Epigenetic Regulation of WNT3A Enhancer during Regeneration of Injured Cortical Neurons

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
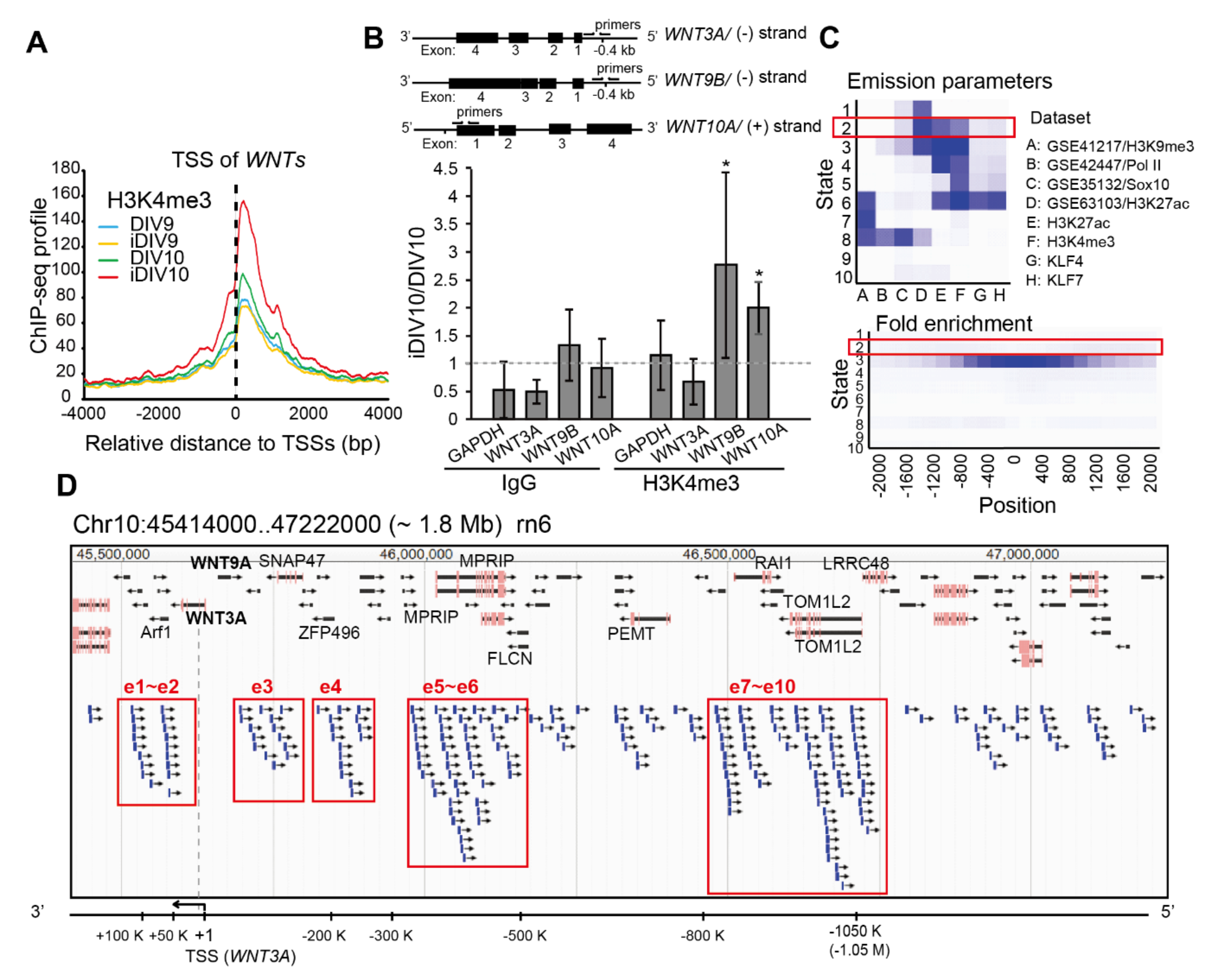
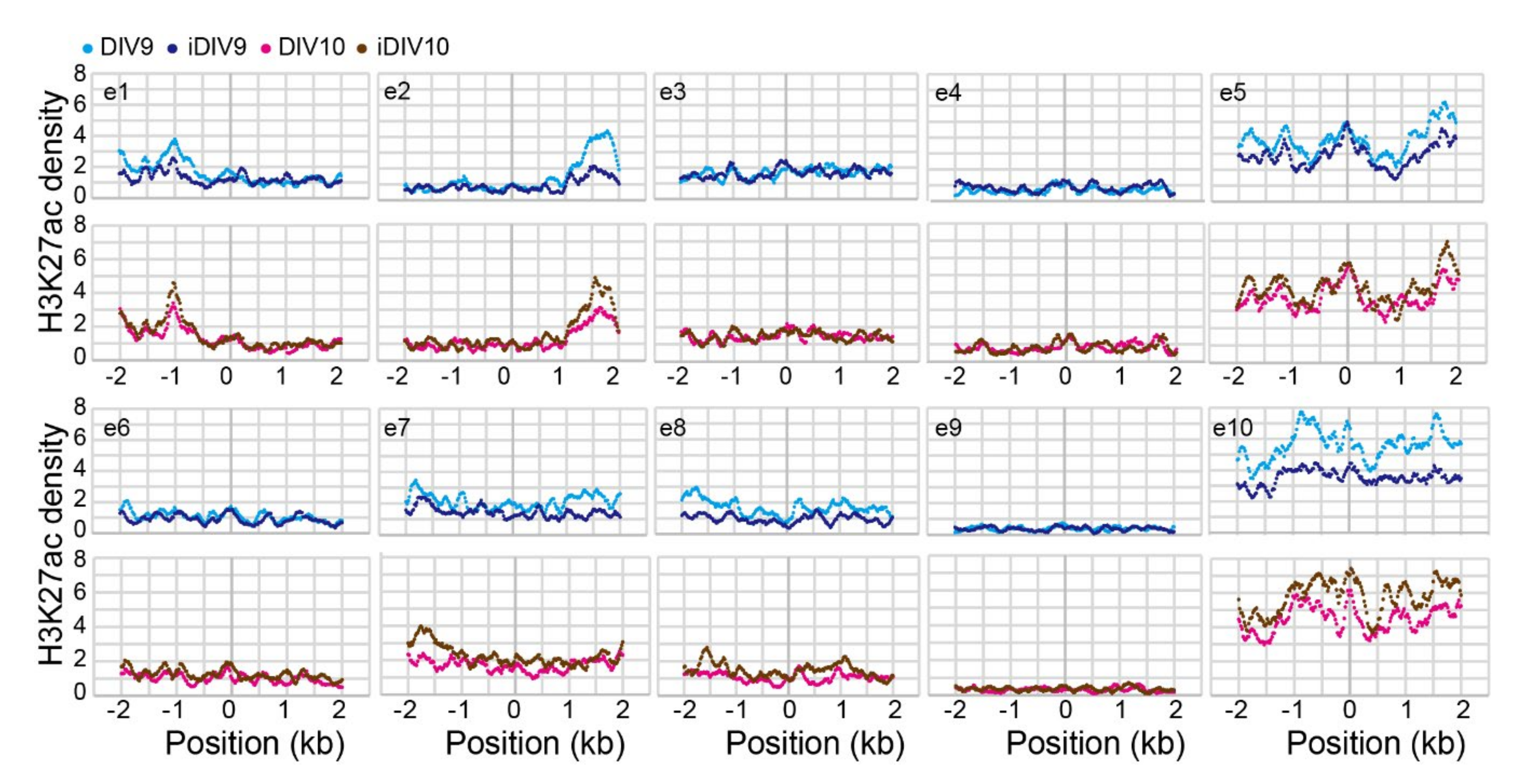
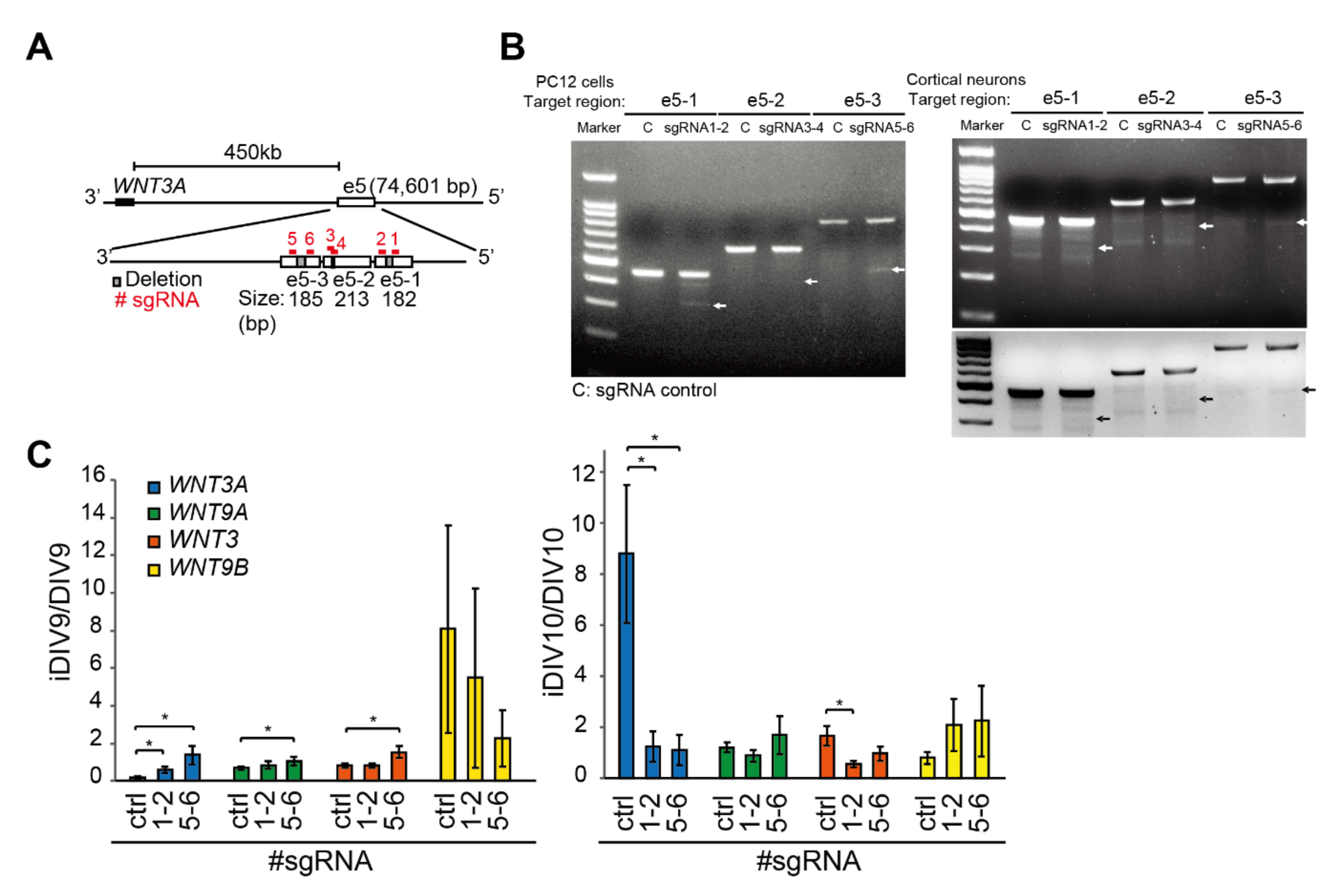
2.1. Identification of Putative Enhancer for WNT3A
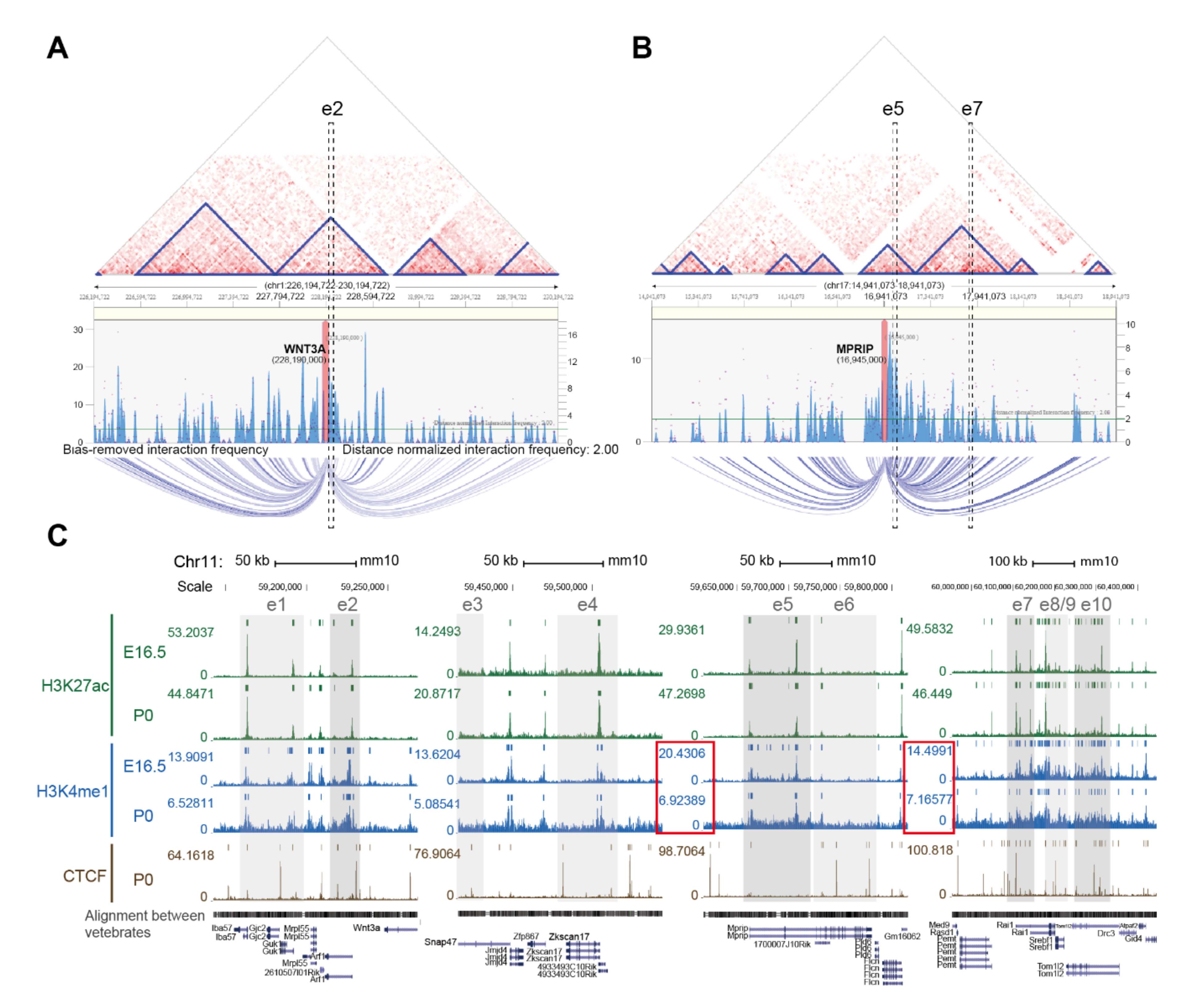
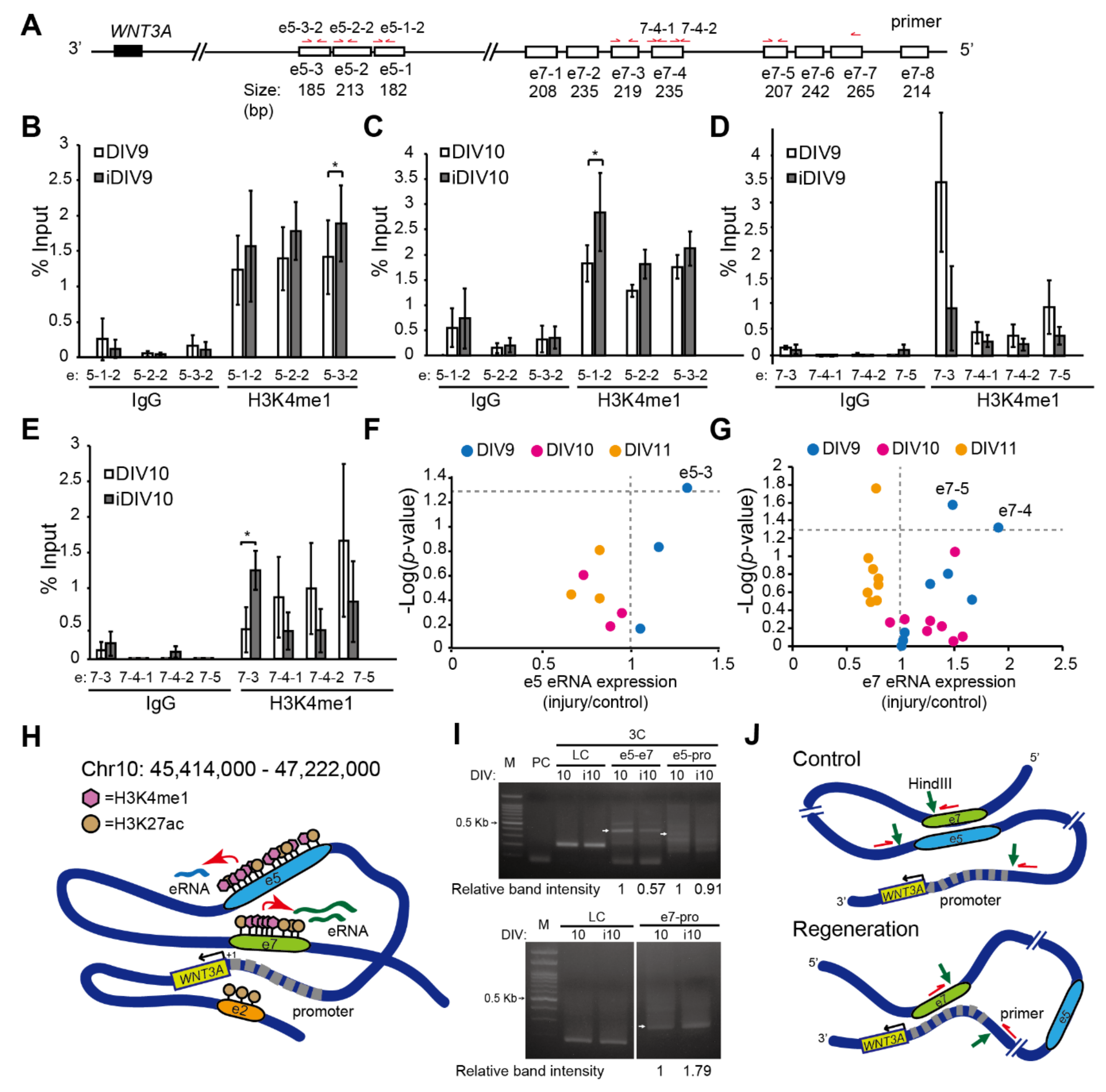
2.2. Enhancer-Mediated WNT3A Expression through Histone Modification and Chromosome Topological Transformation
3. Discussion
4. Materials and Methods
4.1. Animals and Ethics Approval
4.2. Reagents
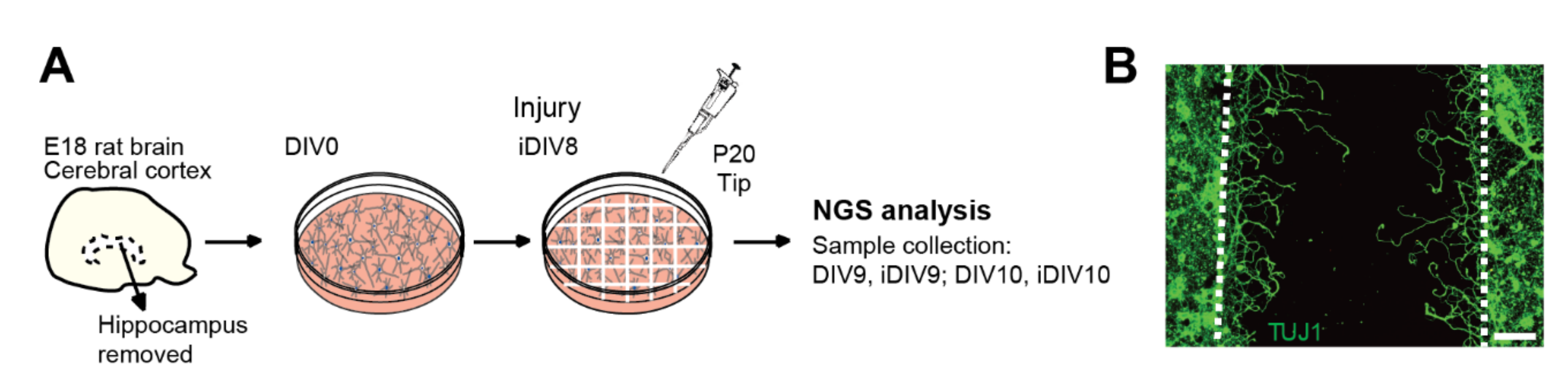
4.3. In Vitro Culture of Primary Neurons and the TBI Model
4.4. ChIP-Seq
4.5. Data Analysis
4.5.1. Aggregation Plot
4.5.2. ChromHMM for Enhancer Prediction
4.5.3. Long-Range Chromatin Interaction Prediction
4.5.4. ENCODE Datasets Processing
4.6. ChIP-qPCR and eRNA Quantification
4.7. Chromosome Conformation Capture (3C) Analysis
4.8. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| TBI | Traumatic brain injury |
| RAG | Regeneration-associated gene |
| H3K27ac | Histone 3 lysine 27 acetylation |
| H3K4me1/3 | Histone3 lysine 4 mono-/tri-methylation |
| ChIP-seq | Chromatin immunoprecipitation-sequencing |
| 3C | Chromosome conformation capture |
| CTCF | CCCTC-binding factor |
| TAD | Topologically associating domain |
| eRNA sgRNA | Enhancer RNA Single guide RNA |
References
- Dewan, M.C.; Rattani, A.; Gupta, S.; Baticulon, R.E.; Hung, Y.C.; Punchak, M.; Agrawal, A.; Adeleye, A.O.; Shrime, M.G.; Rubiano, A.M.; et al. Estimating the global incidence of traumatic brain injury. J. Neurosurg. 2018, 130, 1080–1097. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hegde, M.N. A Coursebook on Aphasia and Other Neurogenic Language Disorders, 3rd ed.; Delmar Cengage Learning: Clifton Park, NY, USA, 2006. [Google Scholar]
