Vasculoprotective Effects of Vildagliptin. Focus on Atherogenesis

 , , ,
, , ,  and
and
Abstract
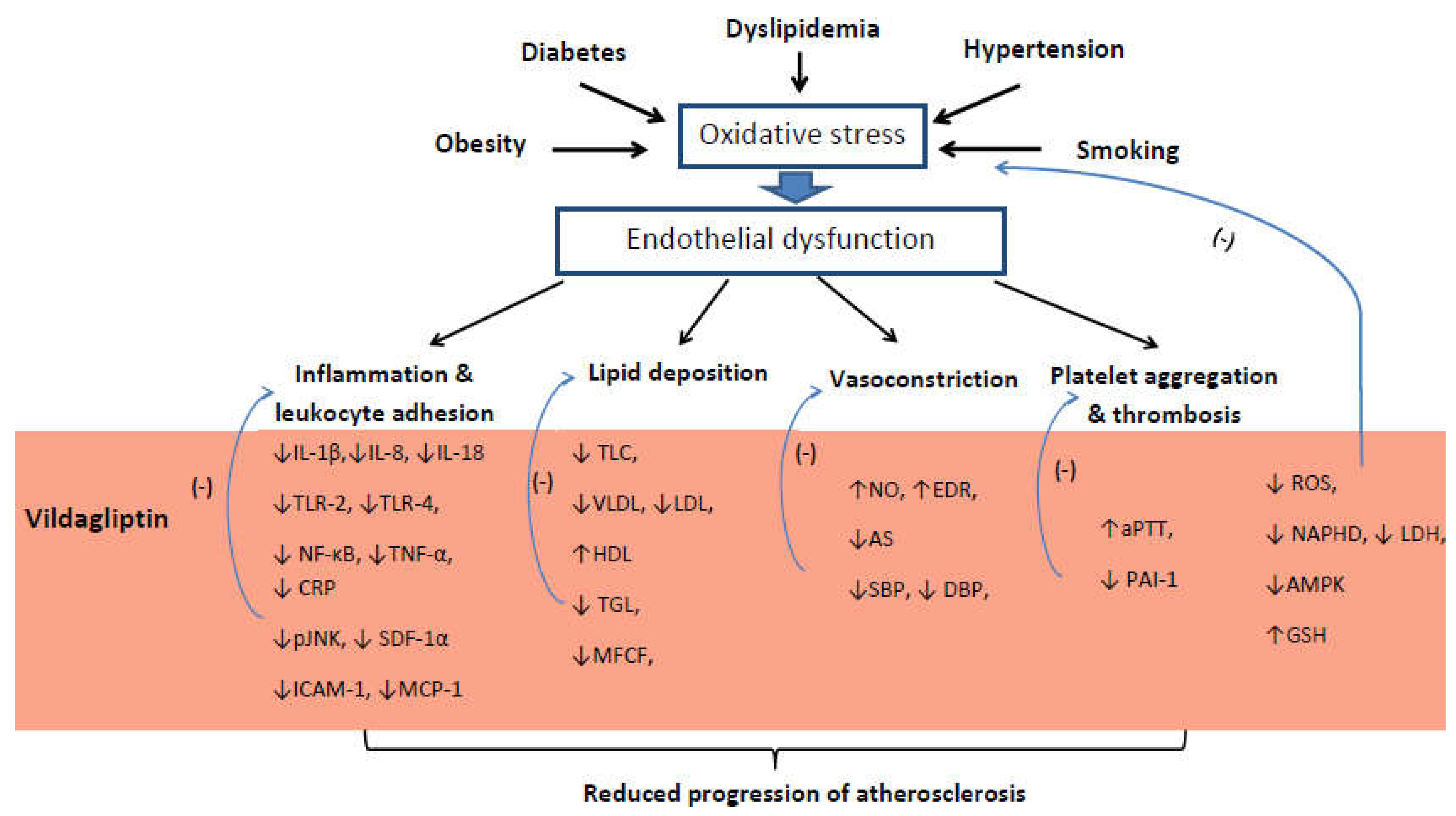
:1. Introduction
2. Vascular Endothelium
3. Inflammation
4. Lipids Metabolism Disorders
5. Vascular Dilatation and Blood Pressure
6. Cardiovascular Outcome
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- World Health Organization Diabetes. Available online: http://www.who.int (accessed on 30 October 2018).
