AHR Signaling Dampens Inflammatory Signature in Neonatal Skin γδ T Cells

 , , , and
, , , and
Abstract
:1. Introduction
2. Results
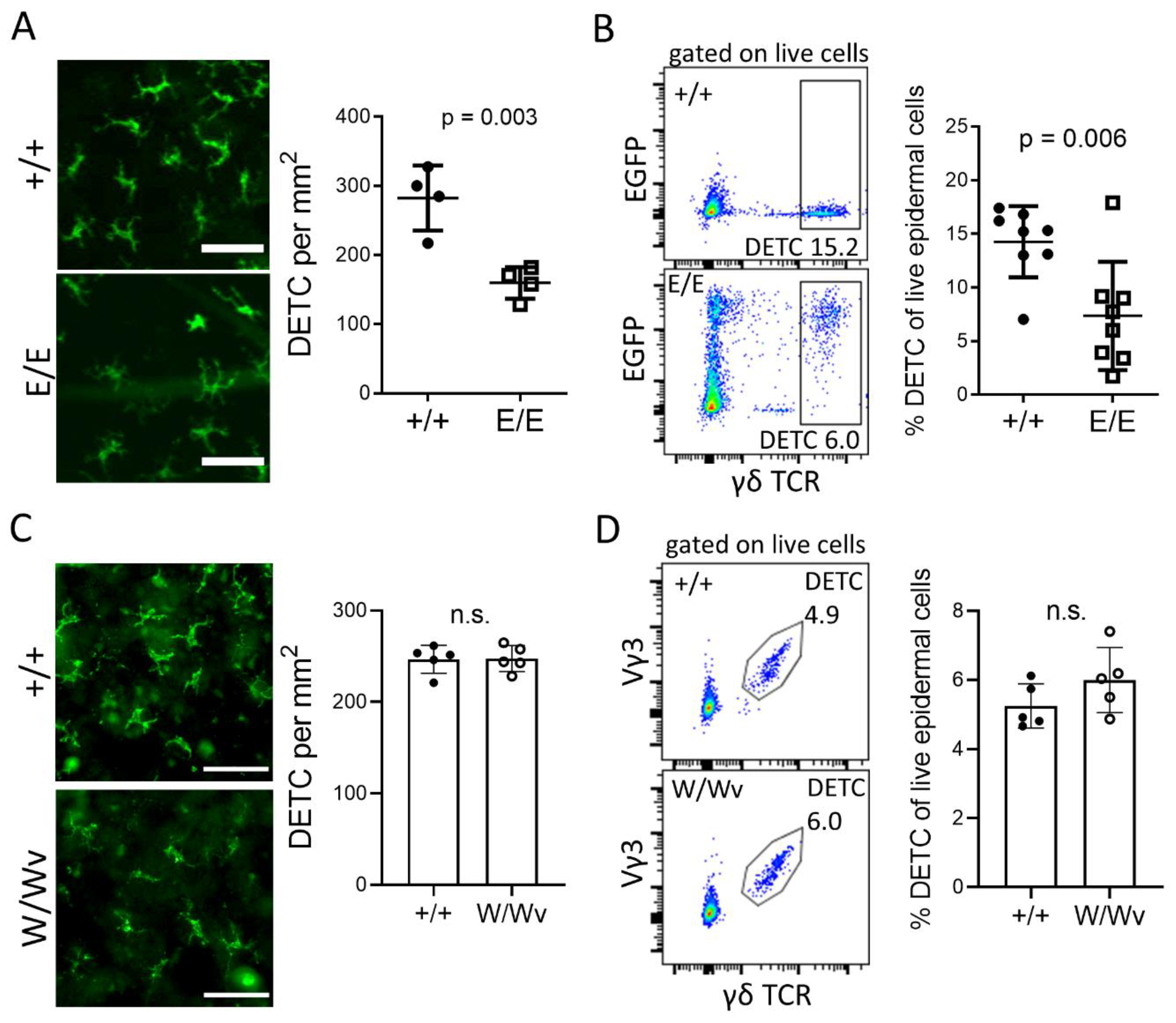
2.1. Establishment of DETC in the Epidermis Depends on AHR and AHRR, But Not on KIT
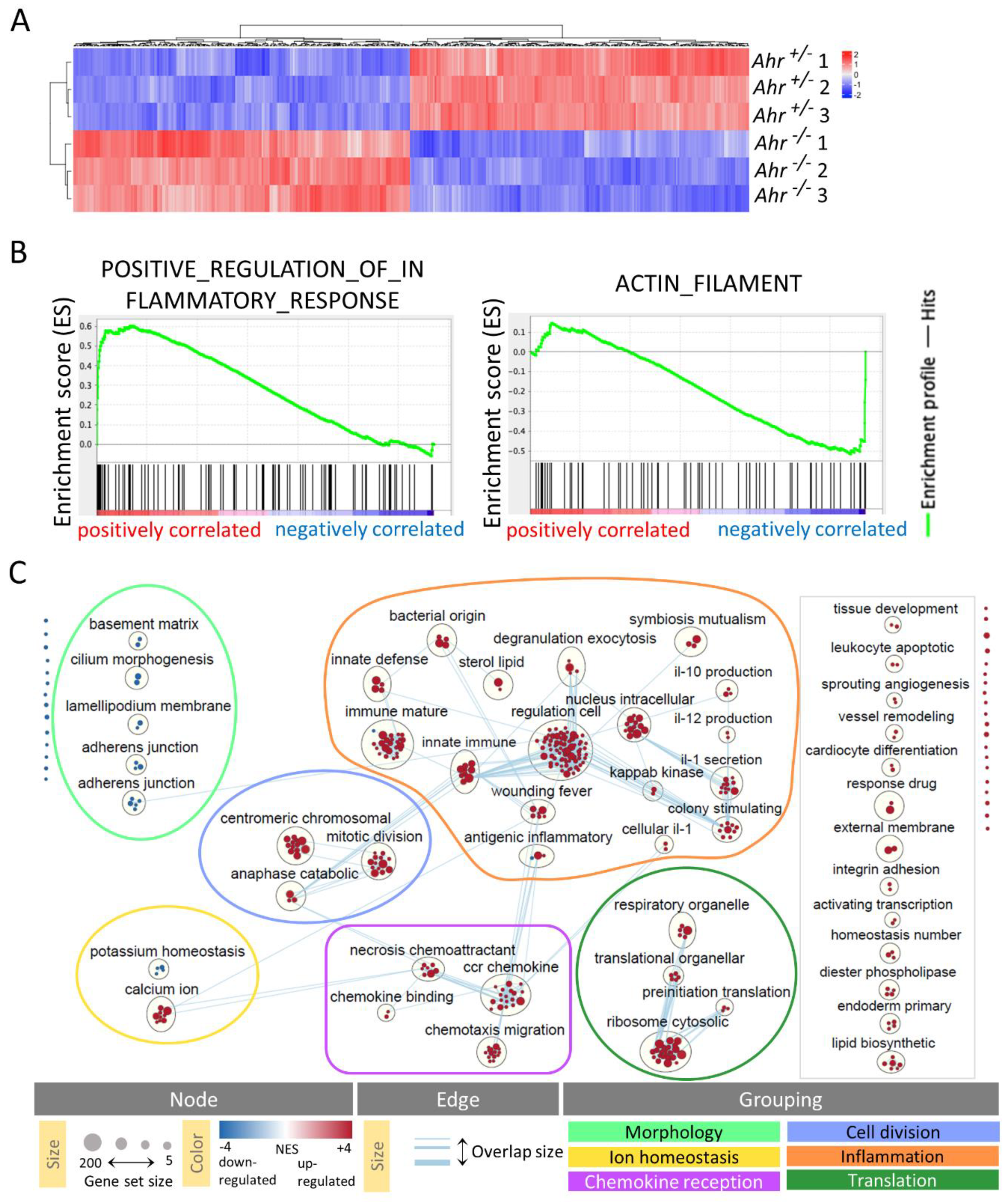
2.2. DETC of Ahr -/- Mice Express More Inflammatory and Less Actin-Modulating Genes
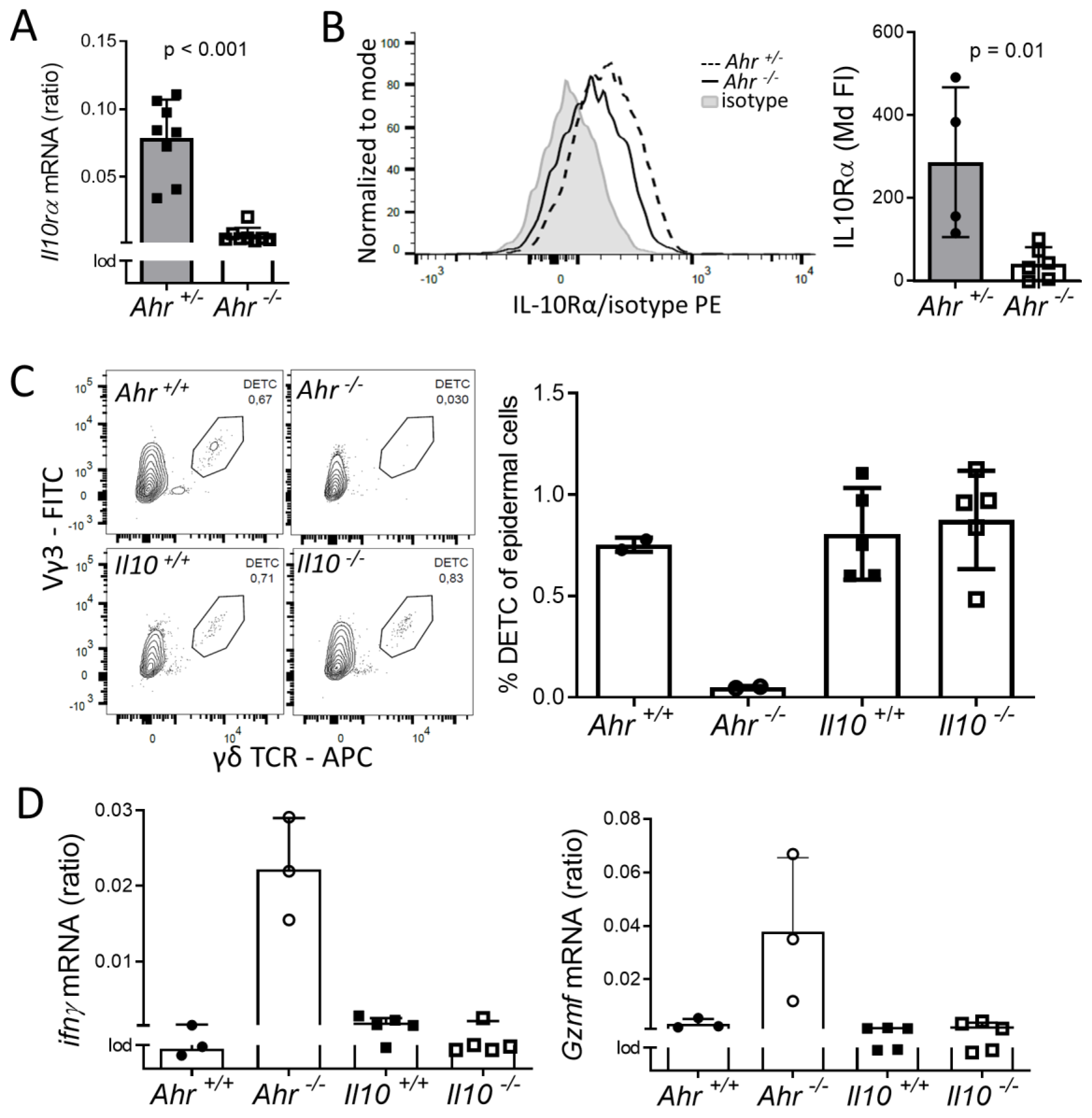
2.3. DETC of Ahr -/- Mice are Inflammatory Active and Have Altered Calcium and F-Actin Levels
2.4. Lack of IL-10-Signaling Does not Affect DETC Homeostasis
3. Discussion
4. Materials and Methods
4.1. Animals
4.2. Immunohistochemistry of Epidermal Ear Sheets
4.3. Isolation of Epidermal Cells
4.4. Flow Cytometry
4.5. Microarray Analysis
4.6. Quantitative Realtime PCR (qPCR)
4.7. Statistics
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| AHR | Aryl hydrocarbon receptor |
| DETC | Dendritic epidermal T cells |
| AHRR | Aryl hydrocarbon receptor repressor |
| GSEA | Gene set enrichment analysis |
| lod | Limit of detection |
| FDR | False discovery rate |
References
- Cruz, M.; Diamond, A.; Russell, A.; Jameson, J. Human ab and gd T Cells in Skin Immunity and Disease. Front. Immunol. 2018, 9, 1304. [Google Scholar] [CrossRef]
- Nielsen, M.; Witherden, D.; Havran, W. γδ T cells in homeostasis and host defence of epithelial barrier tissues. Nat. Rev. Immunol. 2017, 17, 733–745. [Google Scholar] [CrossRef]
- Bos, J.; Teunissen, M.; Cairo, I.; Krieg, S.; Kapsenberg, M.; Das, P.; Borst, J. T-cell receptor gamma delta bearing cells in normal human skin. J. Invest. Derm. 1990, 94, 37–42. [Google Scholar] [CrossRef] [Green Version]
- Kadow, S.; Jux, B.; Zahner, S.P.; Wingerath, B.; Chmill, S.; Clausen, B.E.; Hengstler, J.; Esser, C. Aryl hydrocarbon receptor is critical for homeostasis of invariant gammadelta T cells in the murine epidermis. J. Immunol. 2011, 187, 3104–3110. [Google Scholar] [CrossRef] [Green Version]
- Garman, R.; Doherty, P.; Raulet, D. Diversity, rearrangement, and expression of murine T cell gamma genes. Cell 1986, 45, 733–742. [Google Scholar] [CrossRef]
- Strid, J.; Roberts, S.; Filler, R.; Lewis, J.; Kwong, B.; Schpero, W.; Kaplan, D.; Hayday, A.; Girardi, M. Acute upregulation of an NKG2D ligand promotes rapid reorganization of a local immune compartment with pleiotropic effects on carcinogenesis. Nat. Immunol. 2008, 9, 146–154. [Google Scholar] [CrossRef] [PubMed]
- Strid, J.; Sobelev, O.; Zafirova, B.; Polic, B.; Hayday, A. The Intraepithelial T Cell Repsonses to NKG2D-Ligands Links Lymphoid Stress Surveillance to Atopy. Science 2011, 334, 1293–1297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barbee, S.; Woodward, W.; Turchinovich, G.; Mention, J.-J.; Lewis, J.; Boyden, L.; Lifton, R.; Tigelaar, R.; Hayday, A. Skint-1 is a highly specific unique selecting component for epidermal T cells. Proc. Natl. Acad. Sci. USA 2011, 108, 3330–3335. [Google Scholar] [CrossRef] [Green Version]
- De Creus, A.; Van Beneden, K.; Stevenaert, F.; Debacker, V.; Plum, J.; Leclercq, G. Developmental and functional defects of thymic and epidermal V gamma 3 cells in IL-15-deficient and IFN regulatory factor-1-deficient mice. J. Immunol. 2002, 168, 6486–6493. [Google Scholar] [CrossRef] [Green Version]
