Molecular Mechanisms: Connections between Nonalcoholic Fatty Liver Disease, Steatohepatitis and Hepatocellular Carcinoma

 ,
,  , ,
, ,
Abstract
:1. Introduction
2. Definition of NASH
3. Pathophysiology of NASH
3.1. Obesity
3.2. Apoptosis
3.3. Immune and Inflammatory Pathways
3.4. Intestinal Bacteria
4. NASH and HCC
5. Genetic Factors in Patients with NAFLD, NASH and HCC
5.1. Patatin-Like Phospholipase-3 (PNPLA3)
5.2. Hydroxysteroid 17-Beta Dehydrogenase 13 (HSD17B13)
5.3. Other Genetic Variants
6. Molecular Pathways in Patients with NASH and HCC
7. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| NAFLD | Nonalcoholic fatty liver disease |
| NASH | Nonalcoholic steatohepatitis |
| HCC | Hepatocellular carcinoma |
| SIBO | Small intestinal bacterial overgrowth |
| ALD | Alcoholic liver disease |
| CK-18 | Cytokeratin-18 |
| ER | Endoplasmic reticulum |
| eNOS | Endothelial nitric oxide synthase |
| BNIP3 | Bcl-2-interacting protein-3 |
| LC3B | 1A/1B light chain 3B |
| TRAIL | Tumor necrosis factor-related apoptosis-inducing ligand |
| NK | Natural killer |
| ICAM-1 | Intercellular adhesion molecule 1 |
| sICAM-1 | Soluble ICAM-1 |
| Foxp3 | Forkhead box P3 |
| Treg | Regulatory T cell |
| NAS | NAFLD activity score |
| NLR | NOD-like receptor |
| NLR3 | NLR family pyrin domain-containing 3 |
| IL-1β | Interleukin 1-beta |
| MCD | Methionine and choline-deficient |
| ROS | Reactive oxygen species |
| JAM-A | Junctional adhesion molecule A |
| HFCD | High-fat and cholesterol-deficient |
| LBP | Lipopolysaccharide-binding protein |
| IgG | Immunoglobulin G |
| PNPLA3 | Patatin-like phospholipase-3 |
| JAZF1 | JAZF zinc-finger 1 |
| HSD17B13 | Hydroxysteroid 17-beta dehydrogenase 13 |
| RDH | Retinol dehydrogenase |
| FGF21 | Fibroblast growth factor 21 |
| AR | Androgen receptor |
| CCRK | Cell cycle-related kinase |
| HBV | Hepatitis B virus |
| HCV | Hepatitis C virus |
References
- Kleiner, D.E.; Brunt, E.M.; Wilson, L.A.; Behling, C.; Guy, C.; Contos, M.; Cummings, O.; Yeh, M.; Gill, R.; Chalasani, N.; et al. Nonalcoholic Steatohepatitis Clinical Research Network. Association of Histologic Disease Activity With Progression of Nonalcoholic Fatty Liver Disease. JAMA Netw. Open. 2019, 2, e1912565. [Google Scholar] [CrossRef] [Green Version]
- Angulo, P.; Keach, J.C.; Batts, K.P.; Lindor, K.D. Independent predictors of liver fibrosis in patients with nonalcoholic steatohepatitis. Hepatology 1999, 30, 1356–1362. [Google Scholar] [CrossRef] [PubMed]
- Shimada, M.; Hashimoto, E.; Taniai, M.; Hasegawa, K.; Okuda, H.; Hayashi, N.; Takasaki, K.; Ludwig, J. Hepatocellular carcinoma in patients with non-alcoholic steatohepatitis. J. Hepatol. 2002, 37, 154–160. [Google Scholar] [CrossRef]
- Kanda, T.; Yokosuka, O.; Suzuki, Y. Prolonged hepatitis caused by cytomegalovirus and non-alcoholic steatohepatitis in 16-year-old obese boy. Eur. J. Pediatr. 2005, 164, 212–215. [Google Scholar] [CrossRef] [PubMed]
- Brunt, E.M.; Janney, C.G.; Di Bisceglie, A.M.; Neuschwander-Tetri, B.A.; Bacon, B.R. Nonalcoholic steatohepatitis: A proposal for grading and staging the histological lesions. Am. J. Gastroenterol. 1999, 94, 2467–2474. [Google Scholar] [CrossRef]
- Ludwig, J.; McGill, D.B.; Lindor, K.D. Review: Nonalcoholic steatohepatitis. J. Gastroenterol. Hepatol. 1997, 12, 398–403. [Google Scholar] [CrossRef]
- Ratziu, V. Improving NASH with a little help from thyromimetics. Lancet 2019, 394, 1970–1972. [Google Scholar] [CrossRef]
- Ludwig, J.; Viggiano, T.R.; McGill, D.B.; Oh, B.J. Nonalcoholic steatohepatitis: Mayo Clinic experiences with a hitherto unnamed disease. Mayo Clin. Proc. 1980, 55, 434–438. [Google Scholar]
- Hashimoto, E.; Taniai, M.; Tokushige, K. Characteristics and diagnosis of NAFLD/NASH. J. Gastroenterol. Hepatol. 2013, 28 (Suppl. 4), 64–70. [Google Scholar] [CrossRef] [Green Version]
- Chalasani, N.; Younossi, Z.; Lavine, J.E.; Diehl, A.M.; Brunt, E.M.; Cusi, K.; Charlton, M.; Sanyal, A.J. The diagnosis and management of non-alcoholic fatty liver disease: Practice Guideline by the American Association for the Study of Liver Diseases, American College of Gastroenterology, and the American Gastroenterological Association. Hepatology 2012, 55, 2005–2023. [Google Scholar] [CrossRef]
- Brunner, K.T.; Henneberg, C.J.; Wilechansky, R.M.; Long, M.T. Nonalcoholic Fatty Liver Disease and Obesity Treatment. Curr. Obes. Rep. 2019, 8, 220–228. [Google Scholar] [CrossRef]
