The Respiratory Phenotype of Pompe Disease Mouse Models

 , ,
, , {kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Literature Search
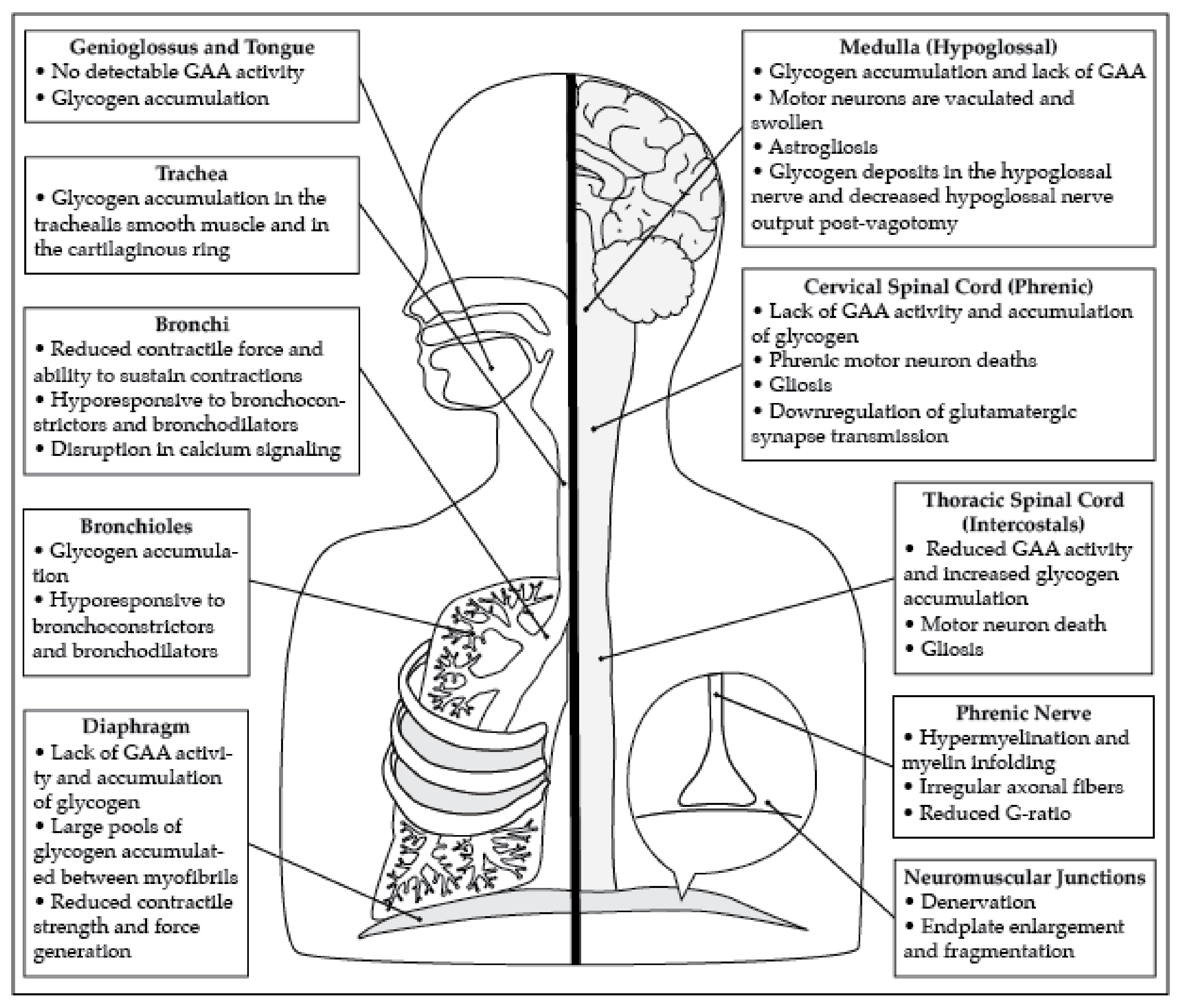
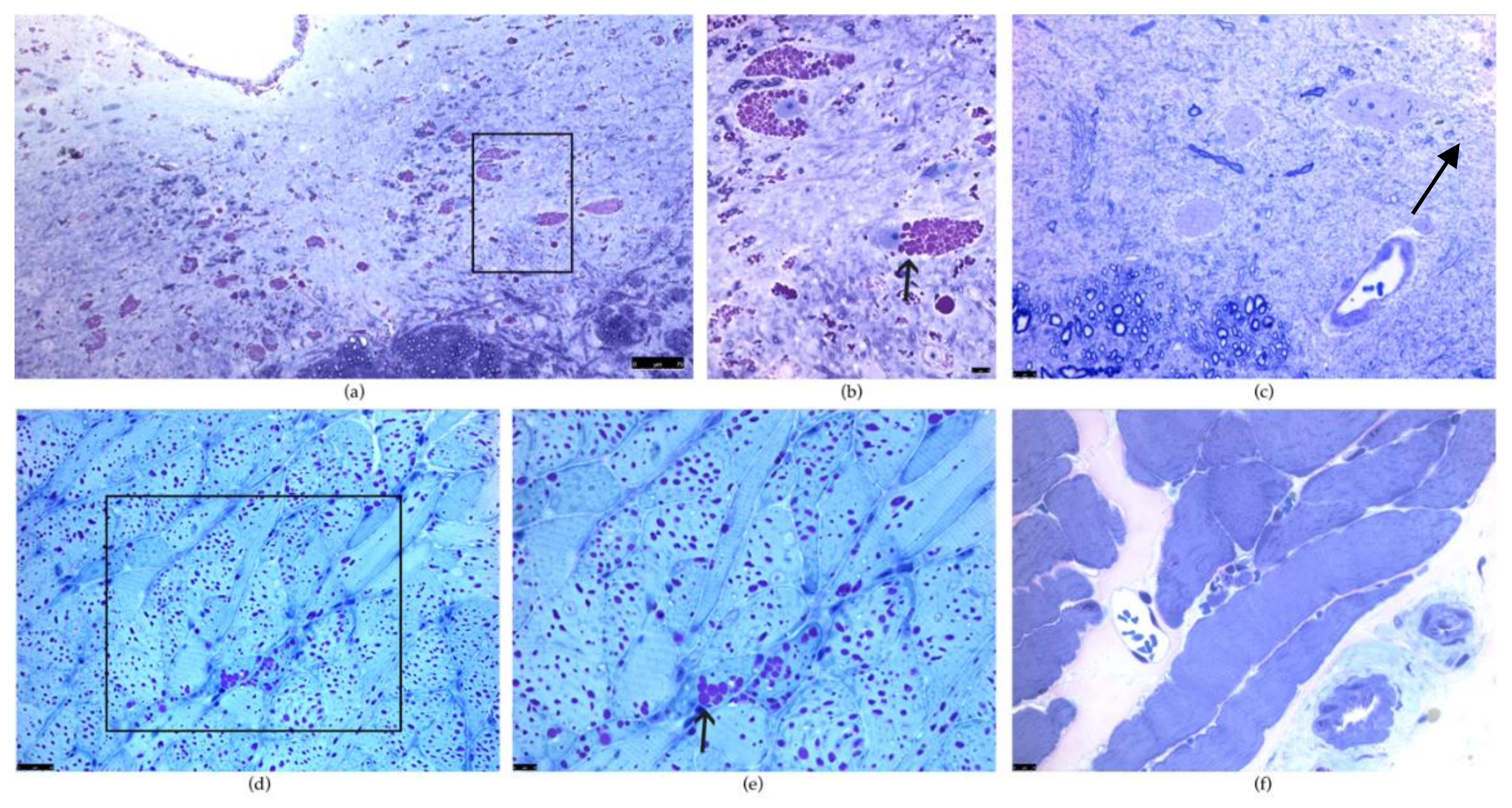
2.2. Diaphragm and Phrenic Motor Neuron Pathology
2.3. Pathology of the Tongue and Hypoglossal Motor Neurons
2.4. Pathology of Airway Smooth Muscle
2.5. The Intercostal Muscles and Thoracic Motor Neuron Pathology
2.6. Additional Neural Control Centers
2.7. Respiratory Pathophysiology in the Gaa−/− Mouse Model
3. Discussion
4. Materials and Methods
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| GAA | acid α-glucosidase |
| IPD | Infantile Onset Pompe Disease |
| LOPD | Late Onset Pompe Disease |
| ERT | Enzyme Replacement Therapy |
| rhGAA | recombinant human GAA |
| hGAA | human GAA |
| XII | Hypoglossal |
| WBPPAS+ | Whole Body PlethysmographyPositive Periodic Acid-Schiff |
| CtB | Cholera Toxin B |
| ChAT | Choline Acetyltransferase |
| IBA-1 | Ionized Calcium Binding Adaptor Molecule 1 |
| GFAP | Glial Fibrillary Acidic Protein |
| TUNEL | Terminal Deoxynucleotidyl Transferase dUTP Nick and Labeling |
| WT | Wild Type |
| CC3 | Cleaved Caspase 3 |
| NTS | Nucleus of the Solitary Tract |
| Te | Expiratory Time |
| TV | Tidal Volume |
| Te | Expiratory Time |
| f | Frequency |
| TV/Ti | Ratio of Tidal Volume to Inspiratory Time |
| VE | Minute Ventilation |
| VE/VCO2 | Ratio of Minute Ventilation to Expired CO2 |
| PIF | Peak Inspiratory Flow |
| PEF | Peak Expiratory Flow |
| PaO2 | Partial Pressure of Oxygen |
References
- Hers, H.G. Alpha-Glucosidase deficiency in generalized glycogenstorage disease (Pompe’s disease). Biochem. J. 1963, 86, 11–16. [Google Scholar] [CrossRef] [PubMed]
- Pompe, J. Over idiopathische hypertrophie van het hart. Ned Tijdschr Geneeskd 1932, 76, 304–311. [Google Scholar]
- Byrne, B.J.; Kishnani, P.S.; Case, L.E.; Merlini, L.; Muller-Felber, W.; Prasad, S.; van der Ploeg, A. Pompe disease: Design, methodology, and early findings from the Pompe Registry. Mol. Genet. Metab. 2011, 103, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Kishnani, P.S.; Howell, R.R. Pompe disease in infants and children. J. Pediatr. 2004, 144, S35–S43. [Google Scholar] [CrossRef] [PubMed]
- Prater, S.N.; Patel, T.T.; Buckley, A.F.; Mandel, H.; Vlodavski, E.; Banugaria, S.G.; Feeney, E.J.; Raben, N.; Kishnani, P.S. Skeletal muscle pathology of infantile Pompe disease during long-term enzyme replacement therapy. Orphanet J. Rare Dis. 2013, 8, 90. [Google Scholar] [CrossRef] [Green Version]
- Kishnani, P.S.; Steiner, R.D.; Bali, D.; Berger, K.; Byrne, B.J.; Case, L.E.; Crowley, J.F.; Downs, S.; Howell, R.R.; Kravitz, R.M.; et al. Pompe disease diagnosis and management guideline. Genet. Med. 2006, 8, 267–288. [Google Scholar] [CrossRef] [Green Version]
- Lim, J.A.; Li, L.; Raben, N. Pompe disease: From pathophysiology to therapy and back again. Front. Aging Neurosci. 2014, 6, 177. [Google Scholar] [CrossRef] [Green Version]
- Kohler, L.; Puertollano, R.; Raben, N. Pompe Disease: From Basic Science to Therapy. Neurotherapeutics 2018, 15, 928–942. [Google Scholar] [CrossRef] [Green Version]
- Kroos, M.A.; Pomponio, R.J.; Hagemans, M.L.; Keulemans, J.L.; Phipps, M.; DeRiso, M.; Palmer, R.E.; Ausems, M.G.; Van der Beek, N.A.; Van Diggelen, O.P.; et al. Broad spectrum of Pompe disease in patients with the same c.-32-13T->G haplotype. Neurology 2007, 68, 110–115. [Google Scholar] [CrossRef]
