Extracellular Vesicle-Induced Classical Complement Activation Leads to Retinal Endothelial Cell Damage via MAC Deposition


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
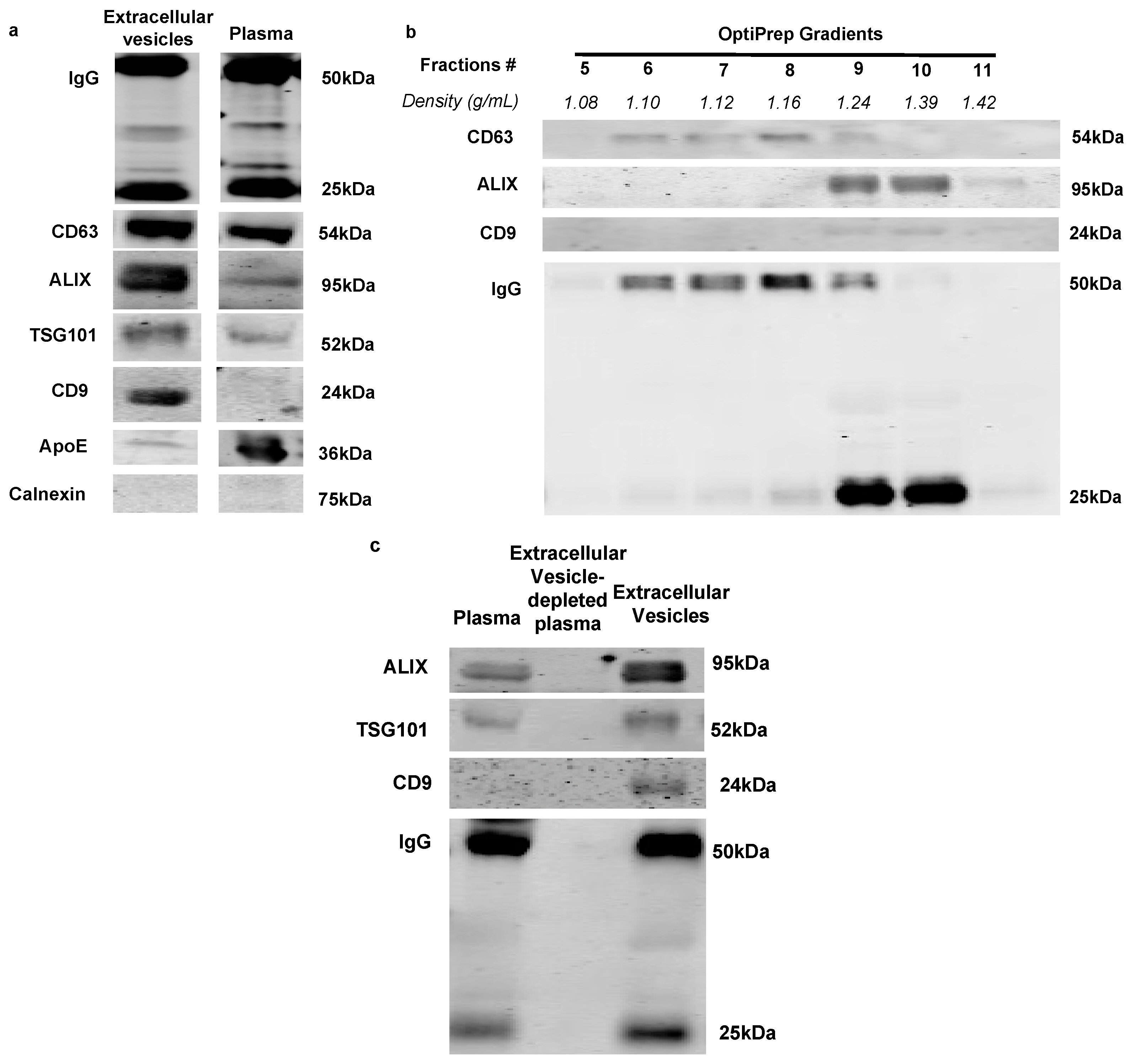
2.1. Immunoglobulins Are Associated with Extracellular Vesicles in Rat Plasma
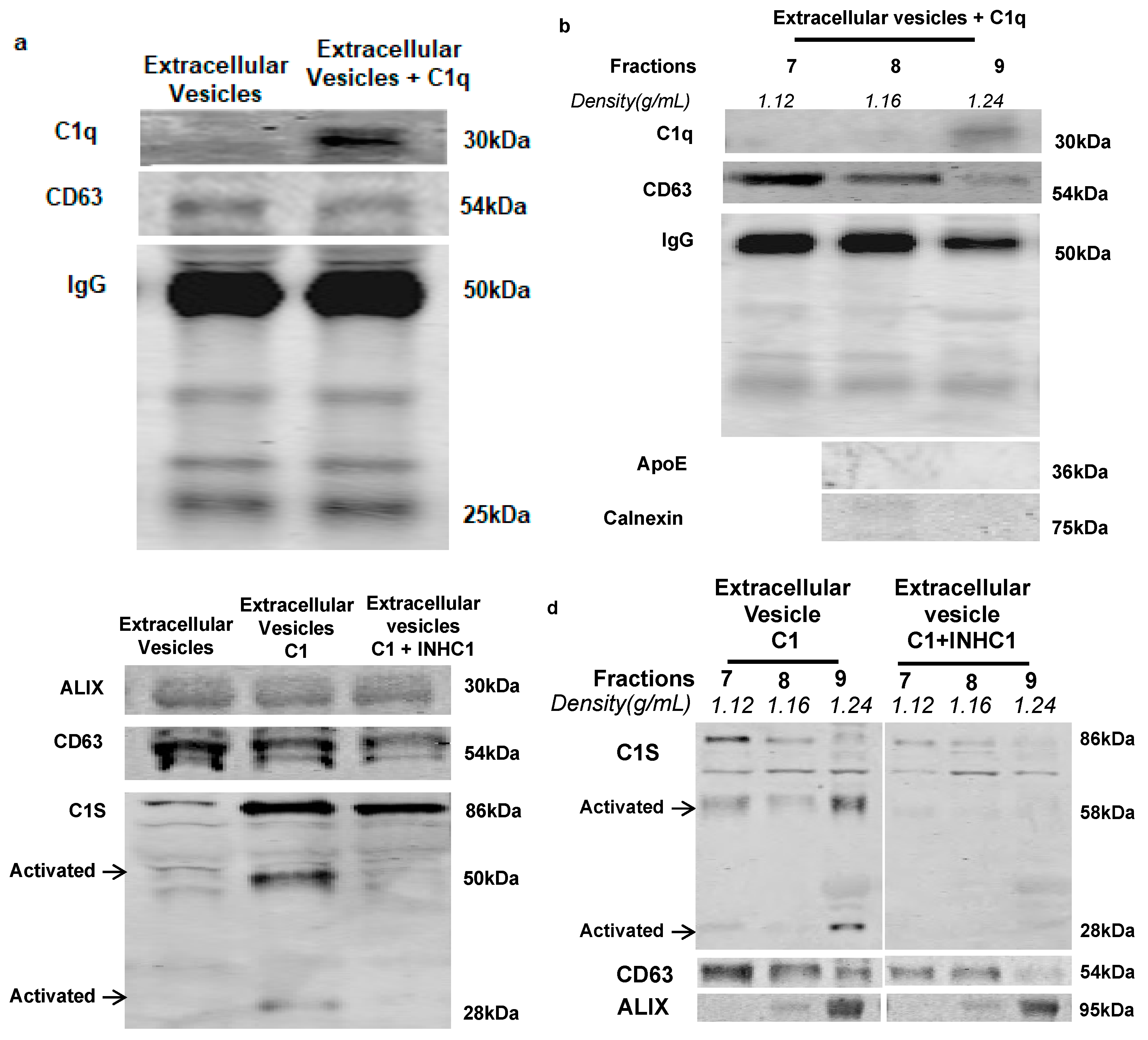
2.2. IgG-Laden Extracellular Vesicles Activate the Classical Complement Protein C1 Complex
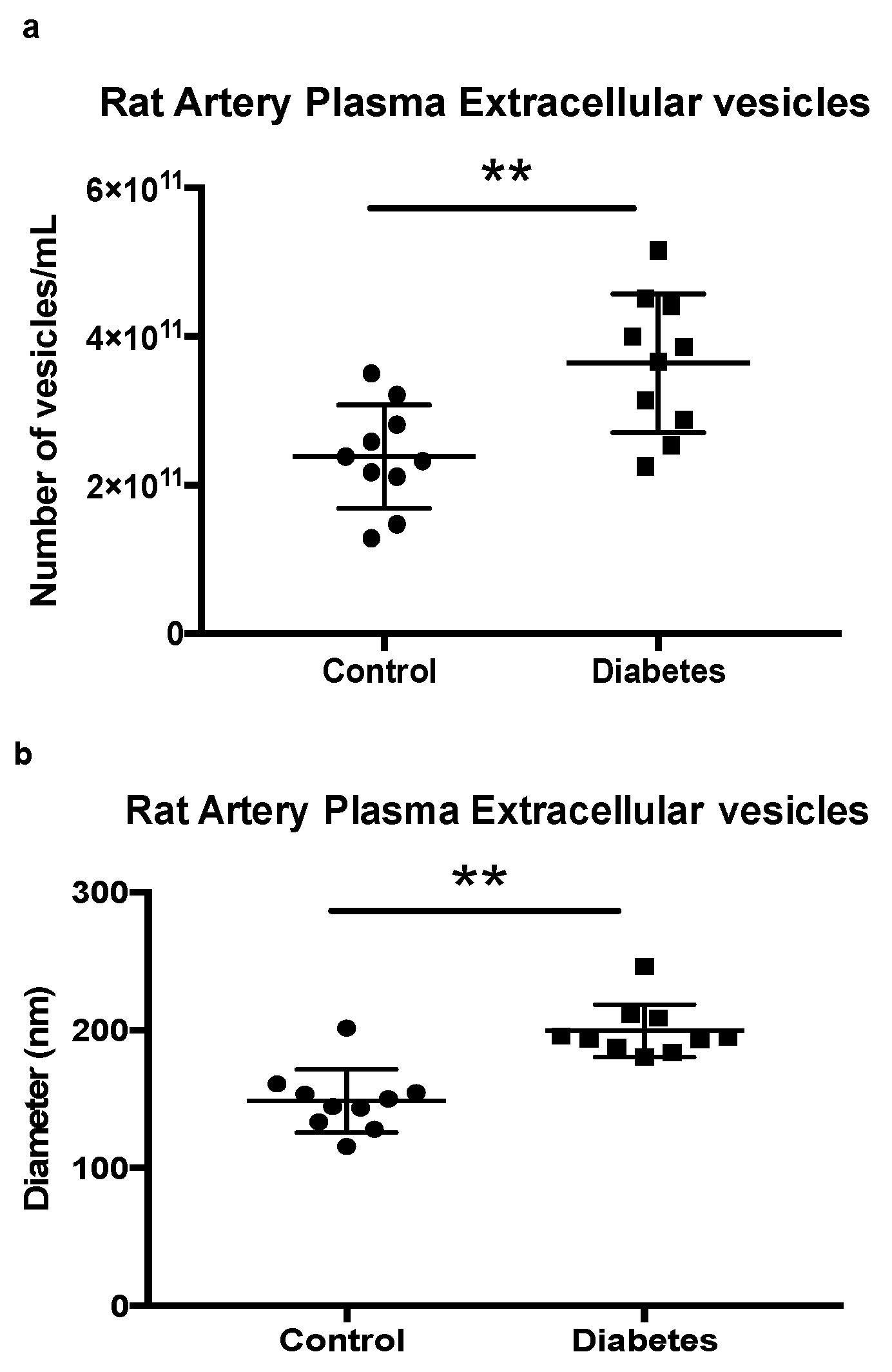
2.3. Quantification of Extracellular Vesicles in Rat Control and Diabetic Artery Plasma
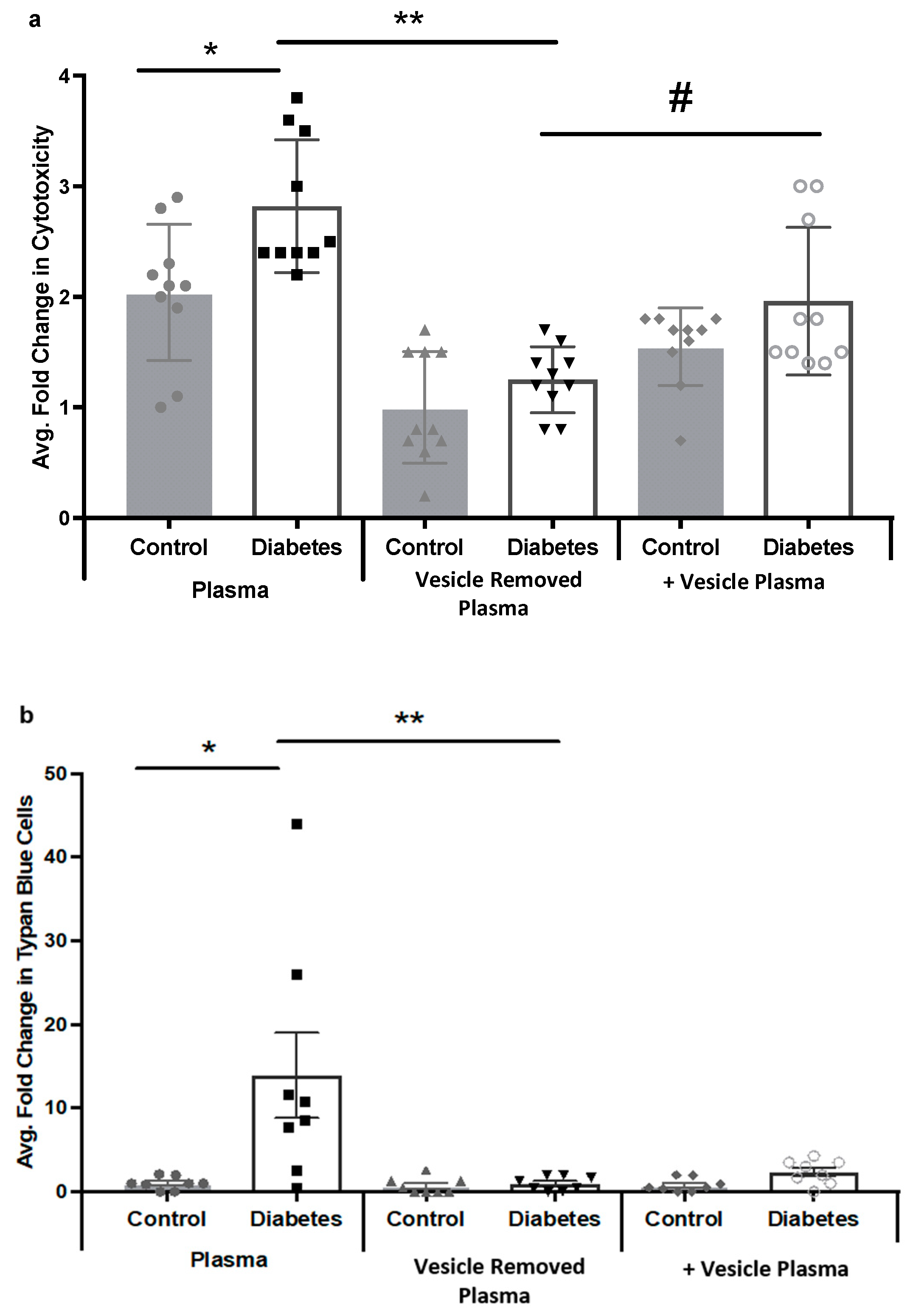
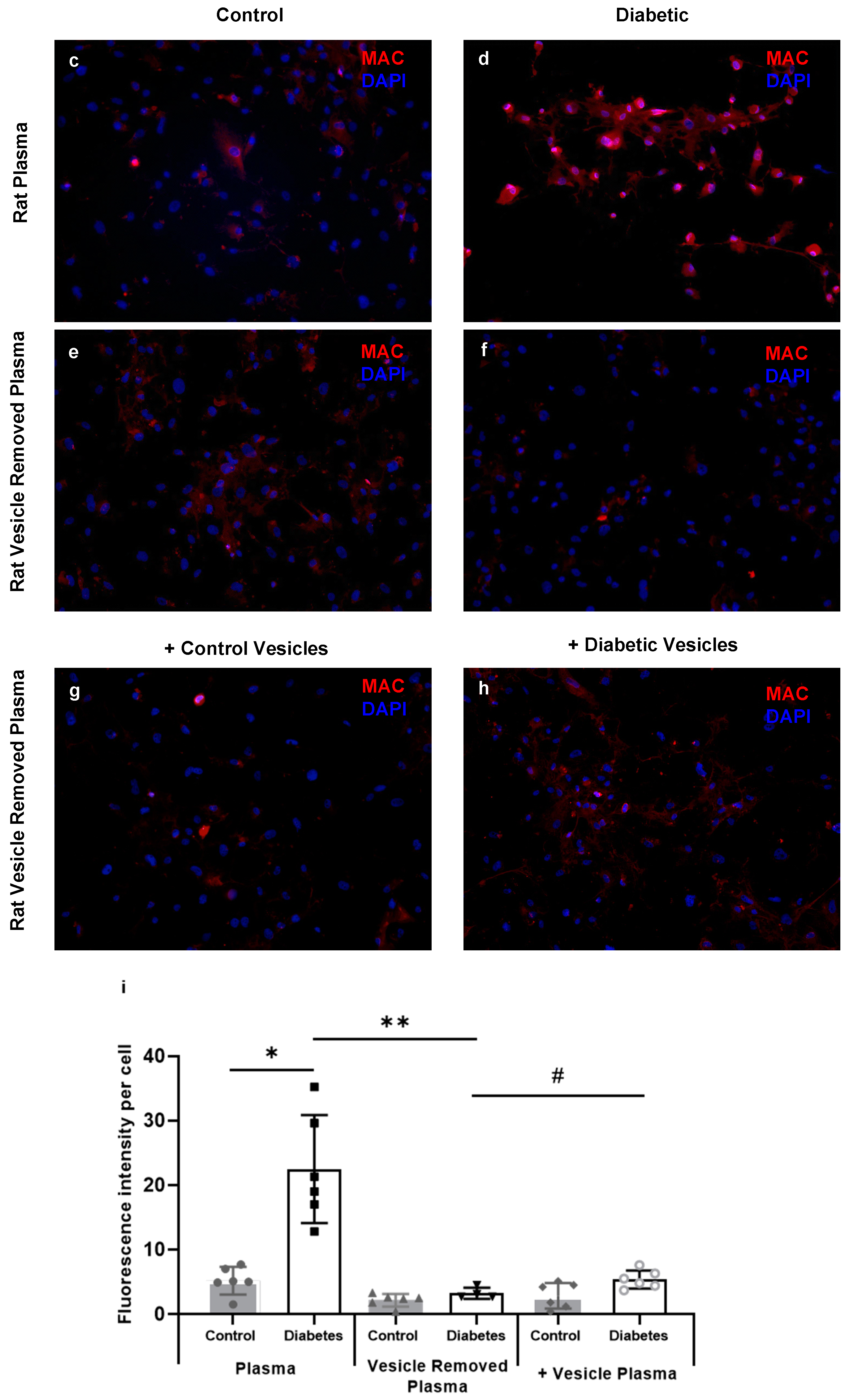
2.4. Diabetic Rat Plasma Extracellular Vesicles Contribute to HREC Cytotoxicity via MAC Deposition
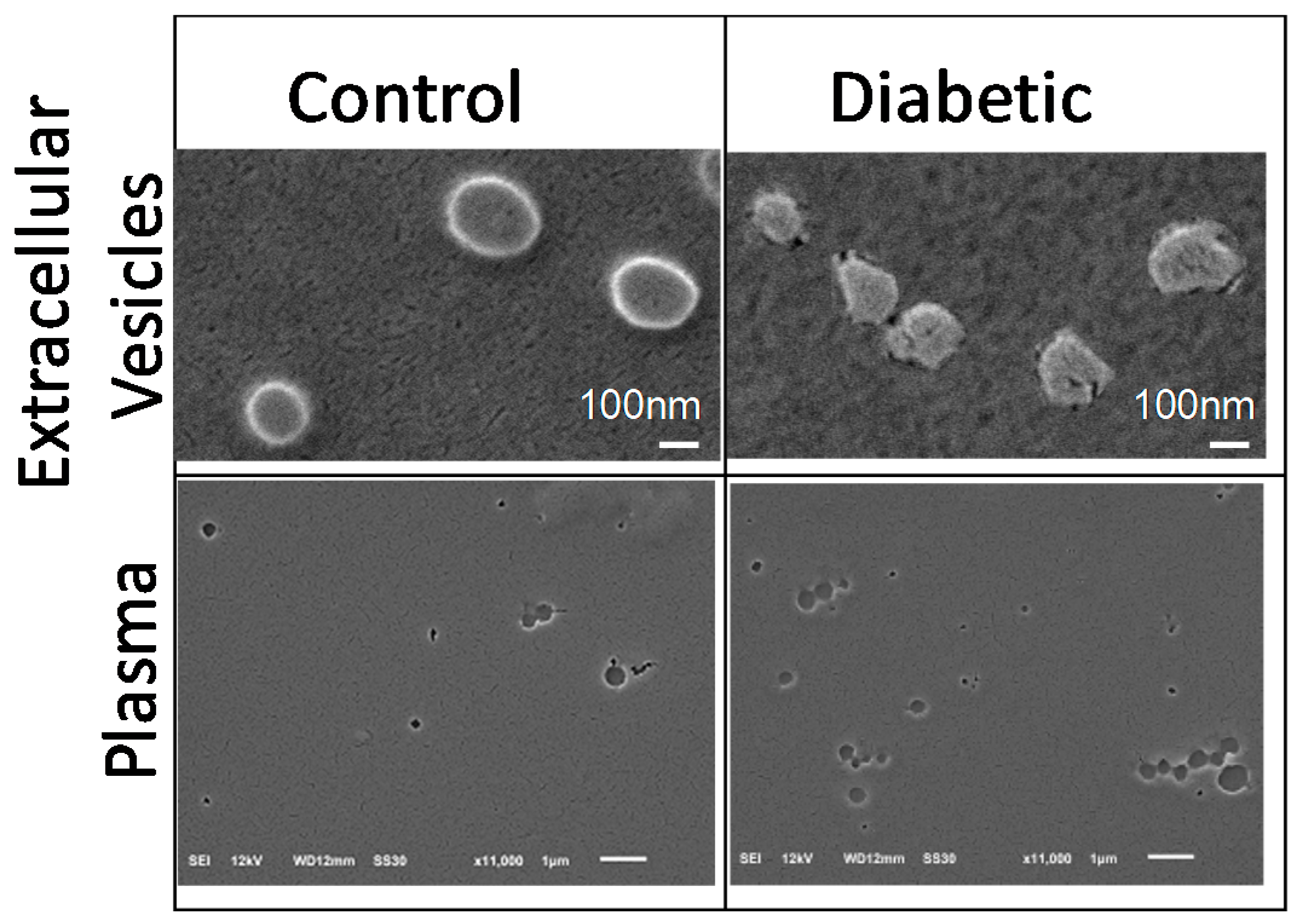
2.5. Scanning Electron Microscopy (SEM) Analysis of Plasma and Extracellular Vesicles
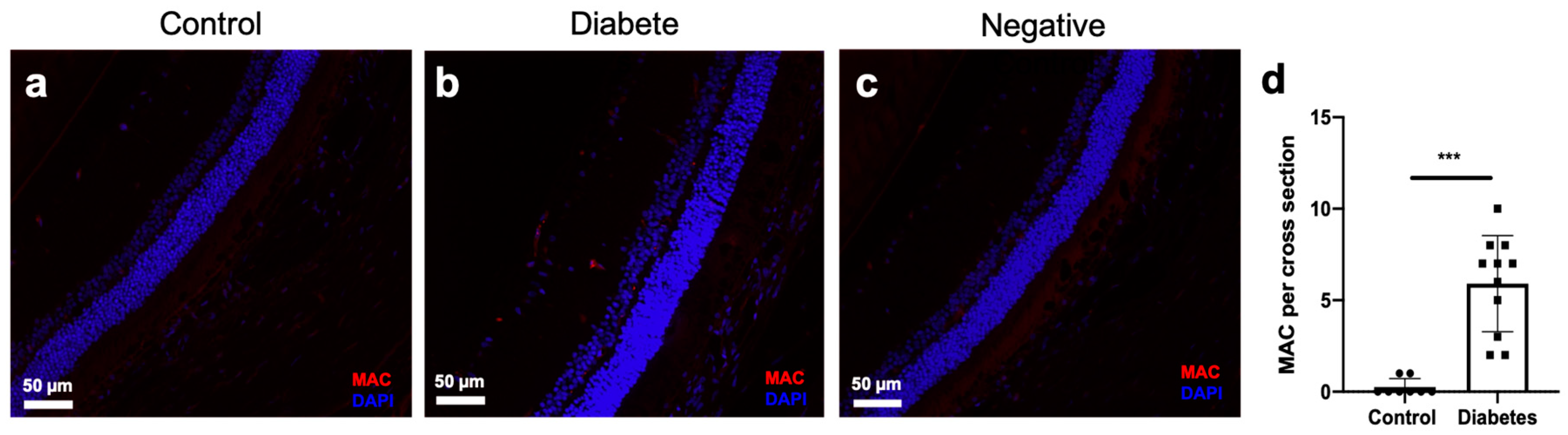
2.6. MAC Deposition in the Retinal Vascular of Diabetic Rats
3. Discussion
4. Materials and Methods
4.1. Animal Studies
4.2. Cell Culture
4.3. Western Blot
4.4. Blood Sample Collection
4.5. Extracellular Vesicle Isolation
4.6. Extracellular Quantification
4.7. OptiPrep Density Gradient Extracellular Vesicle Purification
4.8. C1q Binding Assay
4.9. C1 Activation Assay
4.10. LDH Assay
4.11. Immunocytochemistry
