TGFβ1 Regulates Human RANKL-Induced Osteoclastogenesis via Suppression of NFATc1 Expression

Abstract
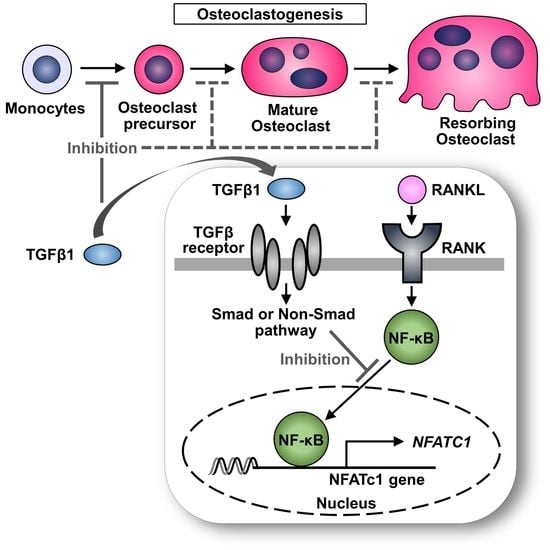
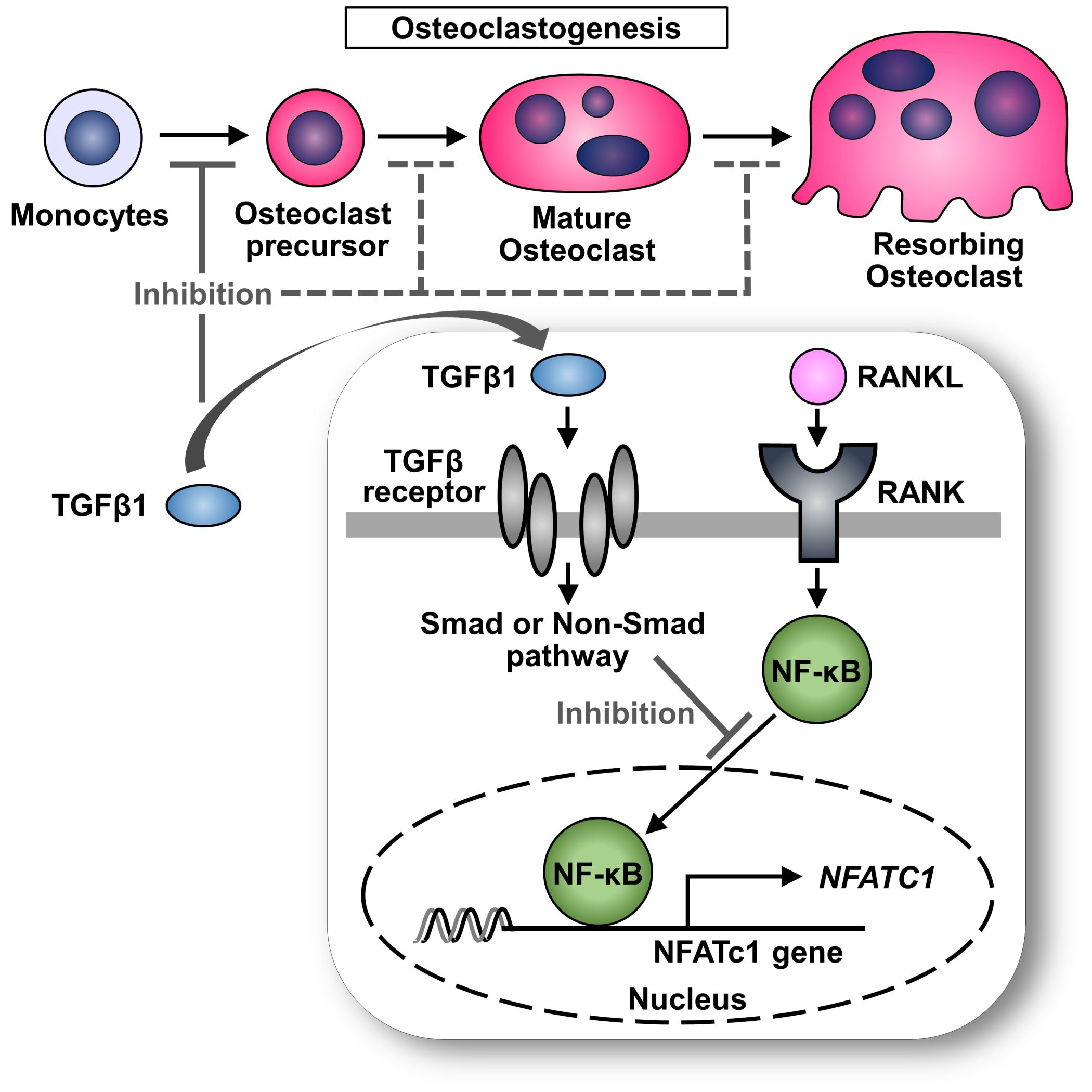
:
1. Introduction
2. Results
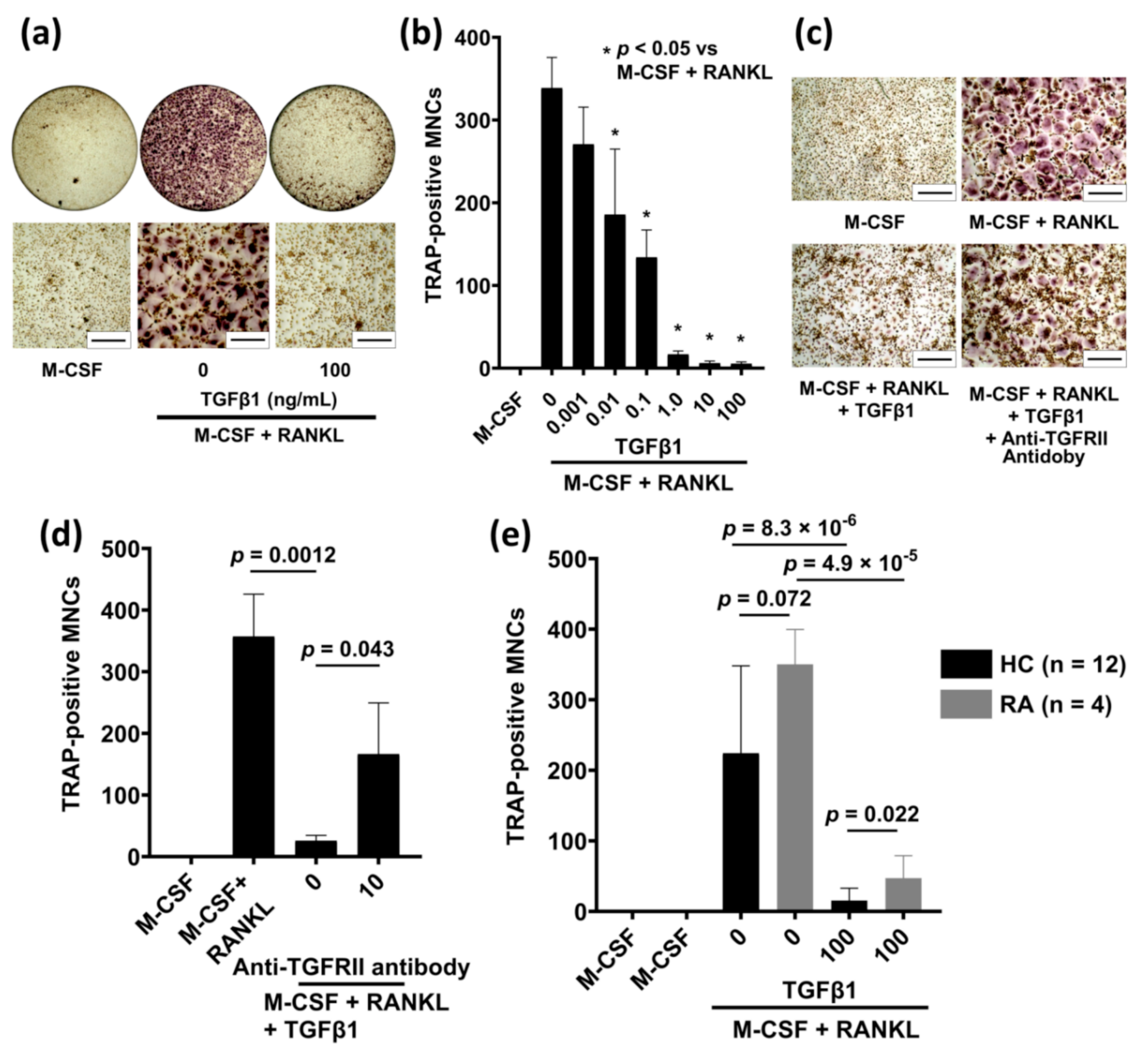
2.1. TGFβ1 Inhibits RANKL-Induced Osteoclastogenesis in Human Peripheral Blood Monocytes (PBMs)
2.2. Anti-TGFBRII Antibody Blocks the Inhibitory Effect of TGFβ1 on RANKL-Induced Osteoclastogenesis
2.3. TGFβ1 Treatment Reduces RANKL-Induced Osteoclastogenesis in Patients with RA
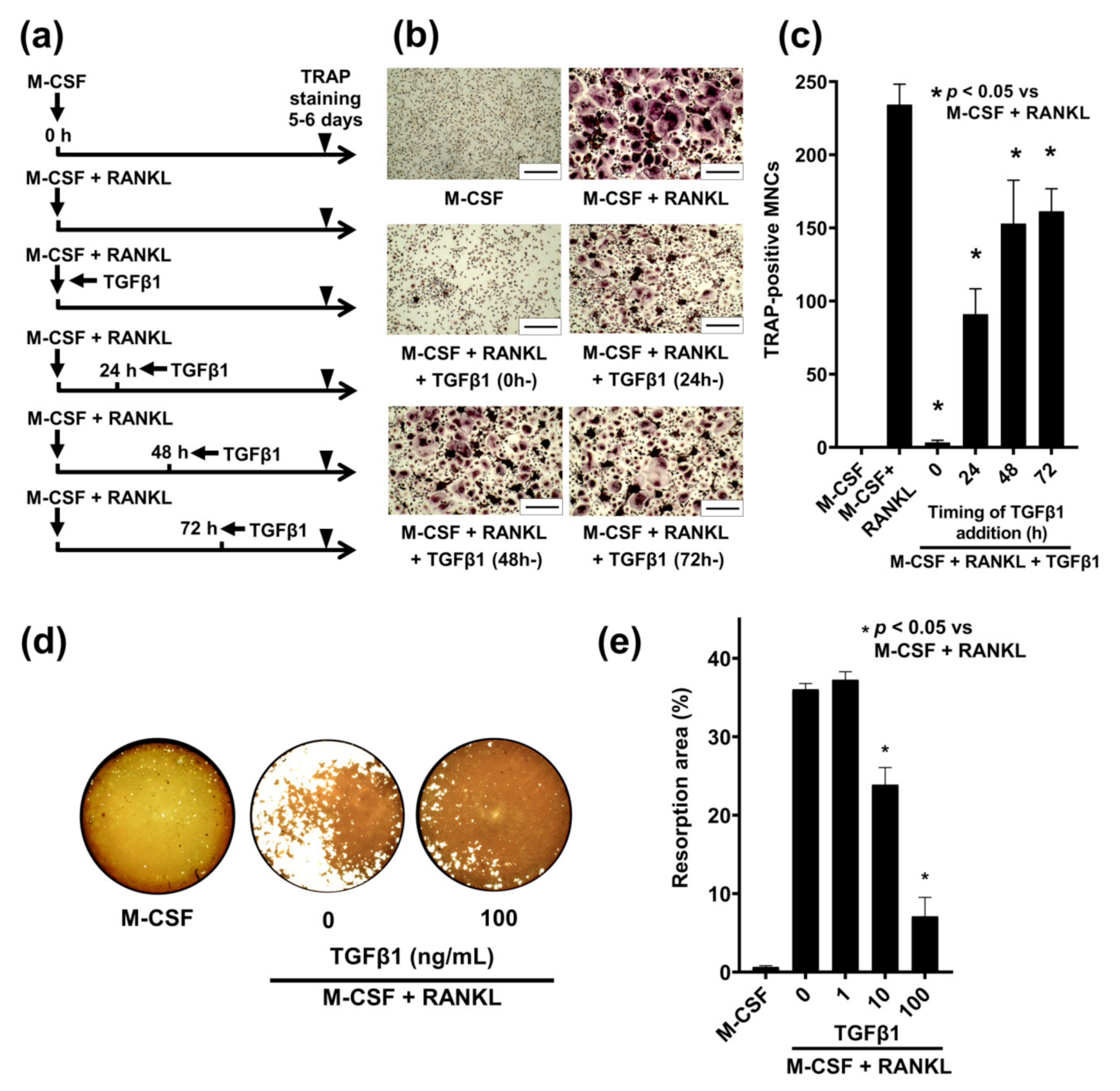
2.4. Time-Dependent Effect of TGFβ1 on RANKL-Induced Osteoclastogenesis in Human PBMs
2.5. TGFβ1 Inhibits RANKL-Mediated Bone Resorption Activity
2.6. Stage-Dependent Differentiation Effect of TGFβ1 on RANKL-Induced Osteoclastogenesis in Human PBMs
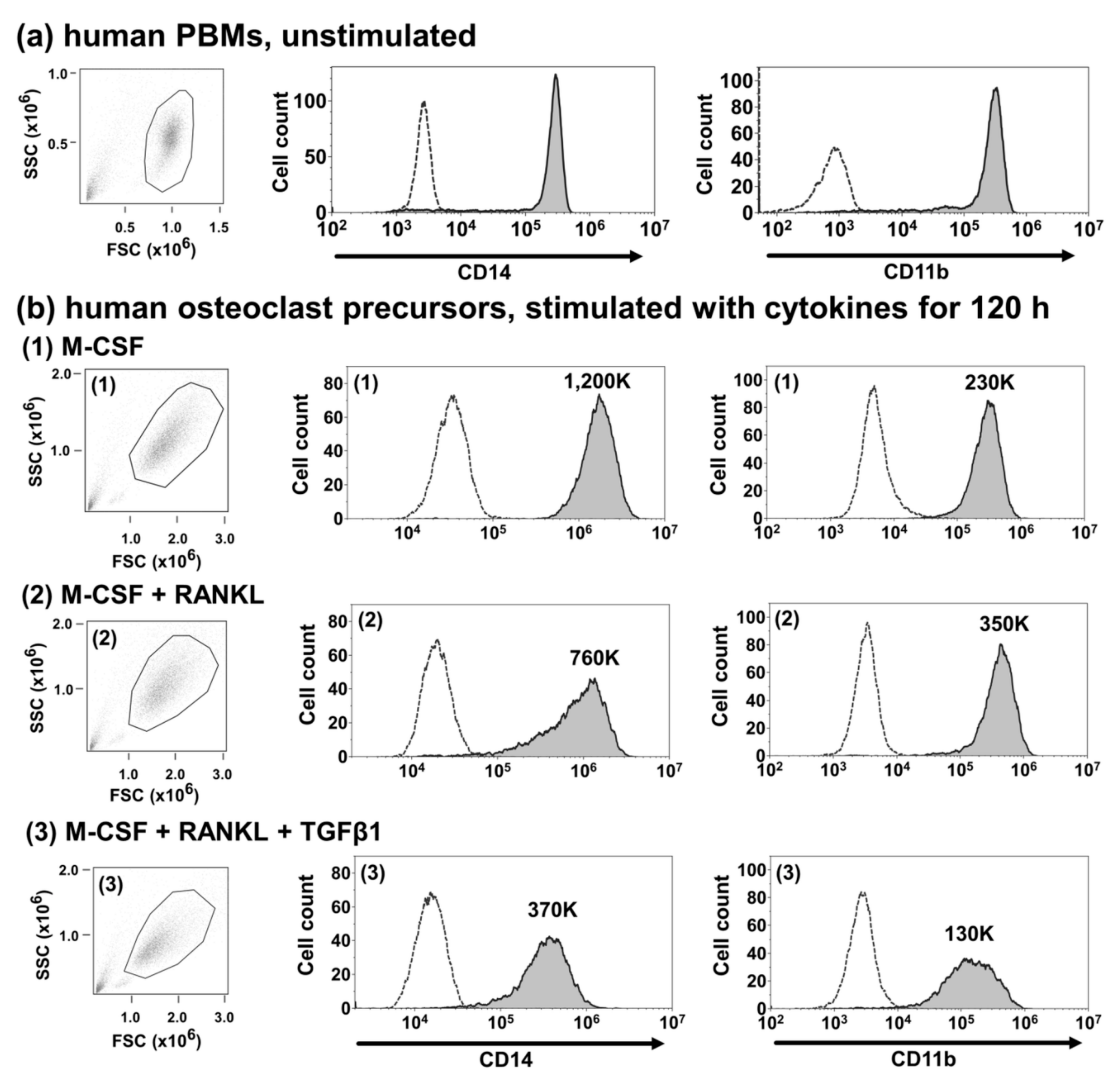
2.7. TGFβ1 Suppresses the Cell Surface Expression of CD14 and CD11b in Human Osteoclast Precursors
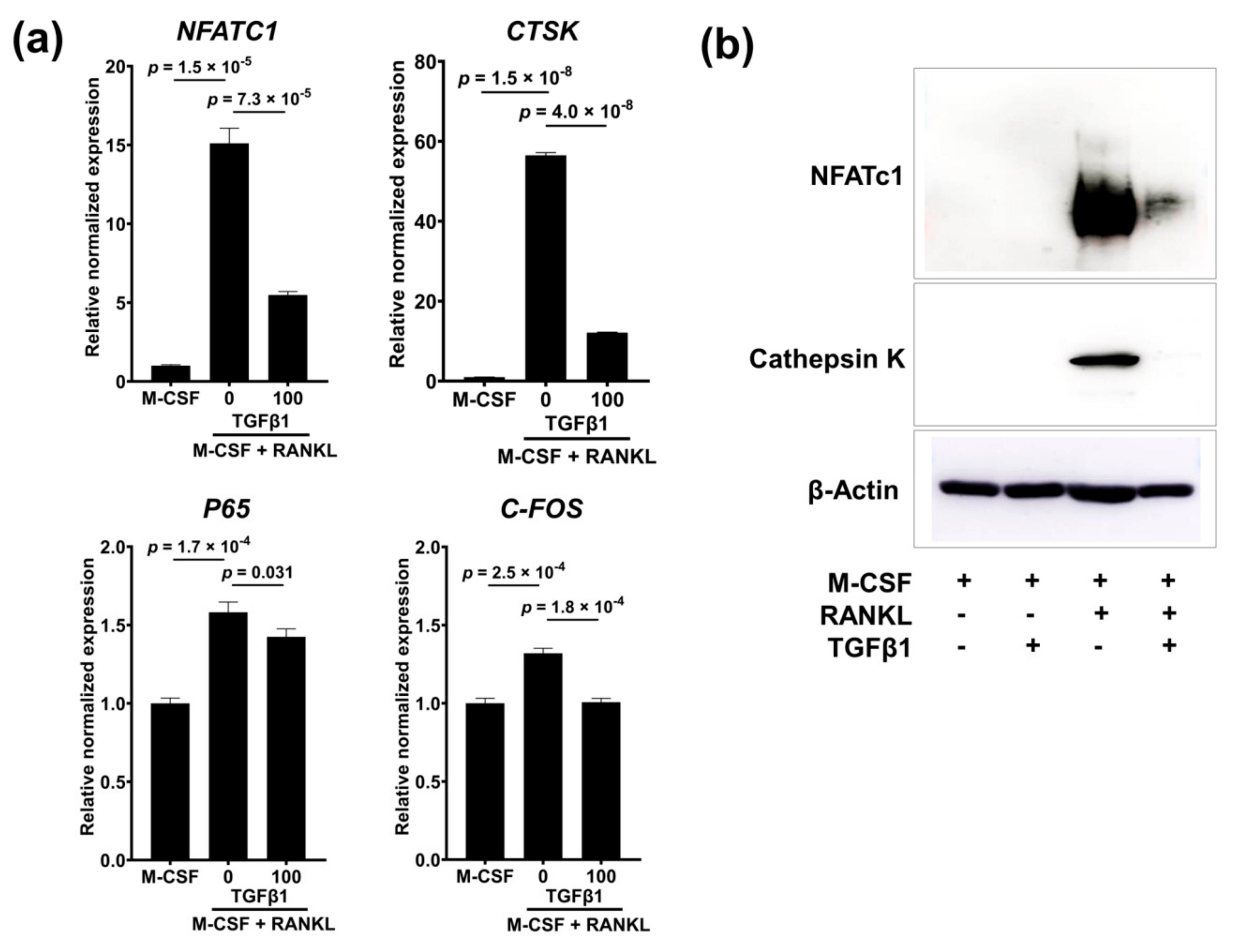
2.8. TGFβ1 Suppresses the Gene and Protein Expression of NFATc1 and Cathepsin K
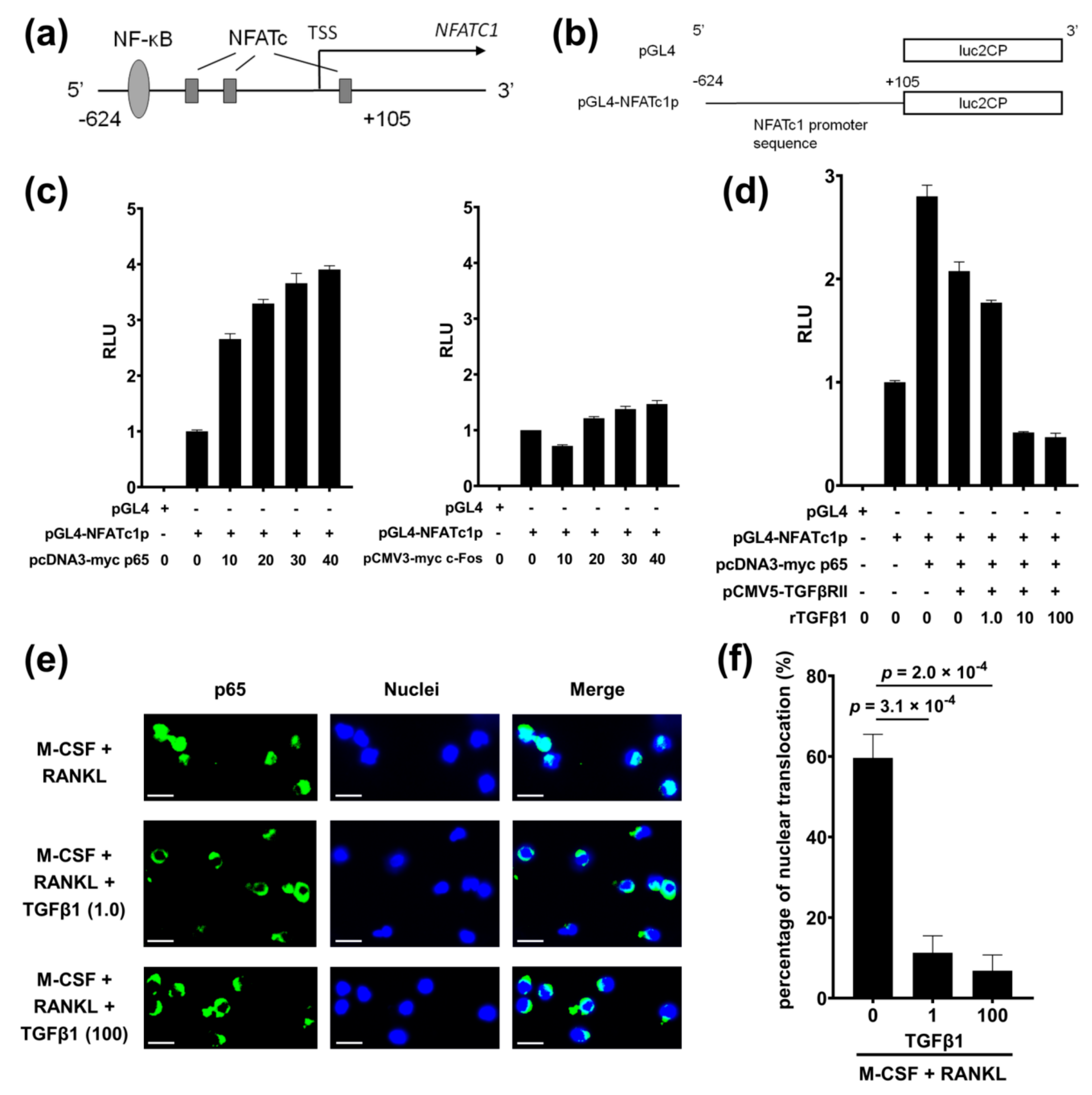
2.9. TGFβ1 Directly Inhibits NFATc1 Promoter Activity
2.10. TGFβ1 Inhibits Nuclear Translocation of p65 Induced by RANKL
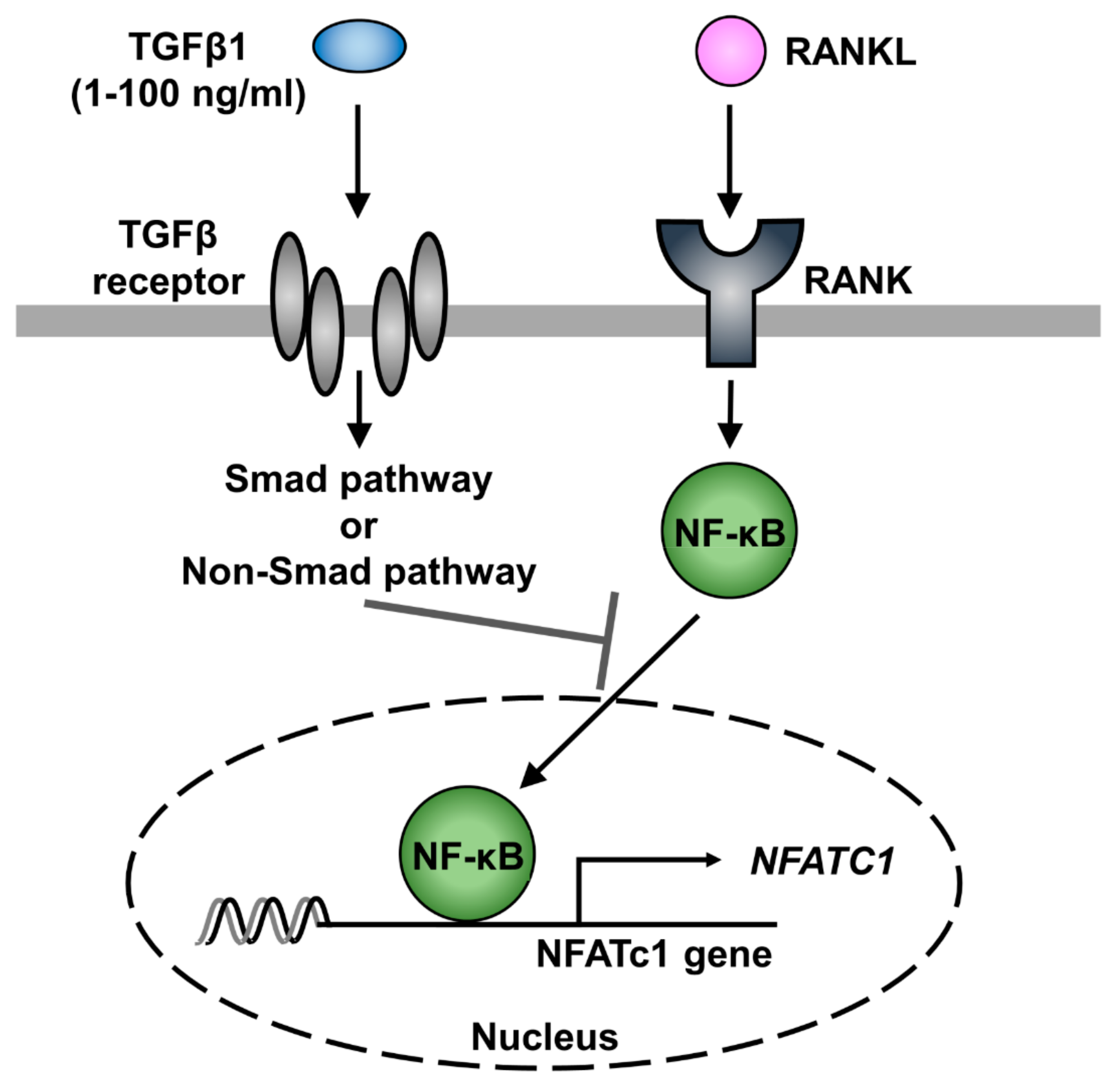
3. Discussion
4. Materials and Methods
4.1. Preparation of PBMs
4.2. In Vitro Osteoclastogenesis
4.3. TGFβ1 Cytotoxicity
4.4. Cell Proliferation Assay
4.5. Preparation of PBMs from Untreated Seropositive Patients with RA and In Vitro Osteoclastogenesis
4.6. TRAP Staining
4.7. In Vitro Assays for Osteoclast Resorption
4.8. Flow Cytometric Analysis
4.9. Real-Time Reverse Transcription PCR (RT-qPCR) Analysis
4.10. Western Blotting
4.11. Plasmid Construction
4.12. Luciferase Assay
4.13. Immunofluorescence Analysis
4.14. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ACTB | beta actin |
| CTSK | cathepsin K |
| HC | healthy control |
| M-CSF | macrophage-colony stimulating factor |
| MFI | mean fluorescence intensity |
| MNC | multinucleated cell |
| NF-κB | nuclear factor kappa-B |
| NFATc1 | nuclear factor of activated T cells, cytoplasmic 1 |
| PCR | polymerase chain reaction |
| PBM | peripheral blood monocyte |
| PBMC | peripheral blood mononuclear cell |
| RA | rheumatoid arthritis |
| RANKL | receptor activator of nuclear factor kappa-B ligand |
| TGFβ1 | transforming growth factor beta1 |