- Zhang, B.; Wu, J.; Jiao, Y.; Bock, C.; Meifang, D.; Chen, B.; Chao, N.; Zhang, W.; Zhuang, Y. Differential Requirements of TCR Signaling in Homeostatic Maintenance and Function of Dendritic Epidermal T Cells. J. Immunol. 2015, 195, 4282–4291. [Google Scholar] [CrossRef] [Green Version]
- Wencker, M.; Turchinovich, G.; Di Marco Barros, R.; Deban, L.; Jandke, A.; Cope, A.; Hayday, A. Innate-like T cells straddle innate and adaptive immunity by altering antigen-receptor responsiveness. Nat. Immunol. 2014, 15, 80–87. [Google Scholar] [CrossRef]
- Gentek, R.; Ghigo, C.; Hoeffel, G.; Jorquera, A.; Msallam, R.; Wienert, S.; Klauschen, F.; Ginhoux, F.; Bajénoff, M. Epidermal γδ T cells originate from yolk sac hematopoiesis and clonally self-renew in the adult. J. Exp. Med. 2018, 215, 1994–3005. [Google Scholar]
- Kiss, E.; Vonarbourg, C.; Kopfmann, S.; Hobeika, E.; Finke, D.; Esser, C.; Diefenbach, A. Natural aryl hydrocarbon receptor ligands control organogenesis of intestinal lymphoid follicles. Science 2011, 334, 1561–1565. [Google Scholar] [CrossRef] [PubMed]
- Gutiérrez-Vázquez, C.; Quintana, F. Regulation of the Immune Response by the Aryl Hydrocarbon Receptor. Immunity 2018, 48, 19–33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Merches, K.; Haarmann-Stemmann, T.; Weighardt, H.; Krutmann, J.; Esser, C. AHR in the skin: From the mediator of chlorachne to a therapeutic panacea? Curr. Opp. Toxicol. 2017, 2, 79–86. [Google Scholar] [CrossRef]
- Haas, K.; Weighardt, H.; Deenen, R.; Köhrer, K.; Clausen, B.; Zahner, S.; Boukamp, P.; Bloch, W.; Krutmann, J.; Esser, C. Aryl hydrocarbon receptor in keratinocytes is essential for murine skin barrier integrity. J. Investig. Dermatol. 2016, 163, 2260–2269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frericks, M.; Temchura, V.; Majora, M.; Stutte, S.; Esser, C. Transcriptional signatures of immune cells in aryl hydrocarbon receptor (AHR)-proficient and AHR-deficient mice. Biol. Chem. 2006, 387, 1219–1226. [Google Scholar] [CrossRef]
- Frericks, M.; Meissner, M.; Esser, C. Microarray analysis of the AHR system: Tissue-specific flexibility in signal and target genes. Toxicol. Appl. Pharm. 2007, 220, 320–332. [Google Scholar] [CrossRef]
- Chodaczek, G.; Papanna, V.; Zal, M.; Zal, T. Body-barrier surveillance by epidermal γδ TCRs. Nat. Immunol. 2012, 13, 272–282. [Google Scholar] [CrossRef]
- Jameson, J.; Ugarte, K.; Chen, N.; Yachi, P.; Fuchs, E.; Boismenu, R.; Havran, W. A role for skin gammadelta T cells in wound repair. Science 2002, 296, 747–749. [Google Scholar] [CrossRef]
- Nielsen, M.M.; Dyring-Andersen, B.; Schmidt, J.D.; Witherden, D.; Lovato, P.; Woetmann, A.; Odum, N.; Poulsen, S.S.; Havran, W.L.; Geisler, C.; et al. NKG2D-dependent activation of dendritic epidermal T cells in contact hypersensitivity. J. Investig. Derm. 2015, 135, 1311–1319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keyes, B.; Liu, S.; Asare, A.; Naik, S.; Levorse, J.; Polak, L.; Lu, C.; Nikolova, M.; Pasolli, H.; Fuchs, E. Impaired Epidermal to Dendritic T Cell Signaling Slows Wound Repair in Aged Skin. Cell 2016, 167, 1323–1338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Girardi, M.; Oppenheim, D.; Steele, C.; Lewis, J.; Glusac, E.; Filler, R.; Hobby, P.; Sutton, B.; Tigelaar, R.; Hayday, A. Regulation of cutaneous malignancy by gammadelta T cells. Science 2001, 294, 605–609. [Google Scholar] [CrossRef] [PubMed]