- Reuser, A.J.; Koster, J.F.; Hoogeveen, A.; Galjaard, H. Biochemical, immunological, and cell genetic studies in glycogenosis type II. Am. J. Hum. Genet. 1978, 30, 132–143. [Google Scholar]
- Slonim, A.E.; Bulone, L.; Ritz, S.; Goldberg, T.; Chen, A.; Martiniuk, F. Identification of two subtypes of infantile acid maltase deficiency. J. Pediatr. 2000, 137, 283–285. [Google Scholar] [CrossRef] [PubMed]
- Beratis, N.G.; LaBadie, G.U.; Hirschhorn, K. Genetic heterogeneity in acid alpha-glucosidase deficiency. Am. J. Hum. Genet. 1983, 35, 21–33. [Google Scholar] [PubMed]
- Van den Hout, H.M.; Hop, W.; van Diggelen, O.P.; Smeitink, J.A.; Smit, G.P.; Poll-The, B.T.; Bakker, H.D.; Loonen, M.C.; de Klerk, J.B.; Reuser, A.J.; et al. The natural course of infantile Pompe’s disease: 20 original cases compared with 133 cases from the literature. Pediatrics 2003, 112, 332–340. [Google Scholar] [CrossRef] [Green Version]
- Fukuhara, Y.; Fuji, N.; Yamazaki, N.; Hirakiyama, A.; Kamioka, T.; Seo, J.H.; Mashima, R.; Kosuga, M.; Okuyama, T. A molecular analysis of the GAA gene and clinical spectrum in 38 patients with Pompe disease in Japan. Mol. Genet. Metab. Rep. 2018, 14, 3–9. [Google Scholar] [CrossRef] [PubMed]
- Wokke, J.H.; Ausems, M.G.; van den Boogaard, M.J.; Ippel, E.F.; van Diggelene, O.; Kroos, M.A.; Boer, M.; Jennekens, F.G.; Reuser, A.J.; Ploos van Amstel, H.K. Genotype-phenotype correlation in adult-onset acid maltase deficiency. Ann. Neurol. 1995, 38, 450–454. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wokke, J.H.; Escolar, D.M.; Pestronk, A.; Jaffe, K.M.; Carter, G.T.; van den Berg, L.H.; Florence, J.M.; Mayhew, J.; Skrinar, A.; Corzo, D.; et al. Clinical features of late-onset Pompe disease: A prospective cohort study. Muscle Nerve 2008, 38, 1236–1245. [Google Scholar] [CrossRef] [PubMed]
- Haley, S.M.; Fragala, M.A.; Skrinar, A.M. Pompe disease and physical disability. Dev. Med. Child Neurol. 2003, 45, 618–623. [Google Scholar] [CrossRef]
- Marsden, D. Infantile onset Pompe disease: A report of physician narratives from an epidemiologic study. Genet. Med. 2005, 7, 147–150. [Google Scholar] [CrossRef] [Green Version]
- Hagemans, M.L.; Winkel, L.P.; Hop, W.C.; Reuser, A.J.; van Doorn, P.A.; van der Ploeg, A.T. Disease severity in children and adults with Pompe disease related to age and disease duration. Neurology 2005, 64, 2139–2141. [Google Scholar] [CrossRef]
- Van den Hout, J.M.; Kamphoven, J.H.; Winkel, L.P.; Arts, W.F.; De Klerk, J.B.; Loonen, M.C.; Vulto, A.G.; Cromme-Dijkhuis, A.; Weisglas-Kuperus, N.; Hop, W.; et al. Long-term intravenous treatment of Pompe disease with recombinant human alpha-glucosidase from milk. Pediatrics 2004, 113, e448–e457. [Google Scholar] [CrossRef] [Green Version]
- Schoser, B.; Hill, V.; Raben, N. Therapeutic approaches in glycogen storage disease type II/Pompe Disease. Neurotherapeutics 2008, 5, 569–578. [Google Scholar] [CrossRef] [PubMed]
- Strothotte, S.; Strigl-Pill, N.; Grunert, B.; Kornblum, C.; Eger, K.; Wessig, C.; Deschauer, M.; Breunig, F.; Glocker, F.X.; Vielhaber, S.; et al. Enzyme replacement therapy with alglucosidase alfa in 44 patients with late-onset glycogen storage disease type 2: 12-month results of an observational clinical trial. J. Neurol. 2010, 257, 91–97. [Google Scholar] [CrossRef] [PubMed]
