In Silico Insights into Protein–Protein Interaction Disruptive Mutations in the PCSK9-LDLR Complex


Abstract
:1. Introduction
2. Results
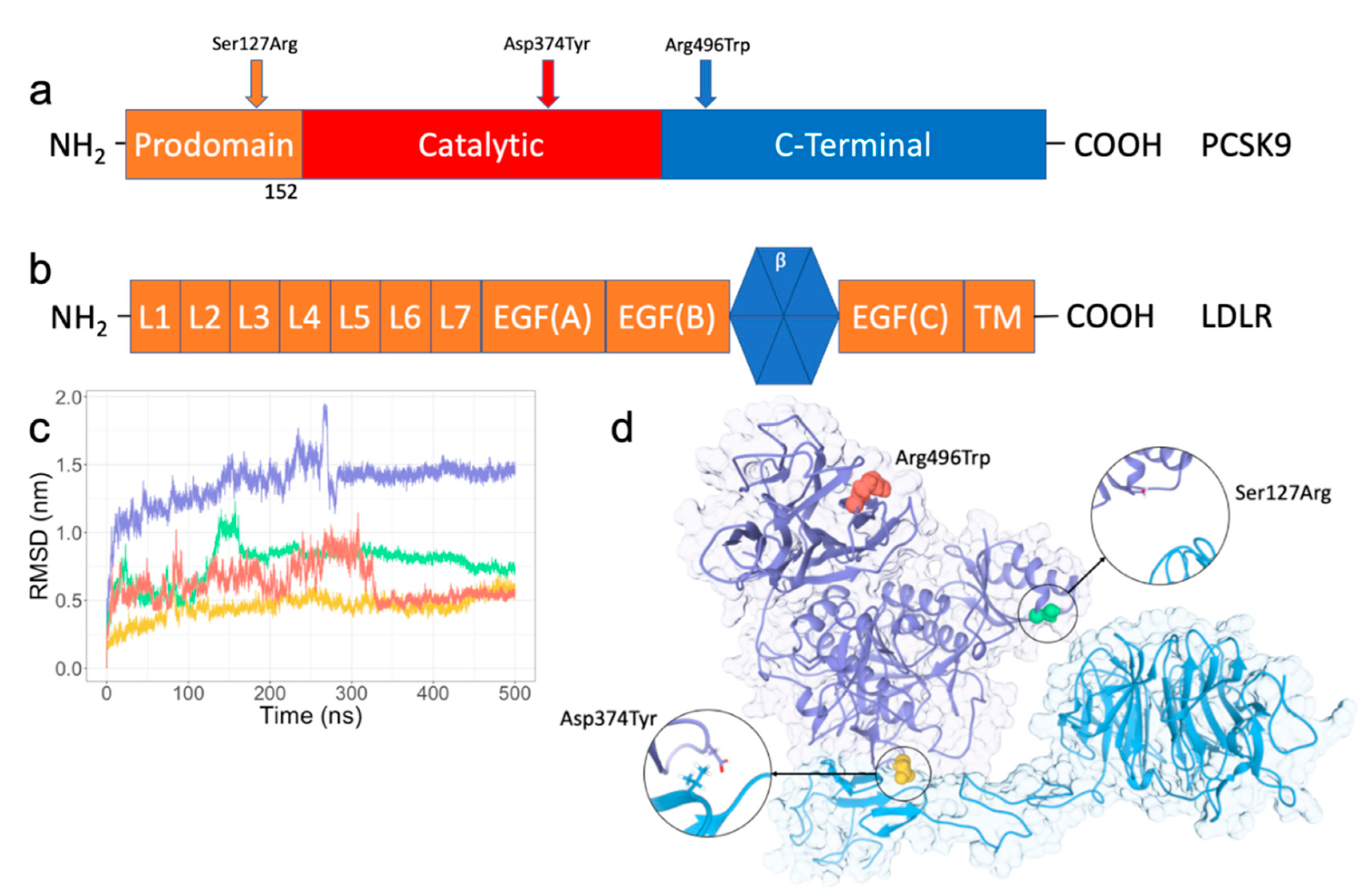
2.1. RMSD/RMSF Analysis of PPI Disruptive Mutations on PCSK9 and LDLR Complex
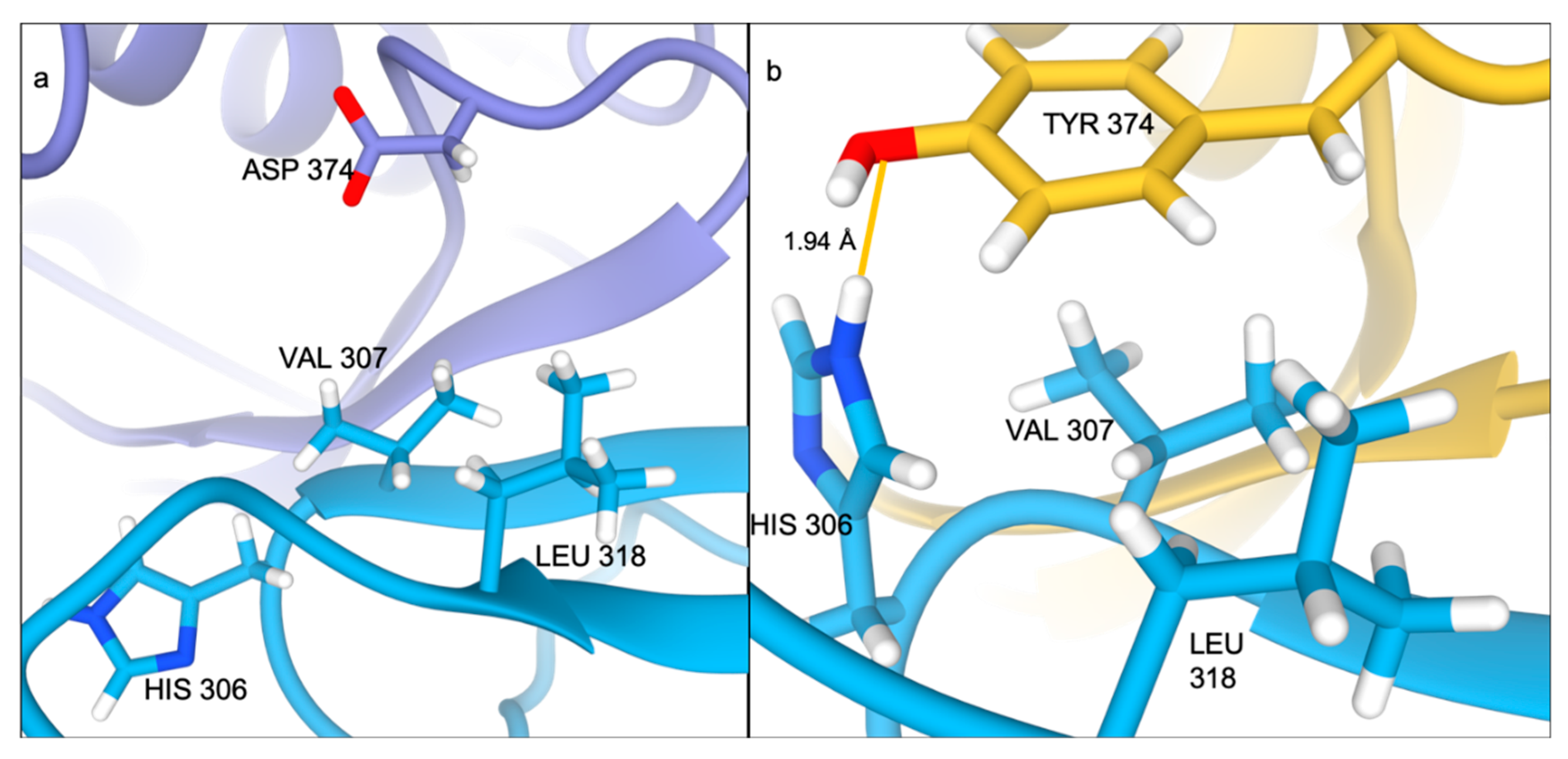
2.2. Ser127Arg/Asp374Tyr Improve the PCSK9-LDLR Interaction by MM/PBSA Analysis
2.3. A direct Contribution by Ser127Arg and Asp374Tyr to the PCSK9-LDLR Interaction by the Domain Decomposition of MM/PBSA
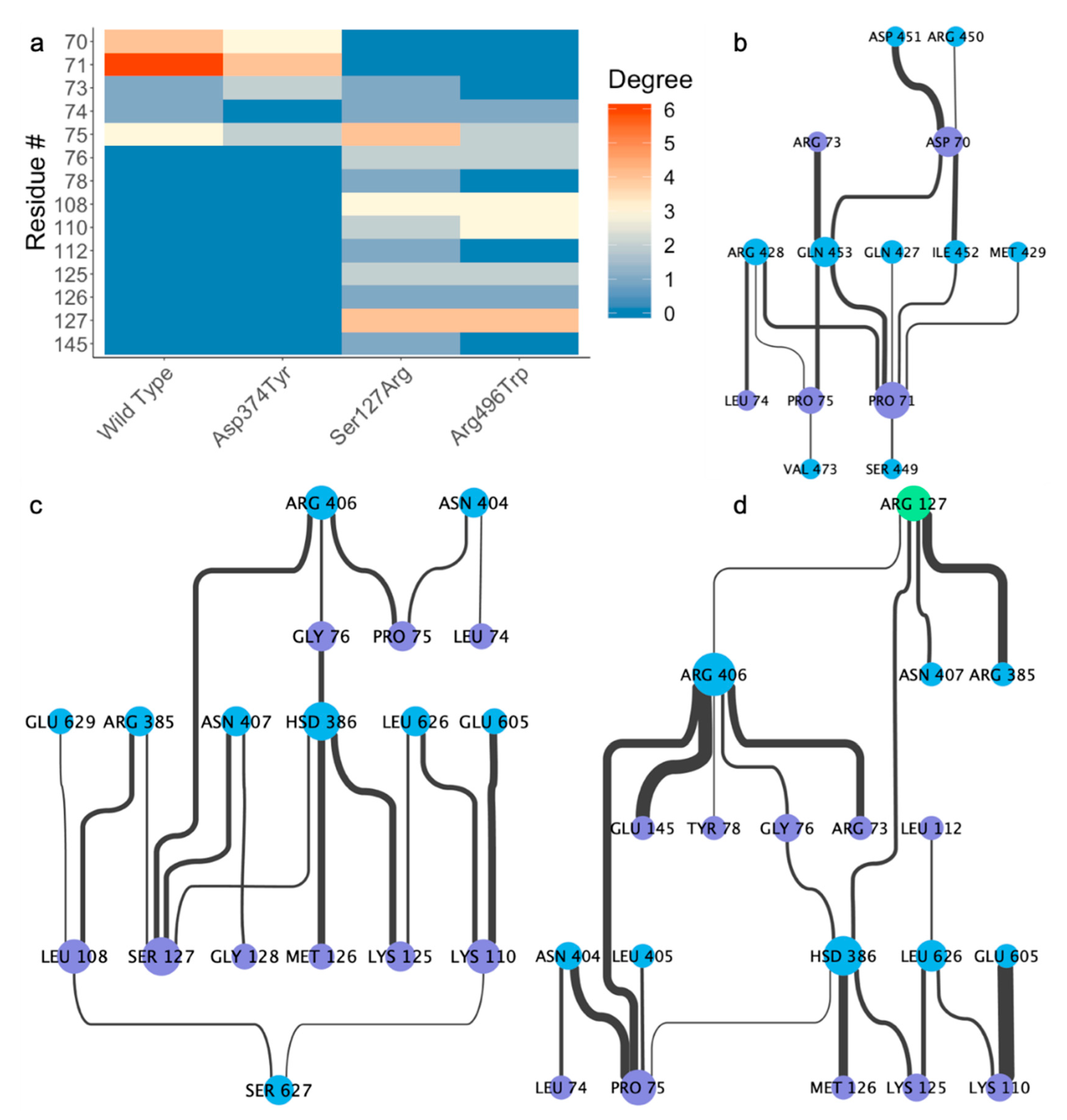
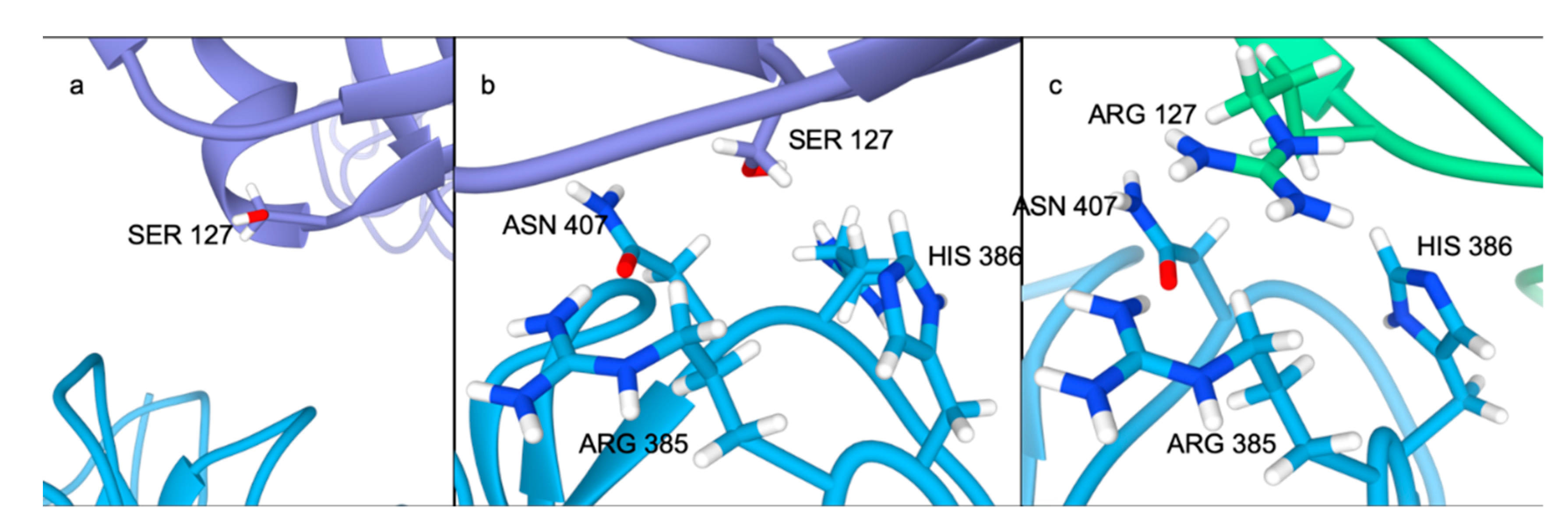
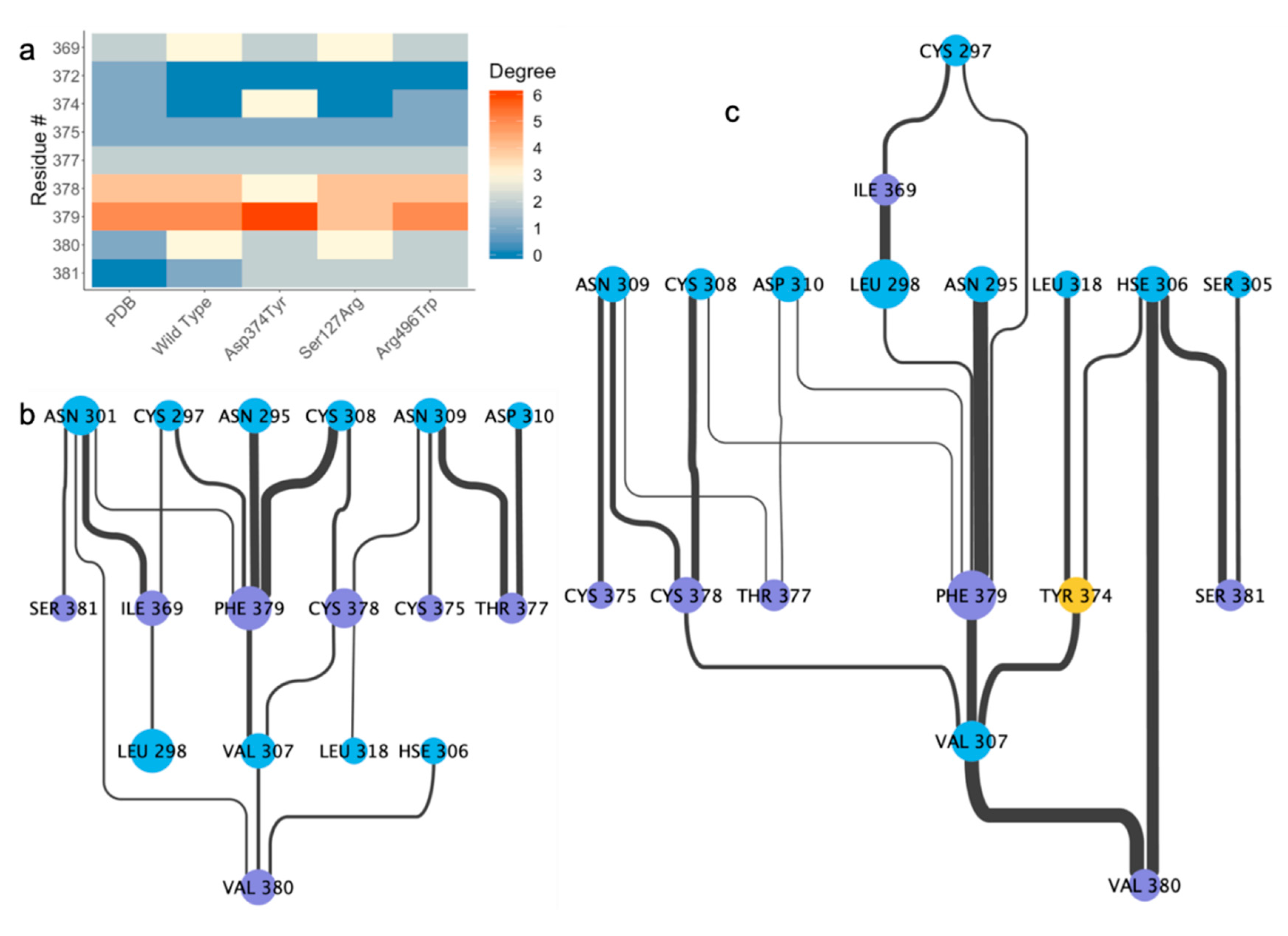
2.4. Residue Interaction Network Analysis Indicates Improved PCSK9/LDLR Binding by Ser127Arg and Asp374Tyr
2.5. Alternative Conformations for the PCSK9/LDLR Interaction
3. Discussion
4. Materials and Methods
4.1. Selection of Disease-Causing Mutations
4.2. System Construction
4.3. Simulation Parameters
4.4. System Analysis
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| APOB | Apolipoprotein B |
| EGFPH | Epidermal growth factor precursor homology domain |
| EFT(A, B, C) | Epidermal growth factor precursor homology domain A, B, C |
| FH | Familial hypercholesterolemia |
| IDL | Intermediate density lipoprotein |
| LDL | Low density lipoprotein |
| LDLR | Low density lipoprotein receptor |
| MD | Molecular dynamics |
| PCSK9 | Proprotein convertase subtilisin/kexin type 9 |
| MM/PBSA | Molecular mechanics/Poisson-Boltzmann surface area |
| VLDL | Very low density lipoprotein |
References
- Hoyert, D.L.; Heron, M.P.; Murphy, S.L.; Kung, H.-C. Deaths: Final Data for 2003. PsycEXTRA Dataset 2013, 68. [Google Scholar]