| TGFBRII | TGFβ1 receptor II |
| TRAP | tartrate-resistant acid phosphatase |
References
- Jacome-Galarza, C.E.; Percin, G.I.; Muller, J.T.; Mass, E.; Lazarov, T.; Eitler, J.; Rauner, M.; Yadav, V.K.; Crozet, L.; Bohm, M.; et al. Developmental origin, functional maintenance and genetic rescue of osteoclasts. Nature 2019, 568, 541–545. [Google Scholar] [CrossRef] [PubMed]
- Schett, G.; Gravallese, E. Bone erosion in rheumatoid arthritis: Mechanisms, diagnosis and treatment. Nat. Rev. Rheumatol. 2012, 8, 656–664. [Google Scholar] [CrossRef] [PubMed]
- Schett, G. Cells of the synovium in rheumatoid arthritis. Osteoclasts. Arthritis Res. Ther. 2007, 9, 203. [Google Scholar] [CrossRef] [Green Version]
- Herman, S.; Krönke, G.; Schett, G. Molecular mechanisms of inflammatory bone damage: Emerging targets for therapy. Trends Mol. Med. 2008, 14, 245–253. [Google Scholar] [CrossRef] [PubMed]
- Harre, U.; Schett, G. Cellular and molecular pathways of structural damage in rheumatoid arthritis. Semin. Immunopathol. 2017, 39, 355–363. [Google Scholar] [CrossRef] [PubMed]
- Takayanagi, H. SnapShot: Osteoimmunology. Cell Metab. 2015, 21, 502. [Google Scholar] [CrossRef] [Green Version]
- Negishi-Koga, T.; Takayanagi, H. Bone cell communication factors and Semaphorins. Bonekey Rep. 2012, 1, 183. [Google Scholar] [CrossRef] [Green Version]
- Charles, J.F.; Aliprantis, A.O. Osteoclasts: More than ‘bone eaters’. Trends Mol. Med. 2014, 20, 449–459. [Google Scholar] [CrossRef] [Green Version]
- Kawai, M.; Mödder, U.I.; Khosla, S.; Rosen, C.J. Emerging therapeutic opportunities for skeletal restoration. Nat. Rev. Drug Discov. 2011, 10, 141–156. [Google Scholar] [CrossRef] [Green Version]
- Tang, Y.; Wu, X.; Lei, W.; Pang, L.; Wan, C.; Shi, Z.; Zhao, L.; Nagy, T.R.; Peng, X.; Hu, J.; et al. TGF-β-1-induced migration of bone mesenchymal stem cells couples bone resorption with formation. Nat. Med. 2009, 15, 757–765. [Google Scholar] [CrossRef] [Green Version]
- Asagiri, M.; Takayanagi, H. The molecular understanding of osteoclast differentiation. Bone 2007, 40, 251–264. [Google Scholar] [CrossRef] [PubMed]