- Van der Ploeg, A.T.; Clemens, P.R.; Corzo, D.; Escolar, D.M.; Florence, J.; Groeneveld, G.J.; Herson, S.; Kishnani, P.S.; Laforet, P.; Lake, S.L.; et al. A randomized study of alglucosidase alfa in late-onset Pompe’s disease. N. Engl. J. Med. 2010, 362, 1396–1406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Angelini, C.; Semplicini, C. Enzyme replacement therapy for Pompe disease. Curr. Neurol. Neurosci. Rep. 2012, 12, 70–75. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.F.; Niu, D.M.; Jeng, M.J.; Lee, Y.S.; Taso, P.C.; Soong, W.J. Late-onset Pompe disease with left-sided bronchomalacia. Respir. Care 2015, 60, e26–e29. [Google Scholar] [CrossRef]
- Byrne, B.J.; Falk, D.J.; Pacak, C.A.; Nayak, S.; Herzog, R.W.; Elder, M.E.; Collins, S.W.; Conlon, T.J.; Clement, N.; Cleaver, B.D.; et al. Pompe disease gene therapy. Hum. Mol. Genet. 2011, 20, R61–R68. [Google Scholar] [CrossRef] [Green Version]
- Fraites, T.J., Jr.; Schleissing, M.R.; Shanely, R.A.; Walter, G.A.; Cloutier, D.A.; Zolotukhin, I.; Pauly, D.F.; Raben, N.; Plotz, P.H.; Powers, S.K.; et al. Correction of the enzymatic and functional deficits in a model of Pompe disease using adeno-associated virus vectors. Mol. Ther. 2002, 5, 571–578. [Google Scholar] [CrossRef]
- Sun, B.; Zhang, H.; Franco, L.M.; Brown, T.; Bird, A.; Schneider, A.; Koeberl, D.D. Correction of glycogen storage disease type II by an adeno-associated virus vector containing a muscle-specific promoter. Mol. Ther. 2005, 11, 889–898. [Google Scholar] [CrossRef]
- Todd, A.G.; McElroy, J.A.; Grange, R.W.; Fuller, D.D.; Walter, G.A.; Byrne, B.J.; Falk, D.J. Correcting Neuromuscular Deficits With Gene Therapy in Pompe Disease. Ann. Neurol. 2015, 78, 222–234. [Google Scholar] [CrossRef] [Green Version]
- McCall, A.L.; Stankov, S.G.; Cowen, G.; Cloutier, D.; Zhang, Z.; Yang, L.; Clement, N.; Falk, D.J.; Byrne, B.J. Reduction of Autophagic Accumulation in Pompe Disease Mouse Model Following Gene Therapy. Curr. Gene. Ther. 2019, 19, 197–207. [Google Scholar] [CrossRef]
- Sandstrom, B.; Westman, J.; Ockerman, P.A. Glycogenosis of the central nervous system in the cat. Acta Neuropathol. 1969, 14, 194–200. [Google Scholar] [CrossRef] [PubMed]
- Matsui, T.; Kuroda, S.; Mizutani, M.; Kiuchi, Y.; Suzuki, K.; Ono, T. Generalized glycogen storage disease in Japanese quail (Coturnix coturnix japonica). Vet. Pathol. 1983, 20, 312–321. [Google Scholar] [CrossRef] [PubMed]
- Howell, J.M.; Dorling, P.R.; Cook, R.D.; Robinson, W.F.; Bradley, S.; Gawthorne, J.M. Infantile and late onset form of generalised glycogenosis type II in cattle. J. Pathol. 1981, 134, 267–277. [Google Scholar] [CrossRef] [PubMed]
- Seppala, E.H.; Reuser, A.J.; Lohi, H. A nonsense mutation in the acid alpha-glucosidase gene causes Pompe disease in Finnish and Swedish Lapphunds. PLoS ONE 2013, 8, e56825. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jolly, R.D.; van-de-Water, N.S.; Richards, R.B.; Dorling, P.R. Generalized glycogenosis in beef shorthorn cattle--heterozygote detection. Aust. J. Exp. Biol. Med. Sci. 1977, 55, 141–150. [Google Scholar] [CrossRef]
- Manktelow, B.W.; Hartley, W.J. Generalized glycogen storage disease in sheep. J. Comp. Pathol. 1975, 85, 139–145. [Google Scholar] [CrossRef]
- Walvoort, H.C. Glycogen storage diseases in animals and their potential value as models of human disease. J. Inherit. Metab. Dis. 1983, 6, 3–16. [Google Scholar] [CrossRef]
- Geel, T.M.; McLaughlin, P.M.; de Leij, L.F.; Ruiters, M.H.; Niezen-Koning, K.E. Pompe disease: Current state of treatment modalities and animal models. Mol. Genet. Metab. 2007, 92, 299–307. [Google Scholar] [CrossRef]