- Ahmad, Z.S.; Andersen, R.L.; Andersen, L.H.; O’Brien, E.C.; Kindt, I.; Shrader, P.; Vasandani, C.; Newman, C.; Degoma, E.M.; Baum, S.J.; et al. US physician practices for diagnosing familial hypercholesterolemia: Data from the CASCADE-FH registry. J. Clin. Lipidol. 2016, 10, 1223–1229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perak, A.M.; Ning, H.; De Ferranti, S.D.; Gooding, H.C.; Wilkins, J.T.; Lloyd-Jones, D. Long-Term Risk of Atherosclerotic Cardiovascular Disease in US Adults With the Familial Hypercholesterolemia Phenotype. Circulation 2016, 134, 9–19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Ferranti, S.D.; Rodday, A.M.; Mendelson, M.; Wong, J.B.; Leslie, L.K.; Sheldrick, R.C. Prevalence of Familial Hypercholesterolemia in the 1999 to 2012 United States National Health and Nutrition Examination Surveys (NHANES)CLINICAL PERSPECTIVE. Circulation 2016, 133, 1067–1072. [Google Scholar] [CrossRef]
- Armitage, J.; Baigent, C.; Barnes, E.; Betteridge, D.J.; Blackwell, L.; Blazing, M.; Bowman, L.; Braunwald, E.; Byington, R.; Cannon, C.; et al. Efficacy and safety of statin therapy in older people: A meta-analysis of individual participant data from 28 randomised controlled trials. Lancet 2019, 393, 407–415. [Google Scholar] [CrossRef] [Green Version]
- Tabas, I.; Williams, K.J.; Borén, J. Subendothelial Lipoprotein Retention as the Initiating Process in Atherosclerosis. Circulation 2007, 116, 1832–1844. [Google Scholar] [CrossRef]
- Shapiro, M.D.; Fazio, S. Apolipoprotein B-containing lipoproteins and atherosclerotic cardiovascular disease. F1000Research 2017, 6, 134. [Google Scholar] [CrossRef] [Green Version]