4.12. Electron Microscopy
4.13. Statistics
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| DR | Diabetic Retinopathy |
| MAC | Membrane Attack Complex |
| HREC | Human Retinal Endothelial Cell |
| RPE | Retinal Pigment Epithelial Cell |
References
- Clark, S.J.; Bishop, P.N. The eye as a complement dysregulation hotspot. Semin. Immunopathol. 2018, 40, 65–74. [Google Scholar] [CrossRef]
- Yanai, R.; Thanos, A.; Connor, K.M. Complement Involvement in Neovascular Ocular Diseases. Adv. Exp. Med. Biol. 2012, 946, 161–183. [Google Scholar] [PubMed]
- Merle, N.S.; Church, S.E.; Fremeaux-Bacchi, V.; Roumenina, L.T. Complement system part I molecular mechanisms of activation and regulation. Front. Immunol. 2015, 6, 1–30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Merle, N.S.; Noe, R.; Halbwachs-Mecarelli, L.; Fremeaux-Bacchi, V.; Roumenina, L.T. Complement system part II: Role in immunity. Front. Immunol. 2015, 6, 1–26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gerl, V.; Bohl, J.; Pitz, S.; Stoffelns, B. Extensive deposits of complement C3d and C5b-9 in the choriocapillaris of eyes of patients with diabetic retinopathy. Investig. Opthalmology Vis. Sci. 2002, 43, 1104–1148. [Google Scholar]