- The Emerging Risk Factors Collaboration. Diabetes mellitus, fasting blood glucose concentration, and risk of vascular disease: A collaborative meta-analysis of 102 prospective studies. Lancet 2010, 375, 2215–2222. [Google Scholar] [CrossRef] [Green Version]
- Croxtall, J.D.; Keam, S.J. Vildagliptin: A Review of its Use in the Management of Type 2 Diabetes Mellitus. Drugs 2008, 68, 2387–2409. [Google Scholar] [CrossRef] [PubMed]
- Thornberry, N.A.; Gallwitz, B. Mechanism of action of inhibitors of dipeptidyl-peptidase-4 (DPP-4). Best Pract. Res. Clin. Endocrinol. Metab. 2009, 23, 479–486. [Google Scholar] [CrossRef] [PubMed]
- Näslund, E.; Gutniak, M.; Skogar, S.; Rössner, S.; Hellström, P.M. Glucagon-like peptide 1 increases the period of postprandial satiety and slows gastric emptying in obese men. Am. J. Clin. Nutr. 1998, 68, 525–530. [Google Scholar] [CrossRef] [PubMed]
- Vella, A. Mechanism of Action of DPP-4 Inhibitors—New Insights. J. Clin. Endocrinol. Metab. 2012, 97, 2626–2628. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choe, E.Y.; Cho, Y.; Choi, Y.; Yun, Y.; Wang, H.J.; Kwon, O.; Lee, B.-W.; Ahn, C.W.; Cha, B.S.; Lee, H.C.; et al. The Effect of DPP-4 Inhibitors on Metabolic Parameters in Patients with Type 2 Diabetes. Diabetes Metab. J. 2014, 38, 211. [Google Scholar] [CrossRef] [Green Version]
- He, Y.-L.; Sadler, B.M.; Sabo, R.; Balez, S.; Wang, Y.; Campestrini, J.; Laurent, A.; Ligueros-Saylan, M.; Howard, D. The Absolute Oral Bioavailability and??Population-Based Pharmacokinetic Modelling of a Novel Dipeptidylpeptidase-IV Inhibitor, Vildagliptin, in Healthy Volunteers. Clin. Pharmacokinet. 2007, 46, 787–802. [Google Scholar] [CrossRef]
- He, Y.-L. Clinical Pharmacokinetics and Pharmacodynamics of Vildagliptin. Clin. Pharmacokinet. 2012, 51, 147–162. [Google Scholar] [CrossRef]
- Dejager, S.; Razac, S.; Foley, J.; Schweizer, A. Vildagliptin in Drug-naïve Patients with Type 2 Diabetes: A 24-Week, Double-blind, Randomized, Placebo-controlled, Multiple-dose Study. Horm Metab Res. 2007, 39, 218–223. [Google Scholar] [CrossRef]
- He, H.; Tran, P.; Yin, H.; Smith, H.; Batard, Y.; Wang, L.; Einolf, H.; Gu, H.; Mangold, J.B.; Fischer, V.; et al. Absorption, Metabolism, and Excretion of [14 C]Vildagliptin, a Novel Dipeptidyl Peptidase 4 Inhibitor, in Humans. Drug Metab Dispos. 2009, 37, 536–544. [Google Scholar] [CrossRef] [Green Version]
- Trevisan, R. The Role of Vildagliptin in the Therapy of Type 2 Diabetic Patients with Renal Dysfunction. Diabetes 2017, 8, 1215–1226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, D.-S.; Lee, E.-S.; Alam, M.M.; Jang, J.-H.; Lee, H.-S.; Oh, H.; Kim, Y.-C.; Manzoor, Z.; Koh, Y.-S.; Kang, D.-G.; et al. Soluble DPP-4 up-regulates toll-like receptors and augments inflammatory reactions, which are ameliorated by vildagliptin or mannose-6-phosphate. Metabolism 2016, 65, 89–101. [Google Scholar] [CrossRef] [PubMed]
- Dei Cas, A.; Spigoni, V.; Cito, M.; Aldigeri, R.; Ridolfi, V.; Marchesi, E.; Marina, M.; Derlindati, E.; Aloe, R.; Bonadonna, R.C.; et al. Vildagliptin, but not glibenclamide, increases circulating endothelial progenitor cell number: A 12-month randomized controlled trial in patients with type 2 diabetes. Cardiovasc. Diabetol. 2017, 16, 27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, M.; Jin, X.; Zhang, Z.; Li, B.; Yang, G. Vildagliptin protects endothelial cells against high glucose-induced damage. Biomed. Pharmacother. 2018, 108, 1790–1796. [Google Scholar] [CrossRef]
