Vallero SG, Monticelli M, Cofano F, Zeppa P, Garbossa D, Pellerino A, Rudà R, et al. NTRK Fusions in Central Nervous System Tumors: A Rare, but Worthy Target. International Journal of Molecular Sciences. 2020; 21(3):753. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21030753
Chicago/Turabian StyleGambella, Alessandro, Rebecca Senetta, Giammarco Collemi, Stefano Gabriele Vallero, Matteo Monticelli, Fabio Cofano, Pietro Zeppa, Diego Garbossa, Alessia Pellerino, Roberta Rudà, and et al. 2020. "NTRK Fusions in Central Nervous System Tumors: A Rare, but Worthy Target" International Journal of Molecular Sciences 21, no. 3: 753. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21030753