- Bijvoet, A.G.; van Hirtum, H.; Vermey, M.; van Leenen, D.; van Der Ploeg, A.T.; Mooi, W.J.; Reuser, A.J. Pathological features of glycogen storage disease type II highlighted in the knockout mouse model. J. Pathol. 1999, 189, 416–424. [Google Scholar] [CrossRef]
- Raben, N.; Nagaraju, K.; Lee, E.; Kessler, P.; Byrne, B.; Lee, L.; LaMarca, M.; King, C.; Ward, J.; Sauer, B.; et al. Targeted disruption of the acid alpha-glucosidase gene in mice causes an illness with critical features of both infantile and adult human glycogen storage disease type II. J. Biol. Chem. 1998, 273, 19086–19092. [Google Scholar] [CrossRef] [Green Version]
- Falk, D.J.; Mah, C.S.; Soustek, M.S.; Lee, K.Z.; Elmallah, M.K.; Cloutier, D.A.; Fuller, D.D.; Byrne, B.J. Intrapleural administration of AAV9 improves neural and cardiorespiratory function in Pompe disease. Mol. Ther. 2013, 21, 1661–1667. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elmallah, M.K.; Falk, D.J.; Nayak, S.; Federico, R.A.; Sandhu, M.S.; Poirier, A.; Byrne, B.J.; Fuller, D.D. Sustained correction of motoneuron histopathology following intramuscular delivery of AAV in pompe mice. Mol. Ther. 2014, 22, 702–712. [Google Scholar] [CrossRef] [PubMed]
- Turner, S.M.F.; Falk, D.J.; Byrne, B.J.; Fuller, D.D. Transcriptome assessment of the Pompe (Gaa−/−) mouse spinal cord indicates widespread neuropathology. Physiol. Genomics. 2016, 48, 785–794. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- ElMallah, M.K.; Pagliardini, S.; Turner, S.M.; Cerreta, A.J.; Falk, D.J.; Byrne, B.J.; Greer, J.J.; Fuller, D.D. Stimulation of Respiratory Motor Output and Ventilation in a Murine Model of Pompe Disease by Ampakines. Am. J. Respir. Cell. Mol. Biol. 2015, 53, 326–335. [Google Scholar] [CrossRef] [PubMed]
- Falk, D.J.; Soustek, M.S.; Todd, A.G.; Mah, C.S.; Cloutier, D.A.; Kelley, J.S.; Clement, N.; Fuller, D.D.; Byrne, B.J. Comparative impact of AAV and enzyme replacement therapy on respiratory and cardiac function in adult Pompe mice. Mol. Ther. Methods.Clin. Dev. 2015, 2, 15007. [Google Scholar] [CrossRef] [PubMed]
- Falk, D.J.; Todd, A.G.; Lee, S.; Soustek, M.S.; ElMallah, M.K.; Fuller, D.D.; Notterpek, L.; Byrne, B.J. Peripheral nerve and neuromuscular junction pathology in Pompe disease. Hum. Mol. Genet. 2015, 24, 625–636. [Google Scholar] [CrossRef] [Green Version]
- Doyle, B.M.; Turner, S.M.F.; Sunshine, M.D.; Doerfler, P.A.; Poirier, A.E.; Vaught, L.A.; Jorgensen, M.L.; Falk, D.J.; Byrne, B.J.; Fuller, D.D. AAV Gene Therapy Utilizing Glycosylation-Independent Lysosomal Targeting Tagged GAA in the Hypoglossal Motor System of Pompe Mice. Mol. Ther. Methods Clin. Dev. 2019, 15, 194–203. [Google Scholar] [CrossRef] [Green Version]
- Turner, S.M.; Hoyt, A.K.; ElMallah, M.K.; Falk, D.J.; Byrne, B.J.; Fuller, D.D. Neuropathology in respiratory-related motoneurons in young Pompe (Gaa(−/−)) mice. Respir. Physiol. Neurobiol. 2016, 227, 48–55. [Google Scholar] [CrossRef] [Green Version]
- DeRuisseau, L.R.; Fuller, D.D.; Qiu, K.; DeRuisseau, K.C.; Donnelly, W.H., Jr.; Mah, C.; Reier, P.J.; Byrne, B.J. Neural deficits contribute to respiratory insufficiency in Pompe disease. Proc. Natl. Acad. Sci. USA 2009, 106, 9419–9424. [Google Scholar] [CrossRef] [Green Version]
- Raben, N.; Lu, N.; Nagaraju, K.; Rivera, Y.; Lee, A.; Yan, B.; Byrne, B.; Meikle, P.J.; Umapathysivam, K.; Hopwood, J.J.; et al. Conditional tissue-specific expression of the acid alpha-glucosidase (GAA) gene in the GAA knockout mice: Implications for therapy. Hum. Mol. Genet. 2001, 10, 2039–2047. [Google Scholar] [CrossRef] [Green Version]