- Kanda, T.; Matsuoka, S.; Yamazaki, M.; Shibata, T.; Nirei, K.; Takahashi, H.; Kaneko, T.; Fujisawa, M.; Higuchi, T.; Nakamura, H.; et al. Apoptosis and non-alcoholic fatty liver diseases. World J. Gastroenterol. 2018, 24, 2661–2672. [Google Scholar] [CrossRef] [PubMed]
- Ganz, M.; Szabo, G. Immune and inflammatory pathways in NASH. Hepatol. Int. 2013, 7 (Suppl. 2), 771–781. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kitade, H.; Chen, G.; Ni, Y.; Ota, T. Nonalcoholic Fatty Liver Disease and Insulin Resistance: New Insights and Potential New Treatments. Nutrients 2017, 9, 387. [Google Scholar] [CrossRef] [Green Version]
- Miele, L.; Valenza, V.; La Torre, G.; Montalto, M.; Cammarota, G.; Ricci, R.; Mascianà, R.; Forgione, A.; Gabrieli, M.L.; Perotti, G.; et al. Increased intestinal permeability and tight junction alterations in nonalcoholic fatty liver disease. Hepatology 2009, 49, 1877–1887. [Google Scholar] [CrossRef]
- Henao-Mejia, J.; Elinav, E.; Jin, C.; Hao, L.; Mehal, W.Z.; Strowig, T.; Thaiss, C.A.; Kau, A.L.; Eisenbarth, S.C.; Jurczak, M.J.; et al. Inflammasome-mediated dysbiosis regulates progression of NAFLD and obesity. Nature 2012, 482, 179–185. [Google Scholar] [CrossRef] [Green Version]
- Le Roy, T.; Llopis, M.; Lepage, P.; Bruneau, A.; Rabot, S.; Bevilacqua, C.; Martin, P.; Philippe, C.; Walker, F.; Bado, A.; et al. Intestinal microbiota determines development of non-alcoholic fatty liver disease in mice. Gut 2013, 62, 1787–1794. [Google Scholar] [CrossRef]
- Younossi, Z.M.; Koenig, A.B.; Abdelatif, D.; Fazel, Y.; Henry, L.; Wymer, M. Global epidemiology of nonalcoholic fatty liver disease-Meta-analytic assessment of prevalence, incidence, and outcomes. Hepatology 2016, 64, 73–84. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Polyzos, S.A.; Kountouras, J.; Mantzoros, C.S. Obesity and nonalcoholic fatty liver disease: From pathophysiology to therapeutics. Metabolism 2019, 92, 82–97. [Google Scholar] [CrossRef]
- Adams, L.A.; Lymp, J.F.; St Sauver, J.; Sanderson, S.O.; Lindor, K.D.; Feldstein, A.; Angulo, P. The natural history of nonalcoholic fatty liver disease: A population-based cohort study. Gastroenterology 2005, 129, 113–121. [Google Scholar] [CrossRef]
- Jelenik, T.; Kaul, K.; Séquaris, G.; Flögel, U.; Phielix, E.; Kotzka, J.; Knebel, B.; Fahlbusch, P.; Hörbelt, T.; Lehr, S.; et al. Mechanisms of Insulin Resistance in Primary and Secondary Nonalcoholic Fatty Liver. Diabetes 2017, 66, 2241–2253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Finck, B.N. Targeting Metabolism, Insulin Resistance, and Diabetes to Treat Nonalcoholic Steatohepatitis. Diabetes 2018, 67, 2485–2493. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wieckowska, A.; Zein, N.N.; Yerian, L.M.; Lopez, A.R.; McCullough, A.J.; Feldstein, A.E. In vivo assessment of liver cell apoptosis as a novel biomarker of disease severity in nonalcoholic fatty liver disease. Hepatology 2006, 44, 27–33. [Google Scholar] [CrossRef]
- Kim, J.Y.; Garcia-Carbonell, R.; Yamachika, S.; Zhao, P.; Dhar, D.; Loomba, R.; Kaufman, R.J.; Saltiel, A.R.; Karin, M. ER Stress Drives Lipogenesis and Steatohepatitis via Caspase-2 Activation of S1P. Cell 2018, 175, 133–145.e15. [Google Scholar] [CrossRef] [Green Version]
- Sheldon, R.D.; Meers, G.M.; Morris, E.M.; Linden, M.A.; Cunningham, R.P.; Ibdah, J.A.; Thyfault, J.P.; Laughlin, M.H.; Rector, R.S. eNOS deletion impairs mitochondrial quality control and exacerbates Western diet-induced NASH. Am. J. Physiol. Endocrinol. Metab. 2019, 317, E605–E616. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Santos, L.; Luka, Z.; Wagner, C.; Fernandez-Alvarez, S.; Lu, S.C.; Mato, J.M.; Martinez-Chantar, M.L.; Beraza, N. Inhibition of natural killer cells protects the liver against acute injury in the absence of glycine N-methyltransferase. Hepatology 2012, 56, 747–759. [Google Scholar] [CrossRef] [Green Version]
- Söderberg, C.; Marmur, J.; Eckes, K.; Glaumann, H.; Sällberg, M.; Frelin, L.; Rosenberg, P.; Stål, P.; Hultcrantz, R. Microvesicular fat, inter cellular adhesion molecule-1 and regulatory T-lymphocytes are of importance for the inflammatory process in livers with non-alcoholic steatohepatitis. APMIS 2011, 119, 412–420. [Google Scholar] [CrossRef] [Green Version]
- Lumeng, C.N.; Saltiel, A.R. Inflammatory links between obesity and metabolic disease. J. Clin. Invest. 2011, 121, 2111–2117. [Google Scholar] [CrossRef] [Green Version]
- Reid, D.T.; Reyes, J.L.; McDonald, B.A.; Vo, T.; Reimer, R.A.; Eksteen, B. Kupffer Cells Undergo Fundamental Changes during the Development of Experimental NASH and Are Critical in Initiating Liver Damage and Inflammation. PLoS ONE 2016, 11, e0159524. [Google Scholar] [CrossRef] [Green Version]
