Overcoming Resistance to FLT3 Inhibitors in the Treatment of FLT3-Mutated AML


Division of Haematology, Department of Medicine, Li Ka Shing Faculty of Medicine, The University of Hong Kong, Hong Kong, China
*
Author to whom correspondence should be addressed.
Int. J. Mol. Sci. 2020, 21(4), 1537; https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21041537
Submission received: 1 February 2020
/
Revised: 21 February 2020
/
Accepted: 21 February 2020
/
Published: 24 February 2020
(This article belongs to the Special Issue Drug Resistance in Hematologic Malignancies)
Abstract
:Acute myeloid leukaemia (AML) carrying internal tandem duplication (ITD) of Fms-Like Tyrosine kinase 3 (FLT3) gene is associated with high risk of relapse and poor clinical outcome upon treatment with conventional chemotherapy. FLT3 inhibitors have been approved for the treatment of this AML subtype but leukaemia relapse remains to be a major cause of treatment failure. Mechanisms of drug resistance have been proposed, including evolution of resistant leukaemic clones; adaptive cellular mechanisms and a protective leukaemic microenvironment. These models have provided important leads that may inform design of clinical trials. Clinically, FLT3 inhibitors in combination with conventional chemotherapy as induction treatment for fit patients; with low-intensity treatment as salvage treatment or induction for unfit patients as well as maintenance treatment with FLT3 inhibitors post HSCT hold promise to improve survival in this AML subtype.
1. Introduction
Acute myeloid leukaemia (AML) is defined pathologically by an abnormal increase in blasts in blood and/or bone marrow (BM). It is a group of heterogeneous diseases with distinct driver events and pathogeneses that may occur at different stages of the haematopoietic hierarchy [1]. Subtypes of AML show different morphologies, immunophenotypes as well as cytogenetic, genetic and clinical features. Conventional chemotherapy, comprising induction and consolidation as well as allogeneic haematopoietic stem cell transplantation (HSCT) are the mainstays of treatment but only 30%–40% of patients can achieve long-term remission. The outcome of elderly patients ineligible for chemotherapy and HSCT is dismal.
Laboratory studies in leukaemia biology in the past few decades have led to identification of molecular targets and development of novel therapeutic strategies. In the past 3 years, eight therapeutic agents have been approved by the U.S. Food and Drug Administration (FDA) for the treatment of different AML subtypes in different clinical contexts (Table 1). In particular, two multi-kinase inhibitors were approved for the treatment of AML carrying gain-of-function mutations in Fms-like tyrosine kinase 3 (FLT3) gene: Midostaurin in combination with conventional induction and consolidation chemotherapy in newly diagnosed patients [2] and gilteritinib monotherapy for relapsed/refractory (R/R) FLT3-mutated patients [3]. Another specific FLT3 inhibitor quizartinib was also approved in Japan for the treatment of R/R patients [4]. However, disease relapse remains an important cause of treatment failure. This review focuses on the potential mechanisms of drug resistance in this AML subtype and strategies that may be exploited to overcome resistance.
2. Fms-Like Tyrosine Kinase 3 (FLT3)
Fms-like tyrosine kinase 3 (FLT3), first identified to be expressed in normal haematopoietic stem and progenitor cells, is one of the most frequently mutated genes in AML. Internal tandem duplication (ITD) is the commonest genetic abnormality and is associated with leucocytosis at diagnosis and high risk of relapse after conventional chemotherapy, particularly those with high ITD allelic ratio [5], large ITD size [6] and multiple ITD clones [7]. FLT3-ITD occurs particularly in AML with normal cytogenetics, where it occurs in up to 40% cases, and those with rare t(6;9) translocation involving DEK/CAN gene fusion, where it occurs in up to 70%–80% [8]. Missense mutations of tyrosine kinase domain (TKD) also occur, albeit infrequently at 5%–10% of AML. Their prognostic impact has remained unclear [9], which could be due to their low incidence of occurrence or modest biologic activities. Mechanistically, both ITD and TKD mutations result in constitutive activation of FLT3 signalling, hence the cellular proliferation, anti-apoptosis and differentiation block that are often seen in FLT3 mutant AML cases [1,10].
Under physiological conditions, FLT3 protein is activated by its ligand (FLT3L) and the binding results in dimerization and conformational changes of FLT3 that expose the phosphorylation sites of its TKD. Subsequent auto-phosphorylation of FLT3 leads to binding of adaptor proteins such as SHP2, Grb2 and SRC family kinases, hence activation of downstream signalling kinases including MAPK/ERK, JAK/STAT and PI3K/AKT/mTOR [11]. While both ITD and TKD mutations result in constitutive activation of FLT3, via MAPK and PI3K pathways, there are significant differences in the activated downstream signalling pathways between them [12]. For instance, FLT3-TKD is associated with activation of SHP1 and SHP2 phosphatases that negatively regulate JAK signalling, whereas STAT5 activation via SRC binding is only seen in FLT3-ITD but not FLT3-TKD or FLT3-WT cells [13].
3. FLT3 Inhibitors
The pathogenetic roles of FLT3-ITD and TKD in AML and the inferior outcome of this AML subtype provide the basis for developing FLT3 inhibitors. Mechanistically FLT3 inhibitors can be categorised into 2 types. Type I inhibitors bind FLT3 in the active conformation near the activation loop or ATP binding site and are effective against both ITD and mutant TKD as exemplified by midostaurin, sunitinib, lestaurtinib, crenolanib and gilteritinib. Type II inhibitors bind FLT3 in the inactive state near the ATP binding domain, targeting FLT3-ITD but not mutant TKD. Examples include sorafenib, ponatinib and quizartinib. In order of their development, FLT3 inhibitors can be categorised into those of first and second generation. First generation FLT3 inhibitors refer to several multi-kinase inhibitors including lestaurtinib, sunitinib, sorafenib and midostaurin that have been evaluated since early 2000. As monotherapy, sorafenib has been widely used as salvage therapy for R/R FLT3-ITD AML with a rate of combined complete remission (CR) and CR with incomplete haematologic recovery (CRi) at about 16%–46%. Responses were typically transient with median duration of 1–3 months, and sorafenib was perceived at best as a bridging therapy to curative allogeneic HSCT. More recent data showed that sorafenib may be effective as maintenance therapy post HSCT, resulting in improved survival of these patients [14,15,16]. Midostaurin showed only modest effect as monotherapy in R/R AML [17]. When used as an adjunct to conventional chemotherapy in newly diagnosed FLT3-mutated AML, midostaurin was shown to prolong overall survival [2], leading to its approval by FDA.
Second generation FLT3 inhibitors refer to new inhibitors with higher specificity and potency against FLT3, including quizartinib, gilteritinib and crenolanib. They have been tested in clinical trials as monotherapy in R/R FLT3-mutated AML with CR/CRi of 23%–57% and median duration of response of 9–20 weeks (Figure 1). Based on an interim analysis of the ADMIRAL trial [3], gilteritinib was granted FDA approval for the treatment of R/R FLT3-mutated AML. In a randomised open-label phase 3 study (QUANTUM-R) [4], quizartinib was shown to improve overall survival compared with salvage chemotherapy in R/R FLT3-mutated AML. Several phase 3 studies were underway including those that evaluate gilteritinib following induction and consolidation chemotherapy (NCT02236013, NCT02310321) as well as allogeneic HSCT (NCT02997202) in newly diagnosed AML; crenolanib in combination with salvage chemotherapy in R/R FLT3-mutated AML (NCT02400281, NCT02298166, NCT03250338), and crenolanib compared with midostaurin in newly diagnosed FLT3-mutated AML when used in conjunction with conventional chemotherapy (NCT03258931); quizartinib in combination with induction chemotherapy in newly diagnosed FLT3-mutated AML (NCT02834390, NCT03723681, NCT02668653, NCT04107727).
A major limitation in the treatment of FLT3-mutated AML by FLT3 inhibitor monotherapy is leukaemia relapse that often occurs within months after initial remission. In most circumstances, this is related to development of drug resistance. The mechanisms are heterogeneous and may involve emergence of clones that are resistant to FLT3 inhibitors being used; protection of leukaemia cells by BM microenvironment; and adaptation of leukaemia cells to survive FLT3 inhibitors. These mechanisms are reviewed in the following sections.
