Developing Computational Model to Predict Protein-Protein Interaction Sites Based on the XGBoost Algorithm

 and
and 
Abstract
:1. Introduction
2. Results
2.1. Evaluation Criteria
2.2. Predictive Performance in Two Balanced Modes
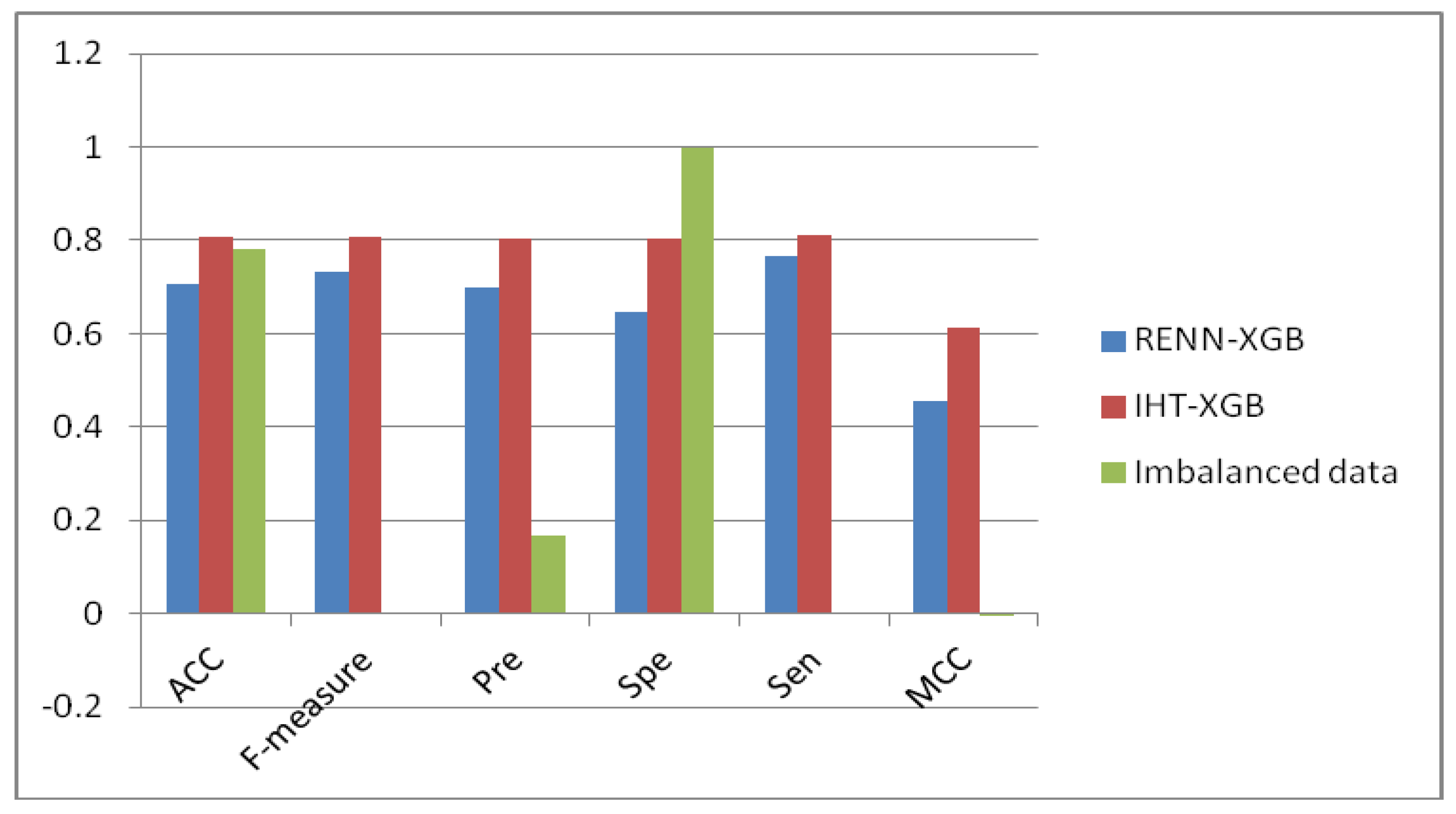
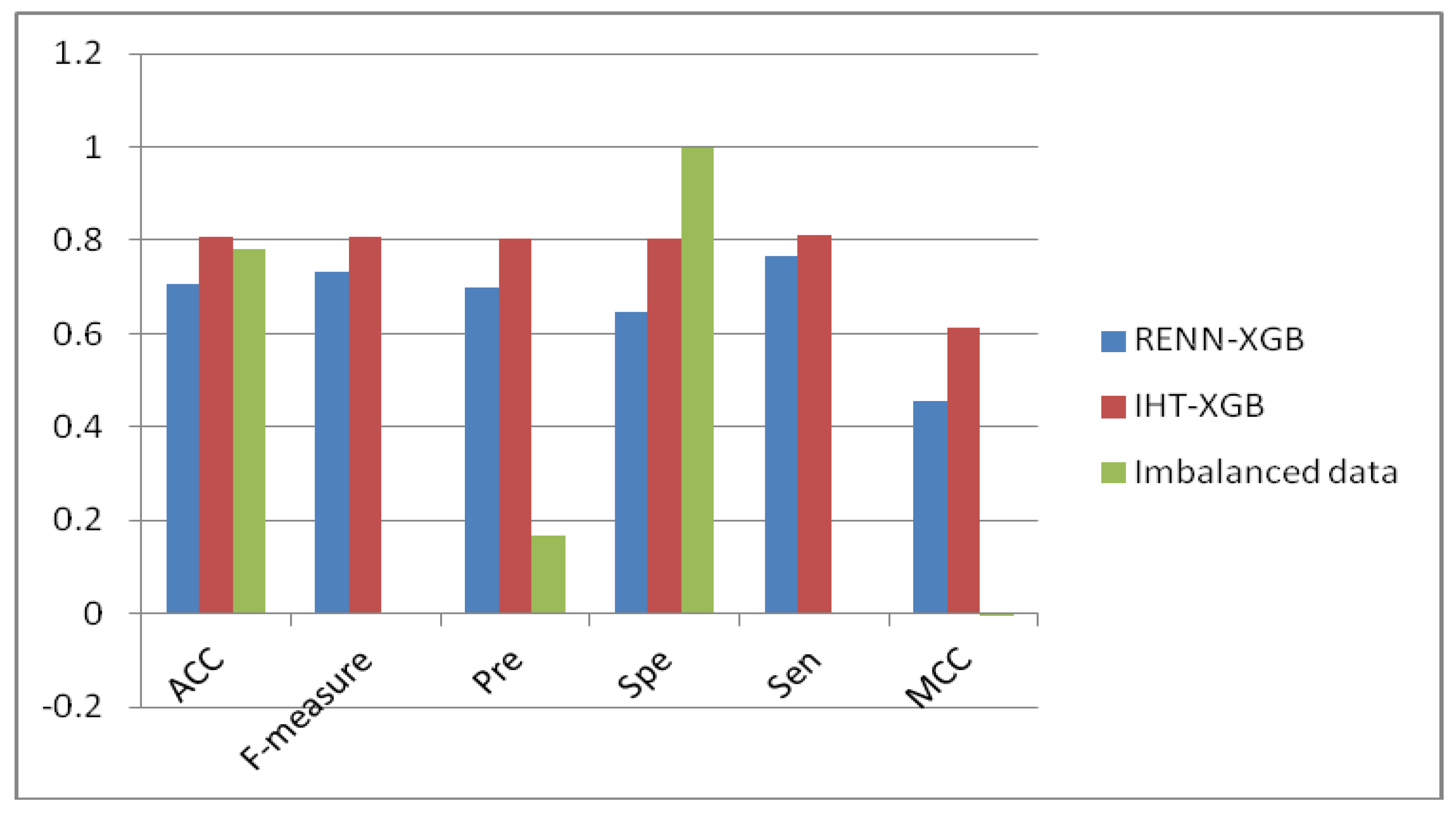
2.3. Comparison of Unbalanced and Balanced Data Sets
3. Discussion
3.1. Comparison with Other Methods
3.2. Prediction Performance in Independent Benchmark Datasets
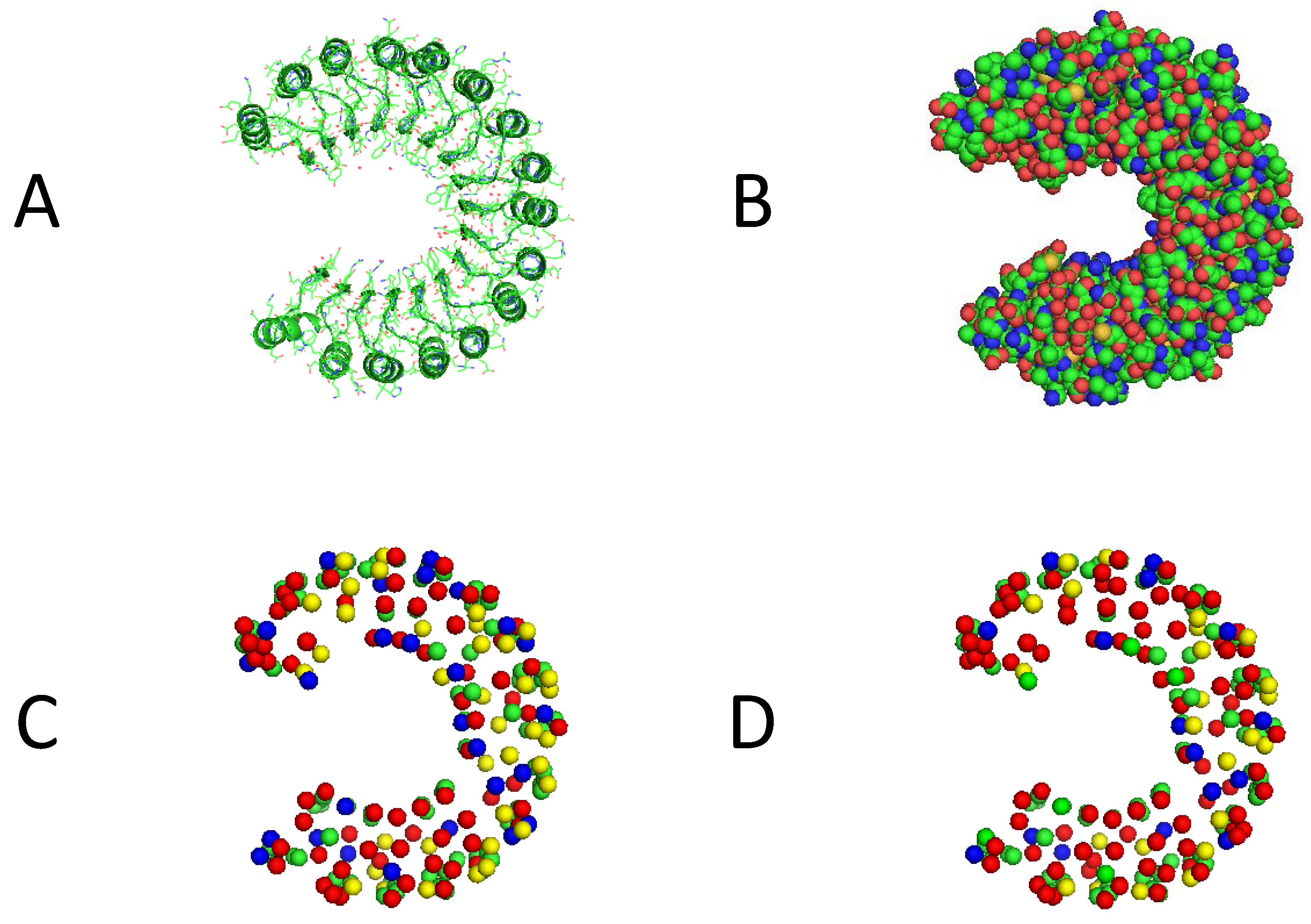
3.3. Visualization of Experimental Results
3.4. Limits of Prediction Validation
4. Materials and Methods
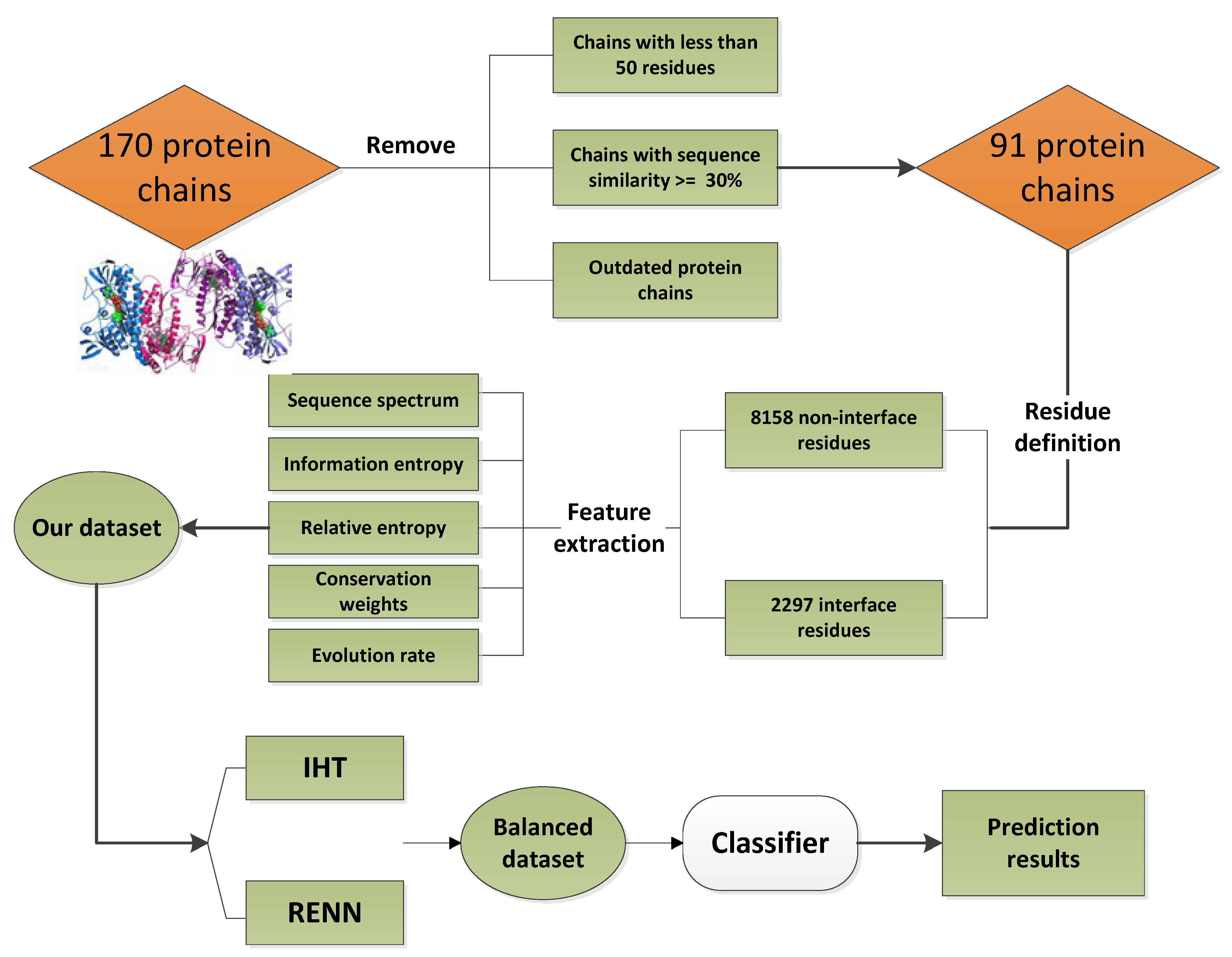
4.1. Dataset
4.2. Feature Extraction
4.3. Unbalanced Data SETS Processing
4.3.1. Instance Hardness Threshold
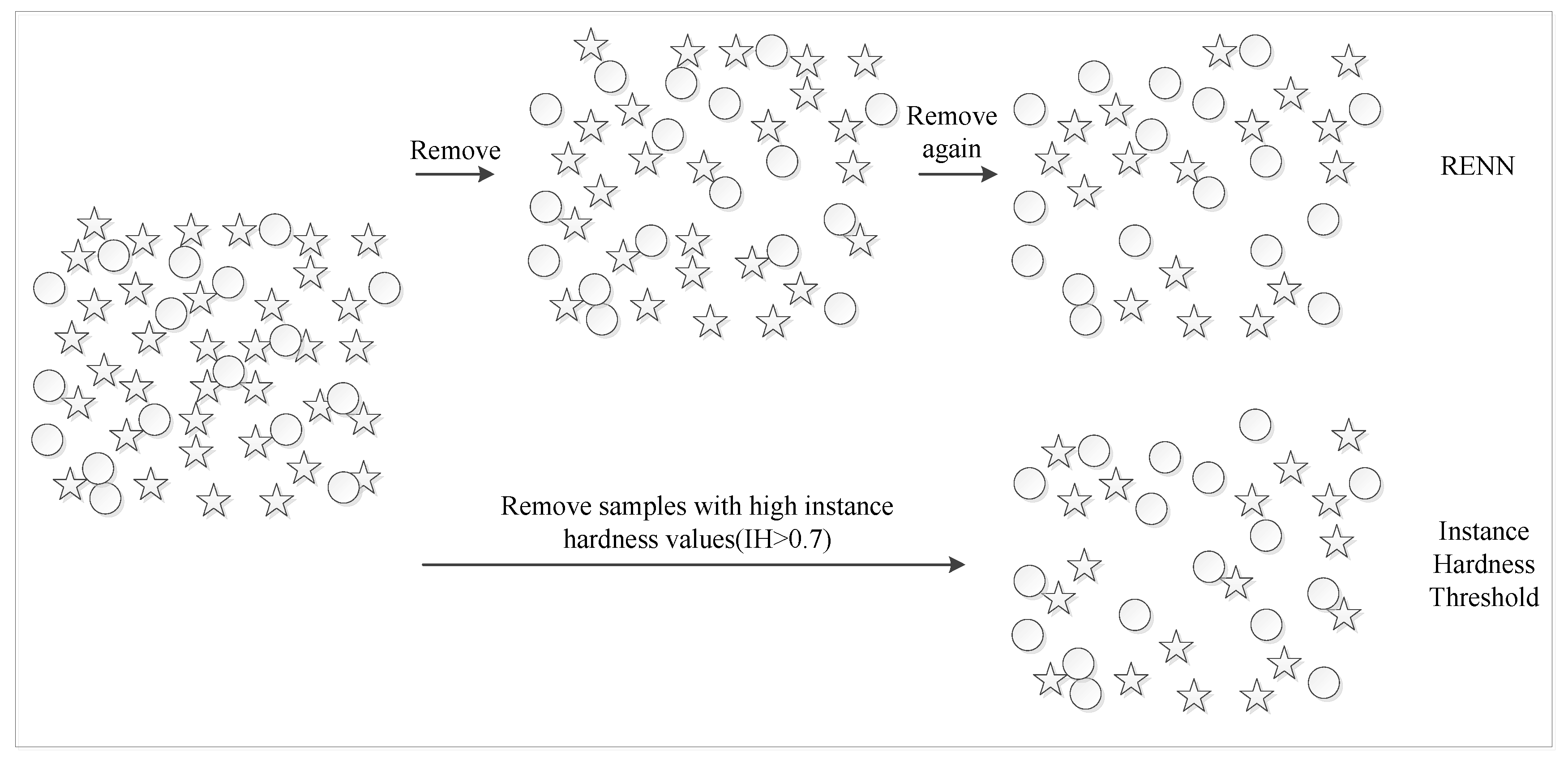
4.3.2. Repeated Edited Nearest Neighbors
| Algorithm 1. RENN algorithm. |
| Input: The original data set D. is the sample in D. |
| For i = 1,2,…,n |
| a. Calculate the Euclidean distances between and other samples in D. b. Get the category information of three samples closest to . c. If two or more nearest samples’ labels are different from , is removed from D. Repeat the above step until cannot be removed. END |
| Output: The balanced data set . |
4.4. XGBoost Algorithm
4.5. Protein Interaction Sites Prediction
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Chelliah, V.; Chen, L.; Blundell, T.L.; Lovell, S.C. Distinguishing structural and functional restraints in evolution in order to identify interaction sites. J. Mol. Biol. 2004, 342, 1487–1504. [Google Scholar] [CrossRef] [PubMed]