- Keeler, A.M.; Zieger, M.; Todeasa, S.H.; McCall, A.L.; Gifford, J.C.; Birsak, S.; Choudhury, S.R.; Byrne, B.J.; Sena-Esteves, M.; ElMallah, M.K. Systemic Delivery of AAVB1-GAA Clears Glycogen and Prolongs Survival in a Mouse Model of Pompe Disease. Hum. Gene Ther. 2019, 30, 57–68. [Google Scholar] [CrossRef] [PubMed]
- Puzzo, F.; Colella, P.; Biferi, M.G.; Bali, D.; Paulk, N.K.; Vidal, P.; Collaud, F.; Simon-Sola, M.; Charles, S.; Hardet, R.; et al. Rescue of Pompe disease in mice by AAV-mediated liver delivery of secretable acid alpha-glucosidase. Sci. Transl. Med. 2017, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matalon, R.; Surendran, S.; Campbell, G.A.; Michals-Matalon, K.; Tyring, S.K.; Grady, J.; Cheng, S.; Kaye, E. Hyaluronidase increases the biodistribution of acid alpha-1,4 glucosidase in the muscle of Pompe disease mice: An approach to enhance the efficacy of enzyme replacement therapy. Biochem. Biophys. Res. Commun. 2006, 350, 783–787. [Google Scholar] [CrossRef] [PubMed]
- Franco, L.M.; Sun, B.; Yang, X.; Bird, A.; Zhang, H.; Schneider, A.; Brown, T.; Young, S.P.; Clay, T.M.; Amalfitano, A.; et al. Evasion of immune responses to introduced human acid alpha-glucosidase by liver-restricted expression in glycogen storage disease type II. Mol. Ther. 2005, 12, 876–884. [Google Scholar] [CrossRef]
- Xu, F.; Ding, E.; Migone, F.; Serra, D.; Schneider, A.; Chen, Y.T.; Amalfitano, A. Glycogen storage in multiple muscles of old GSD-II mice can be rapidly cleared after a single intravenous injection with a modified adenoviral vector expressing hGAA. J. Gene Med. 2005, 7, 171–178. [Google Scholar] [CrossRef]
- Fuller, M.; Duplock, S.; Turner, C.; Davey, P.; Brooks, D.A.; Hopwood, J.J.; Meikle, P.J. Mass spectrometric quantification of glycogen to assess primary substrate accumulation in the Pompe mouse. Anal. Biochem. 2012, 421, 759–763. [Google Scholar] [CrossRef]
- Rucker, M.; Fraites, T.J., Jr.; Porvasnik, S.L.; Lewis, M.A.; Zolotukhin, I.; Cloutier, D.A.; Byrne, B.J. Rescue of enzyme deficiency in embryonic diaphragm in a mouse model of metabolic myopathy: Pompe disease. Development 2004, 131, 3007–3019. [Google Scholar] [CrossRef] [Green Version]
- Farah, B.L.; Madden, L.; Li, S.; Nance, S.; Bird, A.; Bursac, N.; Yen, P.M.; Young, S.P.; Koeberl, D.D. Adjunctive beta2-agonist treatment reduces glycogen independently of receptor-mediated acid alpha-glucosidase uptake in the limb muscles of mice with Pompe disease. FASEB J. 2014, 28, 2272–2280. [Google Scholar] [CrossRef] [Green Version]
- Clayton, N.P.; Nelson, C.A.; Weeden, T.; Taylor, K.M.; Moreland, R.J.; Scheule, R.K.; Phillips, L.; Leger, A.J.; Cheng, S.H.; Wentworth, B.M. Antisense Oligonucleotide-mediated Suppression of Muscle Glycogen Synthase 1 Synthesis as an Approach for Substrate Reduction Therapy of Pompe Disease. Mol. Ther. Nucleic Acids 2014, 3, e206. [Google Scholar] [CrossRef]
- Han, S.O.; Ronzitti, G.; Arnson, B.; Leborgne, C.; Li, S.; Mingozzi, F.; Koeberl, D. Low-Dose Liver-Targeted Gene Therapy for Pompe Disease Enhances Therapeutic Efficacy of ERT via Immune Tolerance Induction. Mol. Ther. Methods Clin. Dev. 2017, 4, 126–136. [Google Scholar] [CrossRef]
- Mah, C.S.; Falk, D.J.; Germain, S.A.; Kelley, J.S.; Lewis, M.A.; Cloutier, D.A.; DeRuisseau, L.R.; Conlon, T.J.; Cresawn, K.O.; Fraites, T.J., Jr.; et al. Gel-mediated delivery of AAV1 vectors corrects ventilatory function in Pompe mice with established disease. Mol. Ther. 2010, 18, 502–510. [Google Scholar] [CrossRef] [Green Version]
