Discovery of a Gatekeeper Residue in the C-Terminal Tail of the Extracellular Signal-Regulated Protein Kinase 5 (ERK5)

 , , ,
, , ,
Abstract
:1. Introduction
2. Results
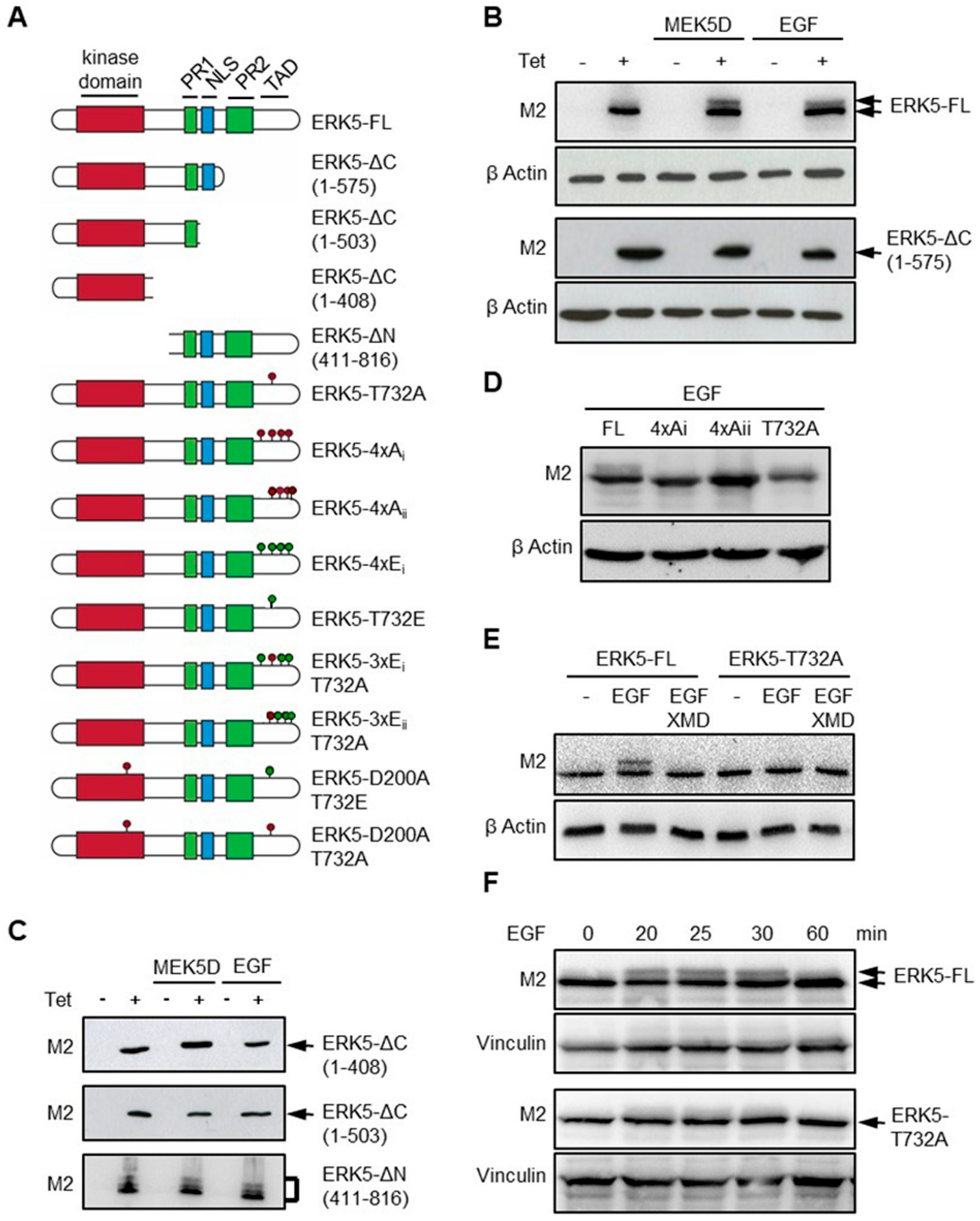
2.1. Thr732 is Critical for C-Terminal Phosphorylation of ERK5
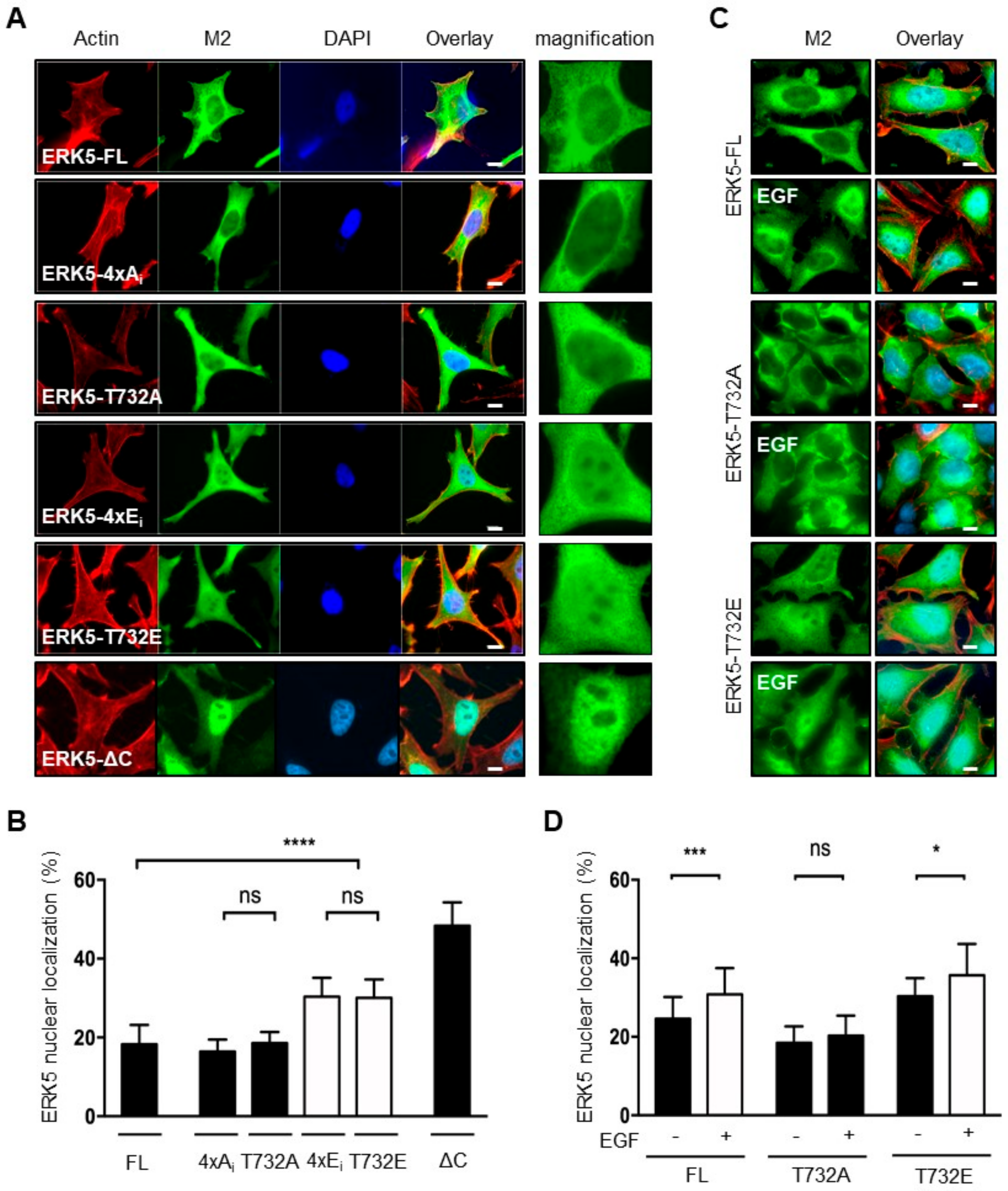
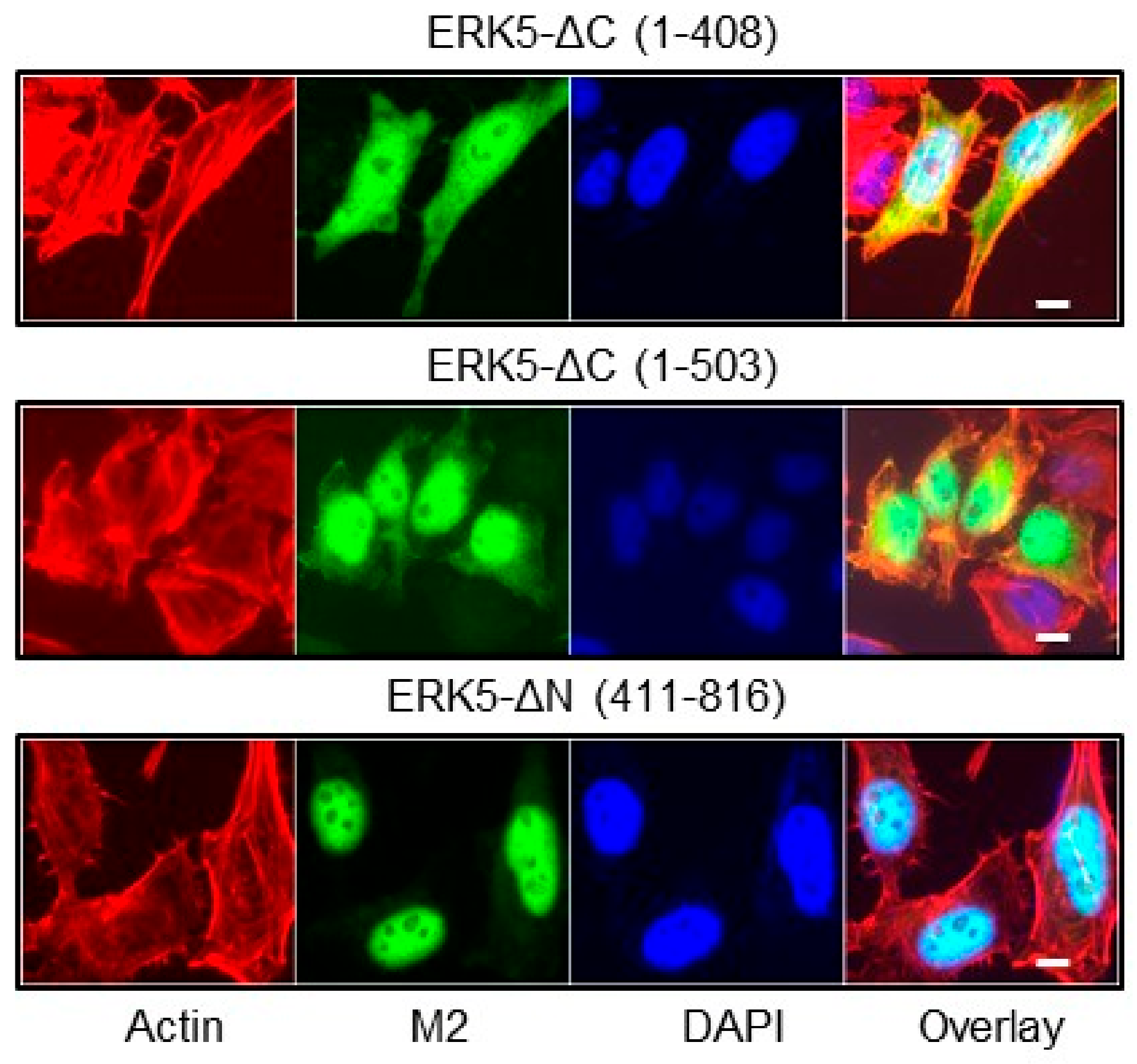
2.2. Phosphorylation at Thr732 Promotes ERK5 Nuclear Translocation
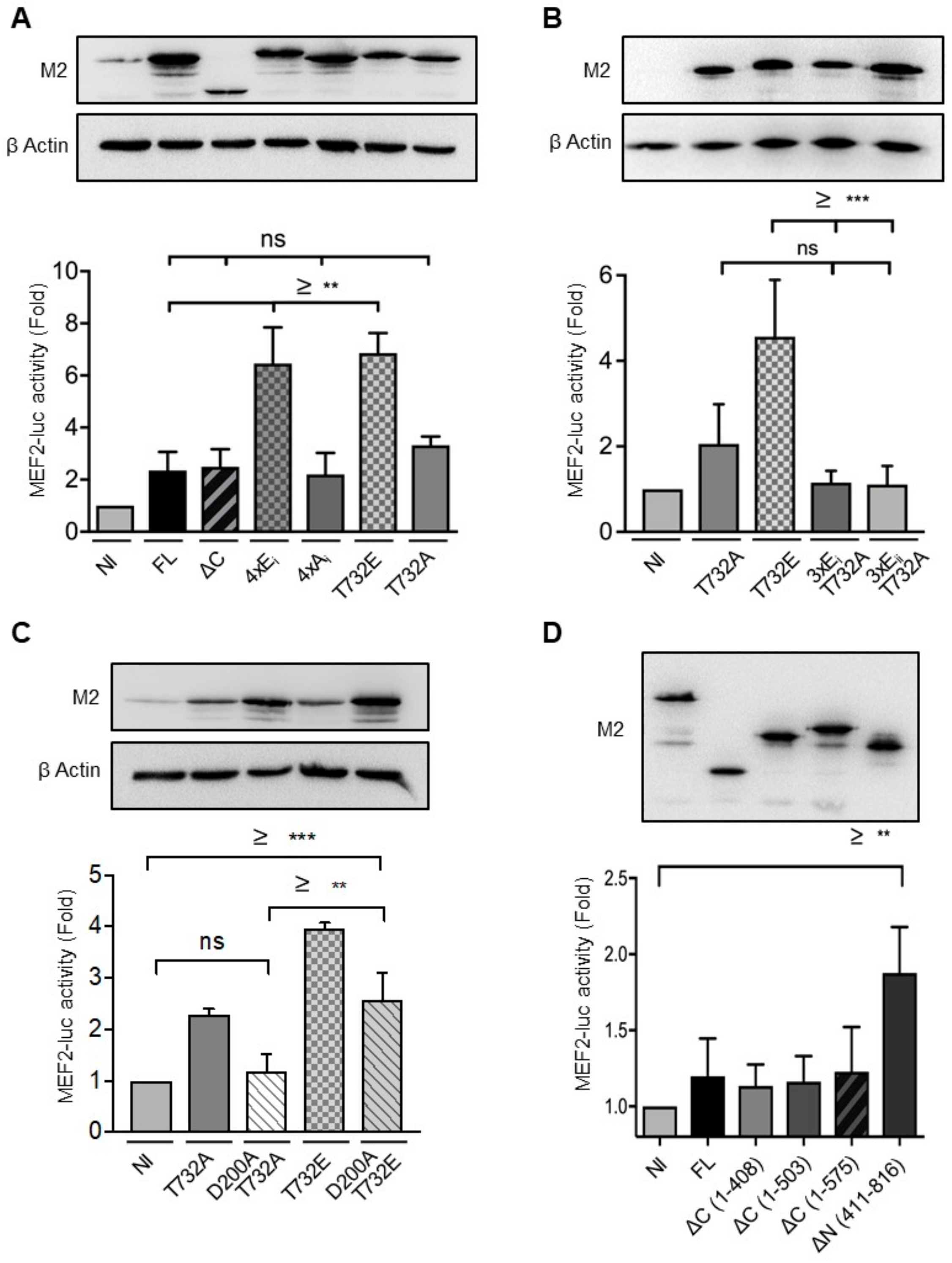
2.3. Phosphorylation at Thr732 Enhances ERK5 Transcriptional Activity
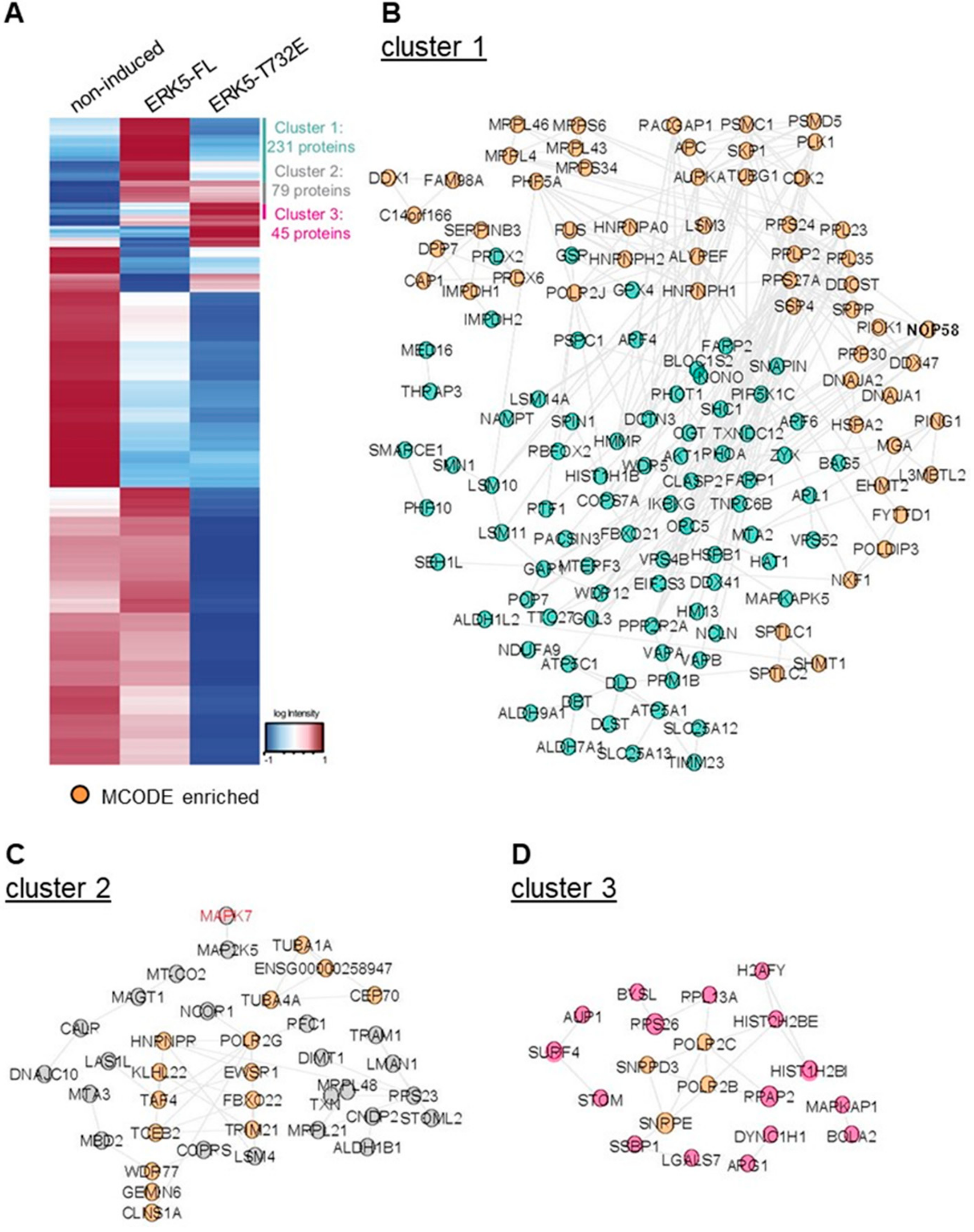
2.4. Phosphorylation at Thr732 Confers Binding Interaction Specificity
3. Discussion
4. Materials and Methods
4.1. Generation of Isogenic Tet-Inducible ERK5 Expressing HeLa Cell Lines
4.2. Immunoblot Analysis
4.3. Luciferase Reporter Assays
4.4. Immunofluorescence
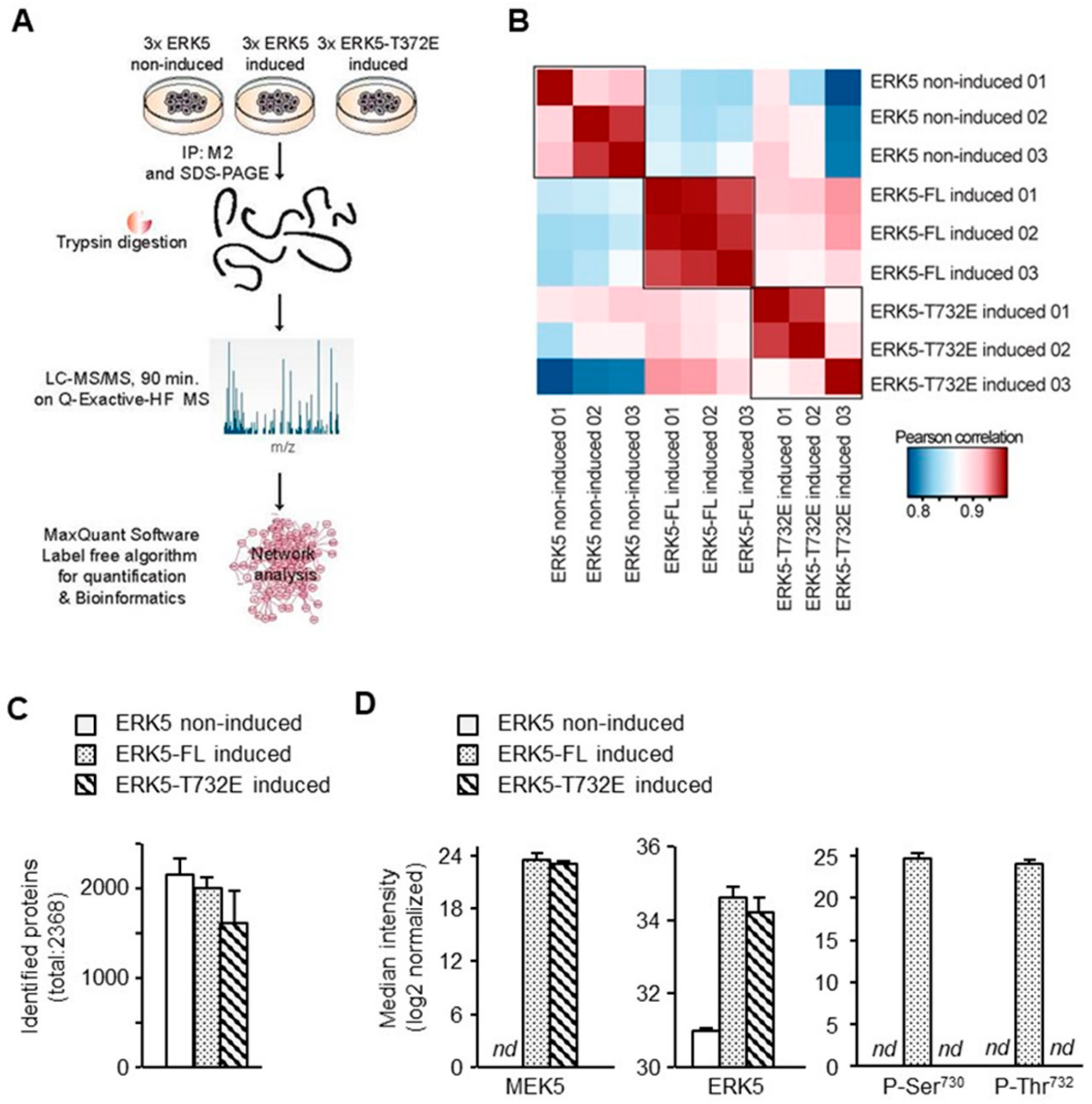
4.5. Mass Spectrometry and Raw Data Analysis
4.6. Data Analysis
4.7. Statistical Analysis
4.8. Data Availability
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ERK5 | extracellular signal-regulated protein kinase 5 |
| MAPK | mitogen-activated protein kinase |
| NLS | nuclear localization signal |
| TAD | transactivation domain |
References
- Cuenda, A.; Lizcano, J.M.; Lozano, J. Editorial: Mitogen Activated Protein Kinases. Front Cell Dev. Biol. 2017, 5, 80. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, M.; Lee, J.D. Role of the BMK1/ERK5 signaling pathway: Lessons from knockout mice. J. Mol. Med. 2004, 82, 800–808. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.D.; Ulevitch, R.J.; Han, J. Primary structure of BMK1: A new mammalian map kinase. Biochem. Biophys. Res. Commun. 1995, 213, 715–724. [Google Scholar] [CrossRef]