- Feingold, K.R.; Grunfeld, C. Introduction to Lipids and Lipoproteins. In Endotext; MDText.com, Inc.: South Dartmouth, MA, USA, 2000. [Google Scholar]
- Segrest, J.P.; Jones, M.K.; De Loof, H.; Dashti, N. Structure of apolipoprotein B-100 in low density lipoproteins. J. Lipid Res. 2001, 42, 1346–1367. [Google Scholar]
- Rudenko, G.; Henry, L.; Henderson, K.; Ichtchenko, K.; Brown, M.S.; Goldstein, J.L.; Deisenhofer, J. Structure of the LDL Receptor Extracellular Domain at Endosomal pH. Science 2002, 298, 2353–2358. [Google Scholar] [CrossRef] [Green Version]
- Fokkema, I.F.A.C.; Taschner, P.; Schaafsma, G.; Celli, J.; Laros, J.F.; Dunnen, J.T.D. LOVD v.2.0: The next generation in gene variant databases. Hum. Mutat. 2011, 32, 557–563. [Google Scholar] [CrossRef]
- Schaefer, J.R.; Kurt, B.; Sattler, A.; Klaus, G.; Soufi, M. Pharmacogenetic aspects in familial hypercholesterolemia with the special focus on FHMarburg (FH p.W556R). Clin. Res. Cardiol. Suppl. 2012, 7, 2–6. [Google Scholar] [CrossRef] [Green Version]
- Abifadel, M.; Varret, M.; Rabès, J.-P.; Allard, D.; Ouguerram, K.; Devillers, M.; Cruaud, C.; Benjannet, S.; Wickham, L.; Erlich, D.; et al. Mutations in PCSK9 cause autosomal dominant hypercholesterolemia. Nat. Genet. 2003, 34, 154–156. [Google Scholar] [CrossRef]
- Zhang, D.-W.; Lagace, T.A.; Garuti, R.; Zhao, Z.; McDonald, M.; Horton, J.D.; Cohen, J.C.; Hobbs, H.H. Binding of Proprotein Convertase Subtilisin/Kexin Type 9 to Epidermal Growth Factor-like Repeat A of Low Density Lipoprotein Receptor Decreases Receptor Recycling and Increases Degradation. J. Boil. Chem. 2007, 282, 18602–18612. [Google Scholar] [CrossRef] [Green Version]