- Nitahara, A.; Shimura, H.; Ito, A.; Tomiyama, K.; Ito, M.; Kawai, K. NKG2D ligation without T cell receptor engagement triggers both cytotoxicity and cytokine production in dendritic epidermal T cells. J. Investig. Derm. 2006, 126, 1052–1058. [Google Scholar] [CrossRef] [Green Version]
- Wang, T.; Langley, K.; Gourley, W.; Klimpel, G. Stem cell factor (SCF) can regulate the activation and expansion of murine intraepithelial lymphocytes. Cytokine 2000, 12, 272–280. [Google Scholar] [CrossRef]
- Mimura, J.; Ema, M.; Sogawa, K.; Fujii-Kuriyama, Y. Identification of a novel mechanism of regulation of Ah (dioxin) receptor function. Genes Dev. 1999, 13, 20–25. [Google Scholar] [CrossRef] [Green Version]
- Brandstätter, O.; Schanz, O.; Vorac, J.; König, J.; Mori, T.; Maruyama, T.; Korkowski, M.; Haarmann-Stemmann, T.; von Smolinski, D.; Schultze, J.; et al. Balancing intestinal and systemic inflammation through cell type-specific expression of the aryl hydrocarbon receptor repressor. Sci. Rep. 2016, 6, 26091. [Google Scholar] [CrossRef]
- Jux, B.; Kadow, S.; Luecke, S.; Rannug, A.; Krutmann, J.; Esser, C. The aryl hydrocarbon receptor mediates UVB radiation-induced skin tanning. J. Investig. Derm. 2011, 131, 203–210. [Google Scholar] [CrossRef] [Green Version]
- Alexander, W.; Lyman, S.; Wagner, E. Expression of functional c-kit receptors rescues the genetic defect of W mutant mast cells. EMBO J. 1991, 10, 3683–3691. [Google Scholar] [CrossRef]
- Reimand, J.; Isserlin, R.; Voisin, V.; Kucera, M.; Tannus-Lopes, C.; Rostamianfar, A.; Wadi, L.; Meyer, M.; Wong, J.; Xu, C.; et al. Pathway enrichment analysis and visualization of omics data using g:Profiler, GSEA, Cytoscape and EnrichmentMap. Nat. Protoc. 2019, 14, 482–517. [Google Scholar] [CrossRef]
- Boismenu, R.; Feng, L.; Xia, Y.; Chang, J.; Havran, W. Chemokine expression by intraepithelial gamma delta T cells. Implications for the recruitment of inflammatory cells to damaged epithelia. J. Immunol. 1996, 157, 985–992. [Google Scholar] [PubMed]
- Subramanian, A.; Tamayo, P.; Mootha, V.; Mukherjee, S.; Ebert, B.; Gillette, M.; Paulovich, A.; Pomeroy, S.; Golub, T.; Lander, E.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, L.; Wu, L.; Wang, S.; Fan, Z. Granzyme F induces a novel death pathway characterized by Bid-independent cytochrome c release without caspase activation. Cell Death Dis. 2009, 16, 1694–1706. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sheppard, K.; Fitz, L.; Lee, J.; Benander, C.; George, J.; Wooters, J.; Qiu, Y.; Jussif, J.; Carter, L.; Wood, C.; et al. PD-1 inhibits T-cell receptor induced phosphorylation of the ZAP70/CD3zeta signalosome and downstream signaling to PKCtheta. FEBS Lett. 2004, 574, 37–41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amarnath, S.; Mangus, C.; Wang, J.; Wei, F.; He, A.; Kapoor, V.; Foley, J.; Massey, P.; Felizardo, T.; Riley, J.; et al. The PDL1-PD1 axis converts human TH1 cells into regulatory T cells. Sci Transl Med. 2011, 3, 111–120. [Google Scholar] [CrossRef] [Green Version]
- Langereis, J.; Pickkers, P.; de Kleijn, S.; Gerretsen, J.; de Jonge, M.; Kox, M. Spleen-derived IFN-γ induces generation of PD-L1+-suppressive neutrophils during endotoxemia. J. Leukoc. Biol. 2017, 106, 1401–1409. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vila-Caballer, M.; González-Granado, J.; Zorita, V.; Abu Nabah, Y.; Silvestre-Roig, C.; Del Monte-Monge, A.; Molina-Sánchez, P.; Ait-Oufella, H.; Andrés-Manzano, M.; Sanz, M.; et al. Disruption of the CCL1-CCR8 axis inhibits vascular Treg recruitment and function and promotes atherosclerosis in mice. J. Mol. Cell. Cardiol. 2019, 153, 154–163. [Google Scholar] [CrossRef]