- Nooren, I.M.; Thornton, J.M. Diversity of protein–protein interactions. EMBO J. 2003, 22, 3486–3492. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, S.; Xia, D.; Su, B.; Chen, P.; Wang, B.; Li, J. A Convolutional Neural Network System to Discriminate Drug-Target Interactions. IEEE/ACM Trans. Comput. Biol. Bioinform. 2019. [Google Scholar] [CrossRef] [PubMed]
- Patel, T.; Pillay, M.; Jawa, R.; Liao, L. Information of binding sites improves prediction of protein-protein interaction. In Proceedings of the 2006 5th International Conference on Machine Learning and Applications (ICMLA’06), Orlando, FL, USA, 14–16 December 2006; pp. 205–212. [Google Scholar]
- Wang, Y.; Mei, C.; Zhou, Y.; Zheng, C.; Zhen, X.; Xiong, Y.; Wang, Y.; Chen, P.; Zhang, J.; Wang, B. Semi-supervised prediction of protein interaction sites from unlabeled sample information. BMC Bioinform. 2019, 20, 699. [Google Scholar] [CrossRef]
- Wang, B.; Wang, L.; Zheng, C.-H.; Xiong, Y. Imbalance Data Processing Strategy for Protein Interaction Sites Prediction. IEEE/ACM Trans. Comput. Biol. Bioinform. 2019. [Google Scholar] [CrossRef]
- Wei, P.J.; Zhang, D.; Xia, J.; Zheng, C.H. LNDriver: Identifying driver genes by integrating mutation and expression data based on gene-gene interaction network. BMC Bioinform. 2016, 17, 467. [Google Scholar] [CrossRef] [Green Version]
- Peng, C.; Liu, C.; Burge, L. DomSVR: Domain boundary prediction with support vector regression from sequence information alone. Amino Acids 2010, 39, 713–726. [Google Scholar]
- Sriwastava, B.K.; Basu, S.; Maulik, U. Protein–Protein interaction site prediction in Homo sapiens and E. coli using an interaction-affinity based membership function in fuzzy SVM. J. Biosci. 2015, 40, 809–818. [Google Scholar] [CrossRef]
- Daberdaku, S.; Ferrari, C. Exploring the potential of 3D Zernike descriptors and SVM for protein–protein interface prediction. BMC Bioinform. 2018, 19, 35. [Google Scholar] [CrossRef]