- Qi, Y.; Du, X.; Yao, X.; Zhao, Y. Vildagliptin inhibits high free fatty acid (FFA)-induced NLRP3 inflammasome activation in endothelial cells. Artif. CellsNanomed. Biotechnol. 2019, 47, 1067–1074. [Google Scholar] [CrossRef] [Green Version]
- Liu, H.; Xiang, H.; Zhao, S.; Sang, H.; Lv, F.; Chen, R.; Shu, Z.; Chen, A.F.; Chen, S.; Lu, H. Vildagliptin improves high glucose-induced endothelial mitochondrial dysfunction via inhibiting mitochondrial fission. J. Cell Mol. Med. 2019, 23, 798–810. [Google Scholar] [CrossRef] [Green Version]
- Seo, M.S.; Li, H.; An, J.R.; Jung, I.D.; Jung, W.-K.; Ha, K.-S.; Han, E.-T.; Hong, S.-H.; Choi, I.-W.; Park, W.S. Vildagliptin, an Anti-diabetic Drug of the DPP-4 Inhibitor, Induces Vasodilation via Kv Channel and SERCA Pump Activation in Aortic Smooth Muscle. Cardiovasc. Toxicol. 2019, 19, 244–254. [Google Scholar] [CrossRef]
- Oeseburg, H.; de Boer, R.A.; Buikema, H.; van der Harst, P.; van Gilst, W.H.; Silljé, H.H.W. Glucagon-Like Peptide 1 Prevents Reactive Oxygen Species–Induced Endothelial Cell Senescence Through the Activation of Protein Kinase A. Arter. Thromb Vasc Biol. 2010, 30, 1407–1414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Terasaki, M.; Nagashima, M.; Watanabe, T.; Nohtomi, K.; Mori, Y.; Miyazaki, A.; Hirano, T. Effects of PKF275-055, a dipeptidyl peptidase–4 inhibitor, on the development of atherosclerotic lesions in apolipoprotein E–null mice. Metabolism 2012, 61, 974–977. [Google Scholar] [CrossRef] [PubMed]
- Terasaki, M.; Nagashima, M.; Nohtomi, K.; Kohashi, K.; Tomoyasu, M.; Sinmura, K.; Nogi, Y.; Katayama, Y.; Sato, K.; Itoh, F.; et al. Preventive Effect of Dipeptidyl Peptidase-4 Inhibitor on Atherosclerosis Is Mainly Attributable to Incretin’s Actions in Nondiabetic and Diabetic Apolipoprotein E-Null Mice. PLoS ONE 2013, 8, e70933. [Google Scholar] [CrossRef] [PubMed]
- Khan, S.; Khan, S.; Panda, B.P.; Akhtar, M.; Najmi, A.K. Potential effects of vildagliptin on biomarkers associated with prothrombosis in diabetes mellitus. Expert Opin. Ther. Targets 2015, 19, 1607–1616. [Google Scholar] [CrossRef] [PubMed]
- Jain, S.; Sharma, B. Neuroprotective effect of selective DPP-4 inhibitor in experimental vascular dementia. Physiol. Behav. 2015, 152, 182–193. [Google Scholar] [CrossRef] [PubMed]
- Koyama, A.; Komori, K.; Otsuka, R.; Kajikuri, J.; Itoh, T. Dipeptidyl peptidase 4 inhibitor reduces intimal hyperplasia in rabbit autologous jugular vein graft under poor distal runoff. J. Vasc. Surg. 2016, 63, 1360–1370. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Q.; Xiao, X.; Zheng, J.; Li, M.; Yu, M.; Ping, F.; Wang, T.; Wang, X. A Possible Mechanism: Vildagliptin Prevents Aortic Dysfunction through Paraoxonase and Angiopoietin-Like 3. BioMed Res. Int. 2018, 2018, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Ji, Y.; Ge, Y.; Xu, X.; Ye, S.; Fan, Y.; Zhang, J.; Mei, L.; Zhang, X.; Ying, L.; Yang, T.; et al. Vildagliptin Reduces Stenosis of Injured Carotid Artery in Diabetic Mouse Through Inhibiting Vascular Smooth Muscle Cell Proliferation via ER Stress/NF-κB Pathway. Front. Pharm. 2019, 10, 142. [Google Scholar] [CrossRef] [PubMed]