4. Clonal Evolution
4.1. Emergence of FL3-TKD Mutations
Initial evidence of emerging FLT3-TKD mutations as a cause of drug resistance to FLT3 inhibitors arose from laboratory studies. In particular, FLT3-TKD mutant clones could be selected from in vitro saturation mutagenesis assay [30] and found in FLT3-ITD AML cell lines treated with increasing dose of FLT3 inhibitors for 6–7 weeks [31]. In clinical practice, a recurrent phenomenon in patients receiving FLT3 inhibitors is the emergence of leukaemia clones carrying FLT3-TKD mutations at relapse. On one hand it has confirmed the on-target effect of FLT3 inhibitors and their anti-leukaemia efficacy, on the other hand it contributes to drug-resistant leukaemia relapse. Mutations may occur at the activation loop (e.g., D835, I836) or gate-keeper site (e.g., F691), and the frequencies and mutation sites depend on the specific FLT3 inhibitors being used (Table 2). Heterogeneity of FLT3 mutant clones and polyclonal architecture with respect to FLT3-ITD and FLT3-TKD have been shown by single-cell sequencing of AML samples from patients who relapsed during quizartinib treatment [32]. In most cases FLT3-TKD mutations were not detectable by molecular means prior to FLT3 inhibitor treatment [10,32]. However, when pre-treatment samples were xenografted into immunodeficient mice, TKD mutant clones could emerge upon engrafting, suggesting that they might exist before treatment and were selected upon continuous exposure to inhibitor ineffective against them [10]. Nevertheless, TKD mutations only occur in 3%–60% of patients who relapsed during or after treatment with FLT3 inhibitors, and the rates are much lower in the new generation of FLT3 inhibitors e.g., crenolanib [33] and gilteritinib [34]. Moreover, in patients who relapsed after quizartinib monotherapy, single-cell analyses showed that TKD mutations occurred in up to 50% of leukaemia cells in individual patients, suggesting that they might not account entirely for the relapse [32]. Furthermore, relapses do occur upon treatment with new generation FLT3 inhibitors (e.g., gilteritinib and crenolanib) that showed inhibitory effects on TKD-mutant FLT3 proteins. Therefore, TKD mutations could only partially explain leukaemia relapse after FLT3 inhibitors. Non-TKD mediated resistance is discussed as followed.
4.2. Emergence of Non-FLT3 Mutations
Non-FLT3 mutant clones have been shown to expand or emerge at relapse in FLT3-ITD AML during FLT3 inhibitor treatment. Next-generation sequencing of paired samples (drug-naïve sensitive and relapse drug-resistant samples) from R/R FLT3-ITD AML patients who relapsed from FLT3 inhibitors crenolanib [33] or gilteritinib [34] demonstrated the emergence or expansion of leukaemia clones either as subclones of the FLT3-ITD clone or new wildtype FLT3 clones. These clones carried mutations of TP53, RAS pathway (NRAS, KRAS, BRAF, PTPN11, CBL), IDH1/2, ASXL1 or TET2. The emergence of these mutations, particularly when they occurred in wildtype FLT3 clones, demonstrated FLT3-independent leukaemia cells that were selected under the pressure of FLT3 inhibitors to which they were resistant. Complete loss of FLT3-ITD clone has been reported in up to 30% of cases [33,34].
5. Adaptive Cellular Mechanisms
In addition to the emergence of resistant clones, FLT3-ITD AML cells may adapt to FLT3 inhibitors and develop cellular mechanisms that circumvent FLT3 blockade. They include upregulation of FLT3 ligand, change in intracellular acidity and upregulation of other protein kinases. These are discussed as follows:
5.1. Upregulation of FLT3 Ligand
Despite the ligand independence of FLT3-ITD signalling, FLT3L has been shown to confer resistance to FLT3 inhibitors in AML [40]. FLT3L exists in soluble and membrane-bound form, the latter being expressed on stromal cells. Serum level of FLT3L was shown to increase after chemotherapy induction [40] and FLT3 inhibitor treatment [23]. FLT3L-mediated resistance to FLT3 inhibitors might be mediated through its binding to FLT3-WT receptor and activation of downstream MAPK pathway [41].
5.2. Increase in Intracellular pH
Microarray analysis of paired FLT3-ITD AML samples before sorafenib treatment and at subsequent relapse showed up-regulation of a gene encoding tescalcin (TESC) [42]. TESC plays a pivotal role in the maturation of sodium/hydrogen exchanger type 1 (NHE1). The latter, when activated, extrudes H+ in exchange for Na+ intake. The resulting intracellular alkalinisation provides proliferative and survival benefits to the blasts, and confers resistance by decreasing intracellular sorafenib concentration via acid–base partitioning.
5.3. Upregulation of other Cooperative Kinases
Upregulation of other oncogenic kinases has been shown in primary FLT3-ITD AML upon development of resistance to FLT3 inhibitors and may play a pathogenetic role. For instance, expression of PIM (proviral integration site for moloney murine leukaemia virus) kinase, a target of FLT3-ITD signalling, has been shown to increase in primary AML cells at resistance to sorafenib [43]. Overexpression of PIM-2 in MOLM-14 (a FLT3-ITD AML cell line) and FLT3-ITD knock-in mouse model has been shown to confer resistance to quizartinib [43]. Also, expression and phosphorylation of AXL receptor tyrosine kinase was increased in PKC412(midostaurin)-resistant primary AML blasts and AML cell line [44] and its upregulation has been implicated in stroma-mediated resistance to quizartinib [45]. Pharmacological inhibition and knockout of AXL have been shown to restore sensitivity to FLT3 inhibitors-resistant AML cell lines [44]. Importantly, AXL is a therapeutic target of gilterinib [46]. Furthermore, the persistence or even up-regulation of PI3K/AKT/mTOR and MAPK/ERK pathways were shown in both FLT3 inhibitors-resistant FLT3-ITD AML cell lines and primary AML blasts despite inhibition of FLT3 phosphorylation by FLT3 inhibitors, suggesting that at resistance, some AML cells become independent of FLT3 signalling [47,48]. The use of inhibitors to target these compensatory pathways in combination with FLT3 inhibitors has been proposed [48] and the clinical benefits remain to be evaluated.
6. Microenvironment Protection
BM niche has been shown to nurture normal haematopoietic stem cell and progenitor cells and maintain steady state haematopoiesis. Mechanistic studies in mouse marrow demonstrated close interaction between osteoblasts and haematopoietic stem and progenitor cells (HSPC), suggesting a physical niche that provides signals to guide HSPC cell-fate decision [49]. Similar niche for AML may exist [50,51] and protect leukaemia cells from the inhibitory effects of FLT3 inhibitors. A number of mechanisms have been proposed (Figure 2).
6.1. Cytokines
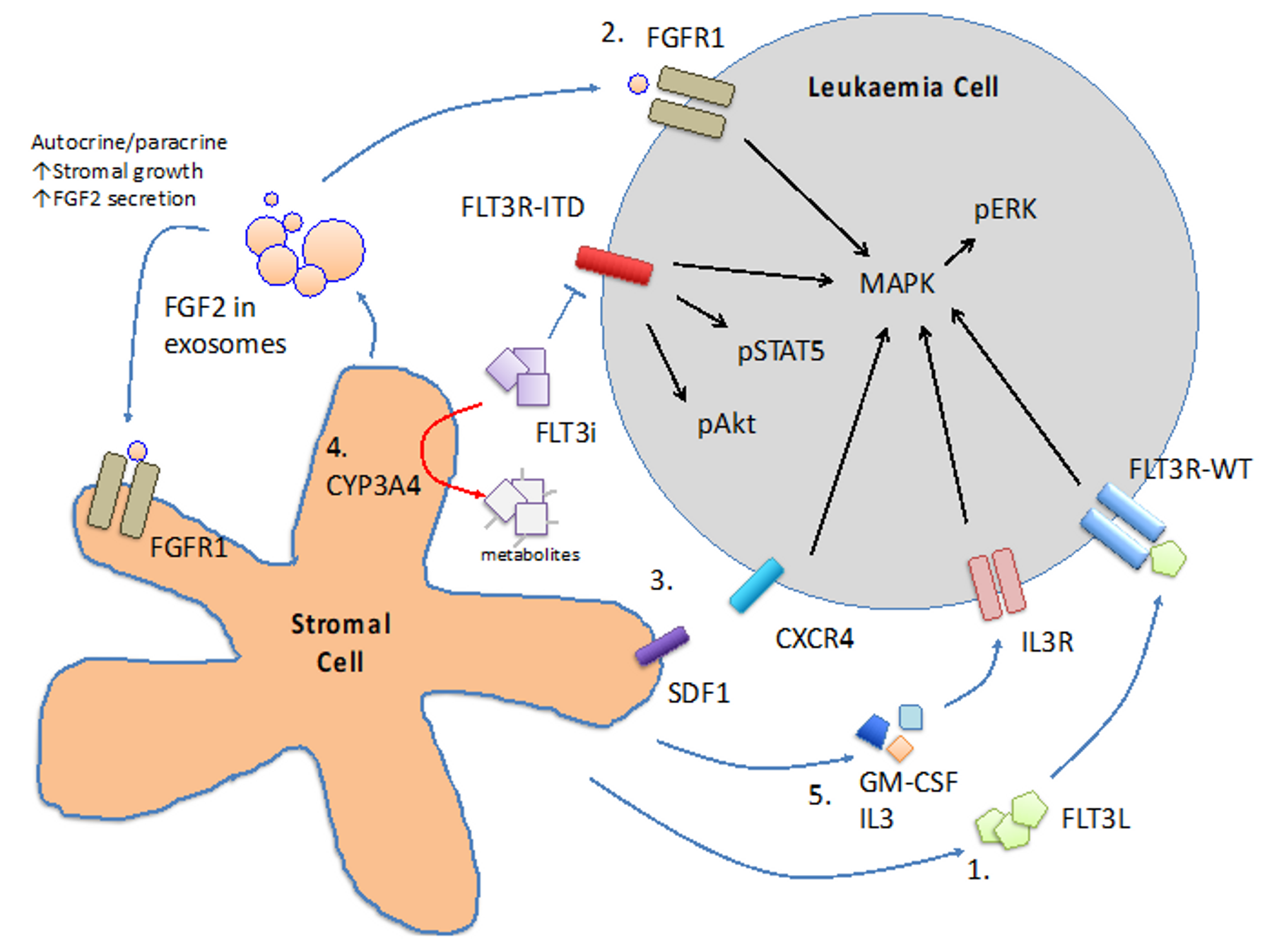
BM stromal cells have been shown to secrete a repertoire of cytokines that protect the blasts from the cytotoxic effect of chemotherapy as well as FLT3 inhibitors. In addition to the FLT3L aforementioned, GM-CSF and IL-3 have been shown to protect primary FLT3-ITD AML cells and MV4-11 (a FLT3-ITD AML cell line) from crenolanib through activation of STAT5 pathway [52]. BM stromal cells also secrete fibroblast growth factor 2 (FGF2) that binds to FGFR1 in FLT3-ITD AML cells and promotes resistance to FLT3 inhibitors via activation of downstream MAPK pathway [53,54]. FGF2 might also bind to FGFR1 on stromal cells in an autocrine and paracrine fashion to stimulate more FGF2 secretion and stromal growth. Intriguingly, quizartinib has been shown to induce FGF2 expression in stromal cells of FLT3-ITD AML patients [53,54] and whether this may account for the subsequent clinical resistance to FLT3 inhibitor would have to be further investigated. Stromal-cell derived factor 1 (SDF-1) and CXCR4 axis plays a pivotal role in BM homing of HSPC. FLT3-ITD AML has been shown to highly express CXCR4 [55], suggesting the SDF-1/CXCR4 axis may play a pathogenetic role in this AML subtype. SDF-1 antagonist has been shown to sensitise FLT3-ITD transduced Ba/F3 cells (a murine pro-B lymphoid cell line) to the inhibitory effects of sorafenib when co-cultured with protective MS5 stromal cell line [56]. Activation of p53 pathway in stromal cells has been shown to reduce SDF-1 expression and abrogates the protective effect of stromal cells [57], providing a potential target for therapeutic intervention.