- Xiong, Y.; Mahmood, A.; Chopp, M. Animal models of traumatic brain injury. Nat. Rev. Neurosci. 2013, 14, 128–142. [Google Scholar] [CrossRef] [Green Version]
- McGuire, J.L.; Ngwenya, L.B.; McCullumsmith, R.E. Neurotransmitter changes after traumatic brain injury: An update for new treatment strategies. Mol. Psychiatry 2018, 24, 995–1012. [Google Scholar] [CrossRef] [PubMed]
- Yiu, G.; He, Z. Glial inhibition of CNS axon regeneration. Nat. Rev. Neurosci. 2006, 7, 617–627. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giger, R.J.; Hollis, E.R., 2nd; Tuszynski, M.H. Guidance molecules in axon regeneration. Cold Spring Harbor Perspect. Biol. 2010, 2, a001867. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, Y.; Tenney, A.P.; Busch, S.A.; Horn, K.P.; Cuascut, F.X.; Liu, K.; He, Z.; Silver, J.; Flanagan, J.G. PTPsigma is a receptor for chondroitin sulfate proteoglycan, an inhibitor of neural regeneration. Science 2009, 326, 592–596. [Google Scholar] [CrossRef] [Green Version]
- Fawcett, J.W.; Asher, R.A. The glial scar and central nervous system repair. Brain Res. Bull. 1999, 49, 377–391. [Google Scholar] [CrossRef]
- Lee, J.K.; Chan, A.F.; Luu, S.M.; Zhu, Y.; Ho, C.; Tessier-Lavigne, M.; Zheng, B. Reassessment of corticospinal tract regeneration in Nogo-deficient mice. J. Neurosci. 2009, 29, 8649–8654. [Google Scholar] [CrossRef]
- Sun, F.; He, Z. Neuronal intrinsic barriers for axon regeneration in the adult CNS. Curr. Opin. Neurobiol. 2010, 20, 510–518. [Google Scholar] [CrossRef] [Green Version]
- Wong, V.S.; Langley, B. Epigenetic changes following traumatic brain injury and their implications for outcome, recovery and therapy. Neurosci. Lett. 2016, 625, 26–33. [Google Scholar] [CrossRef] [Green Version]
