Calcium-Dependent Calpain Activation-Mediated Mitochondrial Dysfunction and Oxidative Stress Are Required for Cytotoxicity of Epinecidin-1 in Human Synovial Sarcoma SW982 Cells


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Epi-1 Induction of Cytotoxicity in Synovial Sarcoma Cells is Dependent on its Folding Structure
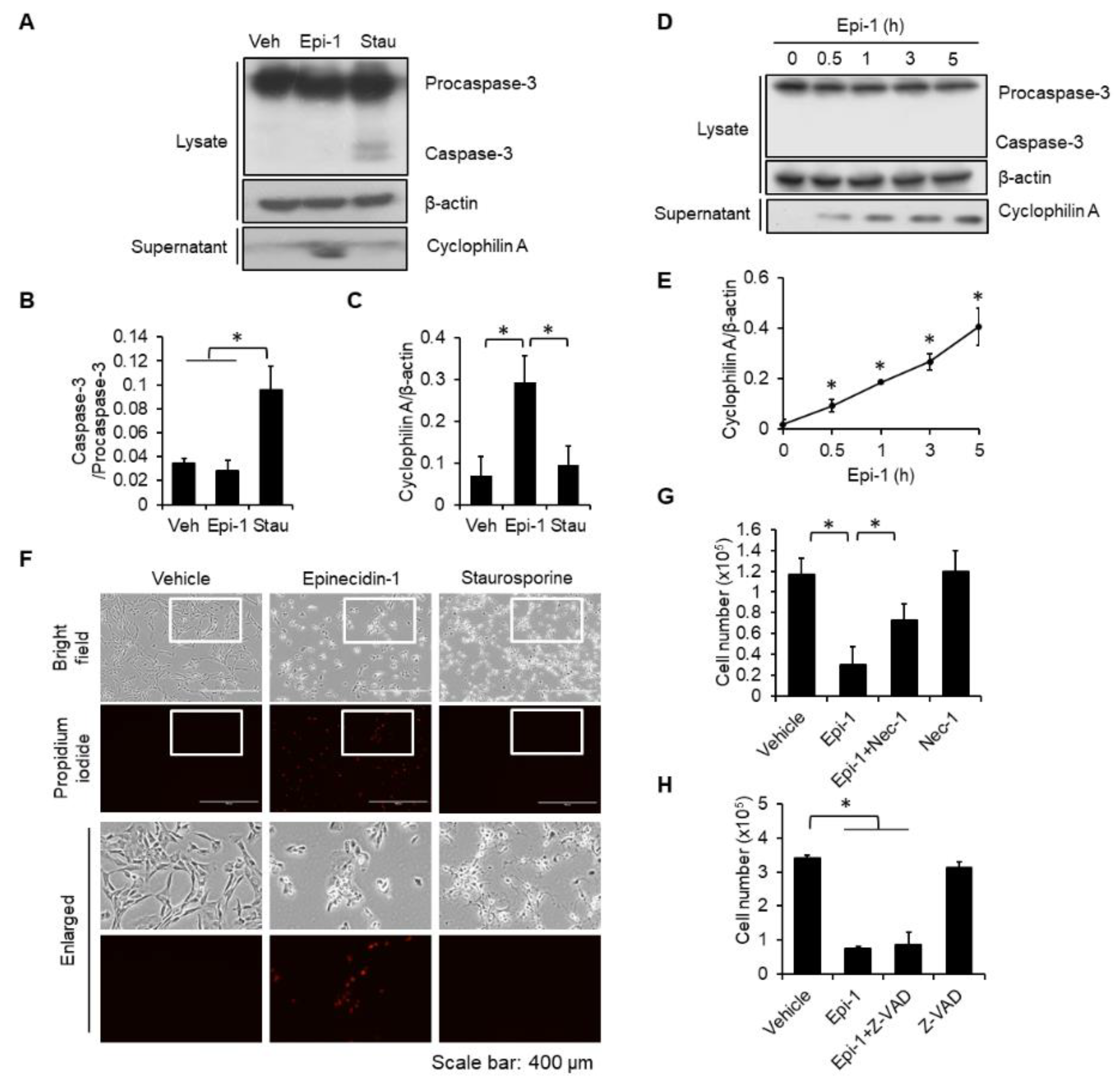
2.2. Epi-1 Triggers Caspase-Independent Cell Death in SW982 Cells
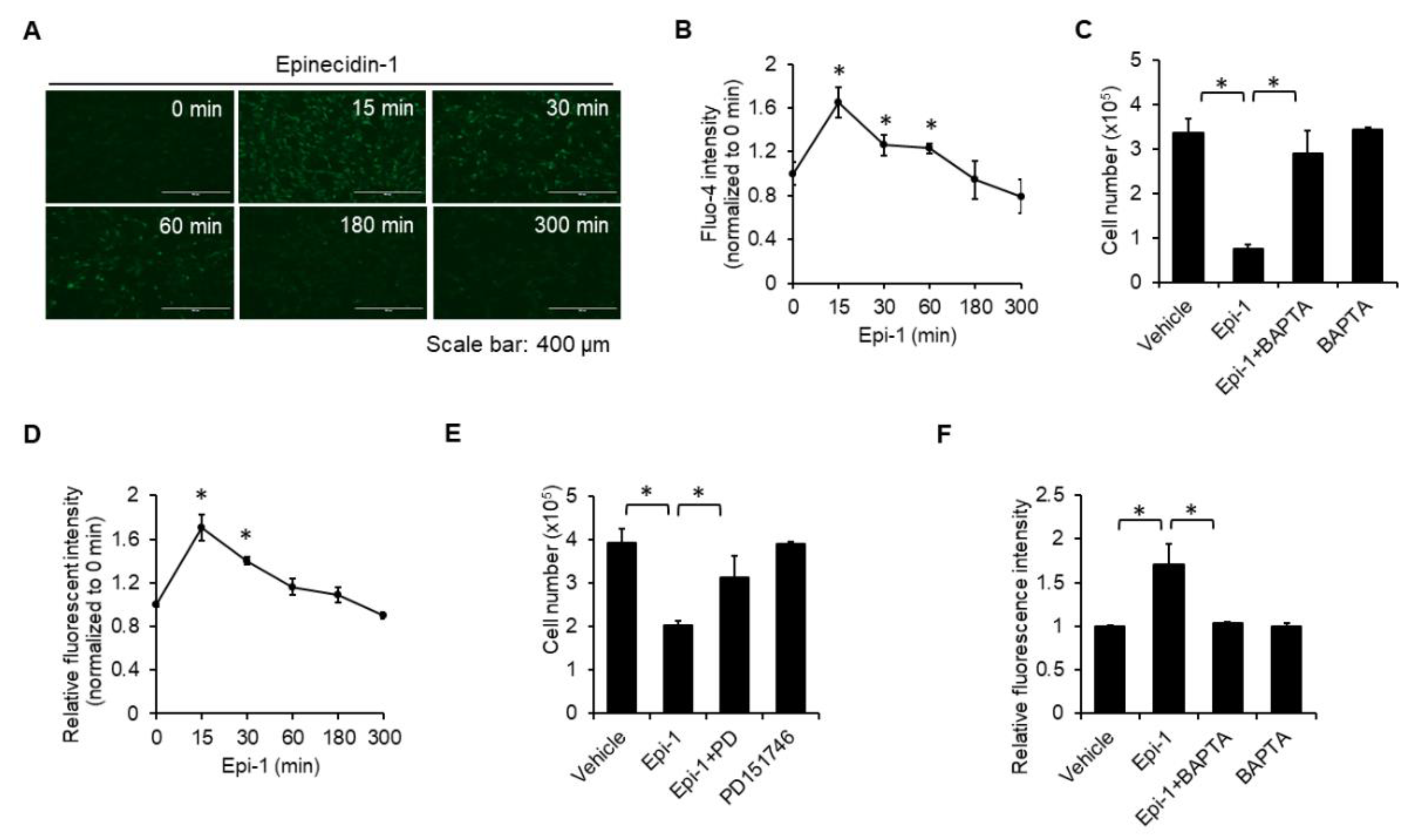
2.3. Calcium and Calpain are Required for Epi-1-Induced Cell Death
2.4. Epi-1 Induces Mitochondrial Hyperpolarization
2.5. Epi-1 Induces Oxidative Stress and Downregulation of Antioxidant Proteins
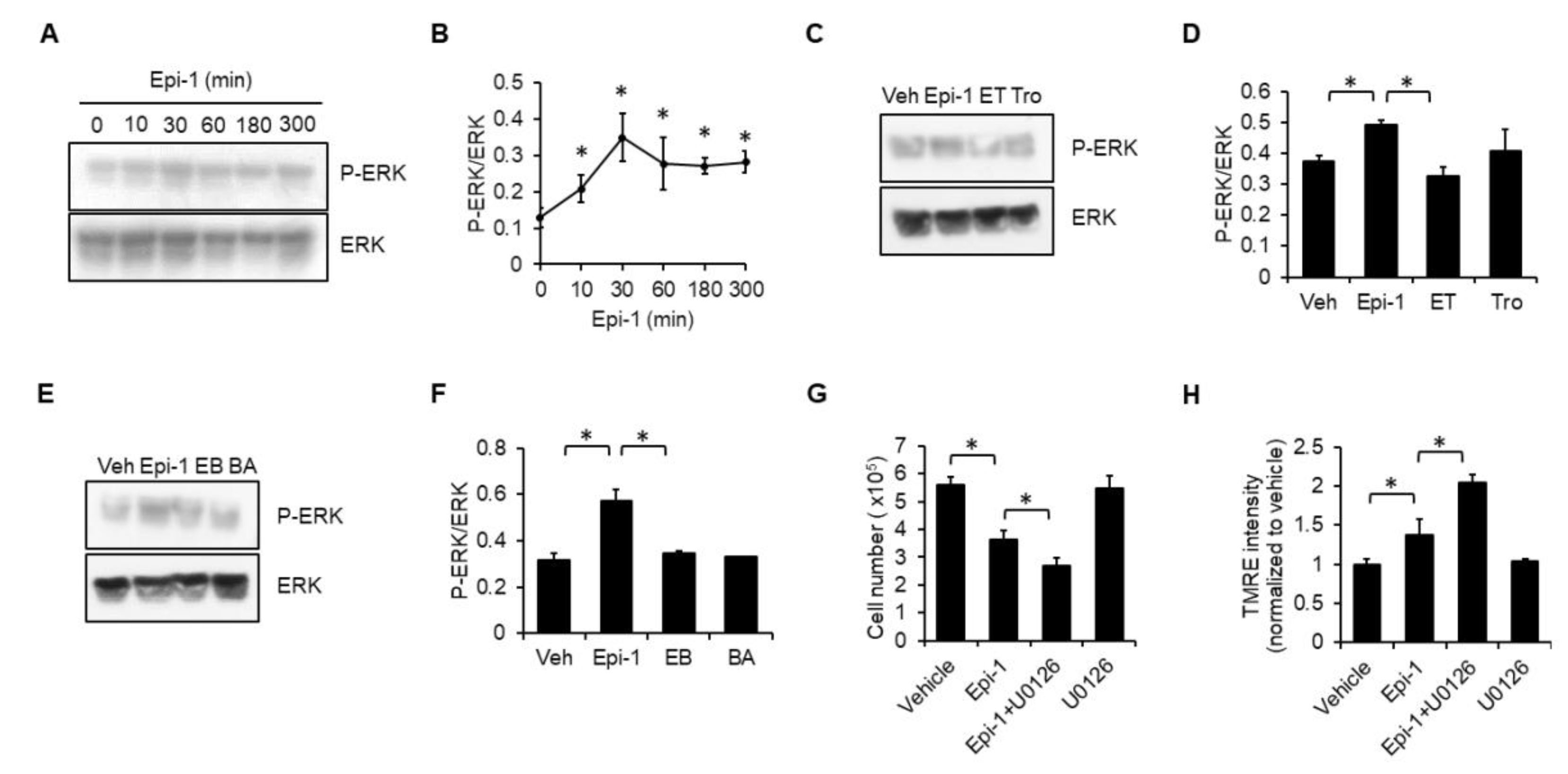
2.6. ERK Plays a Protective Role in Epi-1-Mediated Cell Death
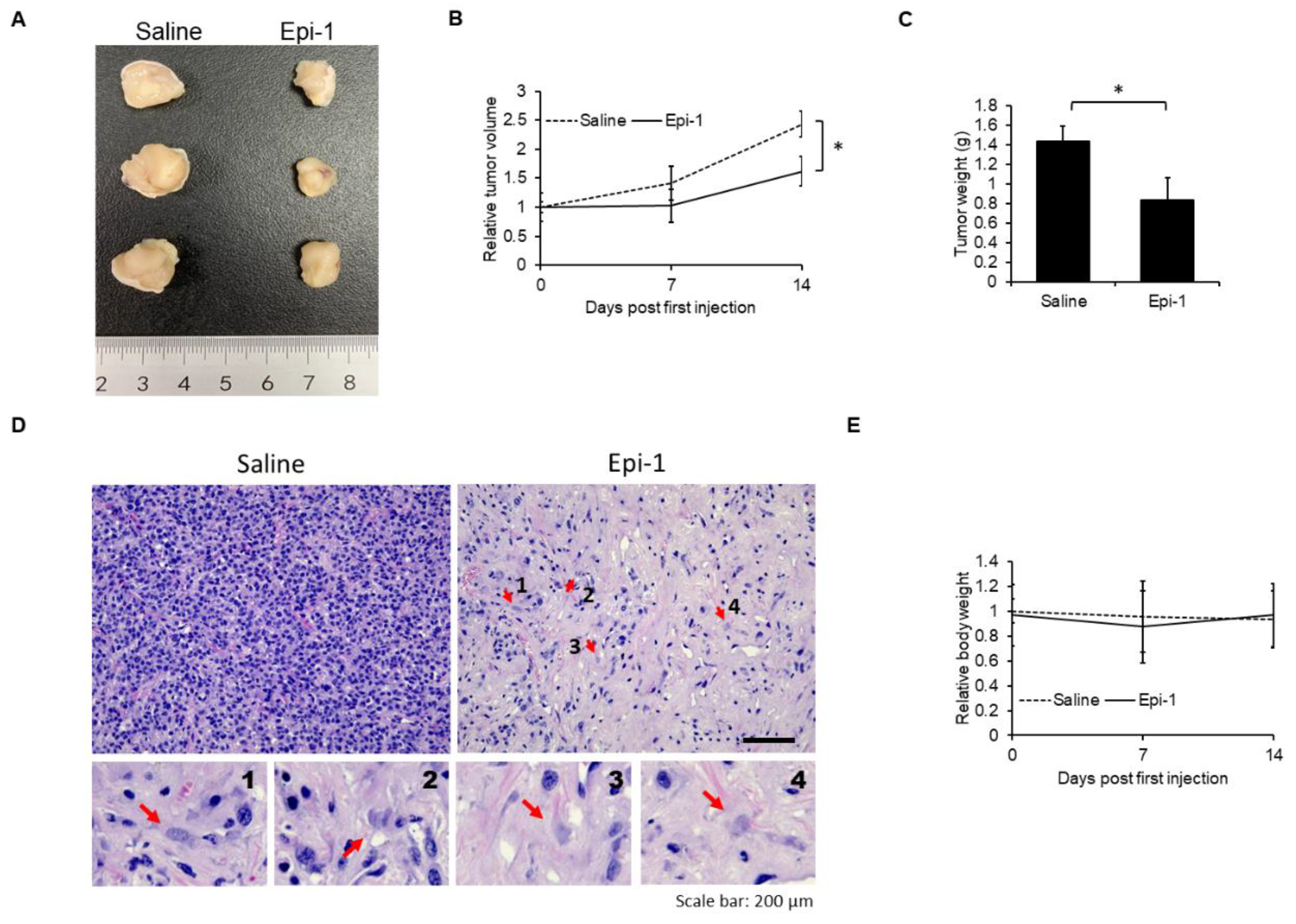
2.7. In Vivo Antisynovial Sarcoma Ability of Epi-1
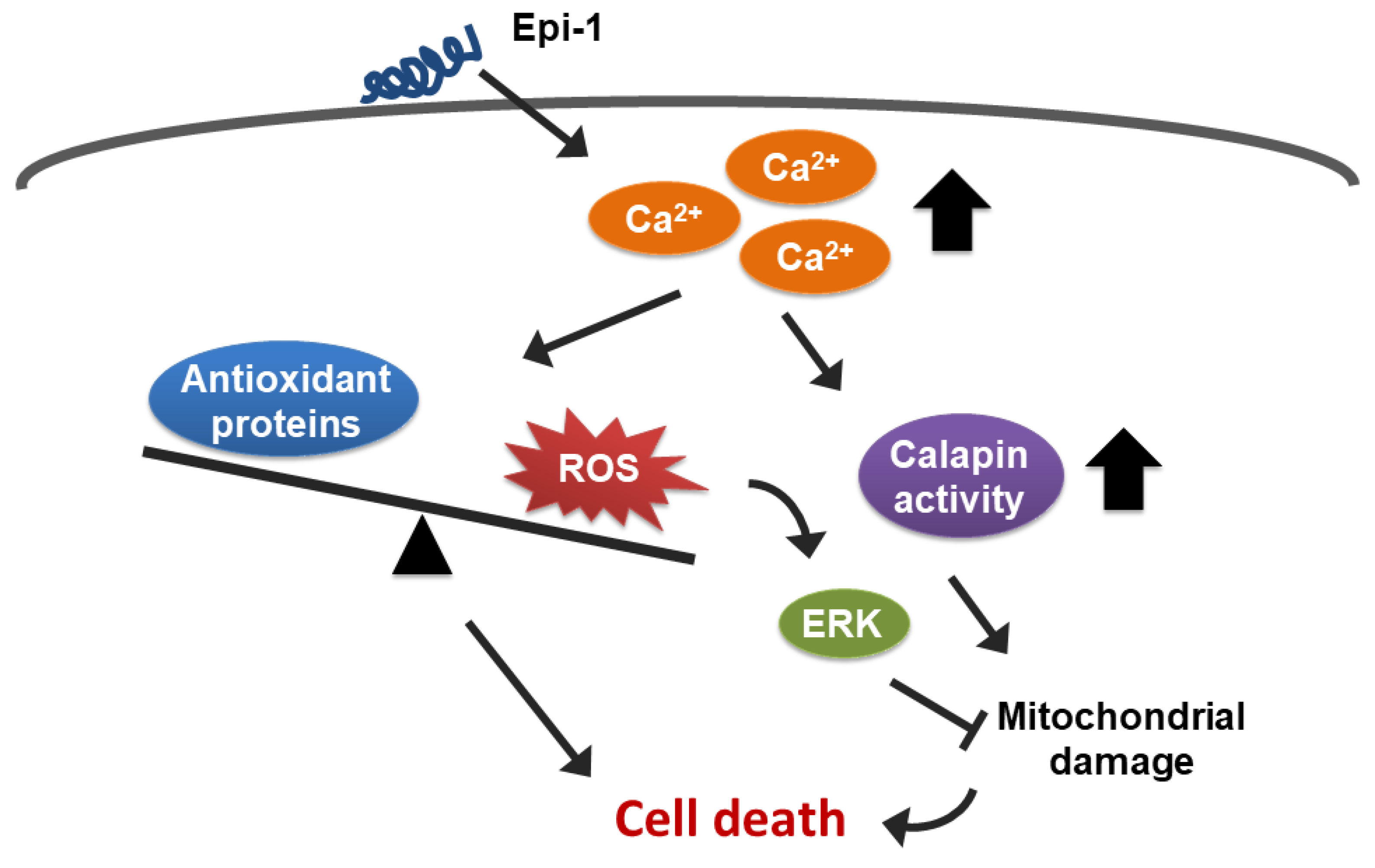
3. Discussion
4. Materials and Methods
4.1. Reagents
4.2. Cell Culture
4.3. Cytotoxic Assay
4.4. Western Blotting
4.5. ROS Measurement
4.6. Calcium and Calpain Activity
4.7. Mitochondrial Function
4.8. Human Synovial Sarcoma Xenograft Nude Mice Model
4.9. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Scheer, M.; Dantonello, T.; Hallmen, E.; Blank, B.; Sparber-Sauer, M.; Vokuhl, C.; Leuschner, I.; Munter, M.W.; von Kalle, T.; Bielack, S.S.; et al. Synovial Sarcoma Recurrence in Children and Young Adults. Ann Surg. Oncol. 2016, 23, 618–626. [Google Scholar] [CrossRef]
