Combined Proteomics and Metabolism Analysis Unravels Prominent Roles of Antioxidant System in the Prevention of Alfalfa (Medicago sativa L.) against Salt Stress

Abstract
:1. Introduction
2. Results
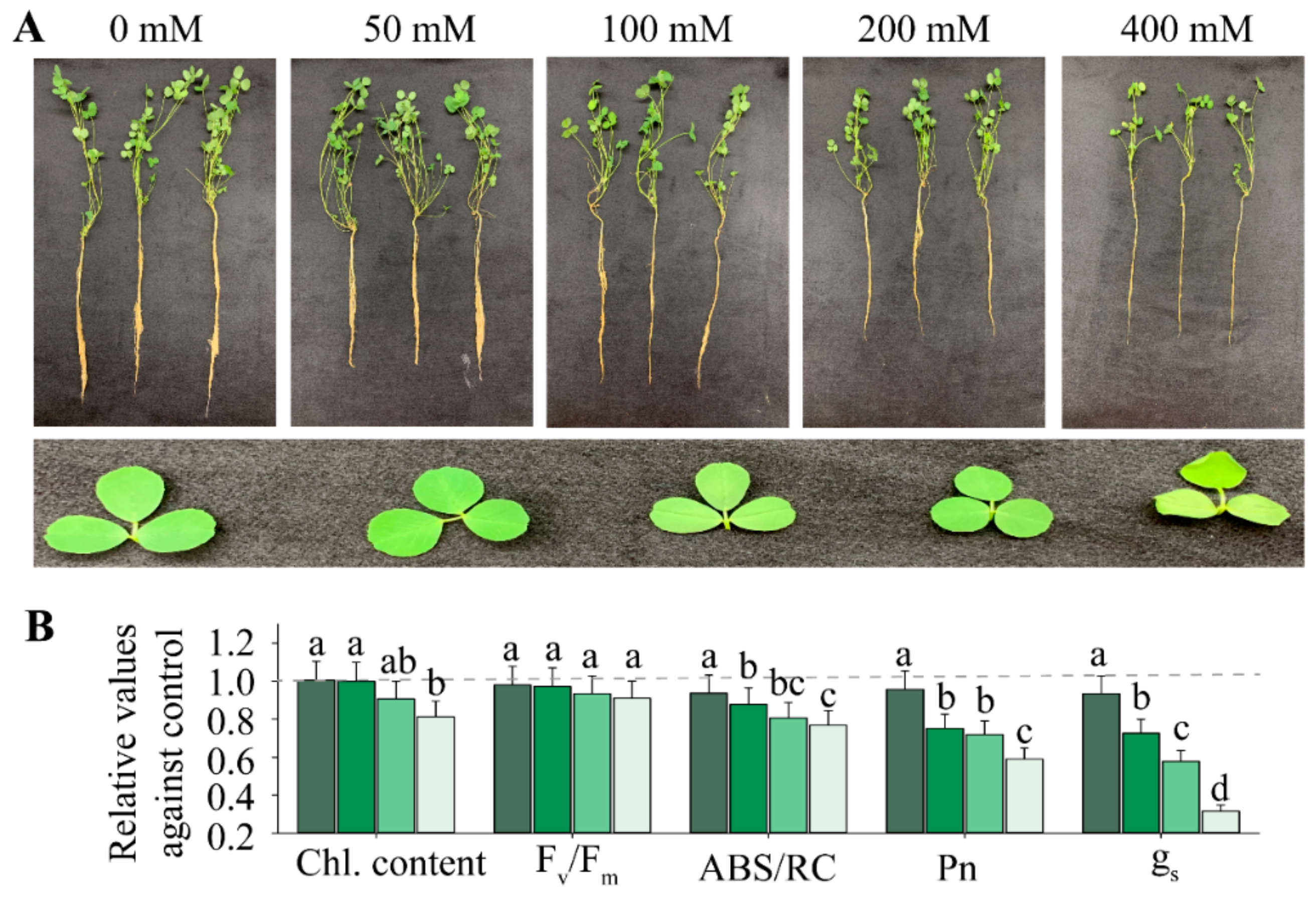
2.1. Physiological Performance of Alfalfa Exposed to Salt Stress
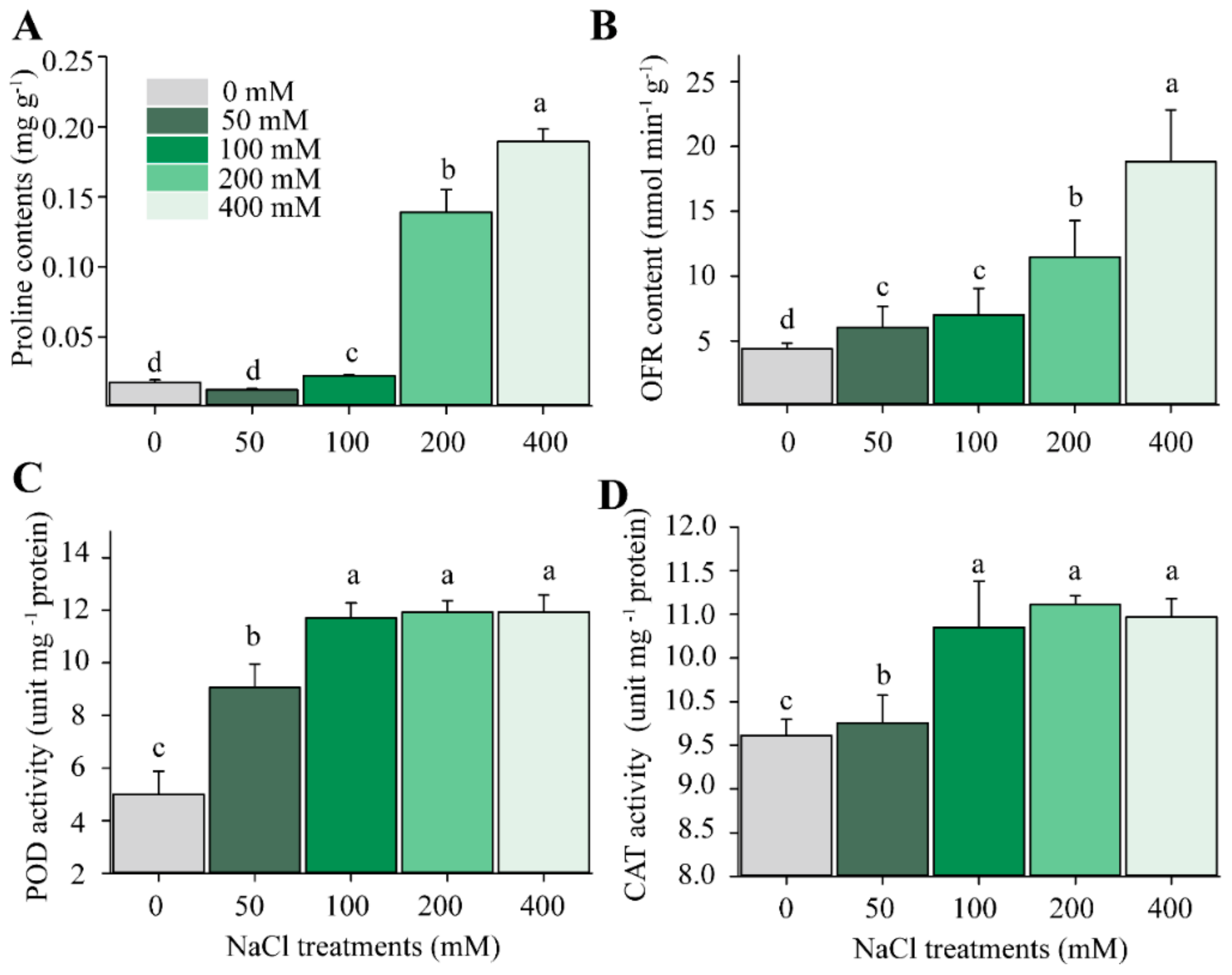
2.2. Changes in H2O2 and Antioxidant Enzyme Activities
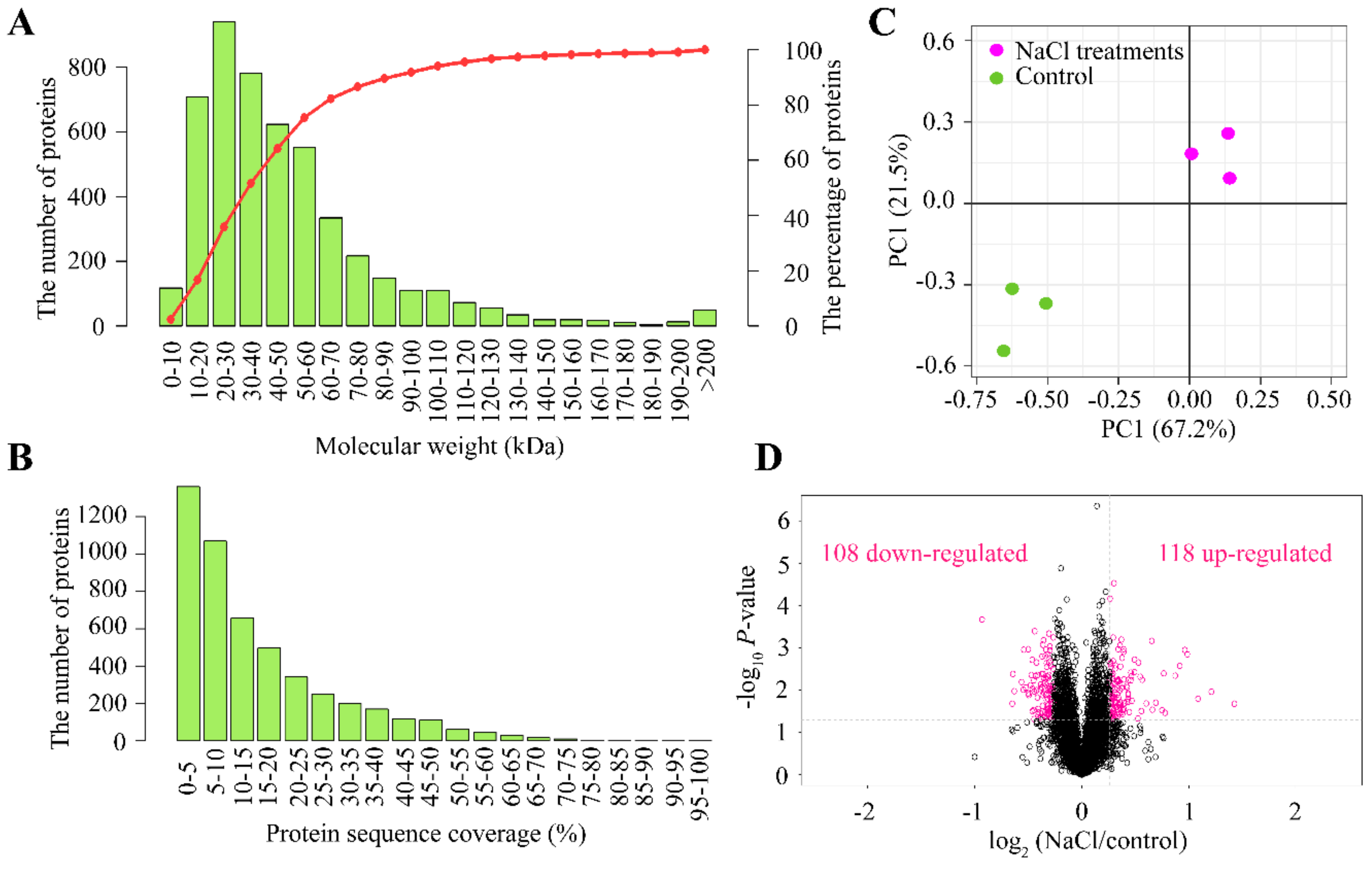
2.3. Identification and Functional Classification of DAPs
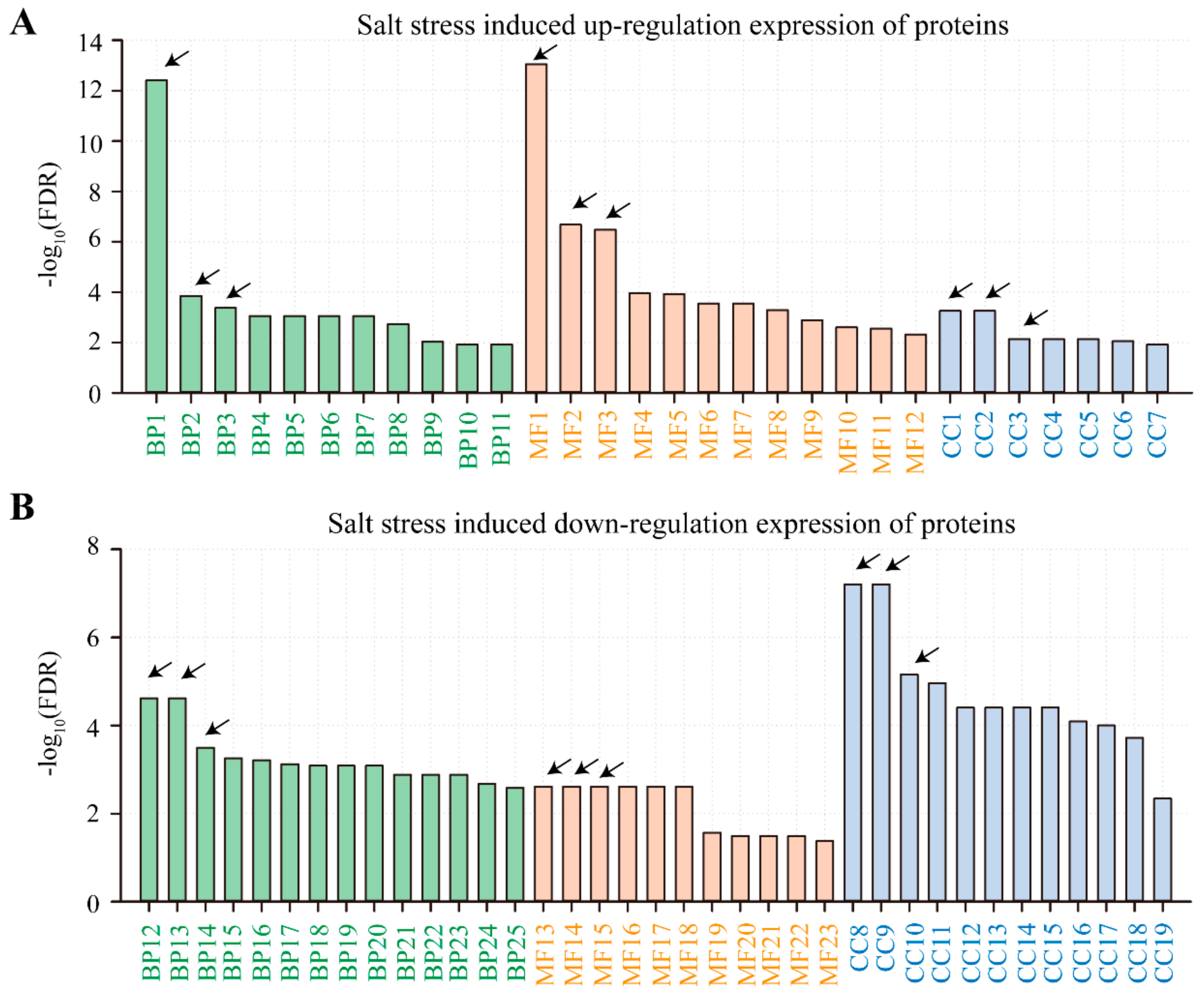
2.4. Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) Analysis
3. Discussion
3.1. Glutathione Metabolism Plays a Crucial Role in Salt Tolerance
3.2. Mechanisms of Proline Stress Protection Coupling with ROS Scavengers
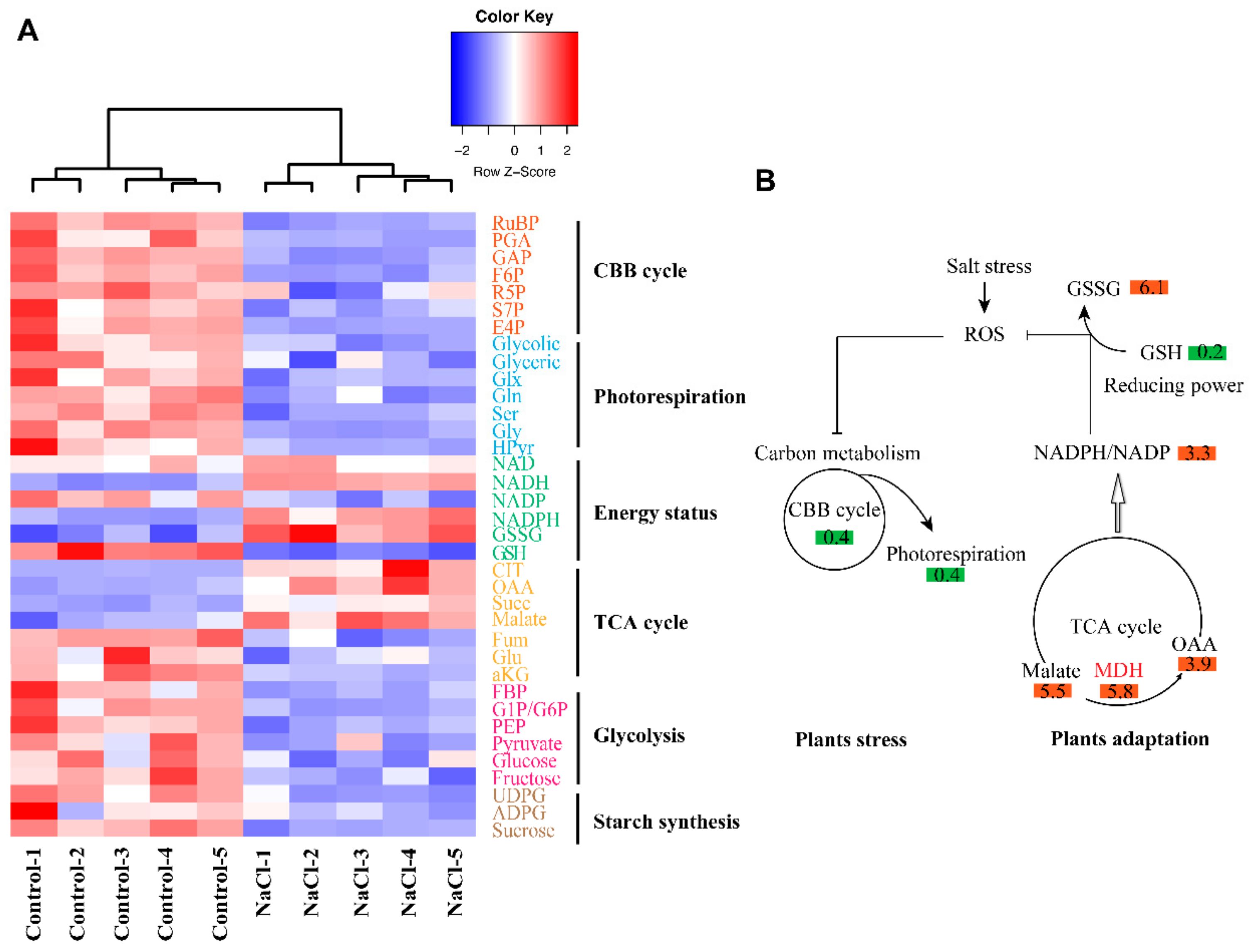
3.3. Inhibition of Photosynthetic Carbon Metabolism by Salt Stress
3.4. Molecular Mechanism of ROS Signaling-Dependent Salt Tolerance
4. Materials and Methods
4.1. Plant Material and Stress Treatments
4.2. Physiological Measurements
4.3. POD, CAT and OFR Activity Analyses
4.4. Proline Determinations
4.5. Metabolic Determinations
4.6. Protein Extraction
4.7. Gene Ontology Information
4.8. Quantitative Real-Time PCR
4.9. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Li, X.H.; Wei, Y.L.; Moore, K.J.; Michaud, R.; Viands, D.R.; Hansen, J.L.; Acharya, A.; Brummer, E.C. Association mapping of biomass yield and stem composition in a tetraploid alfalfa breeding population. Plant Genome 2011, 4, 24–35. [Google Scholar] [CrossRef] [Green Version]