6.2. CYP3A4 in BM Stromal Cells
Cytochrome P450 3A4 (CYP3A4) is important for the metabolism and elimination of drugs and xenobiotics in human body. It is expressed primarily in hepatocytes and BM stromal cells and for the latter it might inactivate tyrosine kinase inhibitors [58] in the BM milieu. Knock-down of CYP3A4 in stromal cells or pharmacologic inhibition of CYP3A4 has been shown to ameliorate the protective effects of stromal cells both in co-culture system and in xenotransplantation model [59].
7. Clinical Strategies to Overcome Drug Resistance in FLT3-ITD AML
The mechanisms aforementioned provide the theoretical bases on which clinical trials can be designed to overcome drug resistance to FLT3 inhibitors. However, the precise mechanisms of drug resistance are likely to vary between patients and multiple mechanisms may co-exist, adding to the complexity of trial design. In clinical practice, a number of strategies have been developed to improve the treatment outcome of FLT3-ITD patients.
7.1. Upfront FLT3 Inhibitors in Combination with Chemotherapy
Until recently, induction chemotherapy, the “7+3” regimen, has been the standard of care for fit patients of all AML subtypes including FLT3-ITD AML. In a Phase III randomised placebo-controlled study, addition of midostaurin to induction and consolidation chemotherapy, followed by maintenance treatment in FLT3-mutated AML was shown to improve overall survival [2]. The results led to FDA approval of midostaurin for the upfront treatment of FLT3-mutated AML. Benefits of sorafenib when added to conventional chemotherapy have also been demonstrated [60,61]. Clinical trials incorporating quizartinib (NCT02668653), crenolanib [62] and gilteritinib (NCT02236013, NCT02310321) to standard chemotherapy are underway.
CPX-351, a liposomal preparation of cytarabine and daunorubicin at a fixed synergistic drug ratio of 5:1, was recently approved as induction therapy in AML with MDS-related changes and therapy-related AML. Patients with this AML subtype receiving CPX-351 showed a superior outcome than those receiving conventional “7+3” [63]. FLT3-ITD AML cells have been shown to be sensitive to this preparation in vitro [64]. The clinical benefits of FLT3 inhibitors in combination with CPX-351 in AML with MRC or tAML carrying FLT3-ITD remain to be investigated.
7.2. Combination of FLT3 Inhibitors with Low Intensity Regimen
FLT3-ITD AML cells have been shown to exhibit high protein synthesis rate to maintain intracellular level of short-lived oncogenic proteins [65]. The addiction to protein synthesis provided a target for therapeutic intervention. A high-throughput ex vivo drug screening using primary AML cells has identified omacetaxine mepesuccinate (OME) as an effective adjunct to FLT3 inhibitors in the treatment of FLT3-ITD AML [65]. OME competes with t-RNA to bind to acceptor (A-site) of eukaryotic ribosome, thereby inhibiting the elongation process of protein synthesis [66]. It suppresses FLT3 downstream signalling via inhibition of the synthesis of FLT3, a short-lived protein. OME showed very acceptable toxicity profile even in the elderly but its monotherapy or combination with cytarabine showed only modest effects on AML generally. However, in combination with sorafenib (SOME), it induced remission (CR/CRi) in 72% of patients with R/R FLT3-ITD AML, with a deeper molecular response and extended response duration (median overall survival and leukaemia-free survival being 43.6 and 22.4 weeks respectively) among responders [20]. A clinical trial evaluating its combination with a more specific and potent FLT3 inhibitor quizartinib (QUIZOM) is in progress (NCT03135054). Preliminary observations showed that it might confer superior response and better survival, and bridge more patients to HSCT compared with SOME [67].
Furthermore, combination of FLT3 inhibitors including sorafenib, quizartinib and gilteritinib with other low intensity treatment including azacytidine or decitabine, the hypomethylating agents, have been shown to be synergistic in laboratories [68,69,70] and appeared to be effective in Phase II clinical trials [19,71,72].
7.3. HSCT with FLT3 Inhibitor as Maintenance Therapy
HSCT is the mainstay of treatment for FLT3-ITD AML in complete remission after induction chemotherapy or salvage treatments including FLT3 inhibitors [73,74,75,76,77]. However, post-HSCT relapse remains a major cause of treatment failure and may occur in up to 75% of patients. Results from clinical trials supported the proposition that maintenance with FLT3 inhibitors post-HSCT could reduce relapse and improve overall survival. In SORMAIN study, sorafenib maintenance significantly prolonged relapse-free survival [16]. Laboratory study showed that sorafenib in the post HSCT setting might increase serum IL-15 from residual FLT3-ITD cells that may enhance activities of allogeneic T-cells and graft-versus leukaemia effect [78]. In RADIUS Trial, patients who received midostaurin maintenance post HSCT and achieved significant FLT3 inhibition (<70% of baseline pFLT3) showed significant improvement in relapse-free and overall survivals compared with those who achieved <30% inhibition or those who received standard of care [79]. Post-HSCT maintenance with quizartinib in FLT3-ITD AML in a phase I study also showed reduced relapse rate [80]. Whether the benefits of FLT3 inhibitor post HSCT are related to its suppressive effects on residual FLT3-ITD AML cells or potentiation on graft-versus-leukaemia effects would have to be further evaluated [81].
8. Conclusions
Despite the availability of effective FLT3 inhibitors in the treatment of FLT3-ITD AML, leukaemia relapse remains to be a major cause of treatment failure. Incorporation of FLT3 inhibitor to upfront induction and consolidation chemotherapy; combination of FLT3 inhibitor with low intensity regimen as salvage treatment and the use of FLT3 inhibitor as post-HSCT maintenance may improve treatment outcome of this AML subtype. Based on findings of laboratory studies, multiple mechanisms of drug resistance have been proposed and it is likely heterogeneous among individual patients. They have provided the important bases for development of clinical trials and might raise the potential need for treatment personalisation.
Author Contributions
Conceptualization, writing—review and editing by S.S.Y.L. and A.Y.H.L. All authors have read and agreed to the published version of the manuscript
Funding
This research received no external funding
Conflicts of Interest
The authors declare no conflict of interest.