- Stacchiotti, S.; Van Tine, B.A. Synovial Sarcoma: Current Concepts and Future Perspectives. J. Clin. Oncol. 2018, 36, 180–187. [Google Scholar] [CrossRef]
- In, G.K.; Hu, J.S.; Tseng, W.W. Treatment of advanced, metastatic soft tissue sarcoma: Latest evidence and clinical considerations. Adv. Med. Oncol. 2017, 9, 533–550. [Google Scholar] [CrossRef]
- Miwa, S.; Yamamoto, N.; Hayashi, K.; Takeuchi, A.; Igarashi, K.; Tsuchiya, H. Therapeutic Targets for Bone and Soft-Tissue Sarcomas. Int. J. Mol. Sci. 2019, 20, 170. [Google Scholar] [CrossRef] [Green Version]
- De Vita, A.; Miserocchi, G.; Recine, F.; Mercatali, L.; Pieri, F.; Medri, L.; Bongiovanni, A.; Cavaliere, D.; Liverani, C.; Spadazzi, C.; et al. Activity of Eribulin in a Primary Culture of Well-Differentiated/Dedifferentiated Adipocytic Sarcoma. Molecules 2016, 21, 1662. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riedel, R.F.; Jones, R.L.; Italiano, A.; Bohac, C.; Thompson, J.C.; Mueller, K.; Khan, Z.; Pollack, S.M.; Van Tine, B.A. Systemic Anti-Cancer Therapy in Synovial Sarcoma: A Systematic Review. Cancers 2018, 10, 417. [Google Scholar] [CrossRef] [Green Version]
- Kawai, A.; Yonemori, K.; Takahashi, S.; Araki, N.; Ueda, T. Systemic Therapy for Soft Tissue Sarcoma: Proposals for the Optimal Use of Pazopanib, Trabectedin, and Eribulin. Adv. Ther. 2017, 34, 1556–1571. [Google Scholar] [CrossRef] [PubMed]
