SIRT2 Affects Primary Cilia Formation by Regulating mTOR Signaling in Retinal Pigmented Epithelial Cells


Abstract
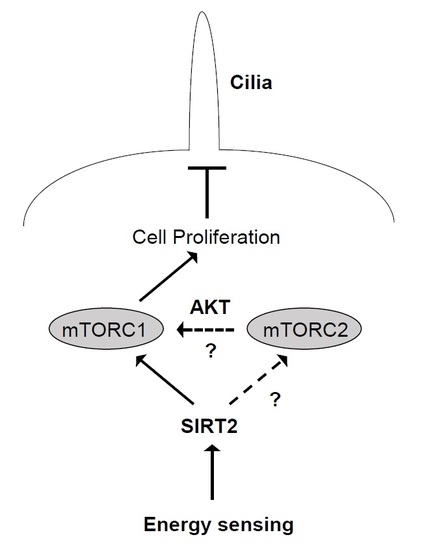
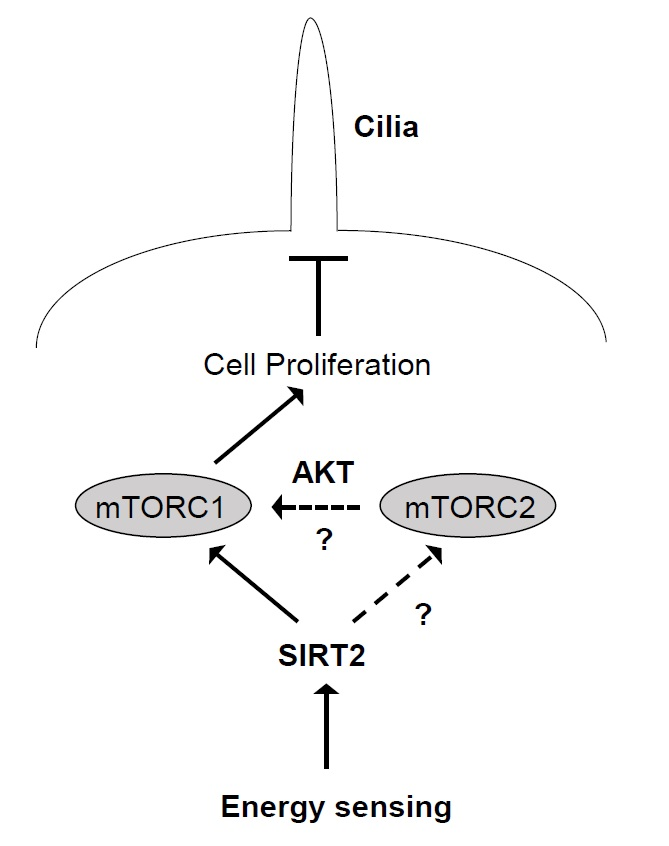
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
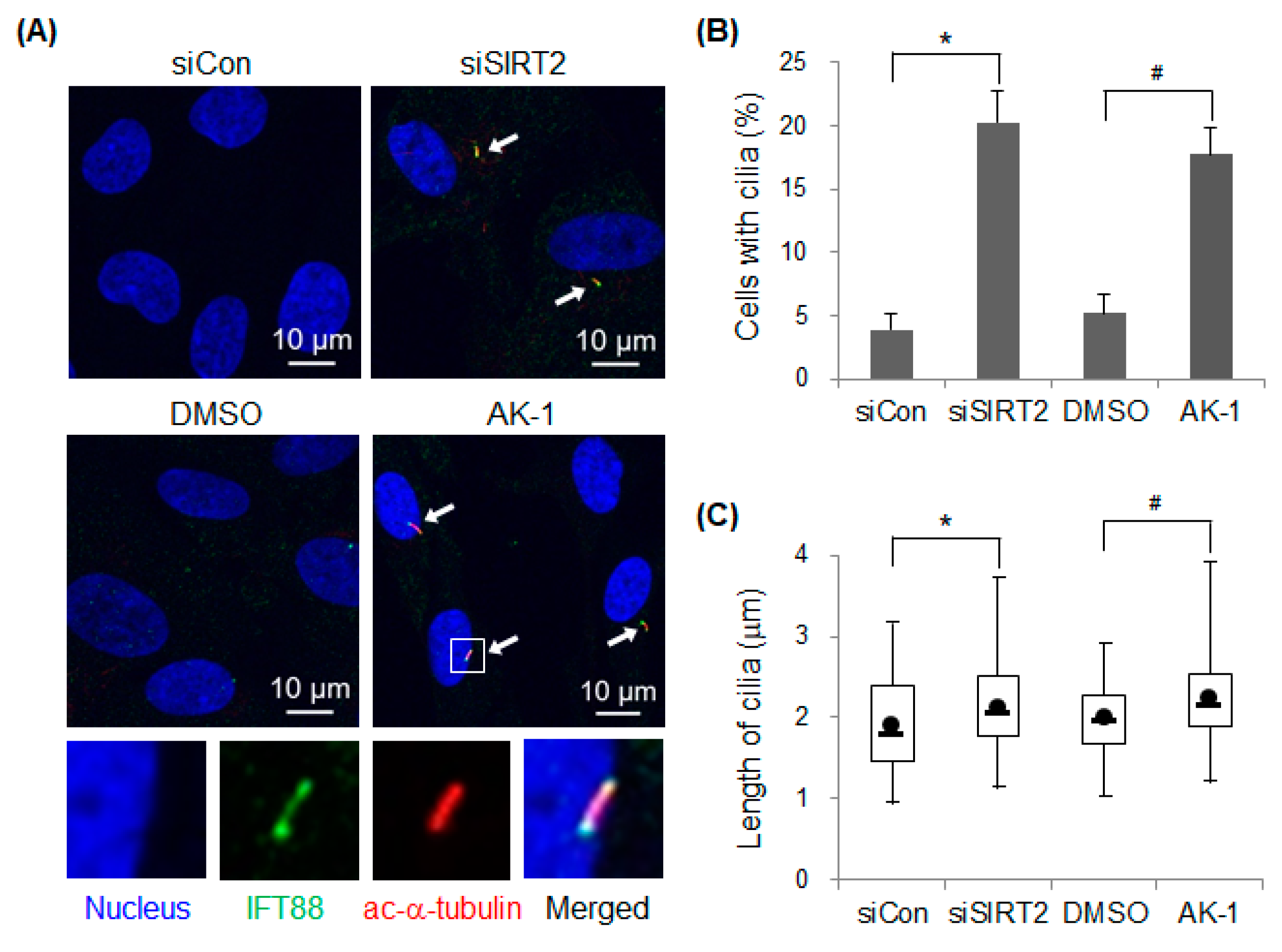
2.1. Suppression of SIRT2 Induces Primary Cilia Formation
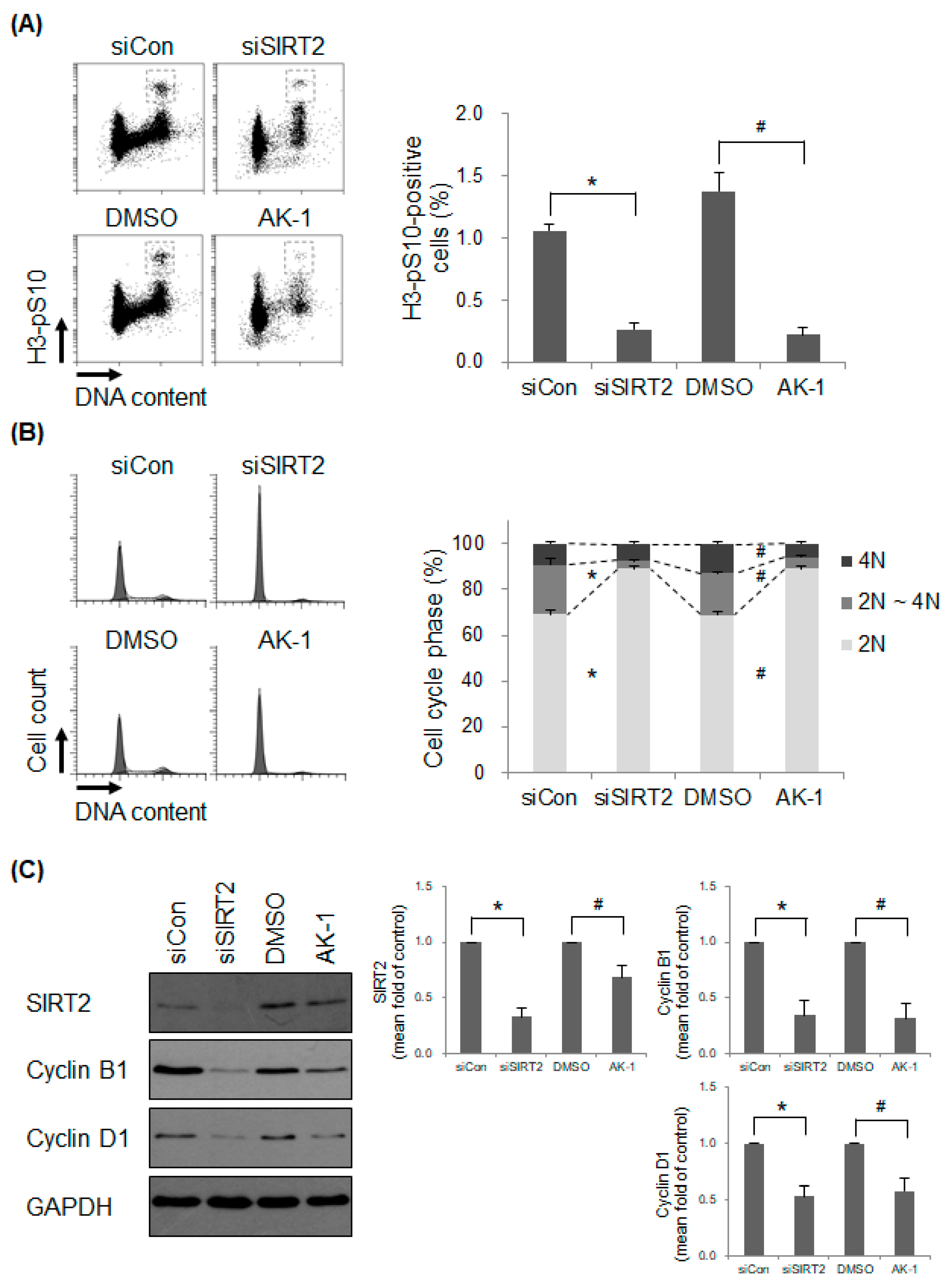
2.2. Suppression of SIRT2 Induces a Non-Proliferating Status
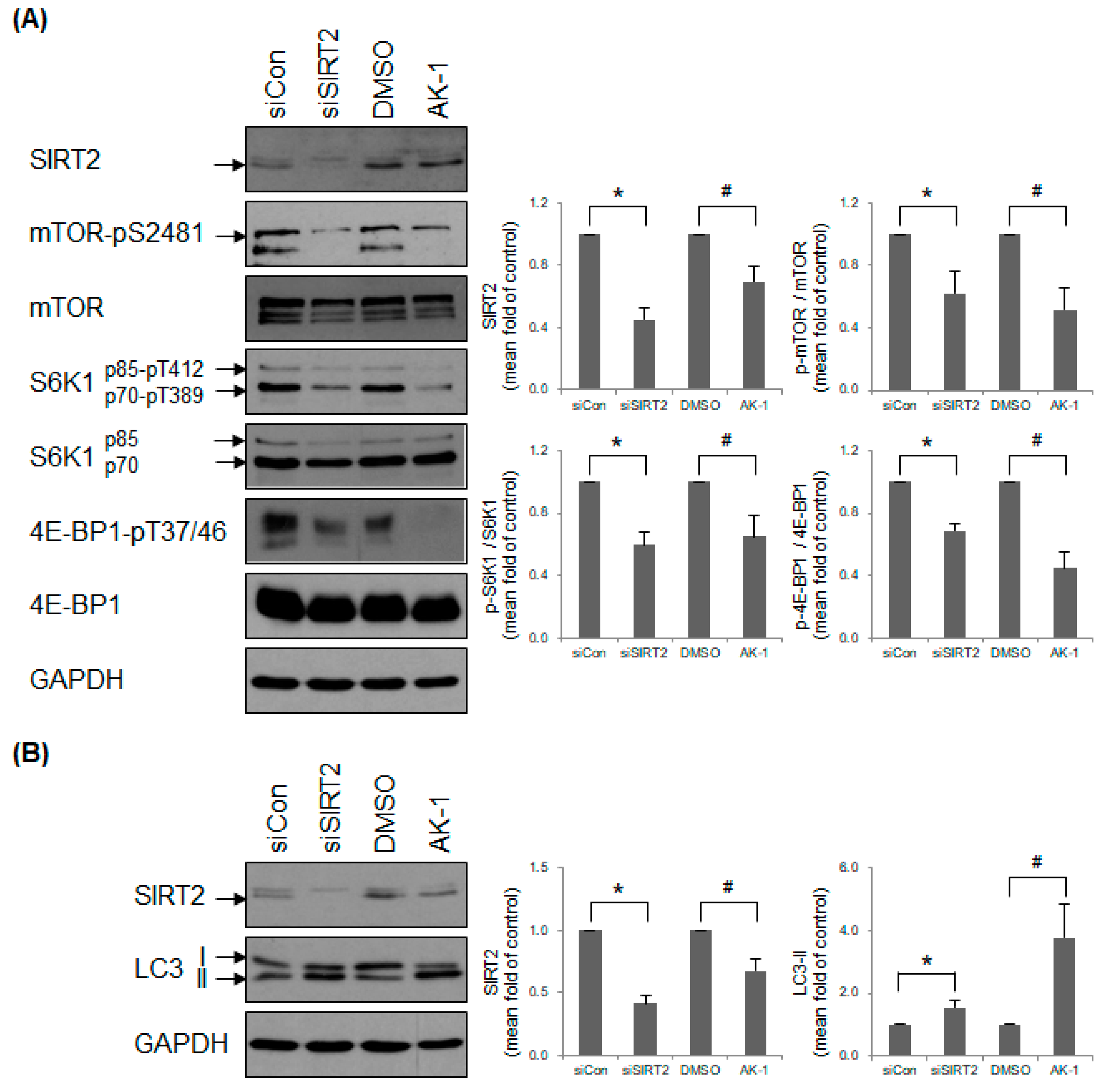
2.3. Suppression of SIRT2 Attenuates mTOR Signaling
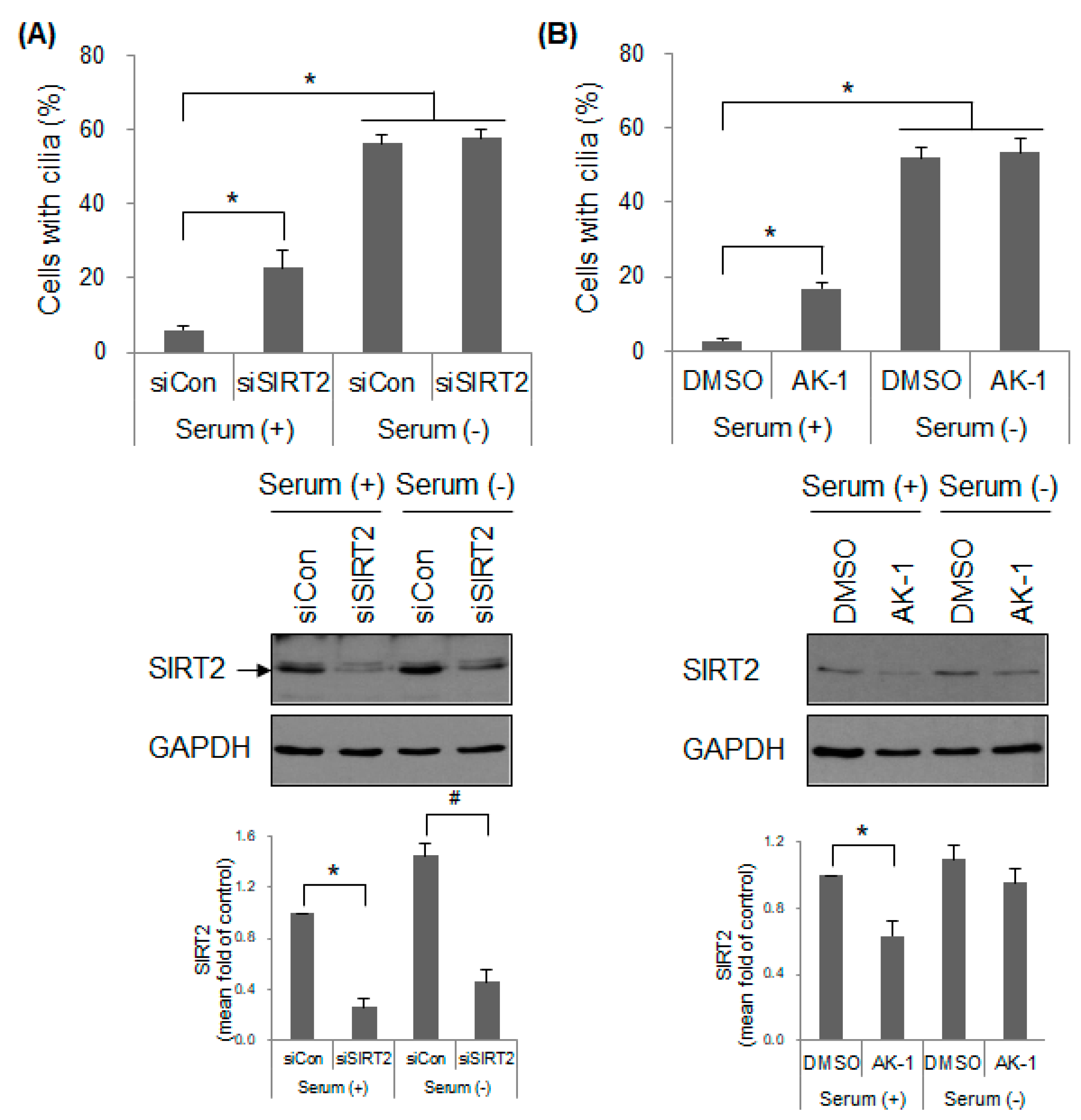
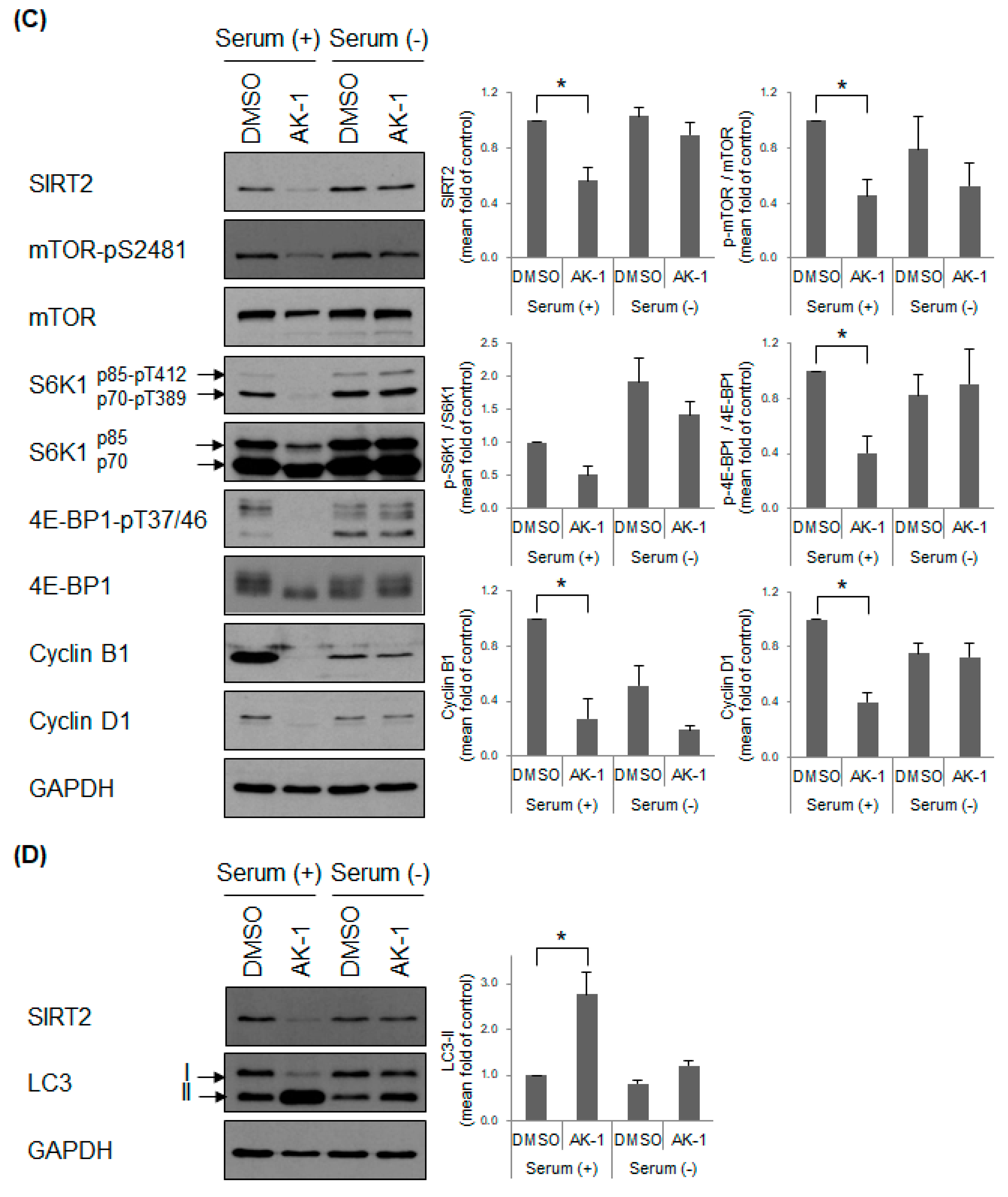
2.4. Suppression of SIRT2 Induces Cilia Formation and Inhibits mTOR Signaling in a Serum-Dependent Manner
2.5. Inhibition of mTOR Induces a Non-Proliferating Status and Increases Primary Cilia Formation
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Drug Treatment
4.3. Small Interfering RNA (siRNA) Transfection
4.4. Immunofluorescence Staining
4.5. Flow Cytometry Analysis
4.6. Western Blotting
4.7. Data and Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| mTOR | Mammalian target of rapamycin |
| S6K1 | Ribosomal protein S6 kinase 1 |
| 4E-BP1 | Eukaryotic initiation factor 4E binding protein 1 |
References
- Milne, J.C.; Denu, J.M. The Sirtuin family: Therapeutic targets to treat diseases of aging. Curr. Opin. Chem. Biol. 2008, 12, 11–17. [Google Scholar] [CrossRef] [PubMed]
- Teng, Y.B.; Jing, H.; Aramsangtienchai, P.; He, B.; Khan, S.; Hu, J.; Lin, H.; Hao, Q. Efficient demyristoylase activity of SIRT2 revealed by kinetic and structural studies. Sci. Rep. 2015, 5, 8529. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gomes, P.; Fleming Outeiro, T.; Cavadas, C. Emerging role of Sirtuin 2 in the regulation of mammalian metabolism. Trends Pharmacol. Sci. 2015, 36, 756–768. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Yang, J.; Hong, T.; Chen, X.; Cui, L. SIRT2: Controversy and multiple roles in disease and physiology. Ageing Res. Rev. 2019, 55, 100961. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.S.; Vassilopoulos, A.; Wang, R.H.; Lahusen, T.; Xiao, Z.; Xu, X.; Li, C.; Veenstra, T.D.; Li, B.; Yu, H.; et al. SIRT2 maintains genome integrity and suppresses tumorigenesis through regulating APC/C activity. Cancer Cell 2011, 20, 487–499. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Kokura, K.; Inoue, T. Stabilization of P/CAF, as a ubiquitin ligase toward MDM2, suppresses mitotic cell death through p53-p21 activation in HCT116cells with SIRT2 suppression. Biochem. Biophys. Res. Commun. 2019, 508, 230–236. [Google Scholar] [CrossRef] [Green Version]
- Inoue, T.; Nakayama, Y.; Yamada, H.; Li, Y.C.; Yamaguchi, S.; Osaki, M.; Kurimasa, A.; Hiratsuka, M.; Katoh, M.; Oshimura, M. SIRT2 downregulation confers resistance to microtubule inhibitors by prolonging chronic mitotic arrest. Cell Cycle 2009, 8, 1279–1291. [Google Scholar] [CrossRef] [Green Version]
- Inoue, T.; Nakayama, Y.; Li, Y.; Matsumori, H.; Takahashi, H.; Kojima, H.; Wanibuchi, H.; Katoh, M.; Oshimura, M. SIRT2 knockdown increases basal autophagy and prevents postslippage death by abnormally prolonging the mitotic arrest that is induced by microtubule inhibitors. FEBS J. 2014, 281, 2623–2637. [Google Scholar] [CrossRef]
- Serrano, L.; Martinez-Redondo, P.; Marazuela-Duque, A.; Vazquez, B.N.; Dooley, S.J.; Voigt, P.; Beck, D.B.; Kane-Goldsmith, N.; Tong, Q.; Rabanal, R.M.; et al. The tumor suppressor SirT2 regulates cell cycle progression and genome stability by modulating the mitotic deposition of H4K20 methylation. Genes Dev. 2013, 27, 639–653. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Park, S.H.; Pantazides, B.G.; Karpiuk, O.; Warren, M.D.; Hardy, C.W.; Duong, D.M.; Park, S.J.; Kim, H.S.; Vassilopoulos, A.; et al. SIRT2 directs the replication stress response through CDK9 deacetylation. Proc. Natl. Acad. Sci. USA 2013, 110, 13546–13551. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Head, P.E.; Daddacha, W.; Park, S.H.; Li, X.; Pan, Y.; Madden, M.Z.; Duong, D.M.; Xie, M.; Yu, B.; et al. ATRIP deacetylation by SIRT2 drives ATR checkpoint activation by promoting binding to RPA-ssDNA. Cell Rep. 2016, 14, 1435–1447. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheon, M.G.; Kim, W.; Choi, M.; Kim, J.E. AK-1, a specific SIRT2 inhibitor, induces cell cycle arrest by downregulating Snail in HCT116 human colon carcinoma cells. Cancer Lett. 2015, 356, 637–645. [Google Scholar] [CrossRef] [PubMed]