- Mah, C.; Pacak, C.A.; Cresawn, K.O.; Deruisseau, L.R.; Germain, S.; Lewis, M.A.; Cloutier, D.A.; Fuller, D.D.; Byrne, B.J. Physiological correction of Pompe disease by systemic delivery of adeno-associated virus serotype 1 vectors. Mol. Ther. 2007, 15, 501–507. [Google Scholar] [CrossRef] [PubMed]
- Qiu, K.; Falk, D.J.; Reier, P.J.; Byrne, B.J.; Fuller, D.D. Spinal delivery of AAV vector restores enzyme activity and increases ventilation in Pompe mice. Mol. Ther. 2012, 20, 21–27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, N.C.; Hwu, W.L.; Muramatsu, S.I.; Falk, D.J.; Byrne, B.J.; Cheng, C.H.; Shih, N.C.; Chang, K.L.; Tsai, L.K.; Chien, Y.H. A Neuron-Specific Gene Therapy Relieves Motor Deficits in Pompe Disease Mice. Mol. Neurobiol. 2018, 55, 5299–5309. [Google Scholar] [CrossRef] [PubMed]
- Lim, J.A.; Yi, H.; Gao, F.; Raben, N.; Kishnani, P.S.; Sun, B. Intravenous Injection of an AAV-PHP.B Vector Encoding Human Acid alpha-Glucosidase Rescues Both Muscle and CNS Defects in Murine Pompe Disease. Mol. Ther. Methods Clin. Dev. 2019, 12, 233–245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, K.Z.; Qiu, K.; Sandhu, M.S.; Elmallah, M.K.; Falk, D.J.; Lane, M.A.; Reier, P.J.; Byrne, B.J.; Fuller, D.D. Hypoglossal neuropathology and respiratory activity in pompe mice. Front. Physiol. 2011, 2, 31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keeler, A.M.; Liu, D.; Zieger, M.; Xiong, L.; Salemi, J.; Bellve, K.; Byrne, B.J.; Fuller, D.D.; ZhuGe, R.; ElMallah, M.K. Airway smooth muscle dysfunction in Pompe (Gaa(−/−)) mice. Am. J. Physiol. Lung Cell Mol. Physiol. 2017, 312, L873–L881. [Google Scholar] [CrossRef] [Green Version]
- Guyenet, P.G.; Stornetta, R.L.; Bochorishvili, G.; Depuy, S.D.; Burke, P.G.; Abbott, S.B. C1 neurons: The body’s EMTs. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2013, 305, R187–R204. [Google Scholar] [CrossRef]
- Rukhadze, I.; Carballo, N.J.; Bandaru, S.S.; Malhotra, A.; Fuller, P.M.; Fenik, V.B. Catecholaminergic A1/C1 neurons contribute to the maintenance of upper airway muscle tone but may not participate in NREM sleep-related depression of these muscles. Respir. Physio.l Neurobiol. 2017, 244, 41–50. [Google Scholar] [CrossRef]
- Zieger, M.; Keeler, A.M.; Flotte, T.R.; ElMallah, M.K. AAV9 gene replacement therapy for respiratory insufficiency in very-long chain acyl-CoA dehydrogenase deficiency. J. Inherit. Metab. Dis. 2019, 42, 870–877. [Google Scholar] [CrossRef]
- Peng, J.; Dalton, J.; Butt, M.; Tracy, K.; Kennedy, D.; Haroldsen, P.; Cahayag, R.; Zoog, S.; O’Neill, C.A.; Tsuruda, L.S. Reveglucosidase alfa (BMN 701), an IGF2-Tagged rhAcid alpha-Glucosidase, Improves Respiratory Functional Parameters in a Murine Model of Pompe Disease. J. Pharmacol. Exp. Ther. 2017, 360, 313–323. [Google Scholar] [CrossRef] [PubMed]
- Han, S.O.; Li, S.; McCall, A.; Arnson, B.; Everitt, J.I.; Zhang, H.; Young, S.P.; ElMallah, M.K.; Koeberl, D.D. Comparisons of Infant and Adult Mice Reveal Age Effects for Liver Depot Gene Therapy in Pompe Disease. Mol. Ther. 2020, 17, 133–142. [Google Scholar] [CrossRef]
- ElMallah, M.K.; Stanley, D.A.; Lee, K.Z.; Turner, S.M.; Streeter, K.A.; Baekey, D.M.; Fuller, D.D. Power spectral analysis of hypoglossal nerve activity during intermittent hypoxia-induced long-term facilitation in mice. J. Neurophysiol. 2016, 115, 1372–1380. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berger, K.I.; Chan, Y.; Rom, W.N.; Oppenheimer, B.W.; Goldring, R.M. Progression from respiratory dysfunction to failure in late-onset Pompe disease. Neuromuscul. Disord. 2016, 26, 481–489. [Google Scholar] [CrossRef] [Green Version]