- Torres-Hernandez, A.; Wang, W.; Nikiforov, Y.; Tejada, K.; Torres, L.; Kalabin, A.; Adam, S.; Wu, J.; Lu, L.; Chen, R.; et al. γδ T cells Promote Steatohepatitis by Orchestrating Innate and Adaptive Immune Programming. Hepatology 2019, 7, 477–494. [Google Scholar] [CrossRef]
- Rabin, R.; Park, M.; Liao, F.; Swofford, R.; Stephany, D.; Farber, J. Chemokine receptor responses on T cells are achieved through regulation of both receptor expression and signaling. J. Immunol. 1999, 162, 3840–3850. [Google Scholar]
- Horrigan, F.; Aldrich, R. Coupling between voltage sensor activation, Ca2+ binding and channel opening in large conductance (BK) potassium channels. J. Gen. Physiol. 2002, 120, 267–305. [Google Scholar] [CrossRef] [Green Version]
- Yasuda-Yamahara, M.; Rogg, M.; Frimmel, J.; Trachte, P.; Helmstaedter, M.; Schroder, P.; Schiffer, M.; Schell, C.; Huber, T. FERMT2 links cortical actin structures, plasma membrane tension and focal adhesion function to stabilize podocyte morphology. Matrix Biol. 2018, 68–69, 263–279. [Google Scholar] [CrossRef] [PubMed]
- Nordenfelt, P.; Elliott, H.L.; Springer, T.A. Coordinated integrin activation by actin-dependent force during T-cell migration. Nat. Commun. 2016, 7, 13119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Swensson, C.; Silverstone, A.; Lai, Z.-W.; Lundberg, K. Dioxin-Induced Adseverin Expression in the Mouse Thymus Is Strictly Regulated and Dependent on the Aryl Hydrocarbon Receptor. Biochem. Biophys. Res. Commun. 2002, 291, 1194–1200. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez Del Castillo, A.; Lemaire, S.; Tchakarov, L.; Jeyapragasan, M.; Doucet, J.; Vitale, M.; Trifaró, J. Chromaffin cell scinderin, a novel calcium-dependent actin filament-severing protein. EMBO J. 1990, 9, 43–52. [Google Scholar] [CrossRef] [PubMed]
- Marks, P.; Arai, M.; Bandura, J.; Kwiatkowski, D. Advillin (p92): A new member of the gelsolin/villin family of actin regulatory proteins. J. Cell Sci. 1998, 111, 2129–2136. [Google Scholar] [PubMed]
- Silacci, P.; Mazzolai, L.; Gauci, C.; Stergiopulos, N.; Yin, H.L.; Hayoz, D. Gelsolin superfamily proteins: Key regulators of cellular functions. Cell. Mol. Life Sci. 2004, 61, 2614–2623. [Google Scholar] [CrossRef] [Green Version]
- Sun, Y.; Boverhof, D.; Burgoon, L.; Fielden, M.; Zacharewski, T. Comparative analysis of dioxin response elements in human, mouse and rat genomic sequences. Nucleic Acids Res. 2004, 32, 4512–4523. [Google Scholar] [CrossRef] [Green Version]
- Chaudhry, A.; Samstein, R.; Treuting, P.; Liang, Y.; Pils, M.; Heinrich, J.; Jack, R.; Wunderlich, F.; Brüning, J.; Müller, W.; et al. Interleukin-10 signaling in regulatory T cells is required for suppression of Th17 cell-mediated inflammation. Immunity 2011, 34, 566–578. [Google Scholar] [CrossRef] [Green Version]
- Schlaak, J.; Hermann, E.; Gallati, H.; Meyer zum Büschenfelde, K.; Fleischer, B. Differential effects of IL-10 on proliferation and cytokine production of human gamma/delta and alpha/beta T cells. Scand. J. Immunol. 1994, 39, 209–215. [Google Scholar] [CrossRef] [PubMed]