- Zhou, G.; Bao, Z.Q.; Dixon, J.E. Components of a new human protein kinase signal transduction pathway. J. Biol. Chem. 1995, 270, 12665–12669. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nithianandarajah-Jones, G.N.; Wilm, B.; Goldring, C.E.; Muller, J.; Cross, M.J. ERK5: Structure, regulation and function. Cell Signal. 2012, 24, 2187–2196. [Google Scholar] [CrossRef] [PubMed]
- Buschbeck, M.; Ullrich, A. The unique C-terminal tail of the mitogen-activated protein kinase ERK5 regulates its activation and nuclear shuttling. J. Biol. Chem. 2005, 280, 2659–2667. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kondoh, K.; Terasawa, K.; Morimoto, H.; Nishida, E. Regulation of nuclear translocation of extracellular signal-regulated kinase 5 by active nuclear import and export mechanisms. Mol. Cell Biol. 2006, 26, 1679–1690. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morimoto, H.; Kondoh, K.; Nishimoto, S.; Terasawa, K.; Nishida, E. Activation of a C-terminal transcriptional activation domain of ERK5 by autophosphorylation. J. Biol. Chem. 2007, 282, 35449–35456. [Google Scholar] [CrossRef] [Green Version]
- Kasler, H.G.; Victoria, J.; Duramad, O.; Winoto, A. ERK5 is a novel type of mitogen-activated protein kinase containing a transcriptional activation domain. Mol. Cell Biol. 2000, 20, 8382–8389. [Google Scholar] [CrossRef] [Green Version]
- Terasawa, K.; Okazaki, K.; Nishida, E. Regulation of c-Fos and Fra-1 by the MEK5-ERK5 pathway. Genes Cells 2003, 8, 263–273. [Google Scholar] [CrossRef]