- Higuchi, T.; Moriyama, M.; Fukushima, A.; Matsumura, H.; Matsuoka, S.; Kanda, T.; Sugitani, M.; Tsunemi, A.; Ueno, T.; Fukuda, N. Association of mRNA expression of iron metabolism-associated genes and progression of non-alcoholic steatohepatitis in rats. Oncotarget 2018, 9, 26183–26194. [Google Scholar] [CrossRef]
- Handa, P.; Morgan-Stevenson, V.; Maliken, B.D.; Nelson, J.E.; Washington, S.; Westerman, M.; Yeh, M.M.; Kowdley, K.V. Iron overload results in hepatic oxidative stress, immune cell activation, and hepatocellular ballooning injury, leading to nonalcoholic steatohepatitis in genetically obese mice. Am. J. Physiol. Gastrointest. Liver Physiol. 2016, 310, G117–G127. [Google Scholar] [CrossRef]
- Wan, X.; Xu, C.; Yu, C.; Li, Y. Role of NLRP3 Inflammasome in the Progression of NAFLD to NASH. Can. J. Gastroenterol. Hepatol. 2016, 2016, 6489012. [Google Scholar] [CrossRef] [Green Version]
- Du, X.; Wu, Z.; Xu, Y.; Liu, Y.; Liu, W.; Wang, T.; Li, C.; Zhang, C.; Yi, F.; Gao, L.; et al. Increased Tim-3 expression alleviates liver injury by regulating macrophage activation in MCD-induced NASH mice. Cell Mol. Immunol. 2019, 16, 878–886. [Google Scholar] [CrossRef]
- Sun, G.; Jin, H.; Zhang, C.; Meng, H.; Zhao, X.; Wei, D.; Ou, X.; Wang, Q.; Li, S.; Wang, T.; et al. OX40 Regulates Both Innate and Adaptive Immunity and Promotes Nonalcoholic Steatohepatitis. Cell Rep. 2018, 25, 3786–3799.e4. [Google Scholar] [CrossRef] [Green Version]
- Wigg, A.J.; Roberts-Thomson, I.C.; Dymock, R.B.; McCarthy, P.J.; Grose, R.H.; Cummins, A.G. The role of small intestinal bacterial overgrowth, intestinal permeability, endotoxaemia, and tumour necrosis factor alpha in the pathogenesis of non-alcoholic steatohepatitis. Gut 2001, 48, 206–211. [Google Scholar] [CrossRef] [Green Version]
- Nair, S.; Cope, K.; Risby, T.H.; Diehl, A.M. Obesity and female gender increase breath ethanol concentration: Potential implications for the pathogenesis of nonalcoholic steatohepatitis. Am. J. Gastroenterol. 2001, 96, 1200–1204. [Google Scholar] [CrossRef]
- Wu, W.C.; Zhao, W.; Li, S. Small intestinal bacteria overgrowth decreases small intestinal motility in the NASH rats. World J. Gastroenterol. 2008, 14, 313–317. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Wu, W.C.; He, C.Y.; Han, Z.; Jin, D.Y.; Wang, L. Change of intestinal mucosa barrier function in the progress of non-alcoholic steatohepatitis in rats. World J. Gastroenterol. 2008, 14, 3254–3258. [Google Scholar] [CrossRef] [PubMed]
- Tsujimoto, T.; Kawaratani, H.; Kitazawa, T.; Uemura, M.; Fukui, H. Innate immune reactivity of the ileum-liver axis in nonalcoholic steatohepatitis. Dig. Dis. Sci. 2012, 57, 1144–1151. [Google Scholar] [CrossRef] [PubMed]
- Wada, N.; Takaki, A.; Ikeda, F.; Yasunaka, T.; Onji, M.; Nouso, K.; Nakatsuka, A.; Wada, J.; Koike, K.; Miyahara, K.; et al. Serum-inducible protein (IP)-10 is a disease progression-related marker for non-alcoholic fatty liver disease. Hepatol. Int. 2017, 11, 115–124. [Google Scholar] [CrossRef]
- Shanab, A.A.; Scully, P.; Crosbie, O.; Buckley, M.; O’Mahony, L.; Shanahan, F.; Gazareen, S.; Murphy, E.; Quigley, E.M. Small intestinal bacterial overgrowth in nonalcoholic steatohepatitis: Association with toll-like receptor 4 expression and plasma levels of interleukin 8. Dig. Dis. Sci. 2011, 56, 1524–1534. [Google Scholar] [CrossRef] [PubMed]
- Rahman, K.; Desai, C.; Iyer, S.S.; Thorn, N.E.; Kumar, P.; Liu, Y.; Smith, T.; Neish, A.S.; Li, H.; Tan, S.; et al. Loss of Junctional Adhesion Molecule A Promotes Severe Steatohepatitis in Mice on a Diet High in Saturated Fat, Fructose, and Cholesterol. Gastroenterology 2016, 151, 733–746.e12. [Google Scholar] [CrossRef] [Green Version]
- Abdelmegeed, M.A.; Banerjee, A.; Jang, S.; Yoo, S.H.; Yun, J.W.; Gonzalez, F.J.; Keshavarzian, A.; Song, B.J. CYP2E1 potentiates binge alcohol-induced gut leakiness, steatohepatitis, and apoptosis. Free Radic. Biol. Med. 2013, 65, 1238–1245. [Google Scholar] [CrossRef]
- Elinav, E.; Strowig, T.; Kau, A.L.; Henao-Mejia, J.; Thaiss, C.A.; Booth, C.J.; Peaper, D.R.; Bertin, J.; Eisenbarth, S.C.; Gordon, J.I.; et al. NLRP6 inflammasome regulates colonic microbial ecology and risk for colitis. Cell 2011, 145, 745–757. [Google Scholar] [CrossRef] [Green Version]