- Chan, J.; Desai, A.K.; Kazi, Z.B.; Corey, K.; Austin, S.; Hobson-Webb, L.D.; Case, L.E.; Jones, H.N.; Kishnani, P.S. The emerging phenotype of late-onset Pompe disease: A systematic literature review. Mol. Genet. Metab. 2017, 120, 163–172. [Google Scholar] [CrossRef]
- ElMallah, M.K.; Desai, A.K.; Nading, E.B.; DeArmey, S.; Kravitz, R.M.; Kishnani, P.S. Pulmonary outcome measures in long-term survivors of infantile Pompe disease on enzyme replacement therapy: A case series. Pediatr. Pulmonol. 2020, 55, 674–681. [Google Scholar] [CrossRef]
- Smith, B.K.; Allen, S.; Mays, S.; Martin, A.D.; Byrne, B.J. Dynamic respiratory muscle function in late-onset Pompe disease. Sci. Rep. 2019, 9, 19006. [Google Scholar] [CrossRef] [Green Version]
- Jones, H.N.; Kuchibhatla, M.; Crisp, K.D.; Hobson Webb, L.D.; Case, L.; Batten, M.T.; Marcus, J.A.; Kravitz, R.M.; Kishnani, P.S. Respiratory muscle training (RMT) in late-onset Pompe disease (LOPD): A protocol for a sham-controlled clinical trial. Mol. Genet. Metab. 2019, 127, 346–354. [Google Scholar] [CrossRef]
- Karam, C.; Dimitrova, D.; Yutan, E.; Chahin, N. Bright tongue sign in patients with late-onset Pompe disease. J. Neurol. 2019, 266, 2518–2523. [Google Scholar] [CrossRef] [Green Version]
- Mellies, U.; Ragette, R.; Schwake, C.; Baethmann, M.; Voit, T.; Teschler, H. Sleep-disordered breathing and respiratory failure in acid maltase deficiency. Neurology 2001, 57, 1290–1295. [Google Scholar] [CrossRef]
- Fuller, D.D.; ElMallah, M.K.; Smith, B.K.; Corti, M.; Lawson, L.A.; Falk, D.J.; Byrne, B.J. The respiratory neuromuscular system in Pompe disease. Respir. Physiol. Neurobiol. 2013, 189, 241–249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Korlimarla, A.; Lim, J.A.; Kishnani, P.S.; Sun, B. An emerging phenotype of central nervous system involvement in Pompe disease: From bench to bedside and beyond. Ann. Transl. Med. 2019, 7, 289. [Google Scholar] [CrossRef] [PubMed]
- McIntosh, P.T.; Hobson-Webb, L.D.; Kazi, Z.B.; Prater, S.N.; Banugaria, S.G.; Austin, S.; Wang, R.; Enterline, D.S.; Frush, D.P.; Kishnani, P.S. Neuroimaging findings in infantile Pompe patients treated with enzyme replacement therapy. Mol. Genet Metab. 2018, 123, 85–91. [Google Scholar] [CrossRef] [PubMed]
- Pena, L.D.; Proia, A.D.; Kishnani, P.S. Postmortem Findings and Clinical Correlates in Individuals with Infantile-Onset Pompe Disease. JIMD Rep. 2015, 23, 45–54. [Google Scholar] [CrossRef] [Green Version]
- McCall, A.L.; Salemi, J.; Bhanap, P.; Strickland, L.M.; Elmallah, M.K. The impact of Pompe disease on smooth muscle: A review. J. Smooth Muscle Res. 2018, 54, 100–118. [Google Scholar] [CrossRef] [Green Version]
- Martin, J.J.; de Barsy, T.; van Hoof, F.; Palladini, G. Pompe’s disease: An inborn lysosomal disorder with storage of glycogen. A study of brain and striated muscle. Acta Neuropathol. 1973, 23, 229–244. [Google Scholar] [CrossRef]
- Van der Walt, J.D.; Swash, M.; Leake, J.; Cox, E.L. The pattern of involvement of adult-onset acid maltase deficiency at autopsy. Muscle Nerve 1987, 10, 272–281. [Google Scholar] [CrossRef]
- Hobson-Webb, L.D.; Proia, A.D.; Thurberg, B.L.; Banugaria, S.; Prater, S.N.; Kishnani, P.S. Autopsy findings in late-onset Pompe disease: A case report and systematic review of the literature. Mol. Genet. Metab. 2012, 106, 462–469. [Google Scholar] [CrossRef]