- Okamoto, K.; Takayanagi, H. Regulation of bone by the adaptive immune system in arthritis. Arthritis Res. Ther. 2011, 13, 219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nicholson, G.C.; Malakellis, M.; Collier, F.M.; Cameron, P.U.; Holloway, W.R.; Gough, T.J.; Gregorio-King, C.; Kirkland, M.A.; Myers, D.E. Induction of osteoclasts from CD14-positive human peripheral blood mononuclear cells by receptor activator of nuclear factor κB ligand (RANKL). Clin. Sci. (Lond.) 2000, 99, 133–140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sørensen, M.G.; Henriksen, K.; Schaller, S.; Henriksen, D.B.; Nielsen, F.C.; Dziegiel, M.H.; Karsdal, M.A. Characterization of osteoclasts derived from CD14+ monocytes isolated from peripheral blood. J. Bone Miner. Metab. 2007, 25, 36–45. [Google Scholar] [CrossRef] [PubMed]
- Goto, T.; Yamaza, T.; Tanaka, T. Cathepsins in the osteoclast. J. Electron Microsc. (Tokyo) 2003, 52, 551–558. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, Y.; Ohira, T. Mechanisms and therapeutic targets for bone damage in rheumatoid arthritis, in particular the RANK-RANKL system. Curr. Opin. Pharmacol. 2018, 40, 110–119. [Google Scholar] [CrossRef] [PubMed]
- Fennen, M.; Pap, T.; Dankbar, B. Smad-dependent mechanisms of inflammatory bone destruction. Arthritis Res. Ther. 2016, 18, 279. [Google Scholar] [CrossRef] [Green Version]
- Kasagi, S.; Chen, W. TGF-beta1 on osteoimmunology and the bone component cells. Cell Biosci. 2013, 3, 4. [Google Scholar] [CrossRef] [Green Version]
- Bonewald, L.F.; Mundy, G.R. Role of transforming growth factor-beta in bone remodeling. Clin. Orthop. Relat. Res. 1990, 261–276. [Google Scholar] [CrossRef]
- Janssens, K.; ten Dijke, P.; Janssens, S.; Van Hul, W. Transforming growth factor-β1 to the bone. Endocr. Rev. 2005, 26, 743–774. [Google Scholar] [CrossRef] [Green Version]
- Galvin, R.J.; Gatlin, C.L.; Horn, J.W.; Fuson, T.R. TGF-β enhances osteoclast differentiation in hematopoietic cell cultures stimulated with RANKL and M-CSF. Biochem. Biophys. Res. Commun. 1999, 265, 233–239. [Google Scholar] [CrossRef] [PubMed]
- Fuller, K.; Lean, J.M.; Bayley, K.E.; Wani, M.R.; Chambers, T.J. A role for TGFbeta(1) in osteoclast differentiation and survival. J. Cell Sci. 2000, 113, 2445–2453. [Google Scholar]
- Kaneda, T.; Nojima, T.; Nakagawa, M.; Ogasawara, A.; Kaneko, H.; Sato, T.; Mano, H.; Kumegawa, M.; Hakeda, Y. Endogenous production of TGF-β is essential for osteoclastogenesis induced by a combination of receptor activator of NF-κB ligand and macrophage-colony-stimulating factor. J. Immunol. 2000, 165, 4254–4263. [Google Scholar] [CrossRef] [Green Version]