- Kapil, S.; Duseja, A.; Sharma, B.K.; Singla, B.; Chakraborti, A.; Das, A.; Ray, P.; Dhiman, R.K.; Chawla, Y. Small intestinal bacterial overgrowth and toll-like receptor signaling in patients with non-alcoholic fatty liver disease. J. Gastroenterol. Hepatol. 2016, 31, 213–221. [Google Scholar] [CrossRef] [PubMed]
- Matsushita, N.; Osaka, T.; Haruta, I.; Ueshiba, H.; Yanagisawa, N.; Omori-Miyake, M.; Hashimoto, E.; Shibata, N.; Tokushige, K.; Saito, K.; et al. Effect of Lipopolysaccharide on the Progression of Non-Alcoholic Fatty Liver Disease in High Caloric Diet-Fed Mice. Scand. J. Immunol. 2016, 83, 109–118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duparc, T.; Plovier, H.; Marrachelli, V.G.; Van Hul, M.; Essaghir, A.; Ståhlman, M.; Matamoros, S.; Geurts, L.; Pardo-Tendero, M.M.; Druart, C.; et al. Hepatocyte MyD88 affects bile acids, gut microbiota and metabolome contributing to regulate glucose and lipid metabolism. Gut 2017, 66, 620–632. [Google Scholar] [CrossRef] [Green Version]
- Sohn, W.; Jun, D.W.; Lee, K.N.; Lee, H.L.; Lee, O.Y.; Choi, H.S.; Yoon, B.C. Lactobacillus paracasei Induces M2-Dominant Kupffer Cell Polarization in a Mouse Model of Nonalcoholic Steatohepatitis. Dig. Dis. Sci. 2015, 60, 3340–3350. [Google Scholar] [CrossRef]
- Mouzaki, M.; Comelli, E.M.; Arendt, B.M.; Bonengel, J.; Fung, S.K.; Fischer, S.E.; McGilvray, I.D.; Allard, J.P. Intestinal microbiota in patients with nonalcoholic fatty liver disease. Hepatology 2013, 58, 120–127. [Google Scholar] [CrossRef] [Green Version]
- Yuan, J.; Baker, S.S.; Liu, W.; Alkhouri, R.; Baker, R.D.; Xie, J.; Ji, G.; Zhu, L. Endotoxemia unrequired in the pathogenesis of pediatric nonalcoholic steatohepatitis. J. Gastroenterol. Hepatol. 2014, 29, 1292–1298. [Google Scholar] [CrossRef]
- Wong, V.W.; Wong, G.L.; Chan, H.Y.; Yeung, D.K.; Chan, R.S.; Chim, A.M.; Chan, C.K.; Tse, Y.K.; Woo, J.; Chu, W.C.; et al. Bacterial endotoxin and non-alcoholic fatty liver disease in the general population: A prospective cohort study. Aliment Pharmacol. Ther. 2015, 42, 731–740. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.Y.; Li, L.; Yu, C.H.; Shen, Z.; Chen, L.H.; Li, Y.M. Effects of probiotics on nonalcoholic fatty liver disease: A meta-analysis. World J. Gastroenterol. 2013, 19, 6911–6918. [Google Scholar] [CrossRef] [PubMed]
- Okubo, H.; Nakatsu, Y.; Sakoda, H.; Kushiyama, A.; Fujishiro, M.; Fukushima, T.; Matsunaga, Y.; Ohno, H.; Yoneda, M.; Kamata, H.; et al. Mosapride citrate improves nonalcoholic steatohepatitis with increased fecal lactic acid bacteria and plasma glucagon-like peptide-1 level in a rodent model. Am. J. Physiol. Gastrointest. Liver Physiol. 2015, 308, G151–G158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakano, M.; Murohisa, T.; Imai, Y.; Tamano, M.; Hiraishi, H. Association Between Appendectomy and Fibrosis Progression in Nonalcoholic Fatty Liver Disease. Gastroenterology Res. 2013, 6, 17–25. [Google Scholar] [CrossRef] [Green Version]
- Mouzaki, M.; Wang, A.Y.; Bandsma, R.; Comelli, E.M.; Arendt, B.M.; Zhang, L.; Fung, S.; Fischer, S.E.; McGilvray, I.G.; Allard, J.P. Bile Acids and Dysbiosis in Non-Alcoholic Fatty Liver Disease. PLoS ONE 2016, 11, e0151829. [Google Scholar] [CrossRef] [Green Version]
- Xie, G.; Wang, X.; Huang, F.; Zhao, A.; Chen, W.; Yan, J.; Zhang, Y.; Lei, S.; Ge, K.; Zheng, X.; et al. Dysregulated hepatic bile acids collaboratively promote liver carcinogenesis. Int. J. Cancer 2016, 139, 1764–1775. [Google Scholar] [CrossRef]
- Nadeem Aslam, M.; Basis, C.M.; Zhang, L.; Zaidi, S.; Varani, J.; Bergin, I.L. Calcium Reduces Liver Injury in Mice on a High-Fat Diet: Alterations in Microbial and Bile Acid Profiles. PLoS ONE 2016, 11, e0166178. [Google Scholar] [CrossRef]
- Pang, J.; Xu, W.; Zhang, X.; Wong, G.L.; Chan, A.W.; Chan, H.Y.; Tse, C.H.; Shu, S.S.; Choi, P.C.; Chan, H.L.; et al. Significant positive association of endotoxemia with histological severity in 237 patients with non-alcoholic fatty liver disease. Aliment Pharmacol. Ther. 2017, 46, 175–182. [Google Scholar] [CrossRef]
- Akiyama, K.; Warabi, E.; Okada, K.; Yanagawa, T.; Ishii, T.; Kose, K.; Tokushige, K.; Ishige, K.; Mizokami, Y.; Yamagata, K.; et al. Deletion of both p62 and Nrf2 spontaneously results in the development of nonalcoholic steatohepatitis. Exp. Anim. 2018, 67, 201–218. [Google Scholar] [CrossRef] [Green Version]
- Da Silva, H.E.; Teterina, A.; Comelli, E.M.; Taibi, A.; Arendt, B.M.; Fischer, S.E.; Lou, W.; Allard, J.P. Nonalcoholic fatty liver disease is associated with dysbiosis independent of body mass index and insulin resistance. Sci. Rep. 2018, 8, 1466. [Google Scholar] [CrossRef] [Green Version]