- Meng, Q.; Zhuang, Y.; Ying, Z.; Agrawal, R.; Yang, X.; Gomez-Pinilla, F. Traumatic Brain Injury Induces Genome-Wide Transcriptomic, Methylomic, and Network Perturbations in Brain and Blood Predicting Neurological Disorders. EBioMedicine 2017, 16, 184–194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagalakshmi, B.; Sagarkar, S.; Sakharkar, A.J. Epigenetic Mechanisms of Traumatic Brain Injuries. Prog. Mol. Biol. Transl. Sci. 2018, 157, 263–298. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.Y.; Liang, M.Z.; Wu, C.C.; Huang, P.Y.; Chen, H.I.; Yet, S.F.; Tsai, J.W.; Kao, C.F.; Chen, L. WNT3A Promotes Neuronal Regeneration upon Traumatic Brain Injury. Int. J. Mol. Sci. 2020, 21, 1463. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wahane, S.; Halawani, D.; Zhou, X.; Zou, H. Epigenetic Regulation of Axon Regeneration and Glial Activation in Injury Responses. Front Genet. 2019, 10, 640. [Google Scholar] [CrossRef]
- Puttagunta, R.; Tedeschi, A.; Soria, M.G.; Hervera, A.; Lindner, R.; Rathore, K.I.; Gaub, P.; Joshi, Y.; Nguyen, T.; Schmandke, A.; et al. PCAF-dependent epigenetic changes promote axonal regeneration in the central nervous system. Nat. Commun. 2014, 5, 3527. [Google Scholar] [CrossRef] [Green Version]
- Chen, K.W.; Chen, L. Epigenetic Regulation of BDNF Gene during Development and Diseases. Int. J. Mol. Sci. 2017, 18, 571. [Google Scholar] [CrossRef]
- Heintzman, N.D.; Stuart, R.K.; Hon, G.; Fu, Y.; Ching, C.W.; Hawkins, R.D.; Barrera, L.O.; Van Calcar, S.; Qu, C.; Ching, K.A.; et al. Distinct and predictive chromatin signatures of transcriptional promoters and enhancers in the human genome. Nat. Genet. 2007, 39, 311–318. [Google Scholar] [CrossRef]
- Venkatesh, I.; Simpson, M.T.; Coley, D.M.; Blackmore, M.G. Epigenetic profiling reveals a developmental decrease in promoter accessibility during cortical maturation in vivo. Neuroepigenetics 2016, 8, 19–26. [Google Scholar] [CrossRef] [Green Version]