- Pender, A.; Davis, E.J.; Chauhan, D.; Messiou, C.; Al-Muderis, O.; Thway, K.; Fisher, C.; Zaidi, S.; Miah, A.; Judson, I.; et al. Poor treatment outcomes with palliative gemcitabine and docetaxel chemotherapy in advanced and metastatic synovial sarcoma. Med. Oncol. 2018, 35, 131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pan, C.Y.; Chen, J.C.; Sheen, J.F.; Lin, T.L.; Chen, J.Y. Epinecidin-1 has immunomodulatory effects, facilitating its therapeutic use in a mouse model of Pseudomonas aeruginosa sepsis. Antimicrob Agents Chemother 2014, 58, 4264–4274. [Google Scholar] [CrossRef] [Green Version]
- Su, B.C.; Chen, J.Y. Antimicrobial Peptide Epinecidin-1 Modulates MyD88 Protein Levels via the Proteasome Degradation Pathway. Mar. Drugs 2017, 15, 362. [Google Scholar] [CrossRef] [Green Version]
- Su, B.C.; Huang, H.N.; Lin, T.W.; Hsiao, C.D.; Chen, J.Y. Epinecidin-1 protects mice from LPS-induced endotoxemia and cecal ligation and puncture-induced polymicrobial sepsis. Biochim Biophys Acta Mol. Basis Dis. 2017, 1863, 3028–3037. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.N.; Chuang, C.M.; Chen, J.Y.; Chieh-Yu, P. Epinecidin-1: A marine fish antimicrobial peptide with therapeutic potential against Trichomonas vaginalis infection in mice. Peptides 2019, 112, 139–148. [Google Scholar] [CrossRef] [PubMed]
- Su, B.C.; Wu, T.H.; Hsu, C.H.; Chen, J.Y. Distribution of positively charged amino acid residues in antimicrobial peptide epinecidin-1 is crucial for in vitro glioblastoma cytotoxicity and its underlying mechanisms. Chem Biol Interact 2019, 315, 108904. [Google Scholar] [CrossRef] [PubMed]