- Bluemel, S.; Wang, L.; Martino, C.; Lee, S.; Wang, Y.; Williams, B.; Horvath, A.; Stadlbauer, V.; Zengler, K.; Schnabl, B. The Role of Intestinal C-type Regenerating Islet Derived-3 Lectins for Nonalcoholic Steatohepatitis. Hepatol. Commun. 2018, 2, 393–406. [Google Scholar] [CrossRef]
- Kazankov, K.; Jørgensen, S.M.D.; Thomsen, K.L.; Møller, H.J.; Vilstrup, H.; George, J.; Schuppan, D.; Grønbæk, H. The role of macrophages in nonalcoholic fatty liver disease and nonalcoholic steatohepatitis. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 145–159. [Google Scholar] [CrossRef] [PubMed]
- Mohs, A.; Kuttkat, N.; Otto, T.; Youssef, S.A.; De Bruin, A.; Trautwein, C. MyD88-dependent signaling in non-parenchymal cells promotes liver carcinogenesis. Carcinogenesis 2019. [Google Scholar] [CrossRef] [PubMed]
- Schwimmer, J.B.; Johnson, J.S.; Angeles, J.E.; Behling, C.; Belt, P.H.; Borecki, I.; Bross, C.; Durelle, J.; Goyal, N.P.; Hamilton, G.; et al. Microbiome Signatures Associated With Steatohepatitis and Moderate to Severe Fibrosis in Children With Nonalcoholic Fatty Liver Disease. Gastroenterology 2019, 157, 1109–1122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mouries, J.; Brescia, P.; Silvestri, A.; Spadoni, I.; Sorribas, M.; Wiest, R.; Mileti, E.; Galbiati, M.; Invernizzi, P.; Adorini, L.; et al. Microbiota-driven gut vascular barrier disruption is a prerequisite for non-alcoholic steatohepatitis development. J. Hepatol. 2019, 71, 1216–1228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kishida, Y.; Okubo, H.; Ohno, H.; Oki, K.; Yoneda, M. Effect of miglitol on the suppression of nonalcoholic steatohepatitis development and improvement of the gut environment in a rodent model. J. Gastroenterol. 2017, 52, 1180–1191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, D.; Pan, Q.; Shen, F.; Cao, H.X.; Ding, W.J.; Chen, Y.W.; Fan, J.G. Total fecal microbiota transplantation alleviates high-fat diet-induced steatohepatitis in mice via beneficial regulation of gut microbiota. Sci. Rep. 2017, 7, 1529. [Google Scholar] [CrossRef] [Green Version]
- García-Lezana, T.; Raurell, I.; Bravo, M.; Torres-Arauz, M.; Salcedo, M.T.; Santiago, A.; Schoenenberger, A.; Manichanh, C.; Genescà, J.; Martell, M.; et al. Restoration of a healthy intestinal microbiota normalizes portal hypertension in a rat model of nonalcoholic steatohepatitis. Hepatology 2018, 67, 1485–1498. [Google Scholar] [CrossRef]
- Zhao, Z.H.; Xin, F.Z.; Xue, Y.; Hu, Z.; Han, Y.; Ma, F.; Zhou, D.; Liu, X.L.; Cui, A.; Liu, Z.; et al. Indole-3-propionic acid inhibits gut dysbiosis and endotoxin leakage to attenuate steatohepatitis in rats. Exp. Mol. Med. 2019, 51, 103. [Google Scholar] [CrossRef] [Green Version]
- Al-Azzawi, Y.; Rouanet, E.; Hendrix, R.J.; Spaho, L.; Malik, H.; Devuni, D.; Szabo, G.; Barnard, G. Segmental Distribution of Hepatocellular Carcinoma Correlates with Microvascular Invasion in Liver Explants Undergoing Transplantation. J. Cancer Epidemiol. 2019, 2019, 8534372. [Google Scholar] [CrossRef] [Green Version]
- Chalasani, N.; Younossi, Z.; Lavine, J.E.; Charlton, M.; Cusi, K.; Rinella, M.; Harrison, S.A.; Brunt, E.M.; Sanyal, A.J. The diagnosis and management of nonalcoholic fatty liver disease: Practice guidance from the American Association for the Study of Liver Diseases. Hepatology 2018, 67, 328–357. [Google Scholar] [CrossRef]
- Younossi, Z.M.; Otgonsuren, M.; Henry, L.; Venkatesan, C.; Mishra, A.; Erario, M.; Hunt, S. Association of nonalcoholic fatty liver disease (NAFLD) with hepatocellular carcinoma (HCC) in the United States from 2004 to 2009. Hepatology 2015, 62, 1723–1730. [Google Scholar] [CrossRef]
- Mittal, S.; El-Serag, H.B.; Sada, Y.H.; Kanwal, F.; Duan, Z.; Temple, S.; May, S.B.; Kramer, J.R.; Richardson, P.A.; Davila, J.A. Hepatocellular Carcinoma in the Absence of Cirrhosis in United States Veterans is Associated With Nonalcoholic Fatty Liver Disease. Clin. Gastroenterol. Hepatol. 2016, 14, 124–131.e1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Enomoto, H.; Ueno, Y.; Hiasa, Y.; Nishikawa, H.; Hige, S.; Takikawa, Y.; Taniai, M.; Ishikawa, T.; Yasui, K.; Takaki, A.; et al. Japan Etiology of Liver Cirrhosis Study Group in the 54th Annual Meeting of JSH. Transition in the etiology of liver cirrhosis in Japan: A nationwide survey. J. Gastroenterol. 2019. [Google Scholar] [CrossRef] [Green Version]