- Fliegauf, M.; Benzing, T.; Omran, H. When cilia go bad: Cilia defects and ciliopathies. Nat. Rev. Mol. Cell Biol. 2007, 8, 880–893. [Google Scholar] [CrossRef] [PubMed]
- Satir, P.; Christensen, S.T. Overview of structure and function of mammalian cilia. Annu. Rev. Physiol. 2007, 69, 377–400. [Google Scholar] [CrossRef] [Green Version]
- Pazour, G.J.; Agrin, N.; Leszyk, J.; Witman, G.B. Proteomic analysis of a eukaryotic cilium. J. Cell Biol. 2005, 170, 103–113. [Google Scholar] [CrossRef] [Green Version]
- Ichikawa, M.; Liu, D.; Kastritis, P.L.; Basu, K.; Hsu, T.C.; Yang, S.; Bui, K.H. Subnanometre-resolution structure of the doublet microtubule reveals new classes of microtubule-associated proteins. Nat. Commun. 2017, 8, 15035. [Google Scholar] [CrossRef]
- Ishikawa, H.; Marshall, W.F. Ciliogenesis: Building the cell’s antenna. Nat. Rev. Mol. Cell Biol. 2011, 12, 222–234. [Google Scholar] [CrossRef]
- Wloga, D.; Joachimiak, E.; Louka, P.; Gaertig, J. Posttranslational modifications of tubulin and cilia. Cold Spring Harb. Perspect. Biol. 2017, 9. [Google Scholar] [CrossRef]
- Gaertig, J.; Wloga, D. Ciliary tubulin and its post-translational modifications. Curr. Top. Dev. Biol. 2008, 85, 83–113. [Google Scholar] [CrossRef]
- Spasic, M.; Jacobs, C.R. Primary cilia: Cell and molecular mechanosensors directing whole tissue function. Semin. Cell Dev. Biol. 2017, 71, 42–52. [Google Scholar] [CrossRef]
- Wheway, G.; Nazlamova, L.; Hancock, J.T. Signaling through the primary cilium. Front. Cell Dev. Biol. 2018, 6, 8. [Google Scholar] [CrossRef] [PubMed]
- Pala, R.; Alomari, N.; Nauli, S.M. Primary cilium-dependent signaling mechanisms. Int. J. Mol. Sci. 2017, 18, 2272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malicki, J.J.; Johnson, C.A. The cilium: Cellular antenna and central processing unit. Trends Cell Biol. 2017, 27, 126–140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, L.; Dynlacht, B.D. The regulation of cilium assembly and disassembly in development and disease. Development 2018, 145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanchez, I.; Dynlacht, B.D. Cilium assembly and disassembly. Nat. Cell Biol. 2016, 18, 711–717. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Izawa, I.; Goto, H.; Kasahara, K.; Inagaki, M. Current topics of functional links between primary cilia and cell cycle. Cilia 2015, 4, 12. [Google Scholar] [CrossRef] [Green Version]