Abbreviations
| AML | Acute myeloid leukaemia |
| CR | Complete remission |
| CRc | Composite complete remission |
| CRi | Complete remission with incomplete haematological response |
| CRp | Complete remission with incomplete platelet count |
| FLT3L | FLT3 ligand |
| HSCT | Haematopoietic stem cell transplantation |
| HSPC | Haematopoietic stem and progenitor cell |
| ITD | Internal tandem duplication |
| OME | Omacetaxine mepesuccinate |
| PR | Partial response |
| R/R | Relapse or refractory |
| TKD | Tyrosine kinase domain |
| TKI | Tyrosine kinase inhibitor |
References
- van Galen, P.; Hovestadt, V.; Wadsworth Ii, M.H.; Hughes, T.K.; Griffin, G.K.; Battaglia, S.; Verga, J.A.; Stephansky, J.; Pastika, T.J.; Lombardi Story, J.; et al. Single-Cell RNA-Seq reveals AML hierarchies relevant to disease progression and immunity. Cell 2019, 176, 1265–1281.e24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stone, R.M.; Mandrekar, S.J.; Sanford, B.L.; Laumann, K.; Geyer, S.; Bloomfield, C.D.; Thiede, C.; Prior, T.W.; Dohner, K.; Marcucci, G.; et al. Midostaurin plus chemotherapy for acute myeloid leukemia with a FLT3 mutation. N. Engl. J. Med. 2017, 377, 454–464. [Google Scholar] [CrossRef] [PubMed]
- Perl, A.E.; Martinelli, G.; Cortes, J.E.; Neubauer, A.; Berman, E.; Paolini, S.; Montesinos, P.; Baer, M.R.; Larson, R.A.; Ustun, C.; et al. Gilteritinib or chemotherapy for relapsed or refractory FLT3-mutated AML. N. Engl. J. Med. 2019, 381, 1728–1740. [Google Scholar] [CrossRef]
- Cortes, J.E.; Khaled, S.; Martinelli, G.; Perl, A.E.; Ganguly, S.; Russell, N.; Kramer, A.; Dombret, H.; Hogge, D.; Jonas, B.A.; et al. Quizartinib versus salvage chemotherapy in relapsed or refractory FLT3-ITD acute myeloid leukaemia (QuANTUM-R): A multicentre, randomised, controlled, open-label, phase 3 trial. Lancet Oncol. 2019, 20, 984–997. [Google Scholar] [CrossRef]
- Santos, F.P.; Jones, D.; Qiao, W.; Cortes, J.E.; Ravandi, F.; Estey, E.E.; Verma, D.; Kantarjian, H.; Borthakur, G. Prognostic value of FLT3 mutations among different cytogenetic subgroups in acute myeloid leukemia. Cancer 2011, 117, 2145–2155. [Google Scholar] [CrossRef] [Green Version]
- Meshinchi, S.; Stirewalt, D.L.; Alonzo, T.A.; Boggon, T.J.; Gerbing, R.B.; Rocnik, J.L.; Lange, B.J.; Gilliland, D.G.; Radich, J.P. Structural and numerical variation of FLT3/ITD in pediatric AML. Blood 2008, 111, 4930–4933. [Google Scholar] [CrossRef] [Green Version]
- Gale, R.E.; Green, C.; Allen, C.; Mead, A.J.; Burnett, A.K.; Hills, R.K.; Linch, D.C. Medical Research Council Adult Leukaemia Working Party. The impact of FLT3 internal tandem duplication mutant level, number, size, and interaction with NPM1 mutations in a large cohort of young adult patients with acute myeloid leukemia. Blood 2008, 111, 2776–2784. [Google Scholar] [CrossRef] [Green Version]
- Papaemmanuil, E.; Gerstung, M.; Bullinger, L.; Gaidzik, V.I.; Paschka, P.; Roberts, N.D.; Potter, N.E.; Heuser, M.; Thol, F.; Bolli, N.; et al. Genomic classification and prognosis in acute myeloid leukemia. N. Engl. J. Med. 2016, 374, 2209–2221. [Google Scholar] [CrossRef]
- Bacher, U.; Haferlach, C.; Kern, W.; Haferlach, T.; Schnittger, S. Prognostic relevance of FLT3-TKD mutations in AML: The combination matters—An analysis of 3082 patients. Blood 2008, 111, 2527–2537. [Google Scholar] [CrossRef]
- Man, C.H.; Fung, T.K.; Ho, C.; Han, H.H.; Chow, H.C.; Ma, A.C.; Choi, W.W.; Lok, S.; Cheung, A.M.; Eaves, C.; et al. Sorafenib treatment of FLT3-ITD(+) acute myeloid leukemia: Favorable initial outcome and mechanisms of subsequent nonresponsiveness associated with the emergence of a D835 mutation. Blood 2012, 119, 5133–5143. [Google Scholar] [CrossRef]
- Takahashi, S. Downstream molecular pathways of FLT3 in the pathogenesis of acute myeloid leukemia: Biology and therapeutic implications. J. Hematol. Oncol. 2011, 4, 13. [Google Scholar] [CrossRef] [Green Version]
- Choudhary, C.; Schwable, J.; Brandts, C.; Tickenbrock, L.; Sargin, B.; Kindler, T.; Fischer, T.; Berdel, W.E.; Muller-Tidow, C.; Serve, H. AML-Associated Flt3 kinase domain mutations show signal transduction differences compared with Flt3 ITD mutations. Blood 2005, 106, 265–273. [Google Scholar] [CrossRef]
- Leischner, H.; Albers, C.; Grundler, R.; Razumovskaya, E.; Spiekermann, K.; Bohlander, S.; Ronnstrand, L.; Gotze, K.; Peschel, C.; Duyster, J. SRC is a signaling mediator in FLT3-ITD-but not in FLT3-TKD-positive AML. Blood 2012, 119, 4026–4033. [Google Scholar] [CrossRef]
- Battipaglia, G.; Ruggeri, A.; Massoud, R.; El Cheikh, J.; Jestin, M.; Antar, A.; Ahmed, S.O.; Rasheed, W.; Shaheen, M.; Belhocine, R.; et al. Efficacy and feasibility of sorafenib as a maintenance agent after allogeneic hematopoietic stem cell transplantation for Fms-like tyrosine kinase 3-mutated acute myeloid leukemia. Cancer 2017, 123, 2867–2874. [Google Scholar] [CrossRef]
- Brunner, A.M.; Li, S.; Fathi, A.T.; Wadleigh, M.; Ho, V.T.; Collier, K.; Connolly, C.; Ballen, K.K.; Cutler, C.S.; Dey, B.R.; et al. Haematopoietic cell transplantation with and without sorafenib maintenance for patients with FLT3-ITD acute myeloid leukaemia in first complete remission. Br. J. Haematol. 2016, 175, 496–504. [Google Scholar] [CrossRef] [Green Version]
- Burchert, A.; Bug, G.; Finke, J.; Stelljes, M.; Rollig, C.; Wäsch, R.; Bornhäuser, M.; Berg, T.; Lang, F.; Ehninger, G.; et al. Sorafenib as maintenance therapy post allogeneic stem cell transplantation for FLT3-ITD positive AML: Results from the randomized, double-blind, placebo-controlled multicentre sormain trial. Blood 2018, 132, 661. [Google Scholar] [CrossRef]
- Fischer, T.; Stone, R.M.; Deangelo, D.J.; Galinsky, I.; Estey, E.; Lanza, C.; Fox, E.; Ehninger, G.; Feldman, E.J.; Schiller, G.J.; et al. Phase IIB trial of oral Midostaurin (PKC412), the FMS-like tyrosine kinase 3 receptor (FLT3) and multi-targeted kinase inhibitor, in patients with acute myeloid leukemia and high-risk myelodysplastic syndrome with either wild-type or mutated FLT3. J. Clin. Oncol. 2010, 28, 4339–4345. [Google Scholar] [CrossRef] [Green Version]
- Borthakur, G.; Kantarjian, H.; Ravandi, F.; Zhang, W.; Konopleva, M.; Wright, J.J.; Faderl, S.; Verstovsek, S.; Mathews, S.; Andreeff, M.; et al. Phase I study of sorafenib in patients with refractory or relapsed acute leukemias. Haematologica 2011, 96, 62–68. [Google Scholar] [CrossRef] [Green Version]
- Ravandi, F.; Alattar, M.L.; Grunwald, M.R.; Rudek, M.A.; Rajkhowa, T.; Richie, M.A.; Pierce, S.; Daver, N.; Garcia-Manero, G.; Faderl, S.; et al. Phase 2 study of azacytidine plus sorafenib in patients with acute myeloid leukemia and FLT-3 internal tandem duplication mutation. Blood 2013, 121, 4655–4662. [Google Scholar] [CrossRef]