- Sairam, R.K.; Tyagi, A. Physiology and molecular biology of salinity stress tolerance in plants. Curr. Sci. 2004, 86, 407–421. [Google Scholar]
- Orsini, F.; D’Urzo, M.D.; Inan, S.; Serra, S.; Oh, D.; Mickelbart, M.V.; Consiglio, F.; Li, X.; Jeong, J.C.; Yun, D.J.; et al. A comparative study of salt tolerance parameters in 11 wild relatives of Arabidopsis thaliana. J. Exp. Bot. 2010, 61, 3787–3798. [Google Scholar] [CrossRef]
- Popova, O.V.; Golldack, D. In the halotolerant Lobularia maritima (Brassicaceae) salt adaptation correlates with activation of the vacuolar H+-ATPase and the vacuolar Na+/H+ antiporter. J. Plant Physiol. 2007, 164, 1278–1288. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J. Plant salt tolerance. Trends Plant Sci. 2001, 6, 66–71. [Google Scholar] [CrossRef]
- Munns, R.; Tester, M. Mechanisms of salinity tolerance. Ann. Rev. Plant Biol. 2008, 59, 651–681. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lei, Y.; Xu, Y.; Hettenhausen, C.; Lu, C.; Shen, G.; Zhang, C.; Li, J.; Song, J.; Lin, H.; Wu, J. Comparative analysis of alfalfa (Medicago sativa L.) leaf transcriptomes reveals genotype-specific salt tolerance mechanisms. BMC Plant Biol. 2018, 18, 35. [Google Scholar] [CrossRef] [Green Version]
- Limami, A.M.; Ricoult, C.; Planchet, E.; González, E.M.; Ladrera, R.; Larrainzar, E.; Arrese-Igor, C.; Merchan, F.; Crespi, M.; Frugier, F.; et al. Response of Medicago truncatula to abiotic stress. In The Medicago truncatula Handbook; Plenum Press: New York, NY, USA, 2006; Available online: http://www.noble.org/MedicagoHandbook.