- Goto, H.; Inoko, A.; Inagaki, M. Cell cycle progression by the repression of primary cilia formation in proliferating cells. Cell. Mol. Life Sci. 2013, 70, 3893–3905. [Google Scholar] [CrossRef] [Green Version]
- Zhou, X.; Fan, L.X.; Li, K.; Ramchandran, R.; Calvet, J.P.; Li, X. SIRT2 regulates ciliogenesis and contributes to abnormal centrosome amplification caused by loss of polycystin-1. Hum. Mol. Genet. 2014, 23, 1644–1655. [Google Scholar] [CrossRef]
- Plotnikova, O.V.; Pugacheva, E.N.; Golemis, E.A. Primary cilia and the cell cycle. Methods Cell Biol. 2009, 94, 137–160. [Google Scholar] [CrossRef] [Green Version]
- Kim, M.S.; Froese, C.D.; Xie, H.; Trimble, W.S. Immunofluorescent staining of septins in primary cilia. Methods Cell Biol. 2016, 136, 269–283. [Google Scholar] [CrossRef]
- Pedersen, L.B.; Rosenbaum, J.L. Intraflagellar transport (IFT) role in ciliary assembly, resorption and signalling. Curr. Top. Dev. Biol. 2008, 85, 23–61. [Google Scholar] [CrossRef] [PubMed]
- Shida, T.; Cueva, J.G.; Xu, Z.; Goodman, M.B.; Nachury, M.V. The major alpha-tubulin K40 acetyltransferase alphaTAT1 promotes rapid ciliogenesis and efficient mechanosensation. Proc. Natl. Acad. Sci. USA 2010, 107, 21517–21522. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guertin, D.A.; Sabatini, D.M. Defining the role of mTOR in cancer. Cancer Cell 2007, 12, 9–22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saxton, R.A.; Sabatini, D.M. mTOR Signaling in Growth, Metabolism, and Disease. Cell 2017, 169, 361–371. [Google Scholar] [CrossRef]
- Kim, J.; Guan, K.L. mTOR as a central hub of nutrient signalling and cell growth. Nat. Cell Biol. 2019, 21, 63–71. [Google Scholar] [CrossRef]
- Kim, L.C.; Cook, R.S.; Chen, J. mTORC1 and mTORC2 in cancer and the tumor microenvironment. Oncogene 2017, 36, 2191–2201. [Google Scholar] [CrossRef] [Green Version]
- Takahashi, K.; Nagai, T.; Chiba, S.; Nakayama, K.; Mizuno, K. Glucose deprivation induces primary cilium formation through mTORC1 inactivation. J. Cell Sci. 2018, 131. [Google Scholar] [CrossRef] [Green Version]