- Liu, Q.; Chen, P.; Wang, B.; Zhang, J.; Li, J. Hot spot prediction in protein-protein interactions by an ensemble system. BMC Syst. Biol. 2018, 12, 132. [Google Scholar] [CrossRef]
- Saethang, T.; Payne, D.M.; Avihingsanon, Y.; Pisitkun, T. A machine learning strategy for predicting localization of post-translational modification sites in protein-protein interacting regions. BMC Bioinform. 2016, 17, 307. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sriwastava, B.K.; Basu, S.; Maulik, U.; Plewczynski, D. PPIcons: Identification of protein-protein interaction sites in selected organisms. J. Mol. Model. 2013, 19, 4059–4070. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, K.; Gao, J.; Shen, S.; Tuszynski, J.A.; Ruan, J.; Hu, G. An accurate method for prediction of protein-ligand binding site on protein surface using SVM and statistical depth function. BioMed Res. Int. 2013, 2013, 409658. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhong, Y.; Guo, Y.; Luo, J.; Pu, X.; Li, M. Effective identification of kinase-specific phosphorylation sites based on domain–domain interactions. Chem. Intell. Lab. Syst. 2014, 136, 97–103. [Google Scholar] [CrossRef]
- Fan, W.; Xu, X.; Shen, Y.; Feng, H.; Li, A.; Wang, M. Prediction of protein kinase-specific phosphorylation sites in hierarchical structure using functional information and random forest. Amino Acids 2014, 46, 1069–1078. [Google Scholar] [CrossRef]
- Hu, S.S.; Peng, C.; Bing, W.; Li, J. Protein binding hot spots prediction from sequence only by a new ensemble learning method. Amino Acids 2017, 49, 1773–1785. [Google Scholar] [CrossRef]