- Bottomley, M.J.; Cirillo, A.; Orsatti, L.; Ruggeri, L.; Fisher, T.S.; Santoro, J.C.; Cummings, R.T.; Cubbon, R.M.; Surdo, P.L.; Calzetta, A.; et al. Structural and Biochemical Characterization of the Wild Type PCSK9-EGF(AB) Complex and Natural Familial Hypercholesterolemia Mutants. J. Boil. Chem. 2008, 284, 1313–1323. [Google Scholar] [CrossRef] [Green Version]
- Kwon, H.J.; Lagace, T.A.; McNutt, M.C.; Horton, J.D.; Deisenhofer, J. Molecular basis for LDL receptor recognition by PCSK9. Proc. Natl. Acad. Sci. USA 2008, 105, 1820–1825. [Google Scholar] [CrossRef] [Green Version]
- Surdo, P.L.; Bottomley, M.J.; Calzetta, A.; Settembre, E.C.; Cirillo, A.; Pandit, S.; Ni, Y.G.; Hubbard, B.; Sitlani, A.; Carfí, A. Mechanistic implications for LDL receptor degradation from the PCSK9/LDLR structure at neutral pH. EMBO Rep. 2011, 12, 1300–1305. [Google Scholar] [CrossRef] [Green Version]
- Leren, T.P. Mutations in the PCSK9 gene in Norwegian subjects with autosomal dominant hypercholesterolemia. Clin. Genet. 2004, 65, 419–422. [Google Scholar] [CrossRef]
- Timms, K.; Wagner, S.; Samuels, M.E.; Forbey, K.; Goldfine, H.; Jammulapati, S.; Skolnick, M.H.; Hopkins, P.N.; Hunt, S.C.; Shattuck, D.M. A mutation in PCSK9 causing autosomal-dominant hypercholesterolemia in a Utah pedigree. Qual. Life Res. 2004, 114, 349–353. [Google Scholar] [CrossRef]
- Cunningham, D.; Danley, D.E.; Geoghegan, K.F.; Griffor, M.C.; Hawkins, J.L.; Subashi, T.A.; Varghese, A.H.; Ammirati, M.J.; Culp, J.S.; Hoth, L.R.; et al. Structural and biophysical studies of PCSK9 and its mutants linked to familial hypercholesterolemia. Nat. Struct. Mol. Boil. 2007, 14, 413–419. [Google Scholar] [CrossRef]