- Zhang, C.; Lam, S.S.Y.; Leung, G.M.K.; Tsui, S.P.; Yang, N.; Ng, N.K.L.; Ip, H.W.; Au, C.H.; Chan, T.L.; Ma, E.S.K.; et al. Sorafenib and omacetaxine mepesuccinate as a safe and effective treatment for acute myeloid leukemia carrying internal tandem duplication of Fms-like tyrosine kinase 3. Cancer 2019, 126, 344–353. [Google Scholar] [CrossRef]
- Stone, R.M.; DeAngelo, D.J.; Klimek, V.; Galinsky, I.; Estey, E.; Nimer, S.D.; Grandin, W.; Lebwohl, D.; Wang, Y.; Cohen, P.; et al. Patients with acute myeloid leukemia and an activating mutation in FLT3 respond to a small-molecule FLT3 tyrosine kinase inhibitor, PKC412. Blood 2005, 105, 54–60. [Google Scholar] [CrossRef]
- Strati, P.; Kantarjian, H.; Ravandi, F.; Nazha, A.; Borthakur, G.; Daver, N.; Kadia, T.; Estrov, Z.; Garcia-Manero, G.; Konopleva, M.; et al. Phase I/II trial of the combination of midostaurin (PKC412) and 5-azacytidine for patients with acute myeloid leukemia and myelodysplastic syndrome. Am. J. Hematol. 2015, 90, 276–281. [Google Scholar] [CrossRef] [Green Version]
- Levis, M.; Ravandi, F.; Wang, E.S.; Baer, M.R.; Perl, A.; Coutre, S.; Erba, H.; Stuart, R.K.; Baccarani, M.; Cripe, L.D.; et al. Results from a randomized trial of salvage chemotherapy followed by lestaurtinib for patients with FLT3 mutant AML in first relapse. Blood 2011, 117, 3294–3301. [Google Scholar] [CrossRef]
- Cortes, J.E.; Kantarjian, H.M.; Kadia, T.M.; Borthakur, G.; Konopleva, M.; Garcia-Manero, G.; Daver, N.G.; Pemmaraju, N.; Jabbour, E.; Estrov, Z. Crenolanib besylate, a type I pan-FLT3 inhibitor, to demonstrate clinical activity in multiply relapsed FLT3-ITD and D835 AML. Am. Soc. Clin. Oncol. J. 2016, 34, 7008. [Google Scholar] [CrossRef]
- Cortes, J.E.; Kantarjian, H.; Foran, J.M.; Ghirdaladze, D.; Zodelava, M.; Borthakur, G.; Gammon, G.; Trone, D.; Armstrong, R.C.; James, J.; et al. Phase I study of quizartinib administered daily to patients with relapsed or refractory acute myeloid leukemia irrespective of FMS-like tyrosine kinase 3-internal tandem duplication status. J. Clin. Oncol. 2013, 31, 3681–3687. [Google Scholar] [CrossRef]
- Cortes, J.E.; Tallman, M.S.; Schiller, G.J.; Trone, D.; Gammon, G.; Goldberg, S.L.; Perl, A.E.; Marie, J.P.; Martinelli, G.; Kantarjian, H.M.; et al. Phase 2b study of 2 dosing regimens of quizartinib monotherapy in FLT3-ITD-mutated, relapsed or refractory AML. Blood 2018, 132, 598–607. [Google Scholar] [CrossRef]
- Cortes, J.; Perl, A.E.; Dohner, H.; Kantarjian, H.; Martinelli, G.; Kovacsovics, T.; Rousselot, P.; Steffen, B.; Dombret, H.; Estey, E.; et al. Quizartinib, an FLT3 inhibitor, as monotherapy in patients with relapsed or refractory acute myeloid leukaemia: An open-label, multicentre, single-arm, phase 2 trial. Lancet Oncol. 2018, 19, 889–903. [Google Scholar] [CrossRef]
- Perl, A.E.; Altman, J.K.; Cortes, J.; Smith, C.; Litzow, M.; Baer, M.R.; Claxton, D.; Erba, H.P.; Gill, S.; Goldberg, S.; et al. Selective inhibition of FLT3 by gilteritinib in relapsed or refractory acute myeloid leukaemia: A multicentre, first-in-human, open-label, phase 1–2 study. Lancet Oncol. 2017, 18, 1061–1075. [Google Scholar] [CrossRef]
- Roboz, G.J.; Rosenblat, T.; Arellano, M.; Gobbi, M.; Altman, J.K.; Montesinos, P.; O’Connell, C.; Solomon, S.R.; Pigneux, A.; Vey, N.; et al. International randomized phase III study of elacytarabine versus investigator choice in patients with relapsed/refractory acute myeloid leukemia. J. Clin. Oncol. 2014, 32, 1919–1926. [Google Scholar] [CrossRef]
- Smith, C.C.; Wang, Q.; Chin, C.S.; Salerno, S.; Damon, L.E.; Levis, M.J.; Perl, A.E.; Travers, K.J.; Wang, S.; Hunt, J.P.; et al. Validation of ITD mutations in FLT3 as a therapeutic target in human acute myeloid leukaemia. Nature 2012, 485, 260–263. [Google Scholar] [CrossRef] [Green Version]
- Moore, A.S.; Faisal, A.; Gonzalez de Castro, D.; Bavetsias, V.; Sun, C.; Atrash, B.; Valenti, M.; de Haven Brandon, A.; Avery, S.; Mair, D.; et al. Selective FLT3 inhibition of FLT3-ITD+ acute myeloid leukaemia resulting in secondary D835Y mutation: A model for emerging clinical resistance patterns. Leukemia 2012, 26, 1462–1470. [Google Scholar] [CrossRef]
- Smith, C.C.; Paguirigan, A.; Jeschke, G.R.; Lin, K.C.; Massi, E.; Tarver, T.; Chin, C.S.; Asthana, S.; Olshen, A.; Travers, K.J.; et al. Heterogeneous resistance to quizartinib in acute myeloid leukemia revealed by single-cell analysis. Blood 2017, 130, 48–58. [Google Scholar] [CrossRef]
- Zhang, H.; Savage, S.; Schultz, A.R.; Bottomly, D.; White, L.; Segerdell, E.; Wilmot, B.; McWeeney, S.K.; Eide, C.A.; Nechiporuk, T.; et al. Clinical resistance to crenolanib in acute myeloid leukemia due to diverse molecular mechanisms. Nat. Commun. 2019, 10, 244. [Google Scholar] [CrossRef] [PubMed]
- McMahon, C.M.; Ferng, T.; Canaani, J.; Wang, E.S.; Morrissette, J.J.; Eastburn, D.J.; Pellegrino, M.; Durruthy-Durruthy, R.; Watt, C.D.; Asthana, S.; et al. Clonal selection with Ras pathway activation mediates secondary clinical resistance to selective FLT3 inhibition in acute myeloid leukemia. Cancer Discov. 2019, 9, 1050–1063. [Google Scholar] [CrossRef] [PubMed]
- Heidel, F.; Solem, F.K.; Breitenbuecher, F.; Lipka, D.B.; Kasper, S.; Thiede, M.H.; Brandts, C.; Serve, H.; Roesel, J.; Giles, F.; et al. Clinical resistance to the kinase inhibitor PKC412 in acute myeloid leukemia by mutation of Asn-676 in the FLT3 tyrosine kinase domain. Blood 2006, 107, 293–300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daver, N.; Cortes, J.; Ravandi, F.; Patel, K.P.; Burger, J.A.; Konopleva, M.; Kantarjian, H. Secondary mutations as mediators of resistance to targeted therapy in leukemia. Blood 2015, 125, 3236–3245. [Google Scholar] [CrossRef] [PubMed]
- Smith, C.C.; Lin, K.; Stecula, A.; Sali, A.; Shah, N.P. FLT3 D835 mutations confer differential resistance to type II FLT3 inhibitors. Leukemia 2015, 29, 2390–2392. [Google Scholar] [CrossRef] [Green Version]
- Smith, C.C.; Lasater, E.A.; Zhu, X.; Lin, K.C.; Stewart, W.K.; Damon, L.E.; Salerno, S.; Shah, N.P. Activity of ponatinib against clinically-relevant AC220-resistant kinase domain mutants of FLT3-ITD. Blood 2013, 121, 3165–3171. [Google Scholar] [CrossRef] [Green Version]
- Smith, C.C.; Zhang, C.; Lin, K.C.; Lasater, E.A.; Zhang, Y.; Massi, E.; Damon, L.E.; Pendleton, M.; Bashir, A.; Sebra, R.; et al. Characterizing and overriding the structural mechanism of the quizartinib-resistant FLT3 “Gatekeeper” F691L mutation with PLX3397. Cancer Discov. 2015, 5, 668–679. [Google Scholar] [CrossRef] [Green Version]