- Atkinson, N.J.; Urwin, P.E. The interaction of plant biotic and abiotic stresses: from genes to the field. J. Exp. Bot. 2012, 63, 3523–3544. [Google Scholar] [CrossRef] [Green Version]
- Gao, Y.; Cui, Y.; Long, R.; Sun, Y.; Zhang, T.; Yang, Q.; Kang, J. Salt-stress induced proteomic changes of two contrasting alfalfa cultivars during germination stage. J. Sci. Food Agric. 2019, 99, 1384–1396. [Google Scholar] [CrossRef]
- Jin, H.; Sun, Y.; Yang, Q.; Chao, Y.; Kang, J.; Jin, H.; Li, Y.; Margaret, G. Screening of genes induced by salt stress from alfalfa. Mol. Biol. Rep. 2010, 37, 745–753. [Google Scholar] [CrossRef]
- Postnikova, O.A.; Shao, J.; Nemchinov, L.G. Analysis of the alfalfa root transcriptome in response to salinity stress. Plant Cell Physiol. 2013, 54, 1041–1055. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.F. Role of silicon in enhancing the resistance of plants to biotic and abiotic stresses. Soil Sci. Plant Nutr. 2004, 50, 11–18. [Google Scholar] [CrossRef]
- Xiong, J.; Sun, Y.; Yang, Q.; Tian, H.; Zhang, H.; Liu, Y.; Chen, M. Proteomic analysis of early salt stress responsive proteins in alfalfa roots and shoots. Proteome Sci. 2017, 15, 19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goez, M.M.; Torres-Madroñero, M.C.; Röthlisberger, S.; Delgado-Trejos, E. Preprocessing of 2-dimensional gel electrophoresis images applied to proteomic analysis: a review. Genom. Proteom. Bioinform. 2018, 16, 63–72. [Google Scholar] [CrossRef] [PubMed]