- Lin, W.J.; Chien, Y.L.; Pan, C.Y.; Lin, T.L.; Chen, J.Y.; Chiu, S.J.; Hui, C.F. Epinecidin-1, an antimicrobial peptide from fish (Epinephelus coioides) which has an antitumor effect like lytic peptides in human fibrosarcoma cells. Peptides 2009, 30, 283–290. [Google Scholar] [CrossRef] [PubMed]
- Su, B.C.; Pan, C.Y.; Chen, J.Y. Antimicrobial Peptide TP4 Induces ROS-Mediated Necrosis by Triggering Mitochondrial Dysfunction in Wild-Type and Mutant p53 Glioblastoma Cells. Cancers 2019, 11, 171. [Google Scholar] [CrossRef] [Green Version]
- Huang, T.C.; Lee, J.F.; Chen, J.Y. Pardaxin, an antimicrobial peptide, triggers caspase-dependent and ROS-mediated apoptosis in HT-1080 cells. Mar Drugs 2011, 9, 1995–2009. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Christofferson, D.E.; Yuan, J. Cyclophilin A release as a biomarker of necrotic cell death. Cell Death Differ. 2010, 17, 1942–1943. [Google Scholar] [CrossRef]
- Bozym, R.A.; Patel, K.; White, C.; Cheung, K.H.; Bergelson, J.M.; Morosky, S.A.; Coyne, C.B. Calcium signals and calpain-dependent necrosis are essential for release of coxsackievirus B from polarized intestinal epithelial cells. Mol. Biol. Cell 2011, 22, 3010–3021. [Google Scholar] [CrossRef] [Green Version]
- Feno, S.; Butera, G.; Reane, D.V.; Rizzuto, R.; Raffaello, A. Crosstalk between Calcium and ROS in Pathophysiological Conditions. Oxid Med. Cell Longev. 2019; Artn 9324018. [Google Scholar]