- van Poppel, P.C.M.; Netea, M.G.; Smits, P.; Tack, C.J. Vildagliptin Improves Endothelium-Dependent Vasodilatation in Type 2 Diabetes. Dia Care 2011, 34, 2072–2077. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Noguchi, K.; Hirota, M.; Miyoshi, T.; Tani, Y.; Noda, Y.; Ito, H.; Nanba, S. Single administration of vildagliptin attenuates postprandial hypertriglyceridemia and endothelial dysfunction in normoglycemic individuals. Exp. Ther. Med. 2015, 9, 84–88. [Google Scholar] [CrossRef] [Green Version]
- Tani, S.; Takahashi, A.; Nagao, K.; Hirayama, A. Effect of Dipeptidyl Peptidase-4 Inhibitor, Vildagliptin on Plasminogen Activator Inhibitor-1 in Patients with Diabetes Mellitus. Am. J. Cardiol. 2015, 115, 454–460. [Google Scholar] [CrossRef]
- Foley, J.E.; Evans, M.; Schweizer, A. Blood pressure and fasting lipid changes after 24 weeks’ treatment with vildagliptin: A pooled analysis in >2,000 previously drug-naïve patients with type 2 diabetes mellitus. VHRM 2016, 12, 337–340. [Google Scholar] [CrossRef] [Green Version]
- Duvnjak, L.; Blaslov, K. Dipeptidyl peptidase-4 inhibitors improve arterial stiffness, blood pressure, lipid profile and inflammation parameters in patients with type 2 diabetes mellitus. Diabetol. Metab. Syndr. 2016, 8, 26. [Google Scholar] [CrossRef] [Green Version]
- Park, K.S.; Kwak, S.; Cho, Y.M.; Park, K.S.; Jang, H.C.; Kim, S.Y.; Jung, H.S. Vildagliptin reduces plasma stromal cell-derived factor-1α in patients with type 2 diabetes compared with glimepiride. J. Diabetes Investig. 2017, 8, 218–226. [Google Scholar] [CrossRef] [PubMed]
- Younis, A.; Eskenazi, D.; Goldkorn, R.; Leor, J.; Naftali-Shani, N.; Fisman, E.Z.; Tenenbaum, A.; Goldenberg, I.; Klempfner, R. The addition of vildagliptin to metformin prevents the elevation of interleukin 1ß in patients with type 2 diabetes and coronary artery disease: A prospective, randomized, open-label study. Cardiovasc. Diabetol. 2017, 16, 69. [Google Scholar] [CrossRef] [Green Version]
- El-Naggar, A.R.; Zaafar, D.; Elyamany, M.; Hassanin, S.; Bassyouni, A.; Abdel-Latif, H. The Role of Vildagliptin in Treating Hypertension Through Modulating Serum VEGF in Diabetic Hypertensive Patients. J. Cardiovasc. Pharm. 2019, 24, 254–261. [Google Scholar] [CrossRef] [PubMed]
- Rajendran, P.; Rengarajan, T.; Thangavel, J.; Nishigaki, Y.; Sakthisekaran, D.; Sethi, G.; Nishigaki, I. The Vascular Endothelium and Human Diseases. Int. J. Biol. Sci. 2013, 9, 1057–1069. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Madden, J.A. Role of the vascular endothelium and plaque in acute ischemic stroke. Neurology 2012, 79, S58–S62. [Google Scholar] [CrossRef] [Green Version]
- Di Meo, S.; Reed, T.T.; Venditti, P.; Victor, V.M. Harmful and Beneficial Role of ROS. Oxidative Med. Cell. Longev. 2016, 2016, 1–3. [Google Scholar] [CrossRef] [Green Version]
- Dey, S.; DeMazumder, D.; Sidor, A.; Foster, D.B.; O’Rourke, B. Mitochondrial ROS Drive Sudden Cardiac Death and Chronic Proteome Remodeling in Heart Failure. Circ. Res. 2018, 123, 356–371. [Google Scholar] [CrossRef]
- Gimbrone, M.A.; García-Cardeña, G. Endothelial Cell Dysfunction and the Pathobiology of Atherosclerosis. Circ. Res. 2016, 118, 620–636. [Google Scholar] [CrossRef] [Green Version]