- Rauniyar, N. Parallel reaction monitoring: A targeted experiment performed using high resolution and high mass accuracy mass spectrometry. Int. J. Mol. Sci. 2015, 16, 28566–28581. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Winterbourn, C.C.; Metodiewa, D. Reactivity of biologically important thiol compounds with superoxide and hydrogen peroxide. Free Radic. Biol. Med. 1999, 27, 322–328. [Google Scholar] [CrossRef]
- Yadav, S.K.; Singla-Pareek, S.L.; Ray, M.; Reddy, M.K.; Sopory, S.K. Methylglyoxal levels in plants under salinity stress are dependent on glyoxalase I and glutathione. Biochem. Biophys. Res. Commun. 2005, 337, 61–67. [Google Scholar] [CrossRef]
- Qu, M.; Chen, G.Y.; Bunce, J.A.; Zhu, X.; Sicher, R.C. Systematic biology analysis on photosynthetic carbon metabolism of maize leaf following sudden heat shock under elevated CO2. Sci. Rep. 2018, 8, 7849. [Google Scholar] [CrossRef]
- Hasanuzzaman, M.; Fujita, M. Exogenous sodium nitroprusside alleviates arsenic-induced oxidative stress in wheat (Triticum aestivum L.) seedlings by enhancing antioxidant defense and glyoxalase system. Ecotoxicology 2013, 22, 584–596. [Google Scholar] [CrossRef]
- Roxas, V.P.; Smith, R.K.; Allen, E.R.; Allen, R.D. Over-expression of glutathione S-transferase/glutathione peroxidase enhances the growth of transgenic tobacco seedlings during stress. Nat. Biotechnol. 1997, 15, 988–991. [Google Scholar] [CrossRef]
- Luo, Y.; Tang, H.; Zhang, Y. Production of reactive oxygen species and antioxidant metabolism about strawberry leaves to low temperatures. J. Agric. Sci. 2011, 3, 89–96. [Google Scholar] [CrossRef]