- Benjannet, S.; Rhainds, D.; Hamelin, J.; Nassoury, N.; Seidah, N.G. The proprotein convertase (PC) PCSK9 is inactivated by furin and/or PC5/6A: Functional consequences of natural mutations and post-translational modifications. J. Biol. Chem. 2006, 281, 30561–30572. [Google Scholar] [CrossRef] [Green Version]
- Pisciotta, L.; Oliva, C.P.; Cefalù, A.B.; Noto, D.; Bellocchio, A.; Fresa, R.; Cantafora, A.; Patel, D.; Averna, M.R.; Tarugi, P.; et al. Additive effect of mutations in LDLR and PCSK9 genes on the phenotype of familial hypercholesterolemia. Atherosclerosis 2006, 186, 433–440. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, S.K.; Foo, A.C.; Matyas, A.; Kosenko, T.; Goto, N.; Vergara-Jaque, A.; Lagace, T.A. A transient amphipathic helix in PCSK9’s prodomain facilitates low-density lipoprotein binding. BioRxiv 2019, 743195. [Google Scholar]
- Deng, S.-J.; Alabi, A.; Gu, H.-M.; Adijiang, A.; Qin, S.; Zhang, D.-W. Identification of amino acid residues in the ligand binding repeats of LDL receptor important for PCSK9 binding. J. Lipid Res. 2019, 60, 516–527. [Google Scholar] [CrossRef] [Green Version]
- Berman, H.M.; Battistuz, T.; Bhat, T.N.; Bluhm, W.F.; Bourne, P.E.; Burkhardt, K.; Feng, Z.; Gilliland, G.L.; Iype, L.; Jain, S.; et al. The Protein Data Bank. Acta Crystallogr. Sect. D Boil. Crystallogr. 2002, 58, 235–242. [Google Scholar] [CrossRef]