- Soliman, G.A.; Acosta-Jaquez, H.A.; Dunlop, E.A.; Ekim, B.; Maj, N.E.; Tee, A.R.; Fingar, D.C. mTOR Ser-2481 autophosphorylation monitors mTORC-specific catalytic activity and clarifies rapamycin mechanism of action. J. Biol. Chem. 2010, 285, 7866–7879. [Google Scholar] [CrossRef] [Green Version]
- Copp, J.; Manning, G.; Hunter, T. TORC-specific phosphorylation of mammalian target of rapamycin (mTOR): Phospho-Ser2481 is a marker for intact mTOR signaling complex 2. Cancer Res. 2009, 69, 1821–1827. [Google Scholar] [CrossRef] [Green Version]
- Dennis, P.B.; Pullen, N.; Kozma, S.C.; Thomas, G. The principal rapamycin-sensitive p70(s6k) phosphorylation sites, T-229 and T-389, are differentially regulated by rapamycin-insensitive kinase kinases. Mol. Cell. Biol. 1996, 16, 6242–6251. [Google Scholar] [CrossRef] [Green Version]
- Pearson, R.B.; Dennis, P.B.; Han, J.W.; Williamson, N.A.; Kozma, S.C.; Wettenhall, R.E.; Thomas, G. The principal target of rapamycin-induced p70s6k inactivation is a novel phosphorylation site within a conserved hydrophobic domain. EMBO J. 1995, 14, 5279–5287. [Google Scholar] [CrossRef] [PubMed]
- Gingras, A.C.; Gygi, S.P.; Raught, B.; Polakiewicz, R.D.; Abraham, R.T.; Hoekstra, M.F.; Aebersold, R.; Sonenberg, N. Regulation of 4E-BP1 phosphorylation: A novel two-step mechanism. Genes Dev. 1999, 13, 1422–1437. [Google Scholar] [CrossRef] [PubMed]
- Showkat, M.; Beigh, M.A.; Andrabi, K.I. mTOR Signaling in Protein Translation Regulation: Implications in Cancer Genesis and Therapeutic Interventions. Mol. Biol. Int. 2014, 2014, 686984. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shah, O.J.; Anthony, J.C.; Kimball, S.R.; Jefferson, L.S. 4E-BP1 and S6K1: Translational integration sites for nutritional and hormonal information in muscle. Am. J. Physiol. Endocrinol. Metab. 2000, 279, E715–E729. [Google Scholar] [CrossRef]
- Averous, J.; Fonseca, B.D.; Proud, C.G. Regulation of cyclin D1 expression by mTORC1 signaling requires eukaryotic initiation factor 4E-binding protein 1. Oncogene 2008, 27, 1106–1113. [Google Scholar] [CrossRef]
- Kim, J.; Kundu, M.; Viollet, B.; Guan, K.L. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat. Cell Biol. 2011, 13, 132–141. [Google Scholar] [CrossRef] [Green Version]
