Augmenting Vacuolar H+-ATPase Function Prevents Cardiomyocytes from Lipid-Overload Induced Dysfunction

 ,
,  and
and 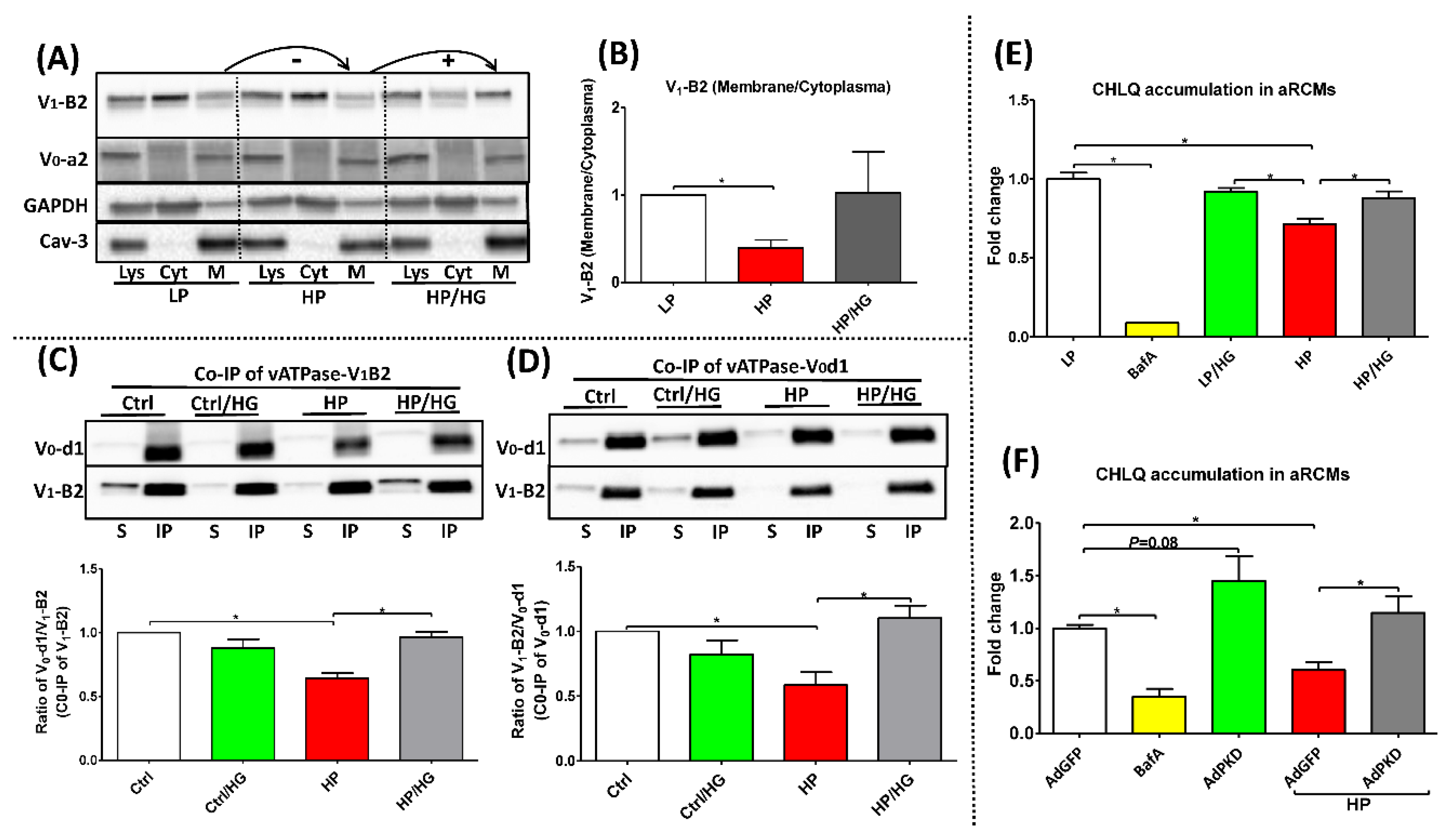{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
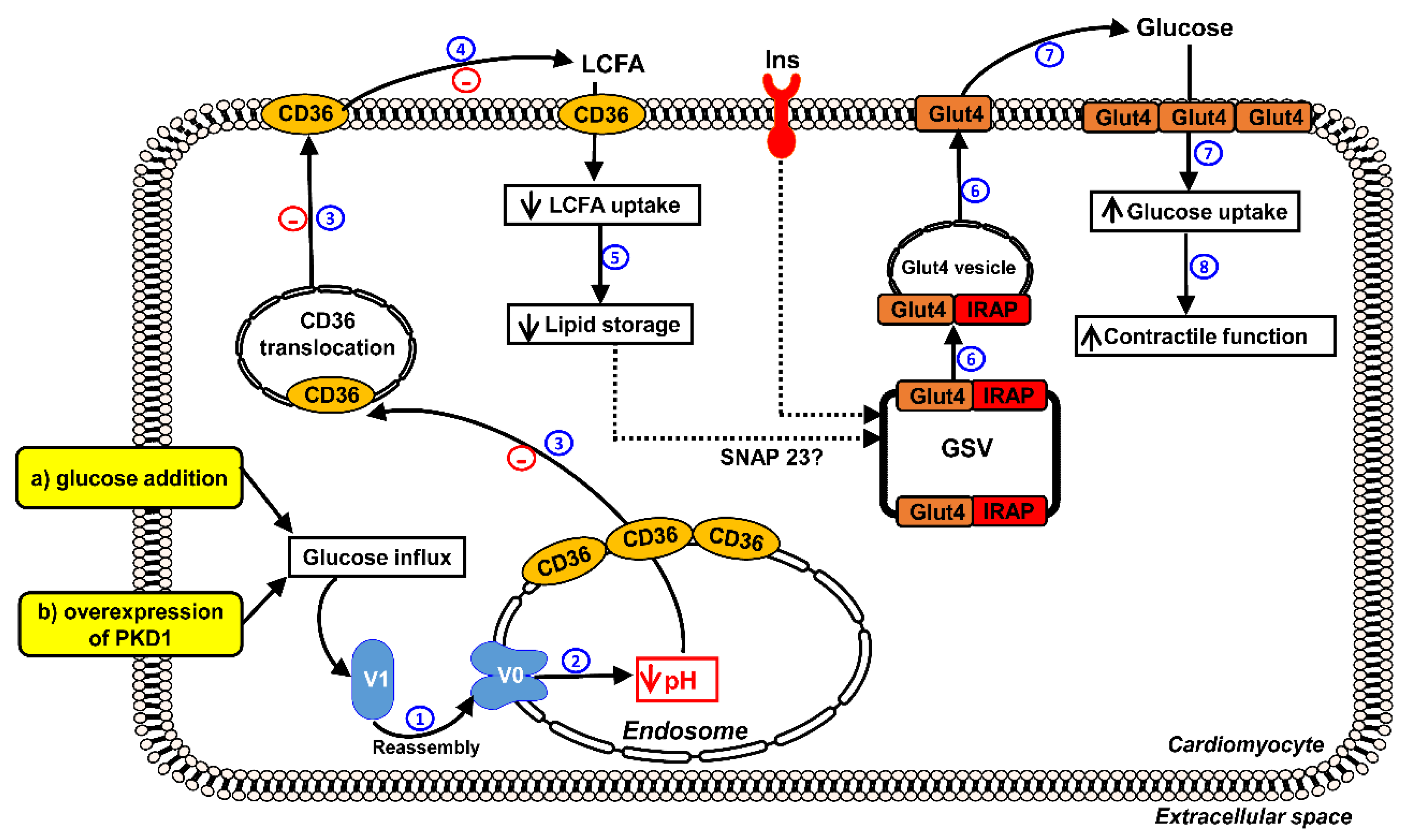
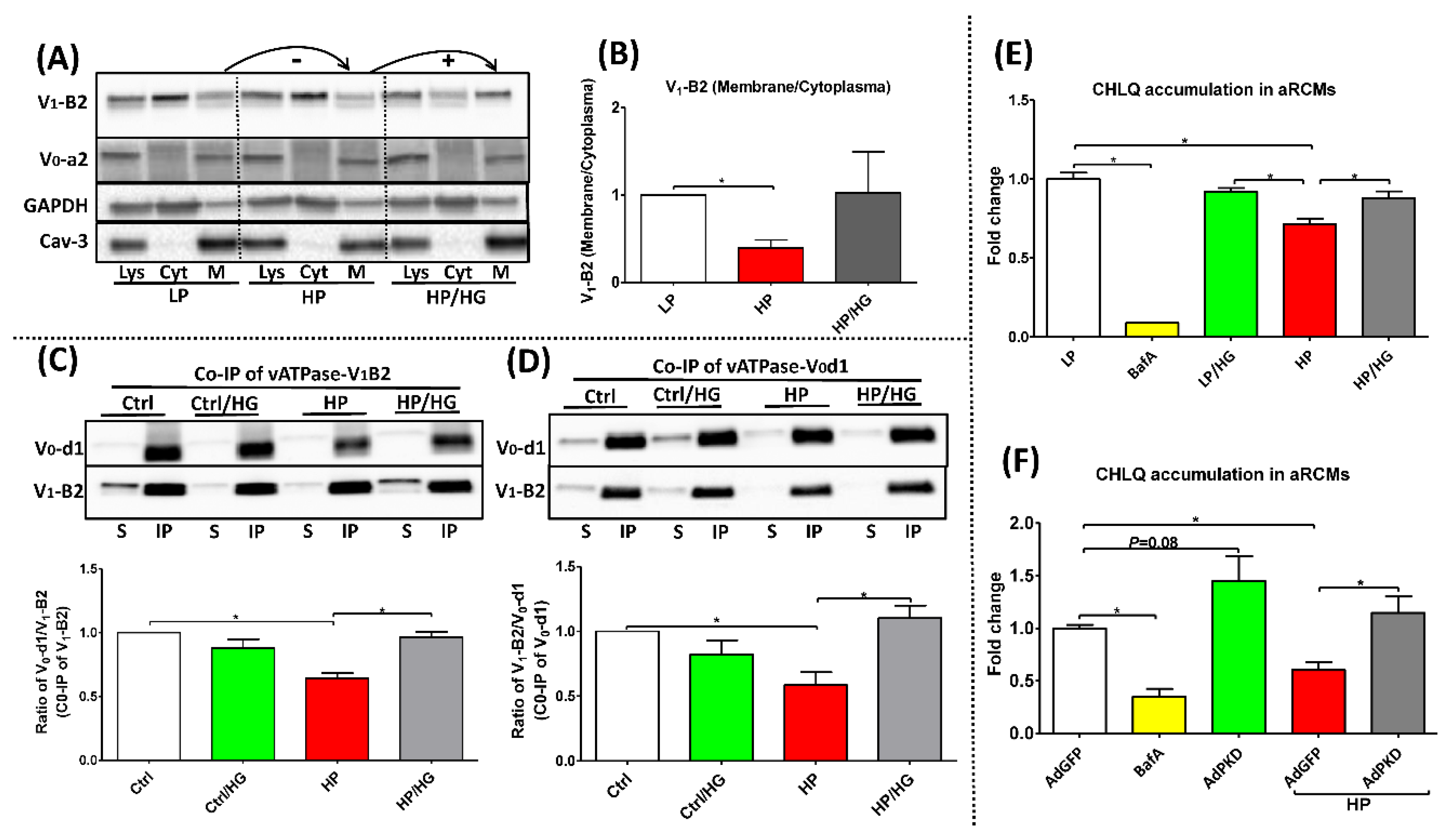
2.1. Forced Glucose Uptake Re-Assembles v-ATPase in Lipid-Overloaded Cardiomyocytes
2.2. Reassembly of V0/V1 Restores Endosomal Acidity in Lipid-overloaded Cardiomyocytes
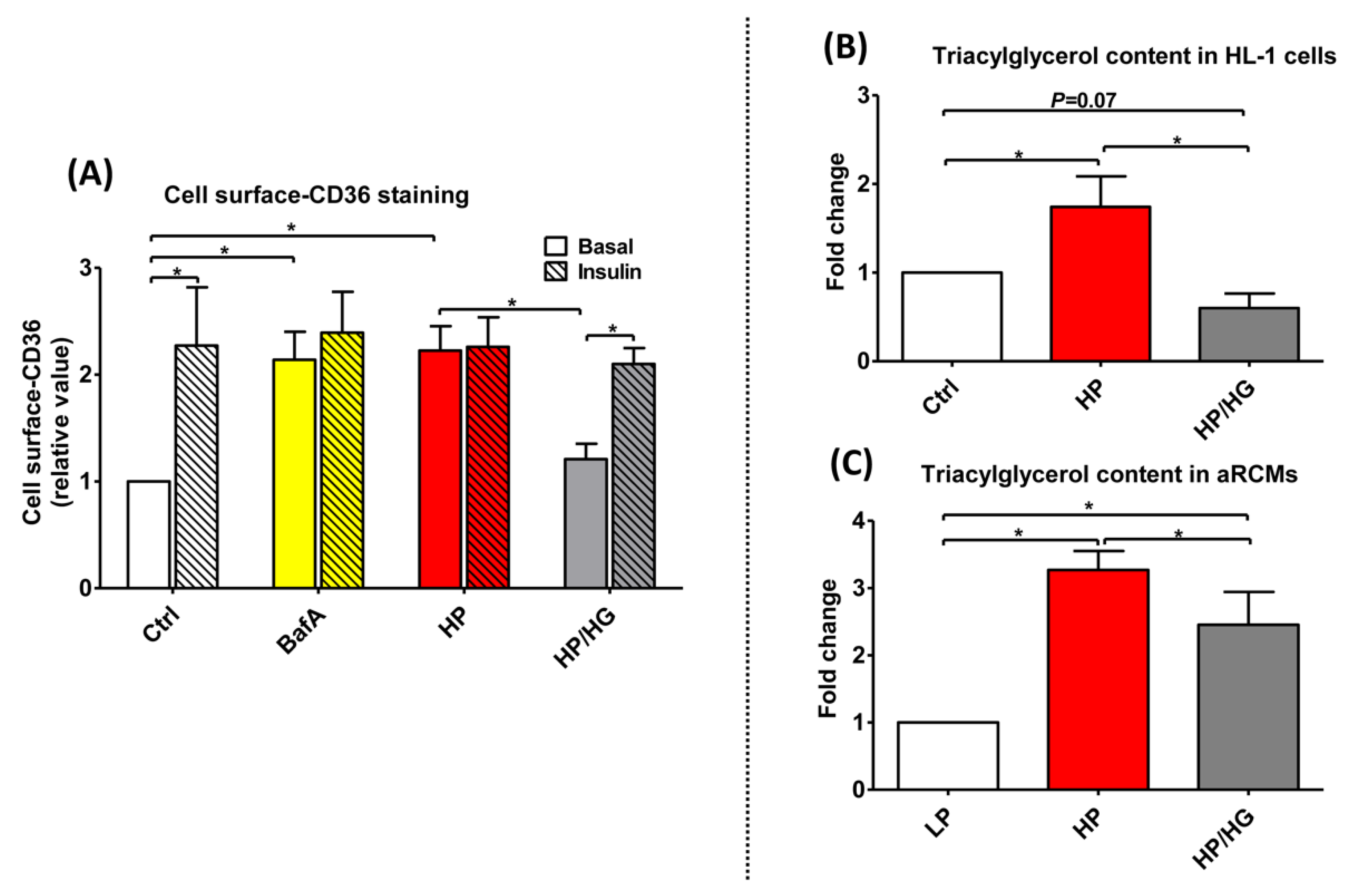
2.3. Increased Endosomal Acidification Induces Endosomal CD36 Retention and Decreases Lipid Accumulation in Lipid-overloaded Cardiomyocytes
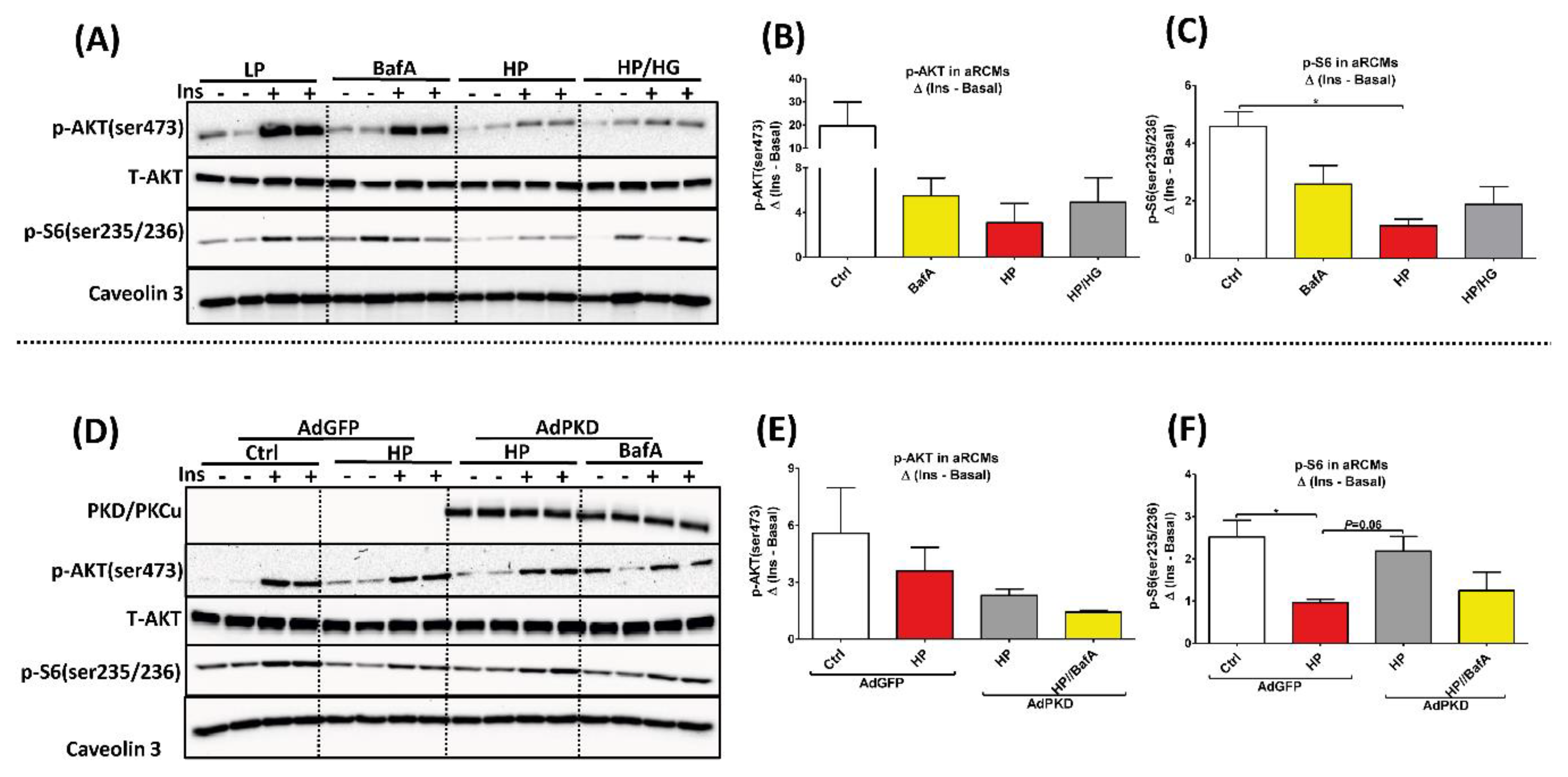
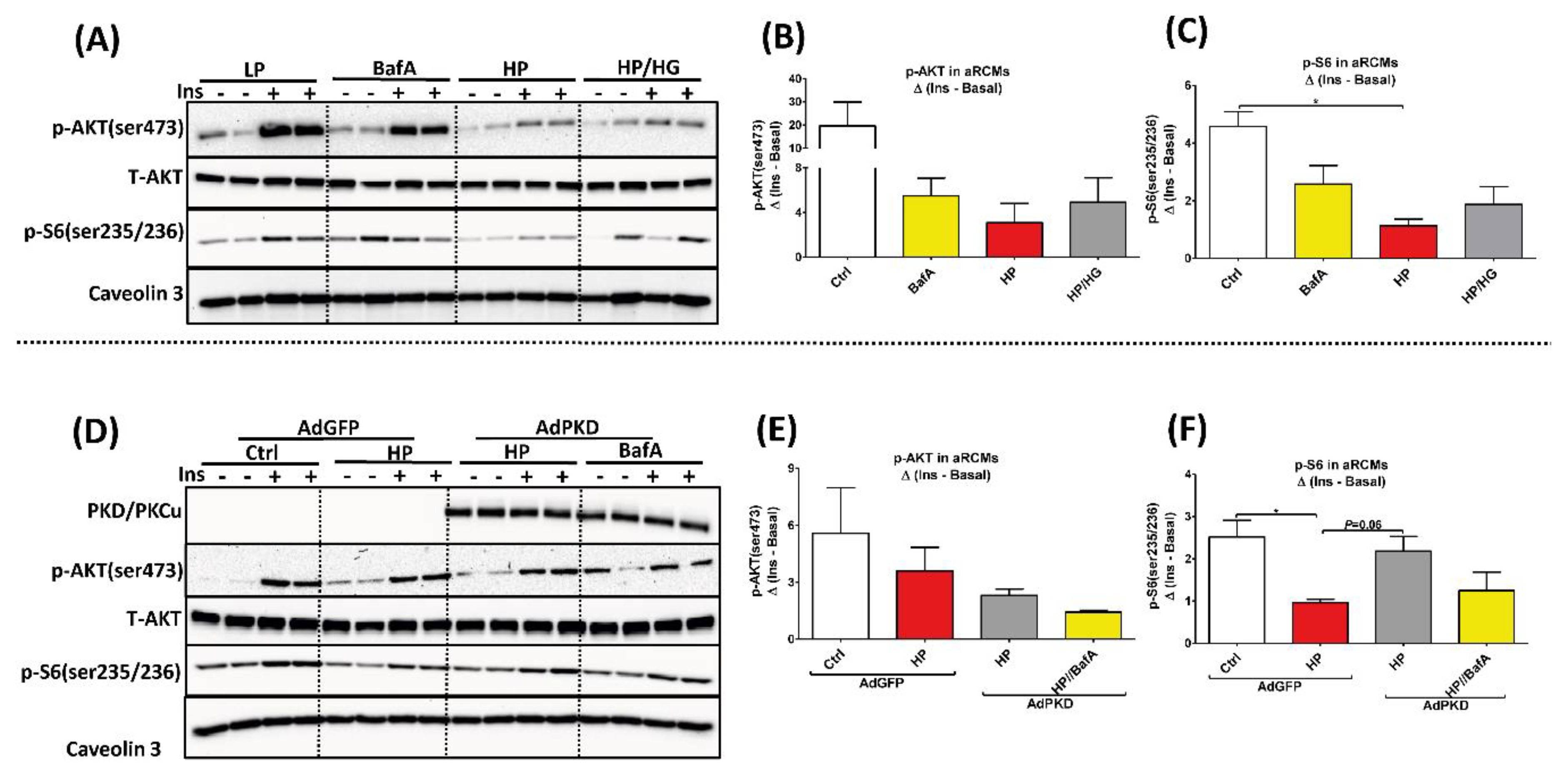
2.4. Reassembly of v-ATPase Does Not Promote Insulin Sensitivity in Lipid-overloaded Cardiomyocytes
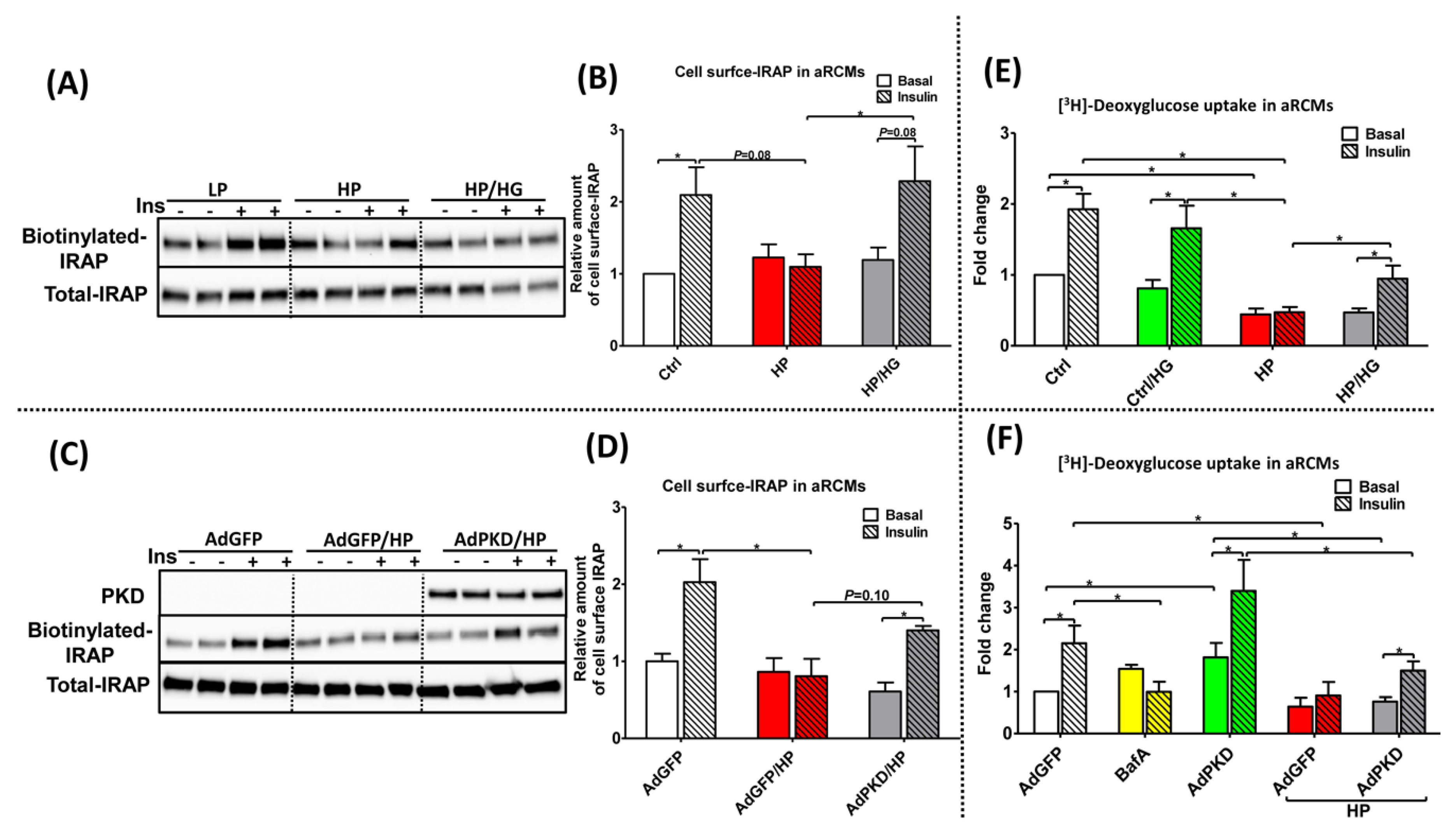
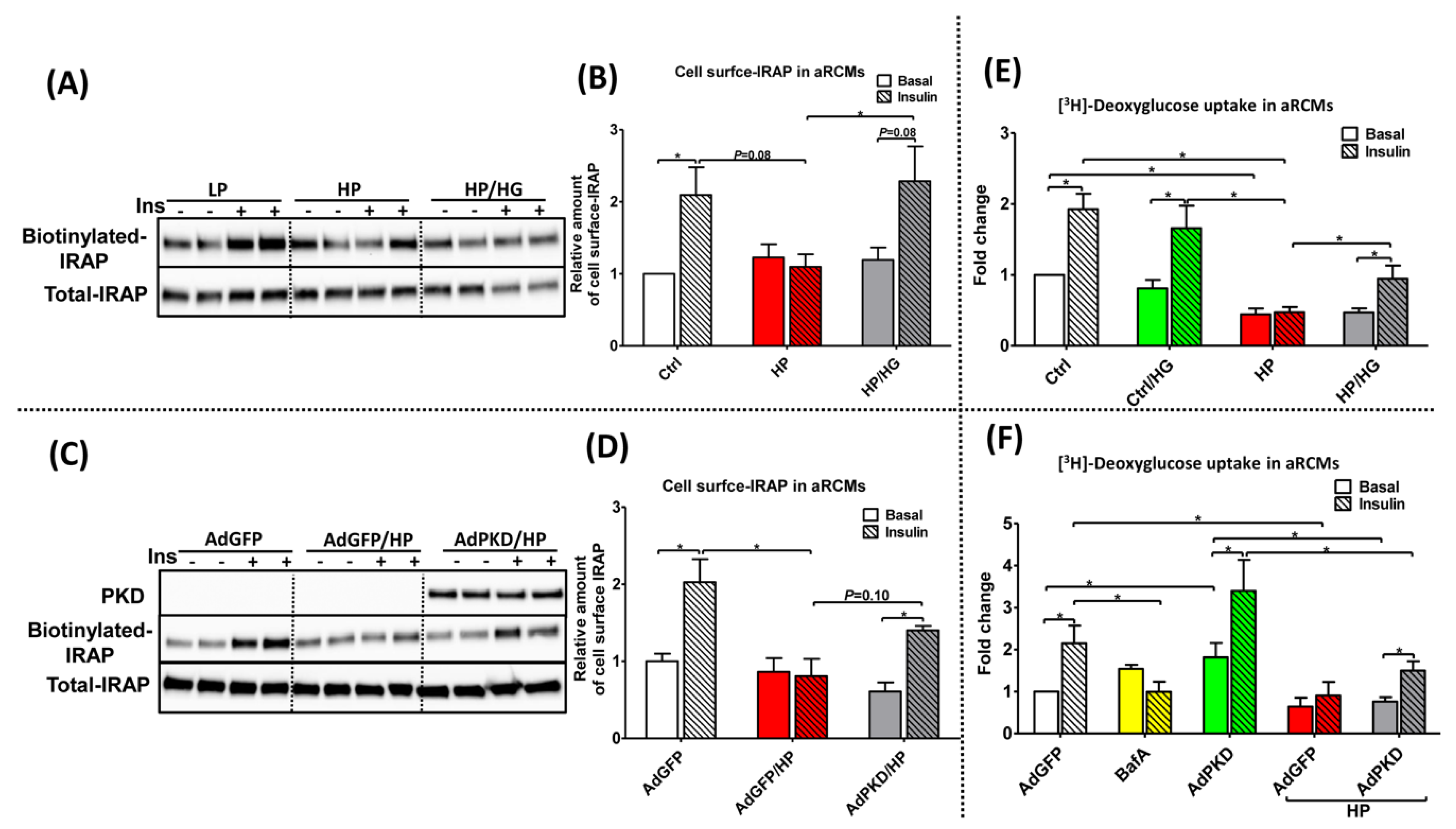
2.5. Reassembly of v-ATPase Improves Insulin Stimulated-GLUT4 Translocation and Glucose Uptake in Lipid-overloaded Cardiomyocytes
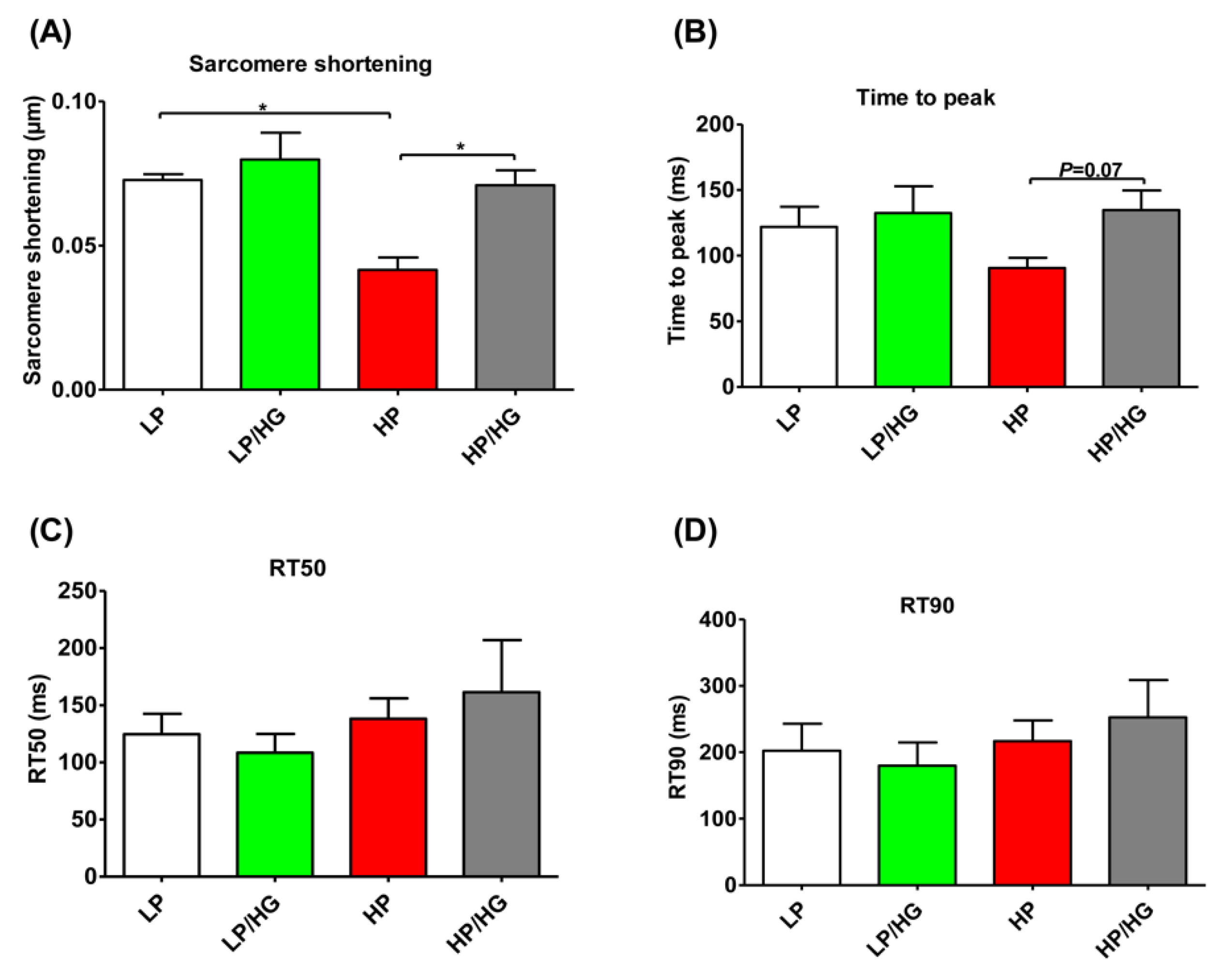
2.6. Reassembly of v-ATPase Restores Contractile Function in Lipid-overloaded Cardiomyocytes
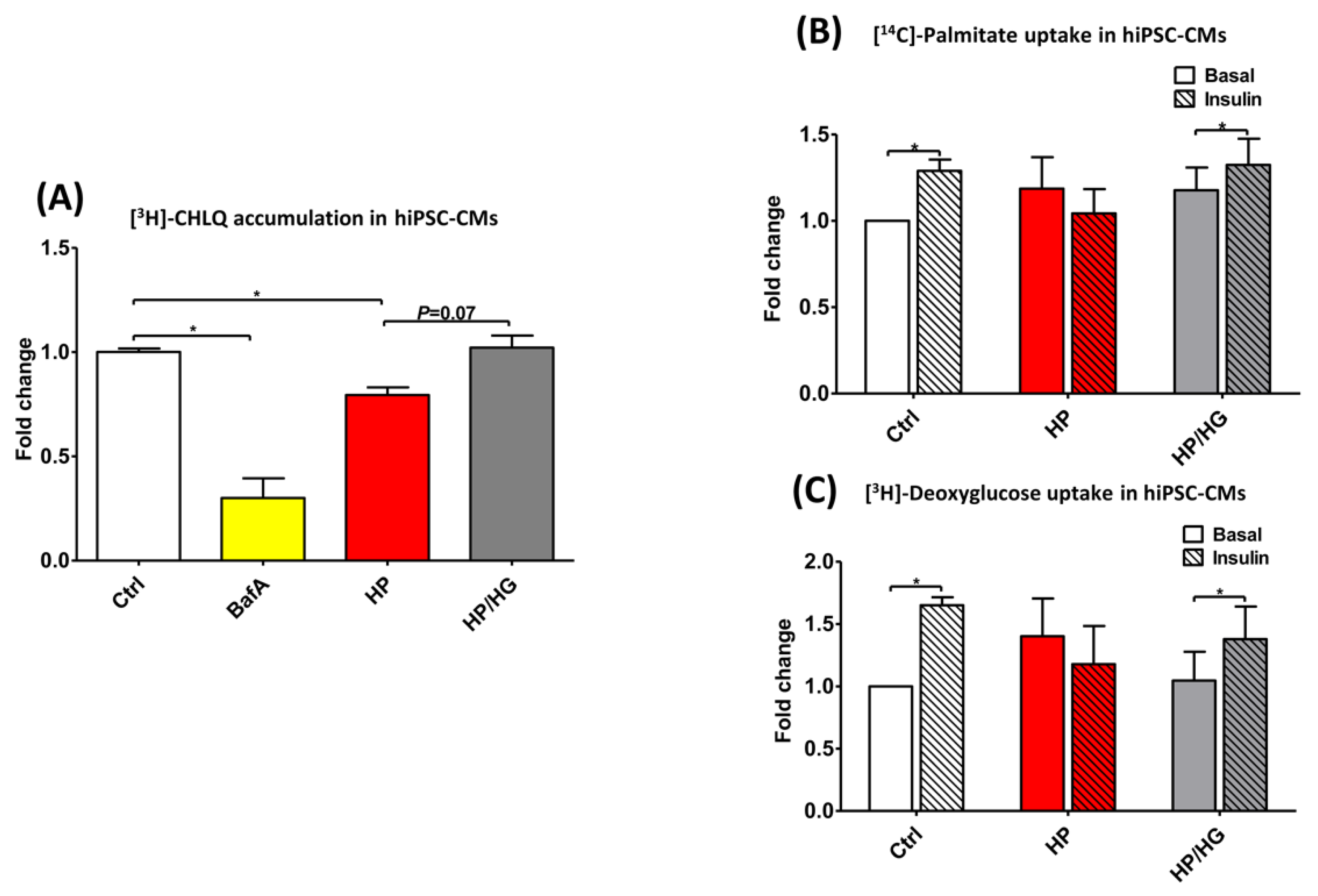
2.7. Restoration of v-ATPase Function Re-balances Energy Substrates in Human iPSC-Cardiomyocytes
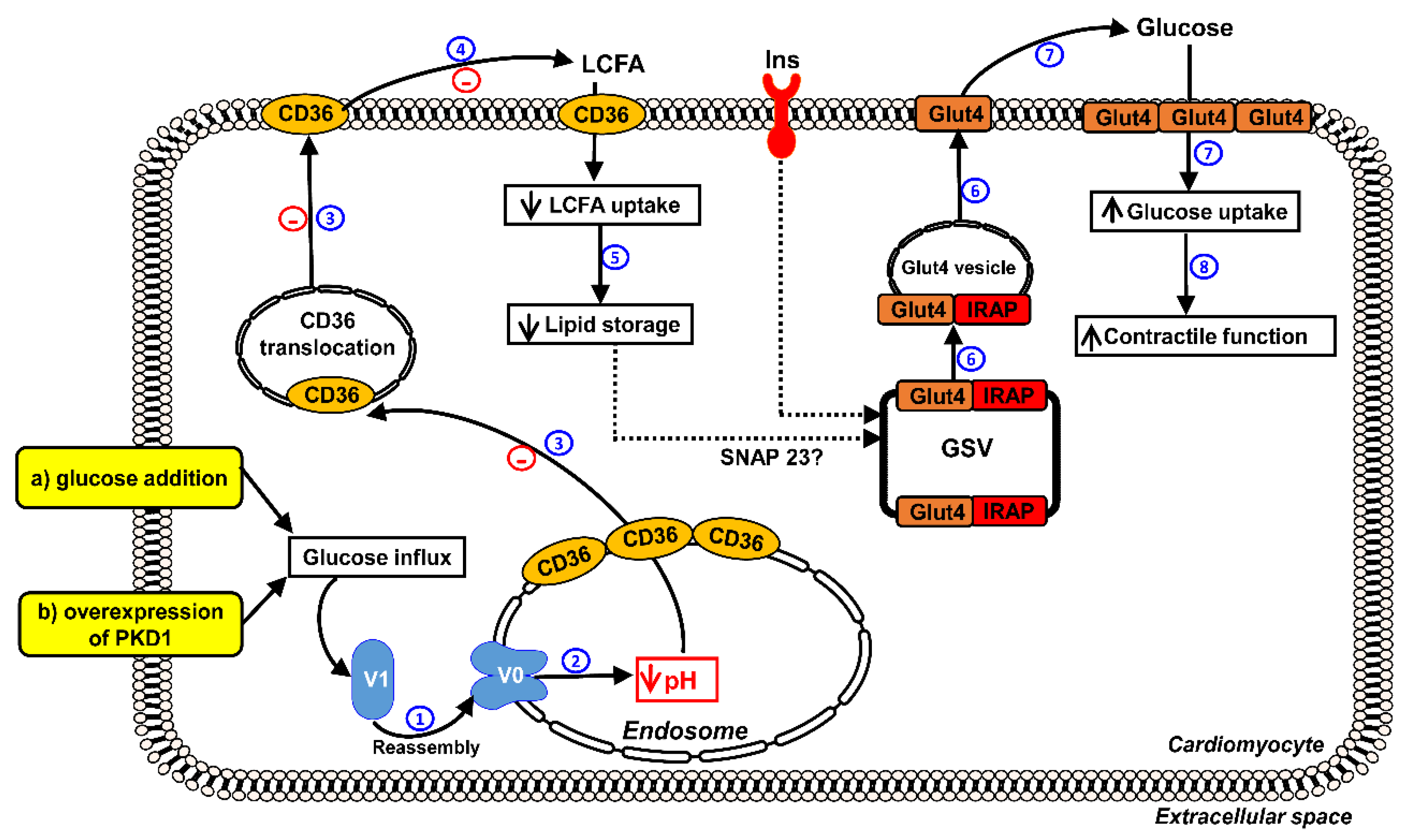
3. Discussion
4. Materials and Methods