- Demicco, E.G.; Maki, R.G.; Lev, D.C.; Lazar, A.J. New therapeutic targets in soft tissue sarcoma. Adv. Anat. Pathol. 2012, 19, 170–180. [Google Scholar] [CrossRef] [Green Version]
- Elmore, S.A.; Dixon, D.; Hailey, J.R.; Harada, T.; Herbert, R.A.; Maronpot, R.R.; Nolte, T.; Rehg, J.E.; Rittinghausen, S.; Rosol, T.J.; et al. Recommendations from the INHAND Apoptosis/Necrosis Working Group. Toxicol. Pathol. 2016, 44, 173–188. [Google Scholar] [CrossRef] [Green Version]
- Smith, M.A.; Schnellmann, R.G. Calpains, mitochondria, and apoptosis. Cardiovasc Res. 2012, 96, 32–37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Villalpando Rodriguez, G.E.; Torriglia, A. Calpain 1 induce lysosomal permeabilization by cleavage of lysosomal associated membrane protein 2. Biochim. Biophys. Acta 2013, 1833, 2244–2253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mandic, A.; Viktorsson, K.; Strandberg, L.; Heiden, T.; Hansson, J.; Linder, S.; Shoshan, M.C. Calpain-mediated Bid cleavage and calpain-independent Bak modulation: Two separate pathways in cisplatin-induced apoptosis. Mol. Cell Biol. 2002, 22, 3003–3013. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verhagen, A.M.; Silke, J.; Ekert, P.G.; Pakusch, M.; Kaufmann, H.; Connolly, L.M.; Day, C.L.; Tikoo, A.; Burke, R.; Wrobel, C.; et al. HtrA2 promotes cell death through its serine protease activity and its ability to antagonize inhibitor of apoptosis proteins. J. Biol. Chem. 2002, 277, 445–454. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aits, S.; Jaattela, M. Lysosomal cell death at a glance. J. Cell Sci. 2013, 126, 1905–1912. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Todkar, K.; Ilamathi, H.S.; Germain, M. Mitochondria and Lysosomes: Discovering Bonds. Front Cell Dev. Biol. 2017, 5, 106. [Google Scholar] [CrossRef] [Green Version]
- Rintanen, N.; Karjalainen, M.; Alanko, J.; Paavolainen, L.; Maki, A.; Nissinen, L.; Lehkonen, M.; Kallio, K.; Cheng, R.H.; Upla, P.; et al. Calpains promote alpha 2 beta 1 integrin turnover in nonrecycling integrin pathway. Mol. Biol. Cell 2012, 23, 448–463. [Google Scholar] [CrossRef]
- Vihinen, P.; Riikonen, T.; Laine, A.; Heino, J. Integrin alpha 2 beta 1 in tumorigenic human osteosarcoma cell lines regulates cell adhesion, migration, and invasion by interaction with type I collagen. Cell Growth Differ. 1996, 7, 439–447. [Google Scholar]
- Kaufmann, S.H.; Earnshaw, W.C. Induction of apoptosis by cancer chemotherapy. Exp. Cell Res. 2000, 256, 42–49. [Google Scholar] [CrossRef]
- Pommier, Y.; Sordet, O.; Antony, S.; Hayward, R.L.; Kohn, K.W. Apoptosis defects and chemotherapy resistance: Molecular interaction maps and networks. Oncogene 2004, 23, 2934–2949. [Google Scholar] [CrossRef] [Green Version]
- Dhotre, K.B.; Adams, S.A.; Hebert, J.R.; Bottai, M.; Heiney, S.P. Oncology Nurses’ Experiences With Patients Who Choose to Discontinue Cancer Chemotherapy. Oncol. Nurs Forum. 2016, 43, 617–623. [Google Scholar] [CrossRef] [PubMed]