- Hoque, M.A.; Banu, M.N.; Nakamura, Y.; Shimoishi, Y.; Murata, Y. Proline and glycine betaine enhance antioxidant and methylglyoxal detoxification systems and reduce NaCl-induced damage in cultured tobacco cells. Plant Physiol. 2008, 165, 813–882. [Google Scholar] [CrossRef] [PubMed]
- Kattab, H. Role of glutathione and polyadenylic acid on the oxidative defense systems of two different cultivars of canola seedlings grown under saline condition. Aust. J. Basic Appl. Sci. 2007, 1, 323–332. [Google Scholar]
- Wang, W.; Vinocur, B.; Shoseyov, O.; Altman, A. Role of plant heat-shock proteins and molecular chaperones in the abiotic stress response. Trends Plant Sci. 2004, 9, 244–252. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Shi, S. Physiological and proteomic responses of contrasting alfalfa (Medicago sativa L.) varieties to PEG-induced osmotic stress. Front. Plant Sci. 2018, 9, 242. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Essemine, J.; Bunce, J.A.; Shang, C.; Zhang, H.; Sun, D.; Chen, G.; Qu, M. Roles of heat shock protein and reprogramming of photosynthetic carbon metabolism in thermotolerance under elevated CO2 in maize. Environ. Exp. Bot. 2019, 168, 103869. [Google Scholar] [CrossRef]
- Law, R.D.; Crafts-Brandner, S.J. Inhibition and acclimation of photosynthesis to heat stress is closely correlated with activation of ribulose-1,5-bisphosphate carboxylase/oxygenase. Plant Physiol. 1999, 120, 173–181. [Google Scholar] [CrossRef] [Green Version]