- Mizushima, N.; Yoshimori, T.; Levine, B. Methods in mammalian autophagy research. Cell 2010, 140, 313–326. [Google Scholar] [CrossRef] [Green Version]
- Klionsky, D.J.; Abdelmohsen, K.; Abe, A.; Abedin, M.J.; Abeliovich, H.; Acevedo Arozena, A.; Adachi, H.; Adams, C.M.; Adams, P.D.; Adeli, K.; et al. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy 2016, 12, 1–222. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Azhar, G.; Wei, J.Y. SIRT2 gene has a classic SRE element, is a downstream target of serum response factor and is likely activated during serum stimulation. PLoS ONE 2017, 12, e0190011. [Google Scholar] [CrossRef] [Green Version]
- Schenone, S.; Brullo, C.; Musumeci, F.; Radi, M.; Botta, M. ATP-competitive inhibitors of mTOR: An update. Curr. Med. Chem. 2011, 18, 2995–3014. [Google Scholar] [CrossRef]
- Thoreen, C.C.; Kang, S.A.; Chang, J.W.; Liu, Q.; Zhang, J.; Gao, Y.; Reichling, L.J.; Sim, T.; Sabatini, D.M.; Gray, N.S. An ATP-competitive mammalian target of rapamycin inhibitor reveals rapamycin-resistant functions of mTORC1. J. Biol. Chem. 2009, 284, 8023–8032. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choo, A.Y.; Yoon, S.O.; Kim, S.G.; Roux, P.P.; Blenis, J. Rapamycin differentially inhibits S6Ks and 4E-BP1 to mediate cell-type-specific repression of mRNA translation. Proc. Natl. Acad. Sci. USA 2008, 105, 17414–17419. [Google Scholar] [CrossRef] [Green Version]
- Feldman, M.E.; Apsel, B.; Uotila, A.; Loewith, R.; Knight, Z.A.; Ruggero, D.; Shokat, K.M. Active-site inhibitors of mTOR target rapamycin-resistant outputs of mTORC1 and mTORC2. PLoS Biol. 2009, 7, e38. [Google Scholar] [CrossRef] [PubMed]
- Sarbassov, D.D.; Guertin, D.A.; Ali, S.M.; Sabatini, D.M. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science 2005, 307, 1098–1101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dibble, C.C.; Cantley, L.C. Regulation of mTORC1 by PI3K signaling. Trends Cell Biol. 2015, 25, 545–555. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, J.; Proud, C.G. Crosstalk between mTOR complexes. Nat. Cell Biol. 2013, 15, 1263–1265. [Google Scholar] [CrossRef]
- Molla-Herman, A.; Ghossoub, R.; Blisnick, T.; Meunier, A.; Serres, C.; Silbermann, F.; Emmerson, C.; Romeo, K.; Bourdoncle, P.; Schmitt, A.; et al. The ciliary pocket: An endocytic membrane domain at the base of primary and motile cilia. J. Cell Sci. 2010, 123, 1785–1795. [Google Scholar] [CrossRef] [Green Version]