- Sandstrom, R.S.; Foret, M.R.; Grow, D.A.; Haugen, E.; Rhodes, C.T.; Cardona, A.E.; Phelix, C.F.; Wang, Y.; Berger, M.S.; Lin, C.H. Epigenetic regulation by chromatin activation mark H3K4me3 in primate progenitor cells within adult neurogenic niche. Sci. Rep. 2014, 4, 5371. [Google Scholar] [CrossRef] [Green Version]
- Santos-Rosa, H.; Schneider, R.; Bannister, A.J.; Sherriff, J.; Bernstein, B.E.; Emre, N.C.; Schreiber, S.L.; Mellor, J.; Kouzarides, T. Active genes are tri-methylated at K4 of histone H3. Nature 2002, 419, 407–411. [Google Scholar] [CrossRef]
- Shlyueva, D.; Stampfel, G.; Stark, A. Transcriptional enhancers: From properties to genome-wide predictions. Nat. Rev. Genet. 2014, 15, 272–286. [Google Scholar] [CrossRef] [PubMed]
- Lam, M.T.; Li, W.; Rosenfeld, M.G.; Glass, C.K. Enhancer RNAs and regulated transcriptional programs. Trends Biochem. Sci. 2014, 39, 170–182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, T.K.; Hemberg, M.; Gray, J.M. Enhancer RNAs: A class of long noncoding RNAs synthesized at enhancers. Cold Spring Harb. Perspect. Biol. 2015, 7, a018622. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ernst, J.; Kellis, M. ChromHMM: Automating chromatin-state discovery and characterization. Nat. Methods 2012, 9, 215–216. [Google Scholar] [CrossRef] [Green Version]
- Yang, D.; Jang, I.; Choi, J.; Kim, M.S.; Lee, A.J.; Kim, H.; Eom, J.; Kim, D.; Jung, I.; Lee, B. 3DIV: A 3D-genome Interaction Viewer and database. Nucleic Acids Res. 2018, 46, D52–D57. [Google Scholar] [CrossRef] [PubMed]