- Sato, T.; Yang, X.; Knapper, S.; White, P.; Smith, B.D.; Galkin, S.; Small, D.; Burnett, A.; Levis, M. FLT3 ligand impedes the efficacy of FLT3 inhibitors in vitro and In Vivo. Blood 2011, 117, 3286–3293. [Google Scholar] [CrossRef] [Green Version]
- Chen, F.; Ishikawa, Y.; Akashi, A.; Naoe, T.; Kiyoi, H. Co-Expression of wild-type FLT3 attenuates the inhibitory effect of FLT3 inhibitor on FLT3 mutated leukemia cells. Oncotarget 2016, 7, 47018–47032. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Man, C.H.; Lam, S.S.; Sun, M.K.; Chow, H.C.; Gill, H.; Kwong, Y.L.; Leung, A.Y. A novel tescalcin-sodium/hydrogen exchange axis underlying sorafenib resistance in FLT3-ITD+ AML. Blood 2014, 123, 2530–2539. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Green, A.S.; Maciel, T.T.; Hospital, M.A.; Yin, C.; Mazed, F.; Townsend, E.C.; Pilorge, S.; Lambert, M.; Paubelle, E.; Jacquel, A.; et al. Pim kinases modulate resistance to FLT3 tyrosine kinase inhibitors in FLT3-ITD acute myeloid leukemia. Sci. Adv. 2015, 1, e1500221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, I.K.; Mundy-Bosse, B.; Whitman, S.P.; Zhang, X.; Warner, S.L.; Bearss, D.J.; Blum, W.; Marcucci, G.; Caligiuri, M.A. Receptor tyrosine kinase Axl is required for resistance of leukemic cells to FLT3-targeted therapy in acute myeloid leukemia. Leukemia 2015, 29, 2382–2389. [Google Scholar] [CrossRef]
- Dumas, P.Y.; Naudin, C.; Martin-Lanneree, S.; Izac, B.; Casetti, L.; Mansier, O.; Rousseau, B.; Artus, A.; Dufossee, M.; Giese, A.; et al. Hematopoietic niche drives FLT3-ITD acute myeloid leukemia resistance to quizartinib via STAT5-and hypoxia-dependent upregulation of AXL. Haematologica 2019, 104, 2017–2027. [Google Scholar] [CrossRef] [Green Version]
- Ueno, Y.; Mori, M.; Kamiyama, Y.; Saito, R.; Kaneko, N.; Isshiki, E.; Kuromitsu, S.; Takeuchi, M. Evaluation of gilteritinib in combination with chemotherapy in preclinical models of FLT3-ITD+ acute myeloid leukemia. Oncotarget 2019, 10, 2530–2545. [Google Scholar] [CrossRef] [Green Version]
- Lindblad, O.; Cordero, E.; Puissant, A.; Macaulay, L.; Ramos, A.; Kabir, N.N.; Sun, J.; Vallon-Christersson, J.; Haraldsson, K.; Hemann, M.T.; et al. Aberrant activation of the PI3K/mTOR pathway promotes resistance to sorafenib in AML. Oncogene 2016, 35, 5119–5131. [Google Scholar] [CrossRef]
- Piloto, O.; Wright, M.; Brown, P.; Kim, K.T.; Levis, M.; Small, D. Prolonged exposure to FLT3 inhibitors leads to resistance via activation of parallel signaling pathways. Blood 2007, 109, 1643–1652. [Google Scholar] [CrossRef] [Green Version]
- Scadden, D.T. Nice neighborhood: Emerging concepts of the stem cell niche. Cell 2014, 157, 41–50. [Google Scholar] [CrossRef] [Green Version]
- Baryawno, N.; Przybylski, D.; Kowalczyk, M.S.; Kfoury, Y.; Severe, N.; Gustafsson, K.; Kokkaliaris, K.D.; Mercier, F.; Tabaka, M.; Hofree, M.; et al. A cellular taxonomy of the bone marrow stroma in homeostasis and leukemia. Cell 1915, 177, 1915–1932.e1916. [Google Scholar] [CrossRef]
- Zambetti, N.A.; Ping, Z.; Chen, S.; Kenswil, K.J.G.; Mylona, M.A.; Sanders, M.A.; Hoogenboezem, R.M.; Bindels, E.M.J.; Adisty, M.N.; Van Strien, P.M.H.; et al. Mesenchymal inflammation drives genotoxic stress in hematopoietic stem cells and predicts disease evolution in human pre-leukemia. Cell Stem Cell 2016, 19, 613–627. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sung, P.J.; Sugita, M.; Koblish, H.; Perl, A.E.; Carroll, M. Hematopoietic cytokines mediate resistance to targeted therapy in FLT3-ITD acute myeloid leukemia. Blood Adv. 2019, 3, 1061–1072. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Traer, E.; Martinez, J.; Javidi-Sharifi, N.; Agarwal, A.; Dunlap, J.; English, I.; Kovacsovics, T.; Tyner, J.W.; Wong, M.; Druker, B.J. FGF2 from marrow microenvironment promotes resistance to FLT3 inhibitors in acute myeloid leukemia. Cancer Res. 2016, 76, 6471–6482. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Javidi-Sharifi, N.; Martinez, J.; English, I.; Joshi, S.K.; Scopim-Ribeiro, R.; Viola, S.K.; Edwards, D.K.T.; Agarwal, A.; Lopez, C.; Jorgens, D.; et al. FGF2-FGFR1 signaling regulates release of Leukemia-Protective exosomes from bone marrow stromal cells. eLife 2019, 8, e40033. [Google Scholar] [CrossRef] [PubMed]
- Fukuda, S.; Broxmeyer, H.E.; Pelus, L.M. Flt3 ligand and the Flt3 receptor regulate hematopoietic cell migration by modulating the SDF-1alpha(CXCL12)/CXCR4 axis. Blood 2005, 105, 3117–3126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeng, Z.; Shi, Y.X.; Samudio, I.J.; Wang, R.Y.; Ling, X.; Frolova, O.; Levis, M.; Rubin, J.B.; Negrin, R.R.; Estey, E.H.; et al. Targeting the leukemia microenvironment by CXCR4 inhibition overcomes resistance to kinase inhibitors and chemotherapy in AML. Blood 2009, 113, 6215–6224. [Google Scholar] [CrossRef] [Green Version]
- Kojima, K.; McQueen, T.; Chen, Y.; Jacamo, R.; Konopleva, M.; Shinojima, N.; Shpall, E.; Huang, X.; Andreeff, M. p53 activation of mesenchymal stromal cells partially abrogates microenvironment-mediated resistance to FLT3 inhibition in AML through HIF-1alpha-mediated down-regulation of CXCL12. Blood 2011, 118, 4431–4439. [Google Scholar] [CrossRef] [Green Version]
- Teo, Y.L.; Ho, H.K.; Chan, A. Metabolism-Related pharmacokinetic drug-drug interactions with tyrosine kinase inhibitors: Current understanding, challenges and recommendations. Br. J. Clin. Pharm. 2015, 79, 241–253. [Google Scholar] [CrossRef] [Green Version]
- Chang, Y.T.; Hernandez, D.; Alonso, S.; Gao, M.; Su, M.; Ghiaur, G.; Levis, M.J.; Jones, R.J. Role of CYP3A4 in bone marrow microenvironment-mediated protection of FLT3/ITD AML from tyrosine kinase inhibitors. Blood Adv. 2019, 3, 908–916. [Google Scholar] [CrossRef]
- Rollig, C.; Serve, H.; Huttmann, A.; Noppeney, R.; Muller-Tidow, C.; Krug, U.; Baldus, C.D.; Brandts, C.H.; Kunzmann, V.; Einsele, H.; et al. Addition of sorafenib versus placebo to standard therapy in patients aged 60 years or younger with newly diagnosed acute myeloid leukaemia (SORAML): A multicentre, phase 2, randomised controlled trial. Lancet Oncol. 2015, 16, 1691–1699. [Google Scholar] [CrossRef]
- Sasaki, K.; Kantarjian, H.M.; Kadia, T.; Patel, K.; Loghavi, S.; Garcia-Manero, G.; Jabbour, E.J.; DiNardo, C.; Pemmaraju, N.; Daver, N.; et al. Sorafenib plus intensive chemotherapy improves survival in patients with newly diagnosed, FLT3-internal tandem duplication mutation-positive acute myeloid leukemia. Cancer 2019, 125, 3755–3766. [Google Scholar] [CrossRef]
- Goldberg, A.D.; Coombs, C.C.; Wang, E.S.; Walter, R.B.; Karanes, C.; Vigil, C.E.; Messahel, B.; Stone, R.M.; Collins, R.H. Younger patients with newly diagnosed FLT3-mutant AML treated with crenolanib plus chemotherapy achieve adequate free crenolanib levels and durable remissions. Blood 2019, 134, 1326. [Google Scholar] [CrossRef]