- Sorokin, S. Centrioles and the formation of rudimentary cilia by fibroblasts and smooth muscle cells. J. Cell Biol. 1962, 15, 363–377. [Google Scholar] [CrossRef] [Green Version]
- Sorokin, S.P. Reconstructions of centriole formation and ciliogenesis in mammalian lungs. J. Cell Sci. 1968, 3, 207–230. [Google Scholar]
- Anwar, T.; Khosla, S.; Ramakrishna, G. Increased expression of SIRT2 is a novel marker of cellular senescence and is dependent on wild type p53 status. Cell Cycle 2016, 15, 1883–1897. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.; Yang, J.; Liao, W.; Liu, X.; Zhang, H.; Wang, S.; Wang, D.; Feng, J.; Yu, L.; Zhu, W.G. Cytosolic FoxO1 is essential for the induction of autophagy and tumour suppressor activity. Nat. Cell Biol. 2010, 12, 665–675. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Livingston, M.J.; Su, Y.; Dong, Z. Reciprocal regulation of cilia and autophagy via the MTOR and proteasome pathways. Autophagy 2015, 11, 607–616. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ran, J.; Yang, Y.; Li, D.; Liu, M.; Zhou, J. Deacetylation of alpha-tubulin and cortactin is required for HDAC6 to trigger ciliary disassembly. Sci. Rep. 2015, 5, 12917. [Google Scholar] [CrossRef] [PubMed]
- Zoncu, R.; Efeyan, A.; Sabatini, D.M. mTOR: From growth signal integration to cancer, diabetes and ageing. Nat. Rev. Mol. Cell Biol. 2011, 12, 21–35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Logue, J.S.; Morrison, D.K. Complexity in the signaling network: Insights from the use of targeted inhibitors in cancer therapy. Genes Dev. 2012, 26, 641–650. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manning, B.D.; Toker, A. AKT/PKB Signaling: Navigating the Network. Cell 2017, 169, 381–405. [Google Scholar] [CrossRef] [Green Version]
- Dowling, R.J.; Topisirovic, I.; Alain, T.; Bidinosti, M.; Fonseca, B.D.; Petroulakis, E.; Wang, X.; Larsson, O.; Selvaraj, A.; Liu, Y.; et al. mTORC1-mediated cell proliferation, but not cell growth, controlled by the 4E-BPs. Science 2010, 328, 1172–1176. [Google Scholar] [CrossRef] [Green Version]