- Diaz-Rodriguez, E.; Pandiella, A. Multisite phosphorylation of Erk5 in mitosis. J. Cell Sci. 2010, 123, 3146–3156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iñesta-Vaquera, F.A.; Campbell, D.G.; Tournier, C.; Gómez, N.; Lizcano, J.M.; Cuenda, A. Alternative ERK5 regulation by phosphorylation during the cell cycle. Cell Signal. 2010, 22, 1829–1837. [Google Scholar] [CrossRef]
- Honda, T.; Obara, Y.; Yamauchi, A.; Couvillon, A.D.; Mason, J.J.; Ishii, K.; Nakahata, N. Phosphorylation of ERK5 on Thr732 Is Associated with ERK5 Nuclear Localization and ERK5-Dependent Transcription. PLoS ONE 2015, 10, e0117914. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Esparís-Ogando, A.; Díaz-Rodríguez, E.; Montero, J.C.; Yuste, L.; Crespo, P.; Pandiella, A. Erk5 participates in neuregulin signal transduction and is constitutively active in breast cancer cells overexpressing ErbB2. Mol. Cell Biol. 2002, 22, 270–285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Montero, J.C.; Ocaña, A.; Abad, M.; Ortiz-Ruiz, M.J.; Pandiella, A.; Esparís-Ogando, A. Expression of Erk5 in early stage breast cancer and association with disease free survival identifies this kinase as a potential therapeutic target. PLoS ONE 2009, 4, e5565. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tusa, I.; Gagliardi, S.; Tubita, A.; Pandolfi, S.; Urso, C.; Borgognoni, L.; Wang, J.; Deng, X.; Gray, N.S.; Stecca, B.; et al. ERK5 is activated by oncogenic BRAF and promotes melanoma growth. Oncogene 2018, 37, 2601–2614. [Google Scholar] [CrossRef] [PubMed]