- Matsuzaki, K. Control of cell selectivity of antimicrobial peptides. Biochim Biophys Acta 2009, 1788, 1687–1692. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, H.N.; Pan, C.Y.; Wu, H.Y.; Chen, J.Y. Antimicrobial peptide Epinecidin-1 promotes complete skin regeneration of methicillin-resistant Staphylococcus aureus-infected burn wounds in a swine model. Oncotarget 2017, 8, 21067–21080. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gong, Y.T.; Fan, Z.Y.; Luo, G.P.; Yang, C.; Huang, Q.Y.; Fan, K.; Cheng, H.; Jin, K.Z.; Ni, Q.X.; YU, X.J.; et al. The role of necroptosis in cancer biology and therapy. Mol. Cancer 2019, 18. [Google Scholar] [CrossRef] [Green Version]
- Ramachandran, S.; Vinitha, A.; Kartha, C.C. Cyclophilin A enhances macrophage differentiation and lipid uptake in high glucose conditions: A cellular mechanism for accelerated macro vascular disease in diabetes mellitus. Cardiovasc. Diabetol. 2016, 15. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.Y.; Lin, W.J.; Wu, J.L.; Her, G.M.; Hui, C.F. Epinecidin-1 peptide induces apoptosis which enhances antitumor effects in human leukemia U937 cells. Peptides 2009, 30, 2365–2373. [Google Scholar] [CrossRef]
- Su, B.C.; Chen, J.Y. Pharmacological inhibition of p38 potentiates antimicrobial peptide TP4-induced cell death in glioblastoma cells. Mol. Cell Biochem. 2019. [Google Scholar] [CrossRef]
- Stalker, T.J.; Gong, Y.; Scalia, R. The calcium-dependent protease calpain causes endothelial dysfunction in type 2 diabetes. Diabetes 2005, 54, 1132–1140. [Google Scholar] [CrossRef] [Green Version]