- Zhang, J.; Gerhardinger, C.; Lorenzi, M. Early Complement Activation and Decreased Levels of Glycosylphosphatidylinositol-Anchored Complement Inhibitors in Human and Experimental Diabetic Retinopathy. Diabetes 2002, 51, 3499–3504. [Google Scholar] [CrossRef] [Green Version]
- Qin, X.; Goldfine, A.; Krumrei, N.; Grubissich, L.; Acosta, J.; Chorev, M.; Hays, A.P.; Halperin, J.A. Glycation Inactivation of the Complement Regulatory Protein Cd59. Diabetes 2004, 53, 2653–2661. [Google Scholar] [CrossRef] [Green Version]
- Acosta, J.; Hettinga, J.; Flückiger, R.; Krumrei, N.; Goldfine, A.; Angarita, L.; Halperin, J. Molecular basis for a link between complement and the vascular complications of diabetes. Proc. Natl. Acad. Sci. USA 2000, 97, 5450–5455. [Google Scholar] [CrossRef]
- Looze, C.; Yui, D.; Leung, L.; Ingham, M.; Kaler, M.; Yao, X. Proteomic profiling of human plasma exosomes identifies PPARγ as an exosome-associated protein. Biochem. Biophys. Res. Commun. 2009, 378, 433–438. [Google Scholar] [CrossRef] [Green Version]
- Kowal, J.; Arras, G.; Colombo, M.; Jouve, M.; Morath, J.P.; Primdal-Bengtson, B. Proteomic comparison defines novel markers to characterize heterogeneous populations of extracellular vesicle subtypes. Proc. Natl. Acad. Sci. USA 2016, 113, E968–E977. [Google Scholar] [CrossRef] [Green Version]