- Mody, N.; Campbell, D.G.; Morrice, N.; Peggie, M.; Cohen, P. An analysis of the phosphorylation and activation of extracellular-signal-regulated protein kinase 5 (ERK5) by mitogen-activated protein kinase kinase 5 (MKK5) in vitro. Biochem. J. 2003, 372, 567–575. [Google Scholar] [CrossRef] [Green Version]
- Yan, C.; Luo, H.; Lee, J.D.; Abe, J.; Berk, B.C. Molecular cloning of mouse ERK5/BMK1 splice variants and characterization of ERK5 functional domains. J. Biol. Chem. 2001, 276, 10870–10878. [Google Scholar] [CrossRef] [Green Version]
- Giurisato, E.; Xu, Q.; Lonardi, S.; Telfer, B.; Russo, I.; Pearson, A.; Finegan, K.G.; Wang, W.; Wang, J.; Gray, N.S.; et al. Myeloid ERK5 deficiency suppresses tumor growth by blocking protumor macrophage polarization via STAT3 inhibition. Proc. Natl. Acad. Sci. USA 2018, 115, E2801–E2810. [Google Scholar] [CrossRef] [Green Version]
- Klein, A.M.; Zaganjor, E.; Cobb, M.H. Chromatin-tethered MAPKs. Curr. Opin. Cell Biol. 2013, 25, 272–277. [Google Scholar] [CrossRef] [Green Version]
- Hu, S.; Xie, Z.; Onishi, A.; Yu, X.; Jiang, L.; Lin, J.; Rho, H.S.; Woodard, C.; Wang, H.; Jeong, J.S.; et al. Profiling the human protein-DNA interactome reveals ERK2 as a transcriptional repressor of interferon signaling. Cell 2009, 139, 610–622. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simone, C.; Forcales, S.V.; Hill, D.A.; Imbalzano, A.N.; Latella, L.; Puri, P.L. p38 pathway targets SWI-SNF chromatin-remodeling complex to muscle-specific loci. Nat. Genet. 2004, 36, 738–743. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suganuma, T.; Mushegian, A.; Swanson, S.K.; Abmayr, S.M.; Florens, L.; Washburn, M.P.; Workman, J.L. The ATAC acetyltransferase complex coordinates MAP kinases to regulate JNK target genes. Cell 2010, 142, 726–736. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, K.Y.; Levin, D.E. Mpk1 MAPK association with the Paf1 complex blocks Sen1-mediated premature transcription termination. Cell 2011, 144, 745–756. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Madak-Erdogan, Z.; Ventrella, R.; Petry, L.; Katzenellenbogen, B.S. Novel roles for ERK5 and cofilin as critical mediators linking ERalpha-driven transcription, actin reorganization, and invasiveness in breast cancer. Mol. Cancer Res. 2014, 12, 714–727. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ralle, T.; Gremmels, D.; Stick, R. Translational control of nuclear lamin B1 mRNA during oogenesis and early development of Xenopus. Mech Dev. 1999, 84, 89–101. [Google Scholar] [CrossRef]
- Nakamura, K.; Johnson, G.L. Noncanonical function of MEKK2 and MEK5 PB1 domains for coordinated extracellular signal-regulated kinase 5 and c-Jun N-terminal kinase signaling. Mol. Cell Biol. 2007, 27, 4566–4577. [Google Scholar] [CrossRef] [Green Version]
- Han, T.H.; Prywes, R. Regulatory role of MEF2D in serum induction of the c-jun promoter. Mol. Cell Biol. 1995, 15, 2907–2915. [Google Scholar] [CrossRef] [Green Version]
- Francavilla, C.; Papetti, M.; Rigbolt, K.T.; Pedersen, A.K.; Sigurdsson, J.O.; Cazzamali, G.; Karemore, G.; Blagoev, B.; Olsen, J.V. Multilayered proteomics reveals molecular switches dictating ligand-dependent EGFR trafficking. Nat. Struct. Mol. Biol. 2016, 23, 608–618. [Google Scholar] [CrossRef] [Green Version]
- Cox, J.; Mann, M. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol. 2008, 26, 1367–1372. [Google Scholar] [CrossRef]