- Yan, T.; Riggs, B.L.; Boyle, W.J.; Khosla, S. Regulation of osteoclastogenesis and RANK expression by TGF-beta1. J. Cell. Biochem. 2001, 83, 320–325. [Google Scholar] [CrossRef] [PubMed]
- Koseki, T.; Gao, Y.; Okahashi, N.; Murase, Y.; Tsujisawa, T.; Sato, T.; Yamato, K.; Nishihara, T. Role of TGF-beta family in osteoclastogenesis induced by RANKL. Cell. Signal. 2002, 14, 31–36. [Google Scholar] [CrossRef]
- Fox, S.W.; Haque, S.J.; Lovibond, A.C.; Chambers, T.J. The possible role of TGF-β-induced suppressors of cytokine signaling expression in osteoclast/macrophage lineage commitment in vitro. J. Immunol. 2003, 170, 3679–3687. [Google Scholar] [CrossRef] [Green Version]
- Fox, S.W.; Evans, K.E.; Lovibond, A.C. Transforming growth factor-beta enables NFATc1 expression during osteoclastogenesis. Biochem. Biophys. Res. Commun. 2008, 366, 123–128. [Google Scholar] [CrossRef] [Green Version]
- Gingery, A.; Bradley, E.W.; Pederson, L.; Ruan, M.; Horwood, N.J.; Oursler, M.J. TGF-beta coordinately activates TAK1/MEK/AKT/NFkB and SMAD pathways to promote osteoclast survival. Exp. Cell Res. 2008, 314, 2725–2738. [Google Scholar] [CrossRef] [Green Version]
- Yasui, T.; Kadono, Y.; Nakamura, M.; Oshima, Y.; Matsumoto, T.; Masuda, H.; Hirose, J.; Omata, Y.; Yasuda, H.; Imamura, T.; et al. Regulation of RANKL-induced osteoclastogenesis by TGF-beta through molecular interaction between Smad3 and Traf6. J. Bone Miner. Res. 2011, 26, 1447–1456. [Google Scholar] [CrossRef]
- Omata, Y.; Yasui, T.; Hirose, J.; Izawa, N.; Imai, Y.; Matsumoto, T.; Masuda, H.; Tokuyama, N.; Nakamura, S.; Tsutsumi, S.; et al. Genomewide comprehensive analysis reveals critical cooperation between Smad and c-Fos in RANKL-induced osteoclastogenesis. J. Bone Miner. Res. 2015, 30, 869–877. [Google Scholar] [CrossRef]
- Itonaga, I.; Sabokbar, A.; Sun, S.G.; Kudo, O.; Danks, L.; Ferguson, D.; Fujikawa, Y.; Athanasou, N.A. Transforming growth factor-β induces osteoclast formation in the absence of RANKL. Bone 2004, 34, 57–64. [Google Scholar] [CrossRef] [PubMed]
- Chenu, C.; Pfeilschifter, J.; Mundy, G.R.; Roodman, G.D. Transforming growth factor beta inhibits formation of osteoclast-like cells in long-term human marrow cultures. Proc. Natl. Acad. Sci. USA 1988, 85, 5683–5687. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takai, H.; Kanematsu, M.; Yano, K.; Tsuda, E.; Higashio, K.; Ikeda, K.; Watanabe, K.; Yamada, Y. Transforming growth factor-beta stimulates the production of osteoprotegerin/osteoclastogenesis inhibitory factor by bone marrow stromal cells. J. Biol. Chem. 1998, 273, 27091–27096. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lari, R.; Fleetwood, A.J.; Kitchener, P.D.; Cook, A.D.; Pavasovic, D.; Hertzog, P.J.; Hamilton, J.A. Macrophage lineage phenotypes and osteoclastogenesis--complexity in the control by GM-CSF and TGF-beta. Bone 2007, 40, 323–336. [Google Scholar] [CrossRef]
- Zhao, H.; Zhang, J.; Shao, H.; Liu, J.; Jin, M.; Chen, J.; Huang, Y. Transforming growth factor β1/Smad4 signaling affects osteoclast differentiation via regulation of miR-155 expression. Mol. Cells 2017, 40, 211–221. [Google Scholar]
- Kale, V.P. Differential activation of MAPK signaling pathways by TGF-β1 forms the molecular mechanism behind its dose-dependent bidirectional effects on hematopoiesis. Stem Cells Dev. 2004, 13, 27–38. [Google Scholar] [CrossRef]
- Kale, V.P.; Vaidya, A.A. Molecular mechanisms behind the dose-dependent differential activation of MAPK pathways induced by transforming growth factor-β1 in hematopoietic cells. Stem Cells Dev. 2004, 13, 536–547. [Google Scholar] [CrossRef]
- Karst, M.; Gorny, G.; Galvin, R.J.; Oursler, M.J. Roles of stromal cell RANKL, OPG, and M-CSF expression in biphasic TGF-beta regulation of osteoclast differentiation. J. Cell. Physiol. 2004, 200, 99–106. [Google Scholar] [CrossRef] [Green Version]
- Karsdal, M.A.; Hjorth, P.; Henriksen, K.; Kirkegaard, T.; Nielsen, K.L.; Lou, H.; Delaisse, J.M.; Foged, N.T. Transforming growth factor-β controls human osteoclastogenesis through the p38 MAPK and regulation of RANK expression. J. Biol. Chem. 2003, 278, 44975–44987. [Google Scholar] [CrossRef] [Green Version]
- Massey, H.M.; Scopes, J.; Horton, M.A.; Flanagan, A.M. Transforming growth factor-beta1 (TGF-beta) stimulates the osteoclast-forming potential of peripheral blood hematopoietic precursors in a lymphocyte-rich microenvironment. Bone 2001, 28, 577–582. [Google Scholar] [CrossRef]
- Massague, J. TGF-beta signal transduction. Annu. Rev. Biochem. 1998, 67, 753–791. [Google Scholar] [CrossRef] [PubMed]
- Blair, H.C.; Zaidi, M. Osteoclastic differentiation and function regulated by old and new pathways. Rev. Endocr. Metab. Disord. 2006, 7, 23–32. [Google Scholar] [CrossRef] [PubMed]