- Guo, H.; Liu, B.; Cai, D.; Lu, T. Predicting protein–protein interaction sites using modified support vector machine. Int. J. Mach. Learn. Cybern. 2018, 9, 393–398. [Google Scholar] [CrossRef]
- Wang, B.; Chen, P.; Wang, P.; Zhao, G.; Zhang, X. Radial basis function neural network ensemble for predicting protein-protein interaction sites in heterocomplexes. Protein Pept. Lett. 2010, 17, 1111–1116. [Google Scholar] [CrossRef]
- Li, H.; Pi, D.; Wang, C. The prediction of protein-protein interaction sites based on RBF classifier improved by SMOTE. Math. Probl. Eng. 2014, 2014, 528767. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.-Y.; Wu, J.; Zhou, Z.-H. Exploratory undersampling for class-imbalance learning. IEEE Trans. Syst. Man Cybern. Part B 2008, 39, 539–550. [Google Scholar]
- Wang, B.; Huang, D.-S.; Jiang, C. A new strategy for protein interface identification using manifold learning method. IEEE Trans. Nanobiosci. 2014, 13, 118–123. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.; Guestrin, C. Xgboost: A scalable tree boosting system. In Proceedings of the 22nd ACM SIGKDD International Conference on Knowledge Discovery and Data Mining (KDD 2016), San Francisco, CA, USA, 13–17 August 2016; pp. 785–794. [Google Scholar]
- Wang, B.; Chen, P.; Huang, D.-S.; Li, J.-J.; Lok, T.-M.; Lyu, M.R. Predicting protein interaction sites from residue spatial sequence profile and evolution rate. Febs Lett. 2006, 580, 380–384. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuo, T.H.; Li, K.B. Predicting Protein-Protein Interaction Sites Using Sequence Descriptors and Site Propensity of Neighboring Amino Acids. Int. J. Mol. Sci. 2016, 17, 1788. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, R.; Jiang, W.; Zhou, Y. Identifying protein-protein interaction sites in transient complexes with temperature factor, sequence profile and accessible surface area. Amino Acids 2010, 38, 263–270. [Google Scholar] [CrossRef] [PubMed]