4.1. Antibodies
4.2. Isolation and Culturing of Primary Rat Cardiomyocytes
4.3. Culturing of HL-1 Cardiomyocytes
4.4. Human Induced Pluripotent Stem Cell (hiPSC) Maintenance and Differentiation into Cardiomyocytes (hiPSC-CMs)
4.5. Adenovirus Amplification
4.6. Measurement of v-ATPase Disassembly/Assembly
4.7. Determination of Content of CD36 at Cell Surface
4.8. Quantification of Triacylglycerol
4.9. Determination of Insulin Sensitivity
4.10. Surface Biotinylation Assay
4.11. Measurement of Substrate Uptake
4.12. Measurement of Cardiomyocytic Contraction Dynamics
4.13. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| aRCMs | adult rat cardiomyocytes |
| hiPSC-CMs | human induced pluripotent stem cells differentiated into cardiomyocytes |
| LCFA | long chain fatty acid |
| v-ATPase | vacuolar H+-ATPase |
| V0-a2 | a2-subunit of the v-ATPase V0 super-complex |
| V0-d1 | d1-subunit of the v-ATPase V0 super-complex |
| V1-B2 | B2-subunit of the v-ATPase V1 super-complex |
| AdPKD | adenoviral overexpression of protein kinase-D1 |
| AdGFP | adenovirus containing Green Fluorescent Protein |
| IRAP | insulin-regulated aminopeptidase |
| IP | immunoprecipitation |
| CHLQ | chloroquine |
References
- Glatz, J.F.; Luiken, J.J.; Bonen, A. Membrane fatty acid transporters as regulators of lipid metabolism: implications for metabolic disease. Physiol. Rev. 2010, 90, 367–417. [Google Scholar] [CrossRef] [Green Version]