- Steinstraesser, L.; Hauk, J.; Jacobsen, F.; Stricker, I.; Steinau, H.U.; Al-Benna, S. Establishment of a synovial sarcoma model in athymic nude mice. In Vivo 2011, 25, 165–169. [Google Scholar]
- Ting, C.H.; Chen, Y.C.; Wu, C.J.; Chen, J.Y. Targeting FOSB with a cationic antimicrobial peptide, TP4, for treatment of triple-negative breast cancer. Oncotarget 2016, 7, 40329–40347. [Google Scholar] [CrossRef]








© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Su, B.-C.; Li, C.-C.; Horng, J.-L.; Chen, J.-Y. Calcium-Dependent Calpain Activation-Mediated Mitochondrial Dysfunction and Oxidative Stress Are Required for Cytotoxicity of Epinecidin-1 in Human Synovial Sarcoma SW982 Cells. Int. J. Mol. Sci. 2020, 21, 2109. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21062109
Su B-C, Li C-C, Horng J-L, Chen J-Y. Calcium-Dependent Calpain Activation-Mediated Mitochondrial Dysfunction and Oxidative Stress Are Required for Cytotoxicity of Epinecidin-1 in Human Synovial Sarcoma SW982 Cells. International Journal of Molecular Sciences. 2020; 21(6):2109. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21062109
Chicago/Turabian StyleSu, Bor-Chyuan, Chao-Chin Li, Jiun-Lin Horng, and Jyh-Yih Chen. 2020. "Calcium-Dependent Calpain Activation-Mediated Mitochondrial Dysfunction and Oxidative Stress Are Required for Cytotoxicity of Epinecidin-1 in Human Synovial Sarcoma SW982 Cells" International Journal of Molecular Sciences 21, no. 6: 2109. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21062109