- Mei, C.; Wang, Y.; Lu, K.; Wang, B.; Chen, P. Unbalance Data Processing Strategy for Protein Interaction Sites Prediction. In Proceedings of the 2018 9th International Conference on Information Technology in Medicine and Education (ITME), Hangzhou, China, 19–21 October 2018; pp. 313–317. [Google Scholar]
- Dhole, K.; Singh, G.; Pai, P.P.; Mondal, S. Sequence-based prediction of protein-protein interaction sites with L1-logreg classifier. J. Theor. Biol. 2014, 348, 47–54. [Google Scholar] [CrossRef]
- Murakami, Y.; Mizuguchi, K. Applying the Naive Bayes classifier with kernel density estimation to the prediction of protein-protein interaction sites. Bioinformatics 2010, 26, 1841–1848. [Google Scholar] [CrossRef]
- Singh, G.; Dhole, K.; Pai, P.P.; Mondal, S. Springs: Prediction of protein-protein interaction sites using artificial neural networks. PeerJ PrePrints 2014, 2, e266v2. [Google Scholar]
- Porollo, A.; Meller, J. Prediction-based fingerprints of protein-protein interactions. Proteins 2007, 66, 630–645. [Google Scholar] [CrossRef]
- Zhang, J.; Kurgan, L. SCRIBER: Accurate and partner type-specific prediction of protein-binding residues from proteins sequences. Bioinformatics 2019, 35, i343–i353. [Google Scholar] [CrossRef] [Green Version]
- Ofran, Y.; Rost, B. ISIS: Interaction sites identified from sequence. Bioinformatics 2007, 23, e13–e16. [Google Scholar] [CrossRef]