- Brauze, D.; Zawierucha, P.; Kiwerska, K.; Bednarek, K.; Oleszak, M.; Rydzanicz, M.; Jarmuz-Szymczak, M. Induction of expression of aryl hydrocarbon receptro-dependent genes in human HepaRG cell line modifierd by shRNA and treated with ß-naphthoflavone. Mol. Cell. Biochem. 2017, 425, 59–75. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, T.; Hoivik, D.; Lee, J.; Safe, S. Interactions of nuclear receptor coactivator/corepressor proteins with the aryl hydrocarbon receptor complex. Arch. Biochem. Biophys. 1999, 367, 250–257. [Google Scholar] [CrossRef] [PubMed]
- Frericks, M.; Burgoon, L.; Zacharewski, T.; Esser, C. Promoter analysis of TCDD-inducible genes in a thymic epithelial cell line indicates the potential for cell-specific transcription factor crosstalk in the AhR response. Toxicol. Appl. Pharm. 2008, 232, 268–279. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Bhattacharya, S.; Zhou, J.; Phadnis-Moghe, A.; Crawford, R.; Kaminski, N. Aryl Hydrocarbon Receptor Activation Suppresses EBF1 and PAX5 and Impairs Human B Lymphopoiesis. J. Immunol. 2017, 199, 3504–3515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hartzell, C.; Jankowska, K.; Burkhardt, J.; Lewis, R. Calcium influx through CRAC channels controls actin organization and dynamics at the immune synapse. eLife 2016, e14850. [Google Scholar] [CrossRef]
- Tsopoulidis, N.; Kaw, S.; Laketa, V.; Kutscheidt, S.; Baarlink, C.; Stolp, B.; Grosse, R.; Fackler, O. T cell receptor-triggered nuclear actin network formation drives CD4+ T cell effector functions. Sci. Immunol. 2019, 4, eaav1987. [Google Scholar] [CrossRef] [Green Version]
- Hayes, S.; Love, P. Distinct Structure and Signalin Potential of the gdTCR Complex. Immunity 2002, 16, 827–838. [Google Scholar] [CrossRef] [Green Version]
- Siegers, G.; Swamy, M.; Fernández-Malavé, E.; Minguet, S.; Rathmann, S.; Guardo, A.; Pérez-Flores, V.; Regueiro, J.; Alarcón, B.; Fisch, P.; et al. Different composition of the human and the mouse gammadelta T cell receptor explains different phenotypes of CD3gamma and CD3delta immunodeficiencies. J. Exp. Med. 2007, 204, 2537–2544. [Google Scholar] [CrossRef]
- Shen, A.; Puente, L.; Ostergaard, H. Tyrosine kinase activity and remodelling of the actin cytoskeleton are co-temporally required for degranulation by cytotoxic T lymphocytes. Immunology 2005, 116, 276–286. [Google Scholar] [CrossRef]
- Cantiello, H. Role of actin filament organisation in CFTR activation. Eur. J. Physiol. 2001, 443, 75–80. [Google Scholar]
- Weber, G.; Menko, A. Actin filament organization regulates the induction of lens cells differentiation and survival. Dev. Biol. 2006, 295, 714–729. [Google Scholar] [CrossRef] [Green Version]
- Chodaczek, G.; Toporkiewicz, M.; Zal, M.A.; Zal, T. Epidermal T Cell Dendrites Serve as Conduits for Bidirectional Trafficking of Granular Cargo. Front. Immunol. 2018, 9, 1430. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Sosa, M.; Elizondo, G.; Lopez-Duran, R.M.; Rivera, I.; Gonzalez, F.J.; Vega, L. Over-production of IFN-gamma and IL-12 in AhR-null mice. FEBS Lett. 2005, 579, 6403–6410. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaplan, D. Ontogeny and function of murine epidermal Langerhans cells. Nat. Immunol. 2017, 18, 1068–1075. [Google Scholar] [CrossRef] [PubMed]
- Moran, A.; Holzapfel, K.; Xing, Y.; Cunningham, N.; Maltzman, J.; Punt, J.; Hogquist, K. T cell receptor signal strength in Treg and iNKT cell development demonstrated by a novel fluorescent reporter mouse. J. Exp. Med. 2011, 208, 1279–1289. [Google Scholar] [CrossRef]