- Cox, J.; Hein, M.Y.; Luber, C.A.; Paron, I.; Nagaraj, N.; Mann, M. Accurate proteome-wide label-free quantification by delayed normalization and maximal peptide ratio extraction, termed MaxLFQ. Mol. Cell Proteomics 2014, 13, 2513–2526. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wettenhall, J.M.; Smyth, G.K. limmaGUI: A graphical user interface for linear modeling of microarray data. Bioinformatics 2004, 20, 3705–3706. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tyanova, S.; Temu, T.; Sinitcyn, P.; Carlson, A.; Hein, M.Y.; Geiger, T.; Mann, M.; Cox, J. The Perseus computational platform for comprehensive analysis of (prote)omics data. Nat. Methods 2016, 9, 731–740. [Google Scholar] [CrossRef] [PubMed]
- Szklarczyk, D.; Gable, A.L.; Lyon, D.; Junge, A.; Wyder, S.; Huerta-Cepas, J.; Simonovic, M.; Doncheva, N.T.; Morris, J.H.; Bork, P.; et al. STRING v11: Protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2019, 47, D607–D613. [Google Scholar] [CrossRef] [PubMed] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Mutant | Details | Reference |
|---|---|---|
| ERK5-FL | [3] | |
| ERK5-ΔC | Deletion of amino acids 576-816 (-TAD) | [9,10] |
| ERK5-ΔC (1-503) | Deletion of amino acids 504-816 (-PR2) | [4] |
| ERK5-ΔC (1-408) | Deletion of amino acids 409-816 (-NLS) | [18] |
| ERK5-ΔN (411-816) | Deletion of amino acids 1-410 | |
| ERK5-T732A | Substitution of Thr732 to Ala | [8,11,12,13,17] |
| ERK5-T732E | Substitution of Thr732 to Glu | [8,11,12,13,17] |
| ERK5-4xAi | Substitution of Ser706, Thr732, Ser753, Ser773 to Ala | [11] |
| ERK5-4xAii | Substitution of Thr732, Ser769, Ser773, Ser775 to Ala | [8] |