- Wingler, A.; Lea, P.J.; Quick, W.P.; Leegood, R.C. Photorespiration: Metabolic pathways and their role in stress protection. Phil. Trans. R. Soc. Lond. B. 2000, 355, 1517–1529. [Google Scholar] [CrossRef] [Green Version]
- Miller, G.; Shulaev, V.; Mittler, R. Reactive oxygen signaling and abiotic stress. Physiol. Plant 2008, 133, 481–489. [Google Scholar] [CrossRef]
- Strasser, B.J.; Strasser, R.J. Measuring fast fluorescence transients to address environmental questions: The JIP-test. Photosynth. Light Biosph. 1995, 5, 977–980. [Google Scholar]
- Zhang, K.P.; Fang, Z.J.; Liang, Y.; Tian, J.C. Genetic dissection of chlorophyll content at different growth stages in common wheat. J. Genet. 2009, 88, 183–189. [Google Scholar] [CrossRef] [PubMed]
- Porra, R.J.; Thompson, W.A.; Kriedemann, P.E. Determination of accurate extinction coefficients and simultaneous equations for assaying chlorophylls a and b extracted with four different solvents: verification of the concentration of chlorophyll standards by atomic absorption spectroscopy. Biochim. Biophys. Acta. 1989, 975, 384–394. [Google Scholar] [CrossRef]
- Chance, B.; Maehly, A.C. Assay of catalase and peroxidase. Meth. Enzymol. 1955, 2, 764–775. [Google Scholar]
- Bates, L.S.; Waldren, R.P.; Teare, I.D. Rapid determination of free proline for water-stress studies. Plant Soil 1973, 39, 205–207. [Google Scholar] [CrossRef]
- Hao, P.; Zhu, J.; Gu, A.; Lv, D.; Ge, P.; Chen, G.; Li, X.; Yan, Y. An integrative proteome analysis of different seedling organs in tolerant and sensitive wheat cultivars under drought stress and recovery. Proteomics 2015, 15, 1544–1563. [Google Scholar] [CrossRef]