- Edelbaum, D.; Mohamadzadeh, M.; Bergstresser, P.; Sugamura, K.; Takashima, A. Interleukin (IL)-15 promotes the growth of murine epidermal gamma delta T cells by a mechanism involving the beta- and gamma c-chains of the IL-2 receptor. J. Investig. Derm. 1995, 105, 837–843. [Google Scholar] [CrossRef] [Green Version]
- Alfei, F.; Kanev, K.; Hofmann, M.; Wu, M.; Ghoneim, H.; Roelli, P.; Utzschneider, D.; von Hoesslin, M.; Cullen, J.; Fan, Y.; et al. TOX reinforces the phenotype and longevity of exhausted T cells in chronic viral infection. Nature 2019, 571, 265–269. [Google Scholar] [CrossRef]
- McMillan, B.; McMillan, S.; Glover, E.; Bradfield, C. 2,3,7,8-Tetrachlorodibenzo-p-dioxin Induces Premature Activation of the KLF2 Regulon during Thymocyte Development. J. Biol. Chem. 2007, 282, 12590–12597. [Google Scholar] [CrossRef] [Green Version]
- Lanis, J.; Alexeev, E.; Curtis, V.; Kitzenberg, D.; Kao, D.; Battista, K.; Gerich, M.; Glover, L.; Kominsky, D.; Colgan, S. Tryptophan metabolite activation of the aryl hydrocarbon receptor regulates IL-10 receptor expression on intestinal epithelia. Mucosal Immunol. 2017, 10, 1133–1144. [Google Scholar] [CrossRef]
- Jin, S.; Koh, S.; Yu, D.; Kim, M.; Yun, H.; Lee, D.; Yoon, H.; Cho, S.; Park, H. Imiquimod-applied Interleukin-10 deficient mice better reflects severe and persistent psoriasis with systemic inflammatory state. Exp. Derm. 2018, 27, 43–49. [Google Scholar] [CrossRef]
- Zaid, A.; Mackay, L.; Rahimpour, A.; Braun, A.; Veldhoen, M.; Carbone, F.; Manton, J.; Heath, W.; Mueller, S. Persistence of skin-resident memory T cells within an epidermal niche. Proc. Natl. Acad. Sci. USA 2014, 111, 5307–5312. [Google Scholar] [CrossRef] [Green Version]
- Schmidt, J.; Su, G.; Reddy, J.; Simon, M.; Bradfield, C. Characterization of a murine Ahr null allele: Involvement of the Ah receptor in hepatic growth and development. Proc. Natl. Acad. Sci. USA 1996, 93, 6731–6733. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jux, B.; Kadow, S.; Esser, C. Langerhans cell maturation and contact hypersensitivity are impaired in aryl hydrocarbon receptor-null mice. J. Immunol. 2009, 182, 6709–6717. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luche, H.; Weber, O.; Nageswara Rao, T.; Blum, C.; Fehling, H.J. Faithful activation of an extra-bright red fluorescent protein in “knock-in” Cre-reporter mice ideally suited for lineage tracing studies. Eur. J. Immunol. 2007, 37, 43–53. [Google Scholar] [CrossRef] [PubMed]
- Kühn, R.; Löhler, J.; Rennick, D.; Rajewsky, K.; Müller, W. Interleukin-10-deficient mice develop chronic enterocolitis. Cell 1993, 75, 163–174. [Google Scholar] [CrossRef]
- Bruesch, I.; Meier, P.; Vital, M.; Pieper, D.H.; Selke, K.; Bohlen, S.; Basic, M.; Meier, M.; Glage, S.; Hundrieser, J.; et al. Analysis of Cdcs1 colitogenic effects in the hematopoietic compartment reveals distinct microbiome interaction and a new subcongenic interval active in T cells. Mucosal Immunol. 2019, 12, 691–702. [Google Scholar] [CrossRef] [PubMed]
- Cossarizza, A.; Chang, H.D.; Radbruch, A.; Acs, A.; Adam, D.; Adam-Klages, S.; Agace, W.W.; Aghaeepour, N.; Akdis, M.; Allez, M.; et al. Guidelines for the use of flow cytometry and cell sorting in immunological studies (second edition). Eur. J. Immunol. 2019, 49, 1457–1973. [Google Scholar] [CrossRef] [Green Version]