- Colombo, M.; Raposo, G.; Théry, C. Biogenesis, Secretion, and Intercellular Interactions of Exosomes and Other Extracellular Vesicles. Annu. Rev. Cell. Dev. Biol. 2014, 30, 255–289. [Google Scholar] [CrossRef] [PubMed]
- Van Niel, G.; D’Angelo, G.R.G. Shedding light on the cell biology of Extracellular Vesicles. Nat. Rev. Mol. Cell. Biol. 2017. accepted. [Google Scholar] [CrossRef]
- Huang, C.; Fisher, K.P.; Hammer, S.S.; Navitskaya, S.; Blanchard, G.J.; Busik, J.V. Plasma Exosomes Contribute to Microvascular Damage in Diabetic Retinopathy by Activating the Classical Complement Pathway. Diabetes 2018, 67, 1639–1649. [Google Scholar] [CrossRef] [Green Version]
- Zubler, R.H.; Lange, G.; Lambert, P.H.; Miescher, P.A. Detection of immune complexes in unheated sera by modified 125I-Clq binding test. Effect of heating on the binding of Clq by immune complexes and application of the test to systemic lupus erythematosus. J. Immunol. 1976, 116, 232–235. [Google Scholar] [PubMed]
- Arlaud, G.J.; Biro, A.; Ling, W.L. Enzymatically modified low-density lipoprotein is recognized by c1q and activates the classical complement pathway. J. Lipids 2011, 2011, 376092. [Google Scholar] [CrossRef] [PubMed]
- Biró, A.; Thielens, N.M.; Cervenák, L.; Prohászka, Z.; Füst, G.; Arlaud, G.J. Modified low density lipoproteins differentially bind and activate the C1 complex of complement. Mol. Immunol. 2007, 44, 1169–1177. [Google Scholar] [CrossRef] [PubMed]