- Schmitt, A.D.; Hu, M.; Jung, I.; Xu, Z.; Qiu, Y.; Tan, C.L.; Li, Y.; Lin, S.; Lin, Y.; Barr, C.L.; et al. A Compendium of Chromatin Contact Maps Reveals Spatially Active Regions in the Human Genome. Cell Rep. 2016, 17, 2042–2059. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fudenberg, G.; Imakaev, M.; Lu, C.; Goloborodko, A.; Abdennur, N.; Mirny, L.A. Formation of Chromosomal Domains by Loop Extrusion. Cell Rep. 2016, 15, 2038–2049. [Google Scholar] [CrossRef] [Green Version]
- Hansen, A.S.; Cattoglio, C.; Darzacq, X.; Tjian, R. Recent evidence that TADs and chromatin loops are dynamic structures. Nucleus 2018, 9, 20–32. [Google Scholar] [CrossRef] [Green Version]
- Fishilevich, S.; Nudel, R.; Rappaport, N.; Hadar, R.; Plaschkes, I.; Iny Stein, T.; Rosen, N.; Kohn, A.; Twik, M.; Safran, M.; et al. GeneHancer: Genome-wide integration of enhancers and target genes in GeneCards. Database (Oxford) 2017, 2017. [Google Scholar] [CrossRef] [Green Version]
- Rosenbloom, K.R.; Sloan, C.A.; Malladi, V.S.; Dreszer, T.R.; Learned, K.; Kirkup, V.M.; Wong, M.C.; Maddren, M.; Fang, R.; Heitner, S.G.; et al. ENCODE data in the UCSC Genome Browser: Year 5 update. Nucleic Acids Res. 2013, 41, D56–D63. [Google Scholar] [CrossRef]
- Casper, J.; Zweig, A.S.; Villarreal, C.; Tyner, C.; Speir, M.L.; Rosenbloom, K.R.; Raney, B.J.; Lee, C.M.; Lee, B.T.; Karolchik, D.; et al. The UCSC Genome Browser database: 2018 update. Nucleic Acids Res. 2018, 46, D762–D769. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kent, W.J.; Sugnet, C.W.; Furey, T.S.; Roskin, K.M.; Pringle, T.H.; Zahler, A.M.; Haussler, D. The human genome browser at UCSC. Genome Res. 2002, 12, 996–1006. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ong, C.T.; Corces, V.G. CTCF: An architectural protein bridging genome topology and function. Nat. Rev. Genet. 