- Team, R.C. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2018. [Google Scholar]
- Kauffmann, A.; Gentleman, R.; Huber, W. arrayQualityMetrics–A bioconductor package for quality assessment of microarray data. Bioinformatics 2009, 25, 415–416. [Google Scholar] [CrossRef] [Green Version]
- Huber, W.; Carey, V.; Gentleman, R.; Anders, S.; Morgan, M.; Carvalho, B.S.; Bravo, H.C.; Davis, S.; Gatto, L.; Girke, T.; et al. Orchestrating high-throughput genomic analysis with Bioconductor. Nat. Methods 2015, 12, 115–121. [Google Scholar] [CrossRef]
- Carvalho, B.; Irizarry, R. A Framework for Oligonucleotide Microarray Preprocessing. Bioinformatics 2010, 26, 2363–2367. [Google Scholar] [CrossRef]
- Ritchie, M.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.; Shi, W.; Smyth, G. limma powers differential expression analyses for RNA-sequencing and microarray. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef]
- Smyth, G. limma: Linear models for microarray data. In Bioinformatics and Computational Biology Solutions Using R and Bioconductor. Statistics for Biology and Health; Gentleman, R., Carey, V., Huber, W., Irizarry, R., Dudoit, S., Eds.; Springer: New York, NY, USA, 2005; pp. 397–420. [Google Scholar] [CrossRef] [Green Version]
- Smyth, G. Linear models and empirical bayes methods for assessing differential expression in microarray experiments. Stat. Appl Genet. Mol. Biol. 2004, 3, 1–25. [Google Scholar] [CrossRef] [PubMed]
- MacDonald, J. Mogene20sttranscriptcluster.db: Affymetrix Mogene20 Annotation Data (Chip mogene20sttranscriptcluster). R package version 8.7.0. Available online: https://bioconductor.org/packages/release/data/annotation/html/mogene20sttranscriptcluster.db.html (accessed on 24 March 2020).
- Benjamini, Y.; Hochberg, Y. Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. J. R. Stat. Soc. Ser. B (Methodol.) 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Wickham, H. Elegant Graphics for Data Analysis. In Ggplot2—Elegant Graphics for Data Analysis; Wickham, H., Ed.; Springer: New York, NY, USA, 2009. [Google Scholar]
- Mootha, V.; Lindgren, C.; Eriksson, K.; Subramanian, A.; Sihag, S.; Lehar, J.; Puigserver, P.; Carlsson, E.; Ridderstråle, M.; Laurila, E.; et al. PGC-1alpha-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat. Genet. 2003, 34, 267–273. [Google Scholar] [CrossRef] [PubMed]
- Merico, D.; Isserlin, R.; Stueker, O.; Emili, A.; Bader, G. Enrichment Map: A Network-Based Method for Gene-Set Enrichment Visualization and Interpretation. PLoS ONE 2010, 5, e13984. [Google Scholar] [CrossRef]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.; Wang, J.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Present in Pathways a | Gene | FC b | p-value c | adjusted. p-value d | XREs e |
|---|---|---|---|---|---|
| 10 | Arhgap6 | −1.716 | 0.000 | 0.008 | 2 |
| 7 | Rhod | −0.998 | 0.006 | 0.149 | 5 |
| 6 | Ptpro | −0.698 | 0.000 | 0.024 | 4 |
| 6 | Phldb2 | −0.459 | 0.029 | 0.332 | 7 |
| 5 | Fermt2 | −4.005 | 0.000 | 0.000 | 10 |
| 5 | Sparc | −3.751 | 0.000 | 0.000 | 4 |
| 4 | Vcl | −1.074 | 0.001 | 0.036 | 1 |
| 4 | Smad3 | −0.406 | 0.004 | 0.131 | 14 |
| 4 | Pdgfa | −0.681 | 0.006 | 0.157 | 12 |
| 4 | Fbln1 | −0.376 | 0.028 | 0.330 | 10 |
| 3 | Nid2 | −3.194 | 0.000 | 0.000 | 5 |
| 3 | Palld | −2.729 | 0.000 | 0.011 | 4 |
| 3 | Prkce | −0.580 | 0.002 | 0.072 | 5 |