- Eswar, N.; Webb, B.; Marti-Renom, M.; Madhusudhan, M.; Eramian, D.; Shen, M.-Y.; Pieper, U.; Sali, A. Comparative Protein Structure Modeling Using Modeller. Curr. Protoc. Bioinform. 2006, 15, 1–6. [Google Scholar] [CrossRef] [Green Version]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [Green Version]
- Søndergaard, C.R.; Olsson, M.H.M.; Rostkowski, M.; Jensen, J.H. Improved Treatment of Ligands and Coupling Effects in Empirical Calculation and Rationalization of p K a Values. J. Chem. Theory Comput. 2011, 7, 2284–2295. [Google Scholar] [CrossRef]
- Jo, S.; Kim, T.; Iyer, V.G.; Im, W. CHARMM-GUI: A web-based graphical user interface for CHARMM. J. Comput. Chem. 2008, 29, 1859–1865. [Google Scholar] [CrossRef]
- Lee, J.; Cheng, X.; Jo, S.; MacKerell, A.D.; Klauda, J.B.; Im, W. CHARMM-GUI Input Generator for NAMD, Gromacs, Amber, Openmm, and CHARMM/OpenMM Simulations using the CHARMM36 Additive Force Field. Biophys. J. 2016, 110, 641. [Google Scholar] [CrossRef]
- Abraham, M.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1–2, 19–25. [Google Scholar] [CrossRef] [Green Version]
- Huang, J.; Rauscher, S.; Nawrocki, G.; Ran, T.; Feig, M.; De Groot, B.L.; Grubmüller, H.; MacKerell, A.D. CHARMM36m: An improved force field for folded and intrinsically disordered proteins. Nat. Methods 2016, 14, 71–73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hess, B.; Bekker, H.; Berendsen, H.J.C.; Fraaije, J.G.E.M. LINCS: A linear constraint solver for molecular simulations. J. Comput. Chem. 1997, 18, 1463–1472. [Google Scholar] [CrossRef]
- Braga, C.; Travis, K. A configurational temperature Nose?-Hoover thermostat. J. Chem. Phys. 2005, 123, 134101. [Google Scholar] [CrossRef] [PubMed]