- de Vries, T.J.; El Bakkali, I.; Kamradt, T.; Schett, G.; Jansen, I.D.C.; D’Amelio, P. What Are the Peripheral Blood Determinants for Increased Osteoclast Formation in the Various Inflammatory Diseases Associated With Bone Loss? Front. Immunol. 2019, 10, 505. [Google Scholar] [CrossRef] [PubMed]
- Hamon, G.; Mulloy, R.H.; Chen, G.; Chow, R.; Birkenmaier, C.; Horn, J.K. Transforming growth factor-β 1 lowers the CD14 content of monocytes. J. Surg. Res. 1994, 57, 574–578. [Google Scholar] [CrossRef] [PubMed]
- Basoni, C.; Nobles, M.; Grimshaw, A.; Desgranges, C.; Davies, D.; Perretti, M.; Kramer, I.M.; Genot, E. Inhibitory control of TGF-β1 on the activation of Rap1, CD11b, and transendothelial migration of leukocytes. FASEB J. 2005, 19, 822–824. [Google Scholar] [CrossRef] [PubMed]
- Park, J.H.; Lee, N.K.; Lee, S.Y. Current Understanding of RANK Signaling in Osteoclast Differentiation and Maturation. Mol. Cells 2017, 40, 706–713. [Google Scholar]
- Moynagh, P.N. The NF-κB pathway. J. Cell Sci. 2005, 118, 4589–4592. [Google Scholar] [CrossRef] [Green Version]
- Yamashita, T.; Yao, Z.; Li, F.; Zhang, Q.; Badell, I.R.; Schwarz, E.M.; Takeshita, S.; Wagner, E.F.; Noda, M.; Matsuo, K.; et al. NF-kappaB p50 and p52 regulate receptor activator of NF-kappaB ligand (RANKL) and tumor necrosis factor-induced osteoclast precursor differentiation by activating c-Fos and NFATc1. J. Biol. Chem. 2007, 282, 18245–18253. [Google Scholar] [CrossRef] [Green Version]
- Udagawa, N.; Takahashi, N.; Akatsu, T.; Tanaka, H.; Sasaki, T.; Nishihara, T.; Koga, T.; Martin, T.J.; Suda, T. Origin of osteoclasts: Mature monocytes and macrophages are capable of differentiating into osteoclasts under a suitable microenvironment prepared by bone marrow-derived stromal cells. Proc. Natl. Acad. Sci. USA. 1990, 87, 7260–7264. [Google Scholar] [CrossRef] [Green Version]
- Kotake, S.; Udagawa, N.; Hakoda, M.; Mogi, M.; Yano, K.; Tsuda, E.; Takahashi, K.; Furuya, T.; Ishiyama, S.; Kim, K.J.; et al. Activated human T cells directly induce osteoclastogenesis from human monocytes: Possible role of T cells in bone destruction in rheumatoid arthritis patients. Arthritis. Rheum. 2001, 44, 1003–1012. [Google Scholar] [CrossRef]
- Kotake, S.; Yago, T.; Kawamoto, M.; Nanke, Y. Effects of NSAIDs on differentiation and function of human and murine osteoclasts –crucial ‘human osteoclastology’. Pharmaceuticals (Basel) 2010, 3, 1394–1410. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nose, M.; Yamazaki, H.; Hagino, H.; Morio, Y.; Hayashi, S.; Teshima, R. Comparison of osteoclast precursors in peripheral blood mononuclear cells from rheumatoid arthritis and osteoporosis patients. J. Bone Miner. Metab. 2009, 27, 57–65. [Google Scholar] [CrossRef] [PubMed]