| 3 | Cd2ap | −0.593 | 0.005 | 0.137 | 5 |
| 3 | Fmn1 | −0.291 | 0.041 | 0.387 | 4 |
| 3 | Aif1 | −0.286 | 0.041 | 0.387 | 6 |
| 2 | Rapgef3 | −1.133 | 0.000 | 0.005 | 4 |
| 2 | Kit | −1.220 | 0.000 | 0.006 | 11 |
| 2 | Ahsg | −0.750 | 0.000 | 0.014 | 4 |
| 2 | Cspg4 | −1.559 | 0.001 | 0.049 | 6 |
| 2 | Bbs2 | −0.704 | 0.001 | 0.059 | 5 |
| 2 | Numbl | −0.451 | 0.005 | 0.133 | 9 |
| 2 | Kifc3 | −0.500 | 0.005 | 0.145 | 4 |
| 2 | Gcnt2 | −0.428 | 0.013 | 0.231 | 2 |
| 2 | Megf9 | −0.346 | 0.015 | 0.246 | 2 |
| 2 | Rab13 | −0.308 | 0.017 | 0.269 | 4 |
| 2 | Shank3 | −0.287 | 0.021 | 0.295 | 4 |
| 2 | Fgd6 | −0.638 | 0.026 | 0.319 | 5 |
| 2 | Aplp1 | −0.351 | 0.036 | 0.365 | 4 |
| 2 | Wdr35 | −0.390 | 0.039 | 0.377 | 7 |
| 2 | Stx2 | −0.492 | 0.042 | 0.393 | 5 |
| 1 | Podn | −2.918 | 0.000 | 0.000 | 6 |
| 1 | Avil | −1.803 | 0.000 | 0.000 | 6 |
| 1 | Kcng3 | −2.935 | 0.000 | 0.000 | 9 |
| 1 | Col27a1 | −2.178 | 0.000 | 0.001 | 10 |
| 1 | Rab38 | −1.393 | 0.000 | 0.001 | 1 |
| 1 | Abi3 | −1.422 | 0.000 | 0.002 | 4 |
| 1 | Mtss1 | −2.123 | 0.000 | 0.002 | 11 |
| 1 | Lrp5 | −1.109 | 0.000 | 0.003 | 8 |
| 1 | Nedd4l | −0.809 | 0.000 | 0.007 | 8 |
| 1 | Il6st | −1.866 | 0.000 | 0.008 | 2 |
| 1 | Islr | −2.173 | 0.000 | 0.009 | 4 |
| 1 | Gng12 | −0.721 | 0.000 | 0.014 | 10 |
| 1 | Il12rb1 | −0.766 | 0.001 | 0.050 | 4 |
| 1 | Nisch | −0.523 | 0.004 | 0.114 | 3 |
| 1 | Snca | −0.382 | 0.004 | 0.128 | 6 |
| 1 | Ptpn13 | −0.575 | 0.005 | 0.145 | 3 |
| 1 | Stx3 | −0.427 | 0.008 | 0.184 | 3 |
| 1 | Tfrc | −0.475 | 0.008 | 0.185 | 11 |
| 1 | Lrp6 | −0.508 | 0.009 | 0.188 | 4 |
| 1 | Pdgfrb | −0.377 | 0.009 | 0.196 | 8 |
| 1 | Bcl11b | −0.444 | 0.012 | 0.223 | 13 |
| 1 | Pard6b | −0.561 | 0.013 | 0.228 | 7 |
| 1 | Cited1 | −0.458 | 0.015 | 0.252 | 13 |
| 1 | Cntnap4 | −0.308 | 0.019 | 0.279 | 8 |
| 1 | Arap1 | −0.395 | 0.019 | 0.281 | 7 |
| 1 | Vil1 | −0.365 | 0.019 | 0.283 | 7 |
| 1 | Stxbp1 | −0.374 | 0.021 | 0.291 | 7 |
| 1 | Kcna6 | −0.309 | 0.022 | 0.298 | 7 |
| 1 | Kcns2 | −0.294 | 0.023 | 0.303 | 6 |
| 1 | Hcn1 | −0.339 | 0.024 | 0.309 | 3 |
| 1 | Cxadr | −1.192 | 0.027 | 0.322 | 9 |
| 1 | Sgk3 | −0.849 | 0.028 | 0.330 | 6 |
| 1 | Ptprm | −0.418 | 0.033 | 0.353 | 5 |
| 1 | Prkcz | −0.335 | 0.033 | 0.354 | 11 |
| 1 | Bsn | −0.315 | 0.047 | 0.409 | 6 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Merches, K.; Schiavi, A.; Weighardt, H.; Steinwachs, S.; Teichweyde, N.; Förster, I.; Hochrath, K.; Schumak, B.; Ventura, N.; Petzsch, P.; et al. AHR Signaling Dampens Inflammatory Signature in Neonatal Skin γδ T Cells. Int. J. Mol. Sci. 2020, 21, 2249. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21062249
Merches K, Schiavi A, Weighardt H, Steinwachs S, Teichweyde N, Förster I, Hochrath K, Schumak B, Ventura N, Petzsch P, et al. AHR Signaling Dampens Inflammatory Signature in Neonatal Skin γδ T Cells. International Journal of Molecular Sciences. 2020; 21(6):2249. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21062249
Chicago/Turabian StyleMerches, Katja, Alfonso Schiavi, Heike Weighardt, Swantje Steinwachs, Nadine Teichweyde, Irmgard Förster, Katrin Hochrath, Beatrix Schumak, Natascia Ventura, Patrick Petzsch, and et al. 2020. "AHR Signaling Dampens Inflammatory Signature in Neonatal Skin γδ T Cells" International Journal of Molecular Sciences 21, no. 6: 2249. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21062249