- Kudo, M.; Izumi, N.; Kubo, S.; Kokudo, N.; Sakamoto, M.; Shiina, S.; Tateishi, R.; Nakashima, O.; Murakami, T.; Matsuyama, Y.; et al. Report of the 20th Nationwide Follow-up Survey of Primary Liver Cancer in Japan. Hepatol. Res. 2019. [Google Scholar] [CrossRef] [Green Version]
- Yatsuji, S.; Hashimoto, E.; Tobari, M.; Taniai, M.; Tokushige, K.; Shiratori, K. Clinical features and outcomes of cirrhosis due to non-alcoholic steatohepatitis compared with cirrhosis caused by chronic hepatitis C. J. Gastroenterol. Hepatol. 2009, 24, 248–254. [Google Scholar] [CrossRef]
- Lee, S.S.; Jeong, S.H.; Byoun, Y.S.; Chung, S.M.; Seong, M.H.; Sohn, H.R.; Min, B.Y.; Jang, E.S.; Kim, J.W.; Park, G.J.; et al. Clinical features and outcome of cryptogenic hepatocellular carcinoma compared to those of viral and alcoholic hepatocellular carcinoma. BMC Cancer 2013, 13, 335. [Google Scholar] [CrossRef] [Green Version]
- Wong, V.W.; Chan, W.K.; Chitturi, S.; Chawla, Y.; Dan, Y.Y.; Duseja, A.; Fan, J.; Goh, K.L.; Hamaguchi, M.; Hashimoto, E.; et al. Asia-Pacific Working Party on Non-alcoholic Fatty Liver Disease guidelines 2017-Part 1: Definition, risk factors and assessment. J. Gastroenterol. Hepatol. 2018, 33, 70–85. [Google Scholar] [CrossRef] [PubMed]
- Dyson, J.; Jaques, B.; Chattopadyhay, D.; Lochan, R.; Graham, J.; Das, D.; Aslam, T.; Patanwala, I.; Gaggar, S.; Cole, M.; et al. Hepatocellular cancer: The impact of obesity, type 2 diabetes and a multidisciplinary team. J. Hepatol. 2014, 60, 110–117. [Google Scholar] [CrossRef]
- European Association for the Study of the Liver (EASL); European Association for the Study of Diabetes (EASD); European Association for the Study of Obesity (EASO). EASL-EASD-EASO Clinical Practice Guidelines for the management of non-alcoholic fatty liver disease. J. Hepatol. 2016, 64, 1388–1402. [Google Scholar] [CrossRef] [PubMed]
- Yasui, K.; Hashimoto, E.; Komorizono, Y.; Koike, K.; Arii, S.; Imai, Y.; Shima, T.; Kanbara, Y.; Saibara, T.; Mori, T.; et al. Japan NASH Study Group, Ministry of Health, Labour, and Welfare of Japan. Characteristics of patients with nonalcoholic steatohepatitis who develop hepatocellular carcinoma. Clin. Gastroenterol. Hepatol. 2011, 9, 428–433, quiz e50. [Google Scholar] [CrossRef] [PubMed]
- Caldwell, S.H.; Harris, D.M.; Patrie, J.T.; Hespenheide, E.E. Is NASH underdiagnosed among African Americans? Am. J. Gastroenterol. 2002, 97, 1496–1500. [Google Scholar] [CrossRef] [PubMed]
- Browning, J.D.; Kumar, K.S.; Saboorian, M.H.; Thiele, D.L. Ethnic differences in the prevalence of cryptogenic cirrhosis. Am. J. Gastroenterol. 2004, 99, 292–298. [Google Scholar] [CrossRef] [PubMed]
- Browning, J.D.; Horton, J.D. Molecular mediators of hepatic steatosis and liver injury. J. Clin. Invest. 2004, 114, 147–152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Romeo, S.; Kozlitina, J.; Xing, C.; Pertsemlidis, A.; Cox, D.; Pennacchio, L.A.; Boerwinkle, E.; Cohen, J.C.; Hobbs, H.H. Genetic variation in PNPLA3 confers susceptibility to nonalcoholic fatty liver disease. Nat. Genet. 2008, 40, 1461–1465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilson, P.A.; Gardner, S.D.; Lambie, N.M.; Commans, S.A.; Crowther, D.J. Characterization of the human patatin-like phospholipase family. J. Lipid. Res. 2006, 47, 1940–1949. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuan, X.; Waterworth, D.; Perry, J.R.; Lim, N.; Song, K.; Chambers, J.C.; Zhang, W.; Vollenweider, P.; Stirnadel, H.; Johnson, T.; et al. Population-based genome-wide association studies reveal six loci influencing plasma levels of liver enzymes. Am. J. Hum. Genet. 2008, 83, 520–528. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Romeo, S.; Sentinelli, F.; Dash, S.; Yeo, G.S.; Savage, D.B.; Leonetti, F.; Capoccia, D.; Incani, M.; Maglio, C.; Iacovino, M.; et al. Morbid obesity exposes the association between PNPLA3 I148M (rs738409) and indices of hepatic injury in individuals of European descent. Int. J. Obes (Lond.) 2010, 34, 190–194. [Google Scholar] [CrossRef] [Green Version]
- Kawaguchi, T.; Sumida, Y.; Umemura, A.; Matsuo, K.; Takahashi, M.; Takamura, T.; Yasui, K.; Saibara, T.; Hashimoto, E.; Kawanaka, M.; et al. Japan Study Group of Nonalcoholic Fatty Liver Disease. Genetic polymorphisms of the human PNPLA3 gene are strongly associated with severity of non-alcoholic fatty liver disease in Japanese. PLoS ONE 2012, 7, e38322. [Google Scholar] [CrossRef] [Green Version]