- Ormazabal, V.; Nair, S.; Elfeky, O.; Aguayo, C.; Salomon, C.; Zuñiga, F.A. Association between insulin resistance and the development of cardiovascular disease. Cardiovasc. Diabetol. 2018, 17, 122. [Google Scholar] [CrossRef] [PubMed]
- Glatz, J.F.; Luiken, J.J. Dynamic role of the transmembrane glycoprotein CD36 (SR-B2) in cellular fatty acid uptake and utilization. J. Lipid Res. 2018, 59, 1084–1093. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Angin, Y.; Steinbusch, L.K.; Simons, P.J.; Greulich, S.; Hoebers, N.T.; Douma, K.; van Zandvoort, M.A.; Coumans, W.A.; Wijnen, W.; Diamant, M.; et al. CD36 inhibition prevents lipid accumulation and contractile dysfunction in rat cardiomyocytes. Biochem. J. 2012, 448, 43–53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steinbusch, L.K.; Wijnen, W.; Schwenk, R.W.; Coumans, W.A.; Hoebers, N.T.; Ouwens, D.M.; Coumans, W.A.; Hoebers, N.T.; Diamant, M.; Bonen, A.; et al. Differential regulation of cardiac glucose and fatty acid uptake by endosomal pH and actin filaments. Am. J. Physiol. Cell Physiol. 2010, 298, C1549–C1559. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Steinbusch, L.K.M.; Nabben, M.; Kapsokalyvas, D.; van Zandvoort, M.; Schonleitner, P.; Antoons, G.; Simons, P.J.; Coumans, W.A.; Geomini, A.; et al. Palmitate-Induced Vacuolar-Type H(+)-ATPase Inhibition Feeds Forward Into Insulin Resistance and Contractile Dysfunction. Diabetes 2017, 66, 1521–1534. [Google Scholar] [CrossRef] [Green Version]