- Parrinello, M. Polymorphic transitions in single crystals: A new molecular dynamics method. J. Appl. Phys. 1981, 52, 7182. [Google Scholar] [CrossRef]
- Verlet, L. Computer “Experiments” on Classical Fluids. I. Thermodynamical Properties of Lennard-Jones Molecules. Phys. Rev. 1967, 159, 98–103. [Google Scholar] [CrossRef]
- Darden, T.; York, D.; Pedersen, L. Particle mesh Ewald: AnN⋅log(N) method for Ewald sums in large systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar] [CrossRef] [Green Version]
- Kumari, R.; Kumar, R.; Lynn, A.; Lynn, A. g_mmpbsa —A GROMACS Tool for High-Throughput MM-PBSA Calculations. J. Chem. Inf. Model. 2014, 54, 1951–1962. [Google Scholar] [CrossRef]
- Baker, N.A.; Sept, D.; Joseph, S.; Holst, M.J.; McCammon, J.A. Electrostatics of nanosystems: Application to microtubules and the ribosome. Proc. Natl. Acad. Sci. USA 2001, 98, 10037–10041. [Google Scholar] [CrossRef] [Green Version]
- Lammi, C.; Sgrignani, J.; Arnoldi, A.; Grazioso, G. Biological Characterization of Computationally Designed Analogs of peptide tVFtsWeeYLDWV (Pep2-8) with Increased PCSK9 Antagonistic Activity. Sci. Rep. 2019, 9, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Morris, J.H.; Huang, C.C.; Babbitt, P.; Ferrin, T. structureViz: Linking Cytoscape and UCSF Chimera. Bioinform. 2007, 23, 2345–2347. [Google Scholar] [CrossRef] [Green Version]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A Software Environment for Integrated Models of Biomolecular Interaction Networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| (kJ/mol) | Wild-type | Asp374Tyr | Arg496Trp | Ser127Arg |
|---|---|---|---|---|