- Kretzschmar, H.A.; Wagner, H.; Hubner, G.; Danek, A.; Witt, T.N.; Mehraein, P. Aneurysms and vacuolar degeneration of cerebral arteries in late-onset acid maltase deficiency. J. Neurol. Sci. 1990, 98, 169–183. [Google Scholar] [CrossRef]
- Martin, J.J.; de Barsy, T.; den Tandt, W.R. Acid maltase deficiency in non-identical adult twins. A morphological and biochemical study. J. Neurol. 1976, 213, 105–118. [Google Scholar] [CrossRef]
- Prigent, H.; Orlikowski, D.; Laforet, P.; Letilly, N.; Falaize, L.; Pellegrini, N.; Annane, D.; Raphael, J.C.; Lofaso, F. Supine volume drop and diaphragmatic function in adults with Pompe disease. Eur. Respir. J. 2012, 39, 1545–1546. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, B.K.; Fuller, D.D.; Martin, A.D.; Lottenberg, L.; Islam, S.; Lawson, L.A.; Onders, R.P.; Byrne, B.J. Diaphragm Pacing as a Rehabilitative Tool for Patients With Pompe Disease Who Are Ventilator-Dependent: Case Series. Phys. Ther. 2016, 96, 696–703. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gambetti, P.; DiMauro, S.; Baker, L. Nervous system in Pompe’s disease. Ultrastructure and biochemistry. J. Neuropathol. Exp. Neurol. 1971, 30, 412–430. [Google Scholar] [CrossRef] [PubMed]
- Teng, Y.T.; Su, W.J.; Hou, J.W.; Huang, S.F. Infantile-onset glycogen storage disease type II (Pompe disease): Report of a case with genetic diagnosis and pathological findings. Chang Gung Med. J. 2004, 27, 379–384. [Google Scholar] [PubMed]
- Mancall, E.L.; Aponte, G.E.; Berry, R.G. Pompe’s Disease (Diffuse Glycogenosis) with Neuronal Storage. J. Neuropathol. Exp. Neurol. 1965, 24, 85–96. [Google Scholar] [CrossRef]
- Margolis, M.L.; Howlett, P.; Goldberg, R.; Eftychiadis, A.; Levine, S. Obstructive sleep apnea syndrome in acid maltase deficiency. Chest 1994, 105, 947–949. [Google Scholar] [CrossRef]
- Kansagra, S.; Austin, S.; Dearmey, S.; Kishnani, P.S.; Kravitz, R.M. Polysomnographic findings in infantile Pompe disease. Am. J. Med. Genet. A 2013. [Google Scholar] [CrossRef]
- Sidman, R.L.; Taksir, T.; Fidler, J.; Zhao, M.; Dodge, J.C.; Passini, M.A.; Raben, N.; Thurberg, B.L.; Cheng, S.H.; Shihabuddin, L.S. Temporal neuropathologic and behavioral phenotype of 6neo/6neo Pompe disease mice. J. Neuropathol. Exp. Neurol. 2008, 67, 803–818. [Google Scholar] [CrossRef] [Green Version]
- Bijvoet, A.G.; van de Kamp, E.H.; Kroos, M.A.; Ding, J.H.; Yang, B.Z.; Visser, P.; Bakker, C.E.; Verbeet, M.P.; Oostra, B.A.; Reuser, A.J.; et al. Generalized glycogen storage and cardiomegaly in a knockout mouse model of Pompe disease. Hum. Mol. Genet. 1998, 7, 53–62. [Google Scholar] [CrossRef]



© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fusco, A.F.; McCall, A.L.; Dhindsa, J.S.; Zheng, L.; Bailey, A.; Kahn, A.F.; ElMallah, M.K. The Respiratory Phenotype of Pompe Disease Mouse Models. Int. J. Mol. Sci. 2020, 21, 2256. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21062256
Fusco AF, McCall AL, Dhindsa JS, Zheng L, Bailey A, Kahn AF, ElMallah MK. The Respiratory Phenotype of Pompe Disease Mouse Models. International Journal of Molecular Sciences. 2020; 21(6):2256. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21062256
Chicago/Turabian StyleFusco, Anna F., Angela L. McCall, Justin S. Dhindsa, Lucy Zheng, Aidan Bailey, Amanda F. Kahn, and Mai K. ElMallah. 2020. "The Respiratory Phenotype of Pompe Disease Mouse Models" International Journal of Molecular Sciences 21, no. 6: 2256. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21062256