- Whitmore, S.S.; Sohn, E.H.; Chirco, K.R.; Drack, A.V.; Stone, E.M.; Tucker, B.A. Complement activation and choriocapillaris loss in early AMD: Implications for pathophysiology and therapy. Prog. Retin Eye Res. 2015, 45, 1–29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benzaquen, L.R. Terminal complement proteins C5b-9 release basic fibroblast growth factor and platelet-derived growth factor from endothelial cells. J. Exp. Med. 1994, 179, 985–992. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, P.; Kaslan, M.; Lee, S.H.; Yao, J.; Gao, Z. Progress in exosome isolation techniques. Theranostics 2017, 7, 789–804. [Google Scholar] [CrossRef]
- Kalra, H.; Adda, C.G.; Liem, M.; Ang, C.S.; Mechler, A.; Simpson, R.J. Comparative proteomics evaluation of plasma exosome isolation techniques and assessment of the stability of exosomes in normal human blood plasma. Proteomics 2013, 13, 3354–3364. [Google Scholar] [CrossRef]
- Jane-wit, D.; Surovtseva, Y.V.; Qin, L.; Li, G.; Liu, R.; Clark, P. Complement membrane attack complexes activate noncanonical NF-κB by forming an Akt + NIK + signalosome on Rab5 + endosomes. Proc. Natl. Acad. Sci. USA 2015, 112, 9686–9691. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Georgiannakis, A.; Burgoyne, T.; Lueck, K.; Futter, C.; Greenwood, J.; Moss, S.E. Retinal Pigment Epithelial Cells Mitigate the Effects of Complement Attack by Endocytosis of C5b-9. J. Immunol. 2015, 195, 3382–3389. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Halperin, J.A.; Taratuska, A.; Nicholson-Weller, A. Terminal complement complex C5b-9 stimulates mitogenesis in 3T3 cells. J. Clin. Investig. 1993, 91, 1974–1978. [Google Scholar] [CrossRef] [Green Version]
- Harshini, C.; Navitskaya, S.; O’reilly, S.; Gallimore, J.; Mize, H.; Beli, E. Role of Acid Sphingomyelinase in Shifting the Balance Between Proinflammatory and Reparative Bone Marrow Cells in Diabetic Retinopathy. Stem Cells 2016, 34, 972–983. [Google Scholar]
- Kim, D.D.; Song, W.C. Membrane complement regulatory proteins. Clin. Immunol. 2006, 118, 127–136. [Google Scholar] [CrossRef] [PubMed]
- Sandrin, M.S.; Mckenzie, I.F.C. Galα(1,3)Gal, the Major Xenoantigen(s) Recognised in Pigs by Human Natural Antibodies. Immunol. Rev. 1994, 141, 169–190. [Google Scholar] [CrossRef]
- Abela, G.S. Cholesterol crystals piercing the arterial plaque and intima trigger local and systemic inflammation. J. Clin. Lipidol. 2010, 4, 156–164. [Google Scholar] [CrossRef]