2014, 15, 234–246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ren, G.; Jin, W.; Cui, K.; Rodrigez, J.; Hu, G.; Zhang, Z.; Larson, D.R.; Zhao, K. CTCF-Mediated Enhancer-Promoter Interaction Is a Critical Regulator of Cell-to-Cell Variation of Gene Expression. Mol Cell 2017, 67, 1049–1058. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ding, M.; Liu, Y.; Liao, X.; Zhan, H.; Liu, Y.; Huang, W. Enhancer RNAs (eRNAs): New Insights into Gene Transcription and Disease Treatment. J. Cancer 2018, 9, 2334–2340. [Google Scholar] [CrossRef] [Green Version]
- Zhu, Y.; Sun, L.; Chen, Z.; Whitaker, J.W.; Wang, T.; Wang, W. Predicting enhancer transcription and activity from chromatin modifications. Nucleic Acids Res. 2013, 41, 10032–10043. [Google Scholar] [CrossRef]
- Meng, H.; Bartholomew, B. Emerging roles of transcriptional enhancers in chromatin looping and promoter-proximal pausing of RNA polymerase II. J. Biol. Chem. 2018, 293, 13786–13794. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Su, Z.; Song, X.; Liang, B.; Zeng, F.; Chang, X.; Huang, D. Enhancer RNA-driven looping enhances the transcription of the long noncoding RNA DHRS4-AS1, a controller of the DHRS4 gene cluster. Sci. Rep. 2016, 6, 20961. [Google Scholar] [CrossRef] [Green Version]
- Rada-Iglesias, A. Is H3K4me1 at enhancers correlative or causative? Nat. Genet. 2018, 50, 4–5. [Google Scholar] [CrossRef]
- Calo, E.; Wysocka, J. Modification of enhancer chromatin: What, how, and why? Mol. Cell 2013, 49, 825–837. [Google Scholar] [CrossRef] [Green Version]
- Hu, D.; Gao, X.; Morgan, M.A.; Herz, H.M.; Smith, E.R.; Shilatifard, A. The MLL3/MLL4 branches of the COMPASS family function as major histone H3K4 monomethylases at enhancers. Mol. Cell Biol. 2013, 33, 4745–4754. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sze, C.C.; Shilatifard, A. MLL3/MLL4/COMPASS Family on Epigenetic Regulation of Enhancer Function and Cancer. Cold Spring Harb. Perspect. Med. 2016, 6, a026427. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.E.; Wang, C.; Xu, S.; Cho, Y.W.; Wang, L.; Feng, X.; Baldridge, A.; Sartorelli, V.; Zhuang, L.; Peng, W.; et al. H3K4 mono- and di-methyltransferase MLL4 is required for enhancer activation during cell differentiation. Elife 2013, 2, e01503. [Google Scholar] [CrossRef] [PubMed]
- Smith, E.; Shilatifard, A. The chromatin signaling pathway: Diverse mechanisms of recruitment of histone-modifying enzymes and varied biological outcomes. Mol. Cell 2010, 40, 689–701. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dorighi, K.M.; Swigut, T.; Henriques, T.; Bhanu, N.V.; Scruggs, B.S.; Nady, N.; Still, C.D., 2nd; Garcia, B.A.; Adelman, K.; Wysocka, J. Mll3 and Mll4 Facilitate Enhancer RNA Synthesis and Transcription from Promoters Independently of H3K4 Monomethylation. Mol. Cell 2017, 66, 568–576. [Google Scholar] [CrossRef] [Green Version]
- Local, A.; Huang, H.; Albuquerque, C.P.; Singh, N.; Lee, A.Y.; Wang, W.; Wang, C.; Hsia, J.E.; Shiau, A.K.; Ge, K.; et al. Identification of H3K4me1-associated proteins at mammalian enhancers. Nat. Genet. 2018, 50, 73–82. [Google Scholar] [CrossRef]
- Chen, C.J.; Shih, C.H.; Chang, Y.J.; Hong, S.J.; Li, T.N.; Wang, L.H.; Chen, L. SH2B1 and IRSp53 proteins promote the formation of dendrites and dendritic branches. J. Biol. Chem. 2015, 290, 6010–6021. [Google Scholar] [CrossRef] [Green Version]
- Shih, C.H.; Chen, C.J.; Chen, L. New function of the adaptor protein SH2B1 in brain-derived neurotrophic factor-induced neurite outgrowth. PLoS ONE 2013, 8, e79619. [Google Scholar] [CrossRef] [Green Version]
- Dahl, J.A.; Collas, P. Q2ChIP, a quick and quantitative chromatin immunoprecipitation assay, unravels epigenetic dynamics of developmentally regulated genes in human carcinoma cells. Stem Cells 2007, 25, 1037–1046. [Google Scholar] [CrossRef]