- Ma, B.; Xiang, Y.; An, L. Structural bases of physiological functions and roles of the vacuolar H(+)-ATPase. Cell. Signal. 2011, 23, 1244–1256. [Google Scholar] [CrossRef]
- Kane, P.M. Targeting reversible disassembly as a mechanism of controlling V-ATPase activity. Curr. Protein. Pept. Sci. 2012, 13, 117–123. [Google Scholar]
- Kane, P.M. The where, when, and how of organelle acidification by the yeast vacuolar H+-ATPase. Microbiol. Mol. Biol. Rev. 2006, 70, 177–191. [Google Scholar] [CrossRef] [Green Version]
- Sautin, Y.Y.; Lu, M.; Gaugler, A.; Zhang, L.; Gluck, S.L. Phosphatidylinositol 3-kinase-mediated effects of glucose on vacuolar H+-ATPase assembly, translocation, and acidification of intracellular compartments in renal epithelial cells. Mol. Cell. Biol. 2005, 25, 575–589. [Google Scholar] [CrossRef] [Green Version]
- Lu, M.; Ammar, D.; Ives, H.; Albrecht, F.; Gluck, S.L. Physical interaction between aldolase and vacuolar H+-ATPase is essential for the assembly and activity of the proton pump. J. Biol. Chem. 2007, 282, 24495–24503. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kane, P.M.; Yamashiro, C.T.; Stevens, T.H. Biochemical characterization of the yeast vacuolar H(+)-ATPase. J. Biol. Chem. 1989, 264, 19236–19244. [Google Scholar] [PubMed]