| van der Waals | −738.7 (3.78) | −544.8 (4.62) | −495.0 (2.97) | −495.8 (3.49) |
| Electrostatic | −227.6 (18.5) | -851.6 (23.3) | 133.5 (16.5) | −391.1 (15.4) |
| Polar Solvation | 1562.6 (24.1) | 1615.5 (26.9) | 878.4 (18.6) | 1138.7 (17.3) |
| Non-polar | −89.7 (0.593) | −76.8 (0.723) | −64.0 (0.507) | −64.2 (0.525) |
| Total | 506.5 (18.7) | 143.0 (12.0) | 453.3 (13.5) | 187.4 (14.2) |
| (kJ/mol) | Wild-type | Asp374Tyr | Arg496Trp | Ser127Arg |
|---|---|---|---|---|
| PCSK9 Prodomain | −147.5 | −124.3 | −281.9 | −417.2 |
| PCSK9 Catalytic | 375.7 | 185.9 | 506.4 | 438.1 |
| Loop | 248.9 | 93.9 | 290.6 | 250.6 |
| LDLR β-propeller | 4.6 | −41.2 | −77.9 | −106.4 |
| Prodomain/β-propeller | −143.0 | −165.5 | −359.8 | −523.6 |
| Catalytic/Loop | 624.6 | 279.8 | 797.0 | 688.7 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Martin, W.R.; Lightstone, F.C.; Cheng, F. In Silico Insights into Protein–Protein Interaction Disruptive Mutations in the PCSK9-LDLR Complex. Int. J. Mol. Sci. 2020, 21, 1550. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21051550
Martin WR, Lightstone FC, Cheng F. In Silico Insights into Protein–Protein Interaction Disruptive Mutations in the PCSK9-LDLR Complex. International Journal of Molecular Sciences. 2020; 21(5):1550. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21051550
Chicago/Turabian StyleMartin, William R., Felice C. Lightstone, and Feixiong Cheng. 2020. "In Silico Insights into Protein–Protein Interaction Disruptive Mutations in the PCSK9-LDLR Complex" International Journal of Molecular Sciences 21, no. 5: 1550. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21051550