- Hou, Q.; de Geest, P.F.G.; Vranken, W.F.; Heringa, J.; Feenstra, K.A. Seeing the trees through the forest: Sequence-based homo- and heteromeric protein-protein interaction sites prediction using random forest. Bioinformatics 2017, 33, 1479–1487. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeng, M.; Zhang, F.; Wu, F.X.; Li, Y.; Wang, J.; Li, M. Protein-protein interaction site prediction through combining local and global features with deep neural networks. Bioinformatics 2020, 36, 1114–1120. [Google Scholar] [CrossRef] [PubMed]
- Wei, Z.-S.; Han, K.; Yang, J.-Y.; Shen, H.-B.; Yu, D.-J. Protein-protein interaction sites prediction by ensembling svm and sample-weighted random forests. Neurocomputing 2016, 193, 201–212. [Google Scholar] [CrossRef]
- Li, Y.; Ilie, L. DELPHI: Accurate deep ensemble model for protein interaction sites prediction. bioRxiv 2020. [Google Scholar] [CrossRef]
- Bonvin, A.M. Flexible protein-protein docking. Curr. Opin. Struct. Biol. 2006, 16, 194–200. [Google Scholar] [CrossRef] [Green Version]
- Ansari, S.; Helms, V. Statistical analysis of predominantly transient protein–protein interfaces. Proteins 2005, 61, 344–355. [Google Scholar] [CrossRef]
- Fariselli, P.; Pazos, F.; Valencia, A.; Casadio, R. Prediction of protein–protein interaction sites in heterocomplexes with neural networks. Eur. J. Biochem. 2002, 269, 1356–1361. [Google Scholar] [CrossRef] [Green Version]
- Glaser, F.; Pupko, T.; Paz, I.; Bell, R.E.; Bechor-Shental, D.; Martz, E.; Ben-Tal, N. ConSurf: Identification of functional regions in proteins by surface-mapping of phylogenetic information. Bioinformatics 2003, 19, 163–164. [Google Scholar] [CrossRef] [Green Version]
- Smith, M.R.; Martinez, T.; Giraud-Carrier, C. An instance level analysis of data complexity. Mach. Learn. 2014, 95, 225–256. [Google Scholar] [CrossRef] [Green Version]