- Lancet, J.E.; Uy, G.L.; Cortes, J.E.; Newell, L.F.; Lin, T.L.; Ritchie, E.K.; Stuart, R.K.; Strickland, S.A.; Hogge, D.; Solomon, S.R.; et al. CPX-351 (cytarabine and daunorubicin) liposome for injection versus conventional cytarabine plus daunorubicin in older patients with newly diagnosed secondary acute myeloid leukemia. J. Clin. Oncol. 2018, 36, 2684–2692. [Google Scholar] [CrossRef] [PubMed]
- Gordon, M.J.; Tardi, P.; Loriaux, M.M.; Spurgeon, S.E.; Traer, E.; Kovacsovics, T.; Mayer, L.D.; Tyner, J.W. CPX-351 exhibits potent and direct ex vivo cytotoxicity against AML blasts with enhanced efficacy for cells harboring the FLT3-ITD mutation. Leuk. Res. 2017, 53, 39–49. [Google Scholar] [CrossRef]
- Lam, S.S.; Ho, E.S.; He, B.L.; Wong, W.W.; Cher, C.Y.; Ng, N.K.; Man, C.H.; Gill, H.; Cheung, A.M.; Ip, H.W.; et al. Homoharringtonine (omacetaxine mepesuccinate) as an adjunct for FLT3-ITD acute myeloid leukemia. Sci. Transl. Med. 2016, 8, 359ra129. [Google Scholar] [CrossRef] [PubMed]
- Gurel, G.; Blaha, G.; Moore, P.B.; Steitz, T.A. U2504 determines the species specificity of the A-site cleft antibiotics: The structures of tiamulin, homoharringtonine, and bruceantin bound to the ribosome. J. Mol. Biol. 2009, 389, 146–156. [Google Scholar] [CrossRef] [Green Version]
- Zhang, C.; Leung, G.M.; Tsui, S.P.; Ip, H.-W.; Ma, E.S.K.; Au, C.H.; Chan, C.T.L.; Ng, K.L.N.; Yip, S.F.; Kho, B.; et al. A phase II single-arm open-labeled study evaluating combination of Quizartinib and Omacetaxine Mepesuccinate (QUIZOM) in newly diagnosed or relapsed/refractory AML carrying FlT3-ITD. Blood 2019, 134, 3825. [Google Scholar] [CrossRef]
- Gill, H.; Man, C.H.; Ip, A.H.; Choi, W.W.; Chow, H.C.; Kwong, Y.L.; Leung, A.Y. Azacitidine as post-remission consolidation for sorafenib-induced remission of Fms-like tyrosine kinase-3 internal tandem duplication positive acute myeloid leukemia. Haematologica 2015, 100, e250–e253. [Google Scholar] [CrossRef] [Green Version]
- Muppidi, M.R.; Portwood, S.; Griffiths, E.A.; Thompson, J.E.; Ford, L.A.; Freyer, C.W.; Wetzler, M.; Wang, E.S. Decitabine and Sorafenib therapy in FLT-3 ITD-mutant acute myeloid leukemia. Clin. Lymphoma Myeloma Leuk. 2015, 15 (Suppl. S73–S79). [Google Scholar] [CrossRef]
- Chang, E.; Ganguly, S.; Rajkhowa, T.; Gocke, C.D.; Levis, M.; Konig, H. The combination of FLT3 and DNA methyltransferase inhibition is synergistically cytotoxic to FLT3/ITD acute myeloid leukemia cells. Leukemia 2016, 30, 1025–1032. [Google Scholar] [CrossRef] [Green Version]
- Swaminathan, M.; Kantarjian, H.M.; Daver, N.; Borthakur, G.; Ohanian, M.; Kadia, T.; DiNardo, C.D.; Jain, N.; Estrov, Z.; Ferrajoli, A.; et al. The combination of Quizartinib with Azacitidine or low dose Cytarabine is highly active in patients (Pts) with FLT3-ITD mutated myeloid leukemias: Interim report of a phase I/II trial. Blood 2017, 130, 723. [Google Scholar] [CrossRef]
- Ohanian, M.; Garcia-Manero, G.; Levis, M.; Jabbour, E.; Daver, N.; Borthakur, G.; Kadia, T.; Pierce, S.; Burger, J.; Richie, M.A.; et al. Sorafenib combined with 5-azacytidine in older patients with untreated FLT3-ITD mutated acute myeloid leukemia. Am. J. Hematol. 2018, 93, 1136–1141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bornhauser, M.; Illmer, T.; Schaich, M.; Soucek, S.; Ehninger, G.; Thiede, C. AML SHG 96 Study Group. Improved outcome after stem-cell transplantation in FLT3/ITD-positive AML. Blood 2007, 109, 2264–2265, author reply 2265. [Google Scholar] [CrossRef] [PubMed]
- Palmieri, S.; Ferrara, F.; Leoni, F.; Ciolli, S.; Pollio, F.; D’Amico, M.R.; Celentano, M.; Viola, A.; Vicari, L.; Izzo, B.; et al. Myeloablative chemotherapy followed by autologous stem cell infusion may overcome the adverse prognostic impact of FLT3 (foetal liver tyrosine kinase 3) mutations in patients with acute myeloid leukaemia and normal karyotype. Hematol. Oncol. 2007, 25, 1–5. [Google Scholar] [CrossRef]
- DeZern, A.E.; Sung, A.; Kim, S.; Smith, B.D.; Karp, J.E.; Gore, S.D.; Jones, R.J.; Fuchs, E.; Luznik, L.; McDevitt, M.; et al. Role of allogeneic transplantation for FLT3/ITD acute myeloid leukemia: Outcomes from 133 consecutive newly diagnosed patients from a single institution. Biol. Blood Marrow Transplant. 2011, 17, 1404–1409. [Google Scholar] [CrossRef] [Green Version]
- Canaani, J.; Labopin, M.; Huang, X.J.; Arcese, W.; Ciceri, F.; Blaise, D.; Irrera, G.; Corral, L.L.; Bruno, B.; Santarone, S.; et al. T-Cell replete haploidentical stem cell transplantation attenuates the prognostic impact of FLT3-ITD in acute myeloid leukemia: A report from the Acute Leukemia Working Party of the European Society for Blood and Marrow Transplantation. Am. J. Hematol. 2018, 93, 736–744. [Google Scholar] [CrossRef] [Green Version]
- Sakaguchi, M.; Yamaguchi, H.; Najima, Y.; Usuki, K.; Ueki, T.; Oh, I.; Mori, S.; Kawata, E.; Uoshima, N.; Kobayashi, Y.; et al. Prognostic impact of low allelic ratio FLT3-ITD and NPM1 mutation in acute myeloid leukemia. Blood Adv. 2018, 2, 2744–2754. [Google Scholar] [CrossRef] [Green Version]
- Mathew, N.R.; Baumgartner, F.; Braun, L.; O’Sullivan, D.; Thomas, S.; Waterhouse, M.; Muller, T.A.; Hanke, K.; Taromi, S.; Apostolova, P.; et al. Sorafenib promotes graft-versus-leukemia activity in mice and humans through IL-15 production in FLT3-ITD-mutant leukemia cells. Nat. Med. 2018, 24, 282–291. [Google Scholar] [CrossRef]
- Maziarz, R.T.; Fernandez, H.; Patnaik, M.M.; Scott, B.L.; Mohan, S.; Deol, A.; Rowley, S.D.; Kim, D.D.; Rajkhowa, T.; Haines, K.; et al. Radius: Midostaurin (mido) plus standard of care (SOC) after allogeneic stem cell transplant (alloSCT) in patients (pts) with FLT3-internal tandem duplication (ITD)-mutated acute myeloid leukemia (AML). Biol. Blood Marrow Transplant. 2019, 25, S11–S12. [Google Scholar] [CrossRef] [Green Version]
- Sandmaier, B.M.; Khaled, S.; Oran, B.; Gammon, G.; Trone, D.; Frankfurt, O. Results of a phase 1 study of quizartinib as maintenance therapy in subjects with acute myeloid leukemia in remission following allogeneic hematopoietic stem cell transplant. Am. J. Hematol. 2018, 93, 222–231. [Google Scholar] [CrossRef]
- Lange, A.; Jaskula, E.; Lange, J.; Dworacki, G.; Nowak, D.; Simiczyjew, A.; Mordak-Domagala, M.; Sedzimirska, M. The sorafenib anti-relapse effect after alloHSCT is associated with heightened alloreactivity and accumulation of CD8+PD-1+ (CD279+) lymphocytes in marrow. PLoS ONE 2018, 13, e0190525. [Google Scholar] [CrossRef] [Green Version]
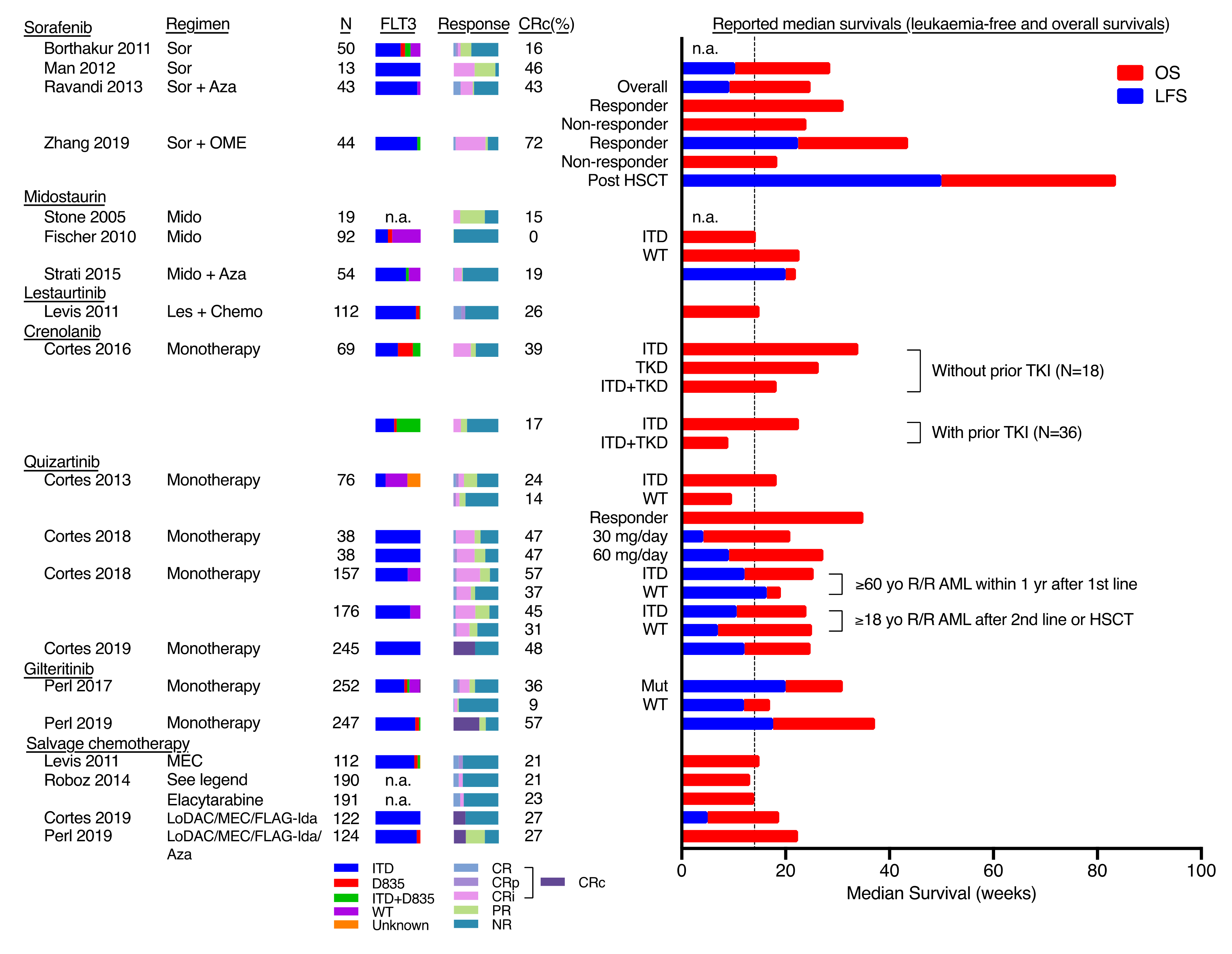
Figure 1.