- Coort, S.L.; Bonen, A.; van der Vusse, G.J.; Glatz, J.F.; Luiken, J.J. Cardiac substrate uptake and metabolism in obesity and type-2 diabetes: role of sarcolemmal substrate transporters. Mol. Cell. Biochem. 2007, 299, 5–18. [Google Scholar] [CrossRef] [PubMed]
- Shao, D.; Tian, R. Glucose transporters in cardiac metabolism and hypertrophy. Compr. Physiol. 2011, 6, 331–351. [Google Scholar]
- Mayor, P.; Maianu, L.; Garvey, W.T. Glucose and insulin chronically regulate insulin action via different mechanisms in BC3H1 myocytes: Effects on glucose transporter gene expression. Diabetes 1992, 41, 274–285. [Google Scholar] [CrossRef]
- Rëgina, A.; Morchoisne, S.; Borson, N.D.; McCall, A.L.; Drewes, L.R.; Roux, F. Factor (s) released by glucose-deprived astrocytes enhance glucose transporter expression and activity in rat brain endothelial cells. Biochim. Biophys. Acta-Mol. Cell Res. 2001, 1540, 233–242. [Google Scholar] [CrossRef] [Green Version]
- Dirkx, E.; Schwenk, R.W.; Coumans, W.A.; Hoebers, N.; Angin, Y.; Viollet, B.; Bonen, A.; van Eys, G.J.; Glatz, J.F.; Luiken, J.J. Protein kinase D1 is essential for contraction-induced glucose uptake but is not involved in fatty acid uptake into cardiomyocytes. J. Biol. Chem. 2012, 287, 5871–5881. [Google Scholar] [CrossRef] [Green Version]