- Matteoni, C.A.; Younossi, Z.M.; Gramlich, T.; Boparai, N.; Liu, Y.C.; McCullough, A.J. Nonalcoholic fatty liver disease: A spectrum of clinical and pathological severity. Gastroenterology 1999, 116, 1413–1419. [Google Scholar] [CrossRef]
- Singal, A.G.; Manjunath, H.; Yopp, A.C.; Beg, M.S.; Marrero, J.A.; Gopal, P.; Waljee, A.K. The effect of PNPLA3 on fibrosis progression and development of hepatocellular carcinoma: A meta-analysis. Am. J. Gastroenterol. 2014, 109, 325–334. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.L.; Patman, G.L.; Leathart, J.B.; Piguet, A.C.; Burt, A.D.; Dufour, J.F.; Day, C.P.; Daly, A.K.; Reeves, H.L.; Anstee, Q.M. Carriage of the PNPLA3 rs738409 C >G polymorphism confers an increased risk of non-alcoholic fatty liver disease associated hepatocellular carcinoma. J. Hepatol. 2014, 61, 75–81. [Google Scholar] [CrossRef] [PubMed]
- Ueyama, M.; Nishida, N.; Korenaga, M.; Korenaga, K.; Kumagai, E.; Yanai, H.; Adachi, H.; Katsuyama, H.; Moriyama, S.; Hamasaki, H.; et al. The impact of PNPLA3 and JAZF1 on hepatocellular carcinoma in non-viral hepatitis patients with type 2 diabetes mellitus. J. Gastroenterol. 2016, 51, 370–379. [Google Scholar] [CrossRef]
- Su, W.; Wang, Y.; Jia, X.; Wu, W.; Li, L.; Tian, X.; Li, S.; Wang, C.; Xu, H.; Cao, J.; et al. Comparative proteomic study reveals 17β-HSD13 as a pathogenic protein in nonalcoholic fatty liver disease. Proc. Natl. Acad. Sci. USA 2014, 111, 11437–11442. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abul-Husn, N.S.; Cheng, X.; Li, A.H.; Xin, Y.; Schurmann, C.; Stevis, P.; Liu, Y.; Kozlitina, J.; Stender, S.; Wood, G.C.; et al. Protein-Truncating HSD17B13 Variant and Protection from Chronic Liver Disease. N. Engl. J. Med. 2018, 378, 1096–1106. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Zhuo, J.Y.; Yang, F.; Liu, Z.K.; Zhou, L.; Xie, H.Y.; Xu, X.; Zheng, S.S. 17-beta-hydroxysteroid dehydrogenase 13 inhibits the progression and recurrence of hepatocellular carcinoma. Hepatobiliary Pancreat. Dis. Int. 2018, 17, 220–226. [Google Scholar] [CrossRef] [PubMed]
- Pirola, C.J.; Garaycoechea, M.; Flichman, D.; Arrese, M.; San Martino, J.; Gazzi, C.; Castaño, G.O.; Sookoian, S. Splice variant rs72613567 prevents worst histologic outcomes in patients with nonalcoholic fatty liver disease. J. Lipid Res. 2019, 60, 176–185. [Google Scholar] [CrossRef] [Green Version]
- Ma, Y.; Belyaeva, O.V.; Brown, P.M.; Fujita, K.; Valles, K.; Karki, S.; de Boer, Y.S.; Koh, C.; Chen, Y.; Du, X.; et al. 17-Beta Hydroxysteroid Dehydrogenase 13 Is a Hepatic Retinol Dehydrogenase Associated With Histological Features of Nonalcoholic Fatty Liver Disease. Hepatology 2019, 69, 1504–1519. [Google Scholar] [CrossRef]
- Yang, J.; Trépo, E.; Nahon, P.; Cao, Q.; Moreno, C.; Letouzé, E.; Imbeaud, S.; Bayard, Q.; Gustot, T.; Deviere, J.; et al. A 17-Beta-Hydroxysteroid Dehydrogenase 13 Variant Protects From Hepatocellular Carcinoma Development in Alcoholic Liver Disease. Hepatology 2019, 70, 231–240. [Google Scholar] [CrossRef]
- Namjou, B.; Lingren, T.; Huang, Y.; Parameswaran, S.; Cobb, B.L.; Stanaway, I.B.; Connolly, J.J.; Mentch, F.D.; Benoit, B.; Niu, X.; et al. GWAS and enrichment analyses of non-alcoholic fatty liver disease identify new trait-associated genes and pathways across eMERGE Network. BMC Med. 2019, 17, 135. [Google Scholar] [CrossRef]
- Ji, F.; Liu, Y.; Hao, J.G.; Wang, L.P.; Dai, M.J.; Shen, G.F.; Yan, X.B. KLB gene polymorphism is associated with obesity and non-alcoholic fatty liver disease in the Han Chinese. Aging (Albany NY) 2019, 11, 7847–7858. [Google Scholar] [CrossRef]
- Xu, M.; Li, Y.; Zhang, S.; Wang, X.; Shen, J.; Zhang, S. Interaction of TM6SF2 E167K and PNPLA3 I148M variants in NAFLD in northeast China. Ann. Hepatol. 2019, 18, 456–460. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Xing, J.; Wang, B.; Wei, C.; Yang, R.; Zhu, Y.; Qiu, H. Correlation Between Adiponectin Gene rs1501299 Polymorphism and Nonalcoholic Fatty Liver Disease Susceptibility: A Systematic Review and Meta-Analysis. Med. Sci. Monit. 2019, 25, 1078–1086. [Google Scholar] [CrossRef] [PubMed]
- Kawaguchi, T.; Shima, T.; Mizuno, M.; Mitsumoto, Y.; Umemura, A.; Kanbara, Y.; Tanaka, S.; Sumida, Y.; Yasui, K.; Takahashi, M.; et al. Risk estimation model for nonalcoholic fatty liver disease in the Japanese using multiple genetic markers. PLoS ONE 2018, 13, e0185490. [Google Scholar] [CrossRef] [PubMed]
- Kanda, T.; Goto, T.; Hirotsu, Y.; Moriyama, M.; Omata, M. Molecular Mechanisms Driving Progression of Liver Cirrhosis towards Hepatocellular Carcinoma in Chronic Hepatitis B and C Infections: A Review. Int. J. Mol. Sci. 2019, 20, 1358. [Google Scholar] [CrossRef] [Green Version]