- Fingar, D.C.; Richardson, C.J.; Tee, A.R.; Cheatham, L.; Tsou, C.; Blenis, J. mTOR controls cell cycle progression through its cell growth effectors S6K1 and 4E-BP1/eukaryotic translation initiation factor 4E. Mol. Cell. Biol. 2004, 24, 200–216. [Google Scholar] [CrossRef] [Green Version]
- Chatterjee, A.; Mukhopadhyay, S.; Tung, K.; Patel, D.; Foster, D.A. Rapamycin-induced G1 cell cycle arrest employs both TGF-beta and Rb pathways. Cancer Lett. 2015, 360, 134–140. [Google Scholar] [CrossRef] [Green Version]
- Reiter, J.F.; Leroux, M.R. Genes and molecular pathways underpinning ciliopathies. Nat. Rev. Mol. Cell Biol. 2017, 18, 533–547. [Google Scholar] [CrossRef]
- Waters, A.M.; Beales, P.L. Ciliopathies: An expanding disease spectrum. Pediatr. Nephrol. 2011, 26, 1039–1056. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fabbri, L.; Bost, F.; Mazure, N.M. Primary cilium in cancer hallmarks. Int. J. Mol. Sci. 2019, 20, 1336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, N.; Berbari, N.F.; Yoder, B.K. Ciliary dysfunction in developmental abnormalities and diseases. Curr. Top. Dev. Biol. 2008, 85, 371–427. [Google Scholar] [CrossRef] [PubMed]
- Dummer, A.; Poelma, C.; DeRuiter, M.C.; Goumans, M.J.; Hierck, B.P. Measuring the primary cilium length: Improved method for unbiased high-throughput analysis. Cilia 2016, 5, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]








© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lim, J.; Son, J.; Ryu, J.; Kim, J.-E. SIRT2 Affects Primary Cilia Formation by Regulating mTOR Signaling in Retinal Pigmented Epithelial Cells. Int. J. Mol. Sci. 2020, 21, 2240. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21062240
Lim J, Son J, Ryu J, Kim J-E. SIRT2 Affects Primary Cilia Formation by Regulating mTOR Signaling in Retinal Pigmented Epithelial Cells. International Journal of Molecular Sciences. 2020; 21(6):2240. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21062240
Chicago/Turabian StyleLim, Jeaho, Juyoung Son, Jaewook Ryu, and Ja-Eun Kim. 2020. "SIRT2 Affects Primary Cilia Formation by Regulating mTOR Signaling in Retinal Pigmented Epithelial Cells" International Journal of Molecular Sciences 21, no. 6: 2240. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21062240