- Dirkx, E.; van Eys, G.J.; Schwenk, R.W.; Steinbusch, L.K.; Hoebers, N.; Coumans, W.A.; Peters, T.; Janssen, B.J.; Brans, B.; Vogg, A.T.; et al. Protein kinase-D1 overexpression prevents lipid-induced cardiac insulin resistance. J. Mol. Cell. Cardiol. 2014, 76, 208–217. [Google Scholar] [CrossRef]
- Kandror, K.V.; Pilch, P.F. gp160, a tissue-specific marker for insulin-activated glucose transport. Proc. Natl. Acad. Sci. USA 1994, 91, 8017–8021. [Google Scholar] [CrossRef] [Green Version]
- Rozengurt, E. Protein kinase D signaling: multiple biological functions in health and disease. Physiology 2011, 26, 23–33. [Google Scholar] [CrossRef] [Green Version]
- Boström, P.; Andersson, L.; Rutberg, M.; Perman, J.; Lidberg, U.; Johansson, B.R.; Fernandez-Rodriguez, J.; Ericson, J.; Nilsson, T.; Borén, J. SNARE proteins mediate fusion between cytosolic lipid droplets and are implicated in insulin sensitivity. Nat. Cell Biol. 2007, 9, 1286–1293. [Google Scholar] [CrossRef] [PubMed]
- Luiken, J.; Van Nieuwenhoven, F.; America, G.; Van der Vusse, G.; Glatz, J. Uptake and metabolism of palmitate by isolated cardiac myocytes from adult rats: involvement of sarcolemmal proteins. J. Lipid Res. 1997, 38, 745–758. [Google Scholar] [PubMed]
- Steinbusch, L.K.; Dirkx, E.; Hoebers, N.T.; Roelants, V.; Foretz, M.; Viollet, B.; Diamant, M.; van Eys, G.; Ouwens, D.M.; Bertrand, L.; et al. Overexpression of AMP-activated protein kinase or protein kinase D prevents lipid-induced insulin resistance in cardiomyocytes. J. Mol. Cell. Cardiol. 2013, 55, 165–173. [Google Scholar] [CrossRef] [PubMed]