| ERK5-4xE | Substitution of Ser706, Thr732, Ser753, Ser773 to Glu | [11] |
| ERK5-4xEii | Substitution of Thr732, Ser769, Ser773, Ser775 to Glu | [8] |
| ERK5-3xEi-T732A | Substitution of Ser706, Ser753, Ser774 to Glu and Thr732 to Ala | [11] |
| ERK5-3xEii-T732A | Substitution of Ser769, Ser773, Ser775 to Glu and Thr732 to Ala | [8] |
| ERK5-D200A-T732E | Substitution of Asp200 to Ala and Thr732 to Glu | [9] |
| ERK5-D200A-T732A | Substitution of Asp200 to Ala and Thr732 to Ala | [9] |
| MEK5DA | Substitution of Ser311 and Thr315 to Asp | [27] |
| ERK5 mutant | Sequence |
|---|---|
| ERK5-FL | GAGAGGATCCATGGCCGAGCCTCTG GAGAGCGGCCGCTCAGGGGTCCTGGAG |
| ERK5- ΔC | GAGAGGATCCATGGCCGAGCCTCTG GAGAGCGGCCGCGGCCATTCGAGTCCA |
| ERK5-ΔC (1-503) | GAGAGGATCCATGGCCGAGCCTCTG GAGAGCGGCCGCAGCCACAGGCTG |
| ERK5-ΔC (1-408) | GAGAGGATCCATGGCCGAGCCTCTG GAGAGCGGCCGCAGGCTCAGGAGC |
| ERK5-ΔN (411-816) | GAGAGGATCCGGCTGTCCAGAT GAGAGCGGCCGCTCAGGGGTCCTGGAG |
| ERK5-T732A | CACTGCCCTTTGGTGCGCCTGAGAACACAGG |
| ERK5-T732E | CCCCACTGCCCTTTGGCTCGCCTGAGAACACAGGG |
| ERK5-4xAi | CACTGCCCTTTGGTGCGCCTGAGAACACAGG CAAGCAGGGAGGCTGCGAGAGAGGCTGAATC CCTGTGTTCTCAGGCGCACCAAAGGGCAGTG GATTCAGCCTCTCTCGCAGCCTCCCTGCTTG |
| ERK5-4xAii | CAGCAAGCAGGGCGGCTGCGAGAGAGGCTGA CAGGATGGCCAGGCAGATGCAGCCTCTCT CACTGCCCTTTGGTGCGCCTGAGAACACAGG |
| ERK5-4xE | GTCAGCAAGCAGGGAGGCCTCGAGAGAGGCTGAATCTGC ATCAGCCACGCCCATGTCGAACTCCTGGTTTAAGAATTCCTCCAG CCCTGTGTTCTCAGGCGAGCCAAAGGGCAGTGGGG CCCCACTGCCCTTTGGCTCGCCTGAGAACACAGGG |
| ERK5-4xEii | CCCCACTGCCCTTTGGCTCGCCTGAGAACACAGGG CCAGTCAGCAAGCAGCTCGGCCTCGAGAGAGGCC TCATCTGCCTGGCCATCCTGTGGCCC |