Results of clinical trials involving Fms-Like Tyrosine kinase 3 (FLT3) inhibitors on relapsed/refractory AML. The reported sample size (N), percentage of FLT3-ITD, D835, ITD/D835 and wildtype (WT), and response rate and duration of reported clinical trials on the use of FLT3 inhibitors on relapsed/refractory (R/R) AML were shown here. The response duration was represented as bar charts (right) for graphical presentation and it was not intended for direct statistical comparison between studies. The vertical dotted line represented the estimated pooled median overall survival in R/R patients treated with salvage chemotherapy (around 14 weeks). CR: complete remission; CRp: complete remission with incomplete platelet count; CRi: complete remission with incomplete haematological recovery; PR: partial response; NR: no response; CRc: composite complete remission rate = CR + CRp + CRi; Sor: sorafenib; Aza: Azacytidine; OME: omacetaxine mepesuccinate; Mido: midostaurin; Les: Lestaurtinib; MEC: mitoxantrone, etoposide, cytarabine; FLAG-Ida: Fludarabine, cytarabine, G-CSF, Idarubicin; LoDAC: low-dose cytarabine; HiDAC: high-dose cytarabine; TKI: tyrosine kinase inhibitors; TKI: tyrosine kinase inhibitor(s). The chemotherapy reported in Roboz et al. (2014) included investigators’ choice among 7 salvage regimens: HiDAC, MEC, FLAG/FLAG-Ida, LoDAC, hypomethylating agents, hydroxyurea, or supportive care. Reference (top to bottom): [18,10,19,20,21,17,22,23,24,25,26,27,4,28,3,23,29,4,3].
Figure 1.
Results of clinical trials involving Fms-Like Tyrosine kinase 3 (FLT3) inhibitors on relapsed/refractory AML. The reported sample size (N), percentage of FLT3-ITD, D835, ITD/D835 and wildtype (WT), and response rate and duration of reported clinical trials on the use of FLT3 inhibitors on relapsed/refractory (R/R) AML were shown here. The response duration was represented as bar charts (right) for graphical presentation and it was not intended for direct statistical comparison between studies. The vertical dotted line represented the estimated pooled median overall survival in R/R patients treated with salvage chemotherapy (around 14 weeks). CR: complete remission; CRp: complete remission with incomplete platelet count; CRi: complete remission with incomplete haematological recovery; PR: partial response; NR: no response; CRc: composite complete remission rate = CR + CRp + CRi; Sor: sorafenib; Aza: Azacytidine; OME: omacetaxine mepesuccinate; Mido: midostaurin; Les: Lestaurtinib; MEC: mitoxantrone, etoposide, cytarabine; FLAG-Ida: Fludarabine, cytarabine, G-CSF, Idarubicin; LoDAC: low-dose cytarabine; HiDAC: high-dose cytarabine; TKI: tyrosine kinase inhibitors; TKI: tyrosine kinase inhibitor(s). The chemotherapy reported in Roboz et al. (2014) included investigators’ choice among 7 salvage regimens: HiDAC, MEC, FLAG/FLAG-Ida, LoDAC, hypomethylating agents, hydroxyurea, or supportive care. Reference (top to bottom): [18,10,19,20,21,17,22,23,24,25,26,27,4,28,3,23,29,4,3].

Figure 2.
Mechanisms involved in microenvironment-mediated resistance to FLT3 inhibitors. 1. FLT3L/FLT3R-WT 2. FGF2/FGFR1 axis 3. SDF1/CXCR4 axis 4. CYP3A4-mediated degradation of FLT3 inhibitors (FLT3i) 5. GM-CSF or IL3/IL3R axis.
Figure 2.
Mechanisms involved in microenvironment-mediated resistance to FLT3 inhibitors. 1. FLT3L/FLT3R-WT 2. FGF2/FGFR1 axis 3. SDF1/CXCR4 axis 4. CYP3A4-mediated degradation of FLT3 inhibitors (FLT3i) 5. GM-CSF or IL3/IL3R axis.


{kind=link}
{kind=link}
Table 1.
Eight FDA-approved therapeutic agents in acute myeloid leukaemia (AML).
| Therapeutic Agents | Indications |
|---|---|
| FLT3 inhibitors | |
| 1. Midostaurin | Newly diagnosed FLT3-mutated AML |
| 2. Gilteritinib | Relapsed/refractory FLT3-mutated AML |
| IDH inhibitors | |
| 3. Ivosidenib | Relapsed/refractory IDH1-mutated AML |
| 4. Enasidenib | Newly diagnosed or relapsed/refractory IDH2-mutated AML |
| BCL2 inhibitor | |
| 5. Venetoclax + hypomethylating agents or LoDAC | Newly diagnosed AML aged ≥ 75 |
| Hedgehog pathway inhibitor | |
| 6. Glasdegib + LoDAC | Newly diagnosed AML aged ≥ 75 |
| Liposomal combination of daunorubicin and cytarabine | |
| 7. CPX-351 | Newly diagnosed AML-MRC and t-AML |
| Antibody-chemotherapy adjunct | |
| 8. Gemtuzumab ozogamicin | Newly diagnosed and relapsed/refractory CD33-positive AML |
MRC: MDS-related changes; t-AML: transformed AML; LoDAC: low-dose cytarabine.
Table 2.
FLT3-TKD mutations conferring clinical resistance to FLT3 inhibitors.
| TKD Mutations | FLT3 Inhibitor Resistance |
|---|---|
| N676K | Resistance to midostaurin [35] |
| D835 I836 Y842 | Resistance to type II FLT3 inhibitors [36,37] (sorafenib, quizartinib, ponatinib) |
| F691L | Resistance to crenolanib, but not to ponatinib [38] and pexidartinib [39] |
| K429E | Resistance to crenolanib [33] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Lam, S.S.Y.; Leung, A.Y.H. Overcoming Resistance to FLT3 Inhibitors in the Treatment of FLT3-Mutated AML. Int. J. Mol. Sci. 2020, 21, 1537. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21041537
AMA Style
Lam SSY, Leung AYH. Overcoming Resistance to FLT3 Inhibitors in the Treatment of FLT3-Mutated AML. International Journal of Molecular Sciences. 2020; 21(4):1537. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21041537
Chicago/Turabian StyleLam, Stephen S.Y., and Anskar Y.H. Leung. 2020. "Overcoming Resistance to FLT3 Inhibitors in the Treatment of FLT3-Mutated AML" International Journal of Molecular Sciences 21, no. 4: 1537. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21041537
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.