- Schwenk, R.W.; Angin, Y.; Steinbusch, L.K.; Dirkx, E.; Hoebers, N.; Coumans, W.A.; Bonen, A.; Broers, J.L.; van Eys, G.J.; Glatz, J.F.; et al. Overexpression of vesicle-associated membrane protein (VAMP) 3, but not VAMP2, protects glucose transporter (GLUT) 4 protein translocation in an in vitro model of cardiac insulin resistance. J. Biol. Chem. 2012, 287, 37530–37539. [Google Scholar] [CrossRef] [Green Version]
- Angin, Y.; Schwenk, R.W.; Nergiz-Unal, R.; Hoebers, N.; Heemskerk, J.W.; Kuijpers, M.J.; Coumans, W.A.; van Zandvoort, M.A.; Bonen, A.; Neumann, D.; et al. Calcium signaling recruits substrate transporters GLUT4 and CD36 to the sarcolemma without increasing cardiac substrate uptake. Am. J. Physiol. Endocrinol. Metab. 2014, 307, E225–E236. [Google Scholar] [CrossRef] [Green Version]
- Luiken, J.J.; Koonen, D.P.; Willems, J.; Zorzano, A.; Becker, C.; Fischer, Y.; Tandon, N.N.; Van Der Vusse, G.J.; Bonen, A.; Glatz, J.F. Insulin stimulates long-chain fatty acid utilization by rat cardiac myocytes through cellular redistribution of FAT/CD36. Diabetes 2002, 51, 3113–3119. [Google Scholar] [CrossRef] [Green Version]