- Li, Y.L.; Weng, J.C.; Hsiao, C.C.; Chou, M.T.; Tseng, C.W.; Hung, J.H. PEAT: An intelligent and efficient paired-end sequencing adapter trimming algorithm. BMC Bioinform. 2015, 16 (Suppl. 1), S2. [Google Scholar] [CrossRef] [Green Version]
- Langmead, B.; Trapnell, C.; Pop, M.; Salzberg, S.L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009, 10, R25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Liu, T.; Meyer, C.A.; Eeckhoute, J.; Johnson, D.S.; Bernstein, B.E.; Nusbaum, C.; Myers, R.M.; Brown, M.; Li, W.; et al. Model-based analysis of ChIP-Seq (MACS). Genome Biol. 2008, 9, R137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Consortium, E.P. An integrated encyclopedia of DNA elements in the human genome. Nature 2012, 489, 57–74. [Google Scholar] [CrossRef] [PubMed]






© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chang, C.-Y.; Hung, J.-H.; Huang, L.-W.; Li, J.; Fung, K.S.; Kao, C.-F.; Chen, L. Epigenetic Regulation of WNT3A Enhancer during Regeneration of Injured Cortical Neurons. Int. J. Mol. Sci. 2020, 21, 1891. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21051891
Chang C-Y, Hung J-H, Huang L-W, Li J, Fung KS, Kao C-F, Chen L. Epigenetic Regulation of WNT3A Enhancer during Regeneration of Injured Cortical Neurons. International Journal of Molecular Sciences. 2020; 21(5):1891. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21051891
Chicago/Turabian StyleChang, Chu-Yuan, Jui-Hung Hung, Liang-Wei Huang, Joye Li, Ka Shing Fung, Cheng-Fu Kao, and Linyi Chen. 2020. "Epigenetic Regulation of WNT3A Enhancer during Regeneration of Injured Cortical Neurons" International Journal of Molecular Sciences 21, no. 5: 1891. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21051891