| ERK5-3xEi-T732A | CACTGCCCTTTGGTGCGCCTGAGAACACAGG GTCAGCAAGCAGGGAGGCCTCGAGAGAGGCTGAATCTGC ATCAGCCACGCCCATGTCGAACTCCTGGTTTAAGAATTCCTCCAG CCCTGTGTTCTCAGGCGAGCCAAAGGGCAGTGGGG |
| ERK5-3xEii-T732A | CACTGCCCTTTGGTGCGCCTGAGAACACAGG CCAGTCAGCAAGCAGCTCGGCCTCGAGAGAGGCC TCATCTGCCTGGCCATCCTGTGGCCC |
| ERK5-D200A-T732E | CACGAGCCATACCAAAGGCACCAATCTTGAGCTCA CCCCACTGCCCTTTGGCTCGCCTGAGAACACAGGG |
| ERK5-D200A-T732A | CACGAGCCATACCAAAGGCACCAATCTTGAGCTCA CACTGCCCTTTGGTGCGCCTGAGAACACAGG |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pearson, A.J.; Fullwood, P.; Toro Tapia, G.; Prise, I.; Smith, M.P.; Xu, Q.; Jordan, A.; Giurisato, E.; Whitmarsh, A.J.; Francavilla, C.; et al. Discovery of a Gatekeeper Residue in the C-Terminal Tail of the Extracellular Signal-Regulated Protein Kinase 5 (ERK5). Int. J. Mol. Sci. 2020, 21, 929. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21030929
Pearson AJ, Fullwood P, Toro Tapia G, Prise I, Smith MP, Xu Q, Jordan A, Giurisato E, Whitmarsh AJ, Francavilla C, et al. Discovery of a Gatekeeper Residue in the C-Terminal Tail of the Extracellular Signal-Regulated Protein Kinase 5 (ERK5). International Journal of Molecular Sciences. 2020; 21(3):929. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21030929
Chicago/Turabian StylePearson, Adam J., Paul Fullwood, Gabriela Toro Tapia, Ian Prise, Michael P. Smith, Qiuping Xu, Allan Jordan, Emanuele Giurisato, Alan J. Whitmarsh, Chiara Francavilla, and et al. 2020. "Discovery of a Gatekeeper Residue in the C-Terminal Tail of the Extracellular Signal-Regulated Protein Kinase 5 (ERK5)" International Journal of Molecular Sciences 21, no. 3: 929. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21030929