- Schwenk, R.W.; Dirkx, E.; Coumans, W.A.; Bonen, A.; Klip, A.; Glatz, J.F.; Luiken, J.J. Requirement for distinct vesicle-associated membrane proteins in insulin- and AMP-activated protein kinase (AMPK)-induced translocation of GLUT4 and CD36 in cultured cardiomyocytes. Diabetologia 2010, 53, 2209–2219. [Google Scholar] [CrossRef] [Green Version]



© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, S.; Wong, L.-Y.; Neumann, D.; Liu, Y.; Sun, A.; Antoons, G.; Strzelecka, A.; Glatz, J.F.C.; Nabben, M.; Luiken, J.J.F.P. Augmenting Vacuolar H+-ATPase Function Prevents Cardiomyocytes from Lipid-Overload Induced Dysfunction. Int. J. Mol. Sci. 2020, 21, 1520. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21041520
Wang S, Wong L-Y, Neumann D, Liu Y, Sun A, Antoons G, Strzelecka A, Glatz JFC, Nabben M, Luiken JJFP. Augmenting Vacuolar H+-ATPase Function Prevents Cardiomyocytes from Lipid-Overload Induced Dysfunction. International Journal of Molecular Sciences. 2020; 21(4):1520. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21041520
Chicago/Turabian StyleWang, Shujin, Li-Yen Wong, Dietbert Neumann, Yilin Liu, Aomin Sun, Gudrun Antoons, Agnieszka Strzelecka, Jan F.C. Glatz, Miranda Nabben, and Joost J.F.P. Luiken. 2020. "Augmenting Vacuolar H+-ATPase Function Prevents Cardiomyocytes from Lipid-Overload Induced Dysfunction" International Journal of Molecular Sciences 21, no. 4: 1520. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21041520