- Durand, M.; Boire, G.; Komarova, S.V.; Dixon, S.J.; Sims, S.M.; Harrison, R.E.; Nabavi, N.; Maria, O.; Manolson, M.F.; Mizianty, M.; et al. The increased in vitro osteoclastogenesis in patients with rheumatoid arthritis is due to increased percentage of precursors and decreased apoptosis—the In Vitro Osteoclast Differentiation in Arthritis (IODA) study. Bone 2011, 48, 588–596. [Google Scholar] [CrossRef] [PubMed]
- Massey, H.M.; Flanagan, A.M. Human osteoclasts derive from CD14-positive monocytes. Br. J. Haematol. 1999, 106, 167–170. [Google Scholar] [CrossRef]
- Sprangers, S.; Schoenmaker, T.; Cao, Y.; Everts, V.; de Vries, T.J. Different Blood-Borne Human Osteoclast Precursors Respond in Distinct Ways to IL-17A. J. Cell Physiol. 2016, 231, 1249–1260. [Google Scholar] [CrossRef]
- Anbazhagan, K.; Duroux-Richard, I.; Jorgensen, C.; Apparailly, F. Transcriptomic network support distinct roles of classical and non-classical monocytes in human. Int. Rev. Immunol. 2014, 33, 470–489. [Google Scholar] [CrossRef]
- Hayashi, H.; Nakahama, K.; Sato, T.; Tuchiya, T.; Asakawa, Y.; Maemura, T.; Tanaka, M.; Morita, M.; Morita, I. The role of Mac-1 (CD11b/CD18) in osteoclast differentiation induced by receptor activator of nuclear factor-kappaB ligand. FEBS Lett. 2008, 582, 3243–3248. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Akiyama, M.; Nakahama, K.; Koshiishi, T.; Takeda, S.; Morita, I. Role of intercellular adhesion molecule-2 in osteoclastogenesis. Genes Cells 2012, 17, 568–575. [Google Scholar] [CrossRef]
- Yang, G.; Chen, X.; Yan, Z.; Zhu, Q.; Yang, C. CD11b promotes the differentiation of osteoclasts induced by RANKL through the spleen tyrosine kinase signalling pathway. J. Cell Mol. Med. 2017, 21, 3445–3452. [Google Scholar] [CrossRef]
- Asagiri, M.; Sato, K.; Usami, T.; Ochi, S.; Nishina, H.; Yoshida, H.; Morita, I.; Wagner, E.F.; Mak, T.W.; Serfling, E.; et al. Autoamplification of NFATc1 expression determines its essential role in bone homeostasis. J. Exp. Med. 2005, 202, 1261–1269. [Google Scholar] [CrossRef] [Green Version]