- Azeem, S.; Li, Z.; Zheng, H.; Lin, W.; Arafat, Y.; Zhang, Z.; Lin, X.; Lin, W. Quantitative proteomics study on Lsi1 in regulation of rice (Oryza sativa L.) cold resistance. Plant Growth Regul. 2016, 78, 307–323. [Google Scholar] [CrossRef]
- Perez-Riverol, Y.; Csordas, A.; Bai, J.; Bernal-Llinares, M.; Hewapathirana, S.; Kundu, D.J.; Inuganti, A.; Griss, J.; Mayer, G.; Eisenacher, M.; et al. The PRIDE database and related tools and resources in 2019: improving support for quantification data. Nucleic Acids Res. 2019, 47, 442–450. [Google Scholar] [CrossRef]
- Cai, H.; Chen, H.; Yi, T.; Daimon, C.M.; Boyle, J.P.; Peers, C.; Maudsley, S.; Martin, B. VennPlex—a novel Venn diagram program for comparing and visualizing datasets with differentially regulated data points. PLoS ONE 2013, 8, e53388. [Google Scholar]
- Ma, Q.; Kang, J.; Long, R.; Zhang, T.; Xiong, J.; Zhang, K.; Wang, T.; Yang, Q.; Sun, Y. Comparative proteomic analysis of alfalfa revealed new salt and drought stress-related factors involved in seed germination. Mol. Biol. Rep. 2017, 44, 261–272. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Gimeno, J.; Eattock, N.; Deynze, A.V.; Blumwald, E. Selection and validation of reference genes for gene expression analysis in switchgrass (Panicum virgatum) using quantitative real-time RT-PCR. PLoS ONE 2014, 9, e91474. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameters | Duration | NaCl | Duration × NaCl |
|---|---|---|---|
| Chl. content (mg.g−1) | 12.033 ** | 2.702 * | 0.083 * |
| Proline content (mg.g−1) | 2.168 | 47.614 *** | 4.822 * |