- Breiman, L. Random forests. Mach. Learn. 2001, 45, 5–32. [Google Scholar] [CrossRef] [Green Version]
- Bahety, A. Extension and evaluation of id3–decision tree algorithm. Entropy 2014, 2, 1–8. [Google Scholar]
- Verdikha, N.A.; Adji, T.B.; Permanasari, A.E. Study of Undersampling Method: Instance Hardness Threshold with Various Estimators for Hate Speech Classification. IJITEE 2018, 2, 39–44. [Google Scholar] [CrossRef]
- Wilson, D.L. Asymptotic properties of nearest neighbor rules using edited data. IEEE Trans. Syst. Man Cybern. 1972, SMC-2, 408–421. [Google Scholar] [CrossRef] [Green Version]
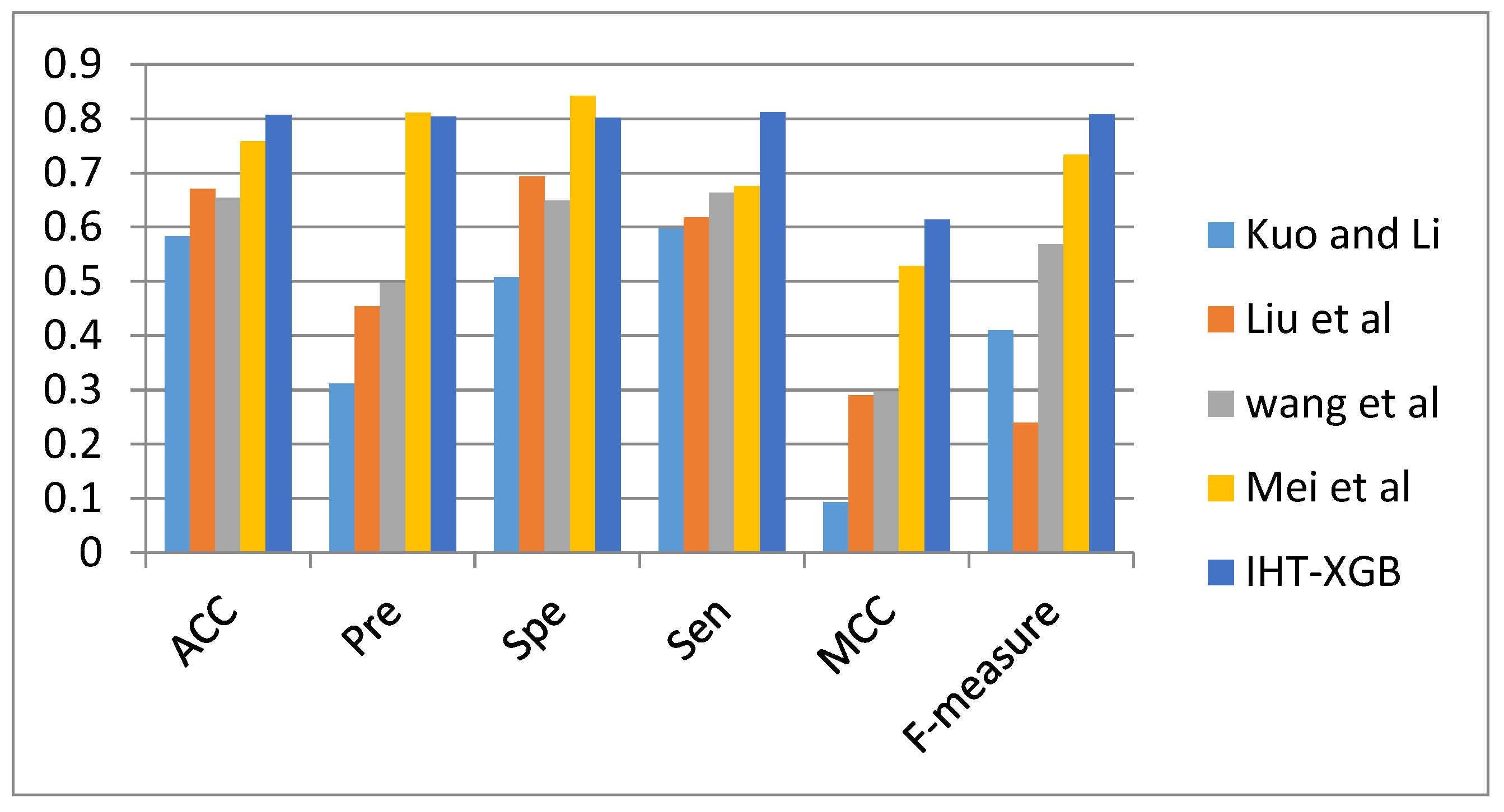




{kind=link}
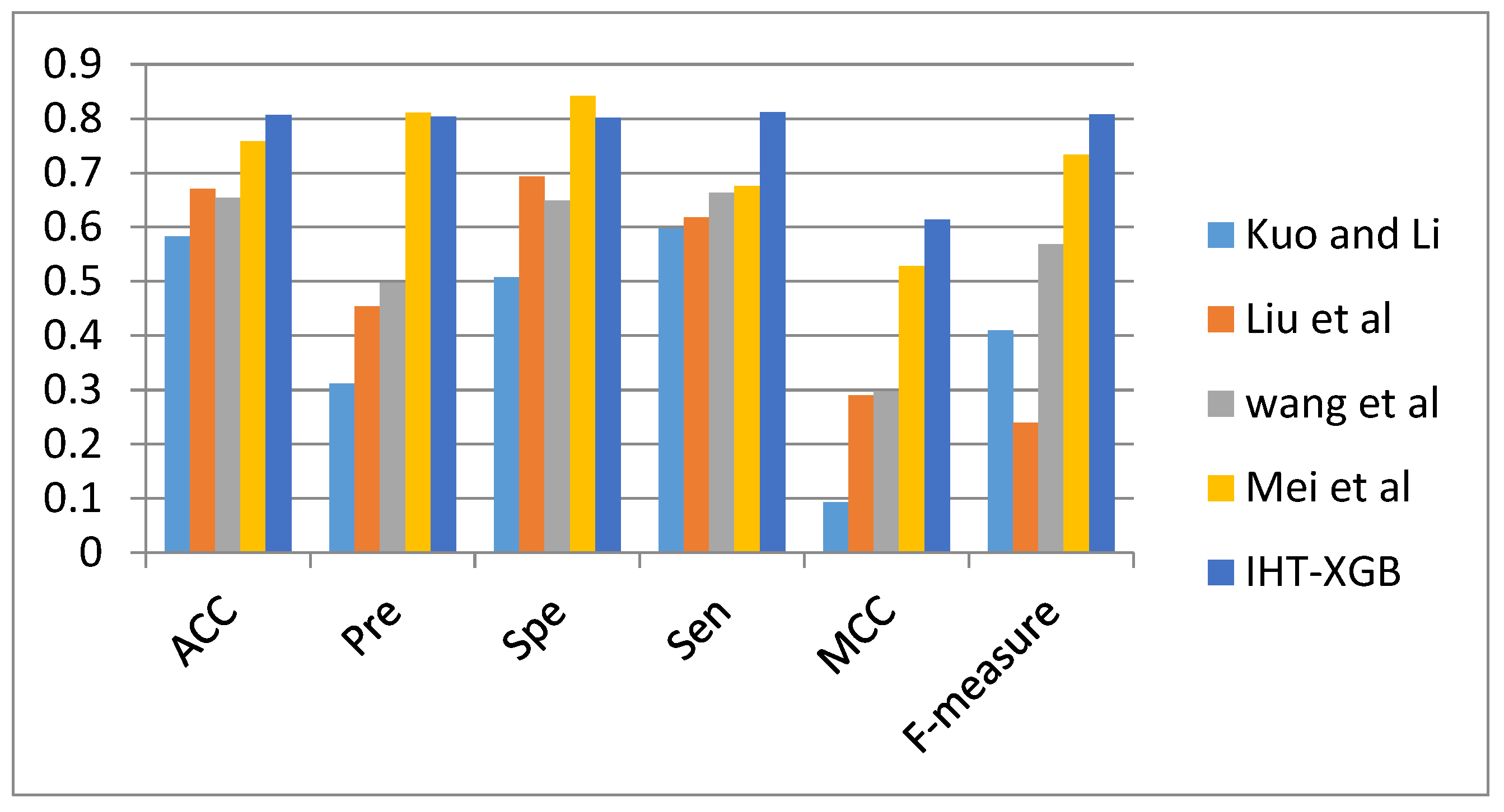{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Samples | ||
|---|---|---|
| Positive | Negative | |
| Original data | 2297 | 8158 |
| RENN | 2297 | 2131 |
| IHT | 2297 | 2297 |
| Acc | F-measure | Pre | Spe | Sen | MCC | |
|---|---|---|---|---|---|---|
| RENN-XGB | 0.707 | 0.731 | 0.699 | 0.645 | 0.765 | 0.454 |
| IHT-XGB | 0.807 | 0.808 | 0.804 | 0.802 | 0.812 | 0.614 |
| Sample | Results | |||||
|---|---|---|---|---|---|---|
| Positive | Negative | TP | TN | FP | FN | |
| Imbalanced data | 2297 | 8158 | 5 | 8151 | 25 | 2249 |
| RENN | 2131 | 2297 | 1758 | 1376 | 755 | 539 |
| IHT | 2291 | 2297 | 1864 | 1844 | 454 | 432 |
| Method | Acc | F-measure | Pre | Spe | Sen | MCC | |
|---|---|---|---|---|---|---|---|
| Dset_186 | SSWRF | 0.679 | 0.386 | 0.322 | 0.697 | 0.581 | 0.234 |
| LORIS | 0.604 | 0.384 | 0.287 | 0.586 | 0.698 | 0.221 | |
| PSIVER | 0.673 | 0.353 | 0.306 | 0.743 | 0.416 | 0.151 | |
| SCRIBER | 0.78 | 0.279 | 0.279 | 0.87 | 0.279 | 0.15 | |
| DELPHI | 0.803 | 0.353 | 0.353 | 0.884 | 0.352 | 0.235 | |