- Sasaki, R.; Kanda, T.; Yokosuka, O.; Kato, N.; Matsuoka, S.; Moriyama, M. Exosomes and Hepatocellular Carcinoma: From Bench to Bedside. Int. J. Mol. Sci. 2019, 20, 1406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, Y.; Xiong, Y.; Sheng, Q.; Zhao, S.; Wattacheril, J.; Flynn, C.R. A micro-RNA expression signature for human NAFLD progression. J. Gastroenterol. 2016, 51, 1022–1030. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, H.; Yang, W.; Tian, Y.; Zeng, X.; Zhou, J.; Mok, M.T.S.; Tang, W.; Feng, Y.; Xu, L.; Chan, A.W.H.; et al. An inflammatory-CCRK circuitry drives mTORC1-dependent metabolic and immunosuppressive reprogramming in obesity-associated hepatocellular carcinoma. Nat. Commun. 2018, 9, 5214. [Google Scholar] [CrossRef]
- Kanda, T.; Steele, R.; Ray, R.; Ray, R.B. Hepatitis C virus core protein augments androgen receptor-mediated signaling. J. Virol. 2008, 82, 11066–11072. [Google Scholar] [CrossRef] [Green Version]
- Gentric, G.; Maillet, V.; Paradis, V.; Couton, D.; L’Hermitte, A.; Panasyuk, G.; Fromenty, B.; Celton-Morizur, S.; Desdouets, C. Oxidative stress promotes pathologic polyploidization in nonalcoholic fatty liver disease. J. Clin. Invest. 2015, 125, 981–992. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuramoto, J.; Arai, E.; Tian, Y.; Funahashi, N.; Hiramoto, M.; Nammo, T.; Nozaki, Y.; Takahashi, Y.; Ito, N.; Shibuya, A.; et al. Genome-wide DNA methylation analysis during non-alcoholic steatohepatitis-related multistage hepatocarcinogenesis: Comparison with hepatitis virus-related carcinogenesis. Carcinogenesis 2017, 38, 261–270. [Google Scholar] [CrossRef]
- Li, C.W.; Chiu, Y.K.; Chen, B.S. Investigating Pathogenic and Hepatocarcinogenic Mechanisms from Normal Liver to HCC by Constructing Genetic and Epigenetic Networks via Big Genetic and Epigenetic Data Mining and Genome-Wide NGS Data Identification. Dis. Markers 2018, 2018, 8635329. [Google Scholar] [CrossRef] [PubMed]
- Alexander, M.; Loomis, A.K.; van der Lei, J.; Duarte-Salles, T.; Prieto-Alhambra, D.; Ansell, D.; Pasqua, A.; Lapi, F.; Rijnbeek, P.; Mosseveld, M.; et al. Risks and clinical predictors of cirrhosis and hepatocellular carcinoma diagnoses in adults with diagnosed NAFLD: Real-world study of 18 million patients in four European cohorts. BMC Med. 2019, 17, 95. [Google Scholar] [CrossRef] [PubMed]
- Desterke, C.; Chiappini, F. Lipid Related Genes Altered in NASH Connect Inflammation in Liver Pathogenesis Progression to HCC: A Canonical Pathway. Int. J. Mol. Sci. 2019, 20, 5594. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anstee, Q.M.; Reeves, H.L.; Kotsiliti, E.; Govaere, O.; Heikenwalder, M. From NASH to HCC: Current concepts and future challenges. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 411–428. [Google Scholar] [CrossRef]


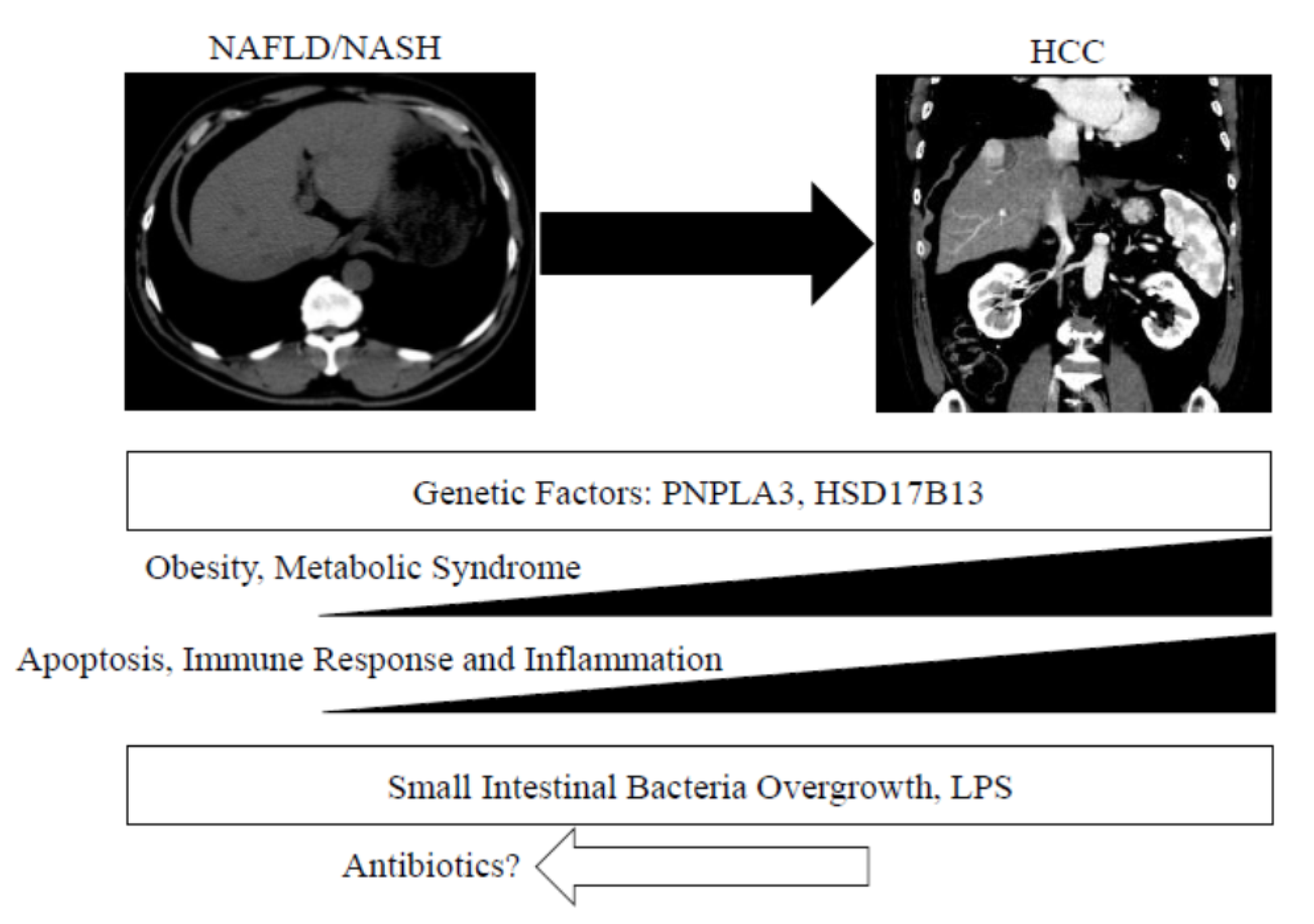{kind=link}
| Factors | Observation |
|---|---|
| Obesity | Hyperinsulinemia, Insulin resistance |
| Apoptosis | ER stress, Oxidative stress |
| Immune and inflammatory pathways | Activation of macrophages, Iron metabolism, |
| Intestinal bacteria | SIBO, Intestinal mucosa barrier malfunction, Intestinal microbiota |
| SNPs | PNPLA3, HSD17B13 |
| Epigenetic alterations | MicroRNA, DNA methylation |
| Molecules |
|---|
| miR-301a-3p, miR-375 [107] |
| CCRK [108] |
| MAML3 [111] |
| DNA methylation of HIST2H2BE, HSPB1, RPL30, ALDOB [112] |
| miR-21, miR-112 [112] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kanda, T.; Goto, T.; Hirotsu, Y.; Masuzaki, R.; Moriyama, M.; Omata, M. Molecular Mechanisms: Connections between Nonalcoholic Fatty Liver Disease, Steatohepatitis and Hepatocellular Carcinoma. Int. J. Mol. Sci. 2020, 21, 1525. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21041525
Kanda T, Goto T, Hirotsu Y, Masuzaki R, Moriyama M, Omata M. Molecular Mechanisms: Connections between Nonalcoholic Fatty Liver Disease, Steatohepatitis and Hepatocellular Carcinoma. International Journal of Molecular Sciences. 2020; 21(4):1525. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21041525
Chicago/Turabian StyleKanda, Tatsuo, Taichiro Goto, Yosuke Hirotsu, Ryota Masuzaki, Mitsuhiko Moriyama, and Masao Omata. 2020. "Molecular Mechanisms: Connections between Nonalcoholic Fatty Liver Disease, Steatohepatitis and Hepatocellular Carcinoma" International Journal of Molecular Sciences 21, no. 4: 1525. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21041525