| Oxygen free radical (OFR; mmol m−2 s−1) | 0.192 | 0.147 * | 0.01 |
| Catalase activity (CAT; mmol m−2 s−1) | 8.212 *** | 1.856 * | 3.207 * |
| Peroxidase (POD; mmol m−2 s−1) | 0.247 | 0.388 * | 6.563 * |
| Fv/Fm | 0.008 | 1.583 * | 0.073 |
| ABS/RC | 0.037 | 5.552 * | 1.244 * |
| Pn (μmol m−2 s−1) | 0.917 | 0.219 ** | 1.015 * |
| gs (mmol m−2 s−1) | 0.451 | 0.089 ** | 0.578 * |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, J.; Essemine, J.; Shang, C.; Zhang, H.; Zhu, X.; Yu, J.; Chen, G.; Qu, M.; Sun, D. Combined Proteomics and Metabolism Analysis Unravels Prominent Roles of Antioxidant System in the Prevention of Alfalfa (Medicago sativa L.) against Salt Stress. Int. J. Mol. Sci. 2020, 21, 909. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21030909
Li J, Essemine J, Shang C, Zhang H, Zhu X, Yu J, Chen G, Qu M, Sun D. Combined Proteomics and Metabolism Analysis Unravels Prominent Roles of Antioxidant System in the Prevention of Alfalfa (Medicago sativa L.) against Salt Stress. International Journal of Molecular Sciences. 2020; 21(3):909. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21030909
Chicago/Turabian StyleLi, Jikai, Jemaa Essemine, Chen Shang, Hailing Zhang, Xiaocen Zhu, Jialin Yu, Genyun Chen, Mingnan Qu, and Dequan Sun. 2020. "Combined Proteomics and Metabolism Analysis Unravels Prominent Roles of Antioxidant System in the Prevention of Alfalfa (Medicago sativa L.) against Salt Stress" International Journal of Molecular Sciences 21, no. 3: 909. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21030909