- Samstad, E.O.; Niyonzima, N.; Nymo, S.; Aune, M.H.; Ryan, L.; Bakke, S.S. Cholesterol Crystals Induce Complement-Dependent Inflammasome Activation and Cytokine Release. J. Immunol. 2014, 192, 2837–2845. [Google Scholar] [CrossRef] [Green Version]
- Opreanu, M.; Tikhonenko, M.; Bozack, S.; Lydic, T.A.; Reid, G.E.; McSorley, K.M. The unconventional role of acid sphingomyelinase in regulation of retinal microangiopathy in diabetic human and animal models. Diabetes 2011, 60, 2370–2378. [Google Scholar] [CrossRef] [Green Version]
- Busik, J.V.; Olson, L.K.; Grant, M.B.; Henry, D.N. Glucose-induced activation of glucose uptake in cells from the inner and outer blood-retinal barrier. Investig. Ophthalmol. Vis. Sci. 2002, 43, 2356–2363. [Google Scholar]
- Théry, C.; Amigorena, S.; Raposo, G.; Clayton, A. Isolation and characterization of exosomes from cell culture supernatants and biological fluids. Curr. Protoc. Cell Biol. 2006, 3, 1–29. [Google Scholar] [CrossRef] [PubMed]
- Bradley, A.J.; Brooks, D.E.; Norris-Jones, R.; Devine, D.V. C1q Binding to liposomes is surface charge dependent and is inhibited by peptides consisting of residues 14-26 of the human C1qA chain in a sequence independent manner. Biochim. Biophys. Acta—Biomembr. 1999, 1418, 19–30. [Google Scholar] [CrossRef] [Green Version]
- Nydegger, U.E.; Lambert, P.H.; Miesghr, P.A.; Gerber, D.I. Circulating Immune Complexes in the Serum in Systemic Lupus Erythematosus and in Carriers of Hepatitis B Antigen. J. Clin. Investig. 1974, 54, 297–309. [Google Scholar] [CrossRef]
- Biro, A.; Ling, W.L.; Arlaud, G.J. Complement protein c1q recognizes enzymatically modified low-density lipoprotein through unesterified fatty acids generated by cholesterol esterase. Biochemistry 2010, 49, 2167–2176. [Google Scholar] [CrossRef] [PubMed]








© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Huang, C.; Fisher, K.P.; Hammer, S.S.; Busik, J.V. Extracellular Vesicle-Induced Classical Complement Activation Leads to Retinal Endothelial Cell Damage via MAC Deposition. Int. J. Mol. Sci. 2020, 21, 1693. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21051693
Huang C, Fisher KP, Hammer SS, Busik JV. Extracellular Vesicle-Induced Classical Complement Activation Leads to Retinal Endothelial Cell Damage via MAC Deposition. International Journal of Molecular Sciences. 2020; 21(5):1693. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21051693
Chicago/Turabian StyleHuang, Chao, Kiera P. Fisher, Sandra S. Hammer, and Julia V. Busik. 2020. "Extracellular Vesicle-Induced Classical Complement Activation Leads to Retinal Endothelial Cell Damage via MAC Deposition" International Journal of Molecular Sciences 21, no. 5: 1693. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21051693