| IHT_XGB | 0.716 | 0.694 | 0.753 | 0.788 | 0.644 | 0.437 | |
| Dset_72 | SSWRF | 0.648 | 0.351 | 0.267 | 0.643 | 0.654 | 0.224 |
| LORIS | 0.614 | 0.324 | 0.238 | 0.610 | 0.631 | 0.177 | |
| PSIVER | 0.661 | 0.278 | 0.25 | 0.693 | 0.465 | 0.135 | |
| SCRIBER | 0.837 | 0.232 | 0.232 | 0.909 | 0.232 | 0.141 | |
| DELPHI | 0.847 | 0.275 | 0.276 | 0.915 | 0.274 | 0.189 | |
| IHT_XGB | 0.702 | 0.689 | 0.721 | 0.741 | 0.663 | 0.405 | |
| Dset_164 | SSWRF | 0.621 | 0.365 | 0.323 | 0.656 | 0.527 | 0.152 |
| LORIS | 0.588 | 0.323 | 0.263 | 0.609 | 0.538 | 0.111 | |
| PSIVER | 0.596 | 0.295 | 0.253 | 0.634 | 0.464 | 0.078 | |
| SCRIBER | 0.756 | 0.327 | 0.327 | 0.851 | 0.327 | 0.179 | |
| DELPHI | 0.758 | 0.332 | 0.332 | 0.852 | 0.332 | 0.184 | |
| IHT_XGB | 0.733 | 0.715 | 0.767 | 0.795 | 0.671 | 0.470 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Deng, A.; Zhang, H.; Wang, W.; Zhang, J.; Fan, D.; Chen, P.; Wang, B. Developing Computational Model to Predict Protein-Protein Interaction Sites Based on the XGBoost Algorithm. Int. J. Mol. Sci. 2020, 21, 2274. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21072274
Deng A, Zhang H, Wang W, Zhang J, Fan D, Chen P, Wang B. Developing Computational Model to Predict Protein-Protein Interaction Sites Based on the XGBoost Algorithm. International Journal of Molecular Sciences. 2020; 21(7):2274. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21072274
Chicago/Turabian StyleDeng, Aijun, Huan Zhang, Wenyan Wang, Jun Zhang, Dingdong Fan, Peng Chen, and Bing Wang. 2020. "Developing Computational Model to Predict Protein-Protein Interaction Sites Based on the XGBoost Algorithm" International Journal of Molecular Sciences 21, no. 7: 2274. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21072274