- Munoz-Valle, J.F.; Torres-Carrillo, N.M.; Guzman-Guzman, I.P.; Torres-Carrillo, N.; Ruiz-Quezada, S.L.; Palafox-Sanchez, C.A.; Rangel-Villalobos, H.; Ramirez-Duenas, M.G.; Parra-Rojas, I.; Fafutis-Morris, M.; et al. The functional class evaluated in rheumatoid arthritis is associated with soluble TGF-beta1 serum levels but not with G915C (Arg25Pro) TGF-beta1 polymorphism. Rheumatol. Int. 2012, 32, 367–372. [Google Scholar] [CrossRef] [PubMed]
- Mieliauskaite, D.; Venalis, P.; Dumalakiene, I.; Venalis, A.; Distler, J. Relationship between serum levels of TGF-beta1 and clinical parameters in patients with rheumatoid arthritis and Sjogren’s syndrome secondary to rheumatoid arthritis. Autoimmunity 2009, 42, 356–358. [Google Scholar] [CrossRef] [PubMed]
- Arnett, F.C.; Edworthy, S.M.; Bloch, D.A.; McShane, D.J.; Fries, J.F.; Cooper, N.S.; Healey, L.A.; Kaplan, S.R.; Liang, M.H.; Luthra, H.S.; et al. The American Rheumatism Association 1987 revised criteria for the classification of rheumatoid arthritis. Arthritis Rheum. 1988, 31, 315–324. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Sense | Anti-Sense |
|---|---|---|
| NFATC1 | 5′-GCATCACAGGGAAGACCGTGTC-3′ | 5′-GAAGTTCAATGTCGGAGTTTCTGAG-3′ |
| CTSK | 5′-AGTTTTACAGCAAAGGTGTG-3′ | 5′-CTTGTTTCCCTTCTGGATTC-3′ |
| P65 | 5′-GAGACATCCTTCCGCAAACT-3′ | 5′-TCCTTCCTGCCCATAATCA-3′ |
| C-FOS | 5′-CAGTTATCTCCAGAAGAAGAAG-3′ | 5′-CTTCTAGTTGGTCTGTCTCC-3′ |
| ACTB | 5′-GACGACATGGAGAAAATCTG-3′ | 5′-ATGATCTGGGTCATCTTCTC-3′ |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tokunaga, T.; Mokuda, S.; Kohno, H.; Yukawa, K.; Kuranobu, T.; Oi, K.; Yoshida, Y.; Hirata, S.; Sugiyama, E. TGFβ1 Regulates Human RANKL-Induced Osteoclastogenesis via Suppression of NFATc1 Expression. Int. J. Mol. Sci. 2020, 21, 800. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21030800
Tokunaga T, Mokuda S, Kohno H, Yukawa K, Kuranobu T, Oi K, Yoshida Y, Hirata S, Sugiyama E. TGFβ1 Regulates Human RANKL-Induced Osteoclastogenesis via Suppression of NFATc1 Expression. International Journal of Molecular Sciences. 2020; 21(3):800. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21030800
Chicago/Turabian StyleTokunaga, Tadahiro, Sho Mokuda, Hiroki Kohno, Kazutoshi Yukawa, Tatsuomi Kuranobu, Katsuhiro Oi, Yusuke Yoshida, Shintaro Hirata, and Eiji Sugiyama. 2020. "TGFβ1 Regulates Human RANKL-Induced Osteoclastogenesis via Suppression of NFATc1 Expression" International Journal of Molecular Sciences 21, no. 3: 800. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21030800