- Su, J.B. Vascular endothelial dysfunction and pharmacological treatment. World J. Cardiol. 2015, 7, 719. [Google Scholar] [CrossRef]
- Park, K.-H.; Park, W.J. Endothelial Dysfunction: Clinical Implications in Cardiovascular Disease and Therapeutic Approaches. J. Korean Med. Sci. 2015, 30, 1213. [Google Scholar] [CrossRef] [Green Version]
- Sharp, W.W.; Fang, Y.H.; Han, M.; Zhang, H.J.; Hong, Z.; Banathy, A.; Morrow, E.; Ryan, J.J.; Archer, S.L. Dynamin-related protein 1 (Drp1)-mediated diastolic dysfunction in myocardial ischemia-reperfusion injury: therapeutic benefits of Drp1 inhibition to reduce mitochondrial fission. Faseb J. 2014, 28, 316–326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hausenloy, D.J.; Whittington, H.J.; Wynne, A.M.; Begum, S.S.; Theodorou, L.; Riksen, N.; Mocanu, M.M.; Yellon, D.M. Dipeptidyl peptidase-4 inhibitors and GLP-1 reduce myocardial infarct size in a glucose-dependent manner. Cardiovasc. Diabetol. 2013, 12, 154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chinda, K.; Sanit, J.; Chattipakorn, S.; Chattipakorn, N. Dipeptidyl peptidase-4 inhibitor reduces infarct size and preserves cardiac function via mitochondrial protection in ischaemia–reperfusion rat heart. Diabetes Vasc. Dis. Res. 2014, 11, 75–83. [Google Scholar] [CrossRef] [Green Version]
- Bayrami, G.; Karimi, P.; Agha-Hosseini, F.; Feyzizadeh, S.; Badalzadeh, R. Effect of Ischemic Postconditioning on Myocardial Function and Infarct Size Following Reperfusion Injury in Diabetic Rats Pretreated With Vildagliptin. J. Cardiovasc. Pharm. 2018, 23, 174–183. [Google Scholar] [CrossRef] [PubMed]
- McMurray, J.J.V.; Ponikowski, P.; Bolli, G.B.; Lukashevich, V.; Kozlovski, P.; Kothny, W.; Lewsey, J.D.; Krum, H. Effects of Vildagliptin on Ventricular Function in Patients with Type 2 Diabetes Mellitus and Heart Failure. JACC Heart Fail. 2018, 6, 8–17. [Google Scholar] [CrossRef]
- Loboda, A.; Damulewicz, M.; Pyza, E.; Jozkowicz, A.; Dulak, J. Role of Nrf2/HO-1 system in development, oxidative stress response and diseases: An evolutionarily conserved mechanism. Cell. Mol. Life Sci. 2016, 73, 3221–3247. [Google Scholar] [CrossRef] [Green Version]
- Werner, N.; Kosiol, S.; Schiegl, T.; Ahlers, P.; Walenta, K.; Link, A.; Böhm, M.; Nickenig, G. Circulating Endothelial Progenitor Cells and Cardiovascular Outcomes. N. Engl. J. Med. 2005, 353, 999–1007. [Google Scholar] [CrossRef]
- Wei, D.; Wang, G.; Tang, C.; Qiu, J.; Zhao, J.; Gregersen, H.; Deng, L. Upregulation of SDF-1 is Associated with Atherosclerosis Lesions Induced by LDL Concentration Polarization. Ann. Biomed. Eng. 2012, 40, 1018–1027. [Google Scholar] [CrossRef]
- Zhong, J.; Rajagopalan, S. Dipeptidyl Peptidase-4 Regulation of SDF-1/CXCR4 Axis: Implications for Cardiovascular Disease. Front. Immunol. 2015, 6, 477. [Google Scholar] [CrossRef] [Green Version]
- Deshane, J.; Chen, S.; Caballero, S.; Grochot-Przeczek, A.; Was, H.; Li Calzi, S.; Lach, R.; Hock, T.D.; Chen, B.; Hill-Kapturczak, N.; et al. Stromal cell–derived factor 1 promotes angiogenesis via a heme oxygenase 1–dependent mechanism. J. Exp. Med. 2007, 204, 605–618. [Google Scholar] [CrossRef]
- Hristov, M.; Zernecke, A.; Bidzhekov, K.; Liehn, E.A.; Shagdarsuren, E.; Ludwig, A.; Weber, C. Importance of CXC Chemokine Receptor 2 in the Homing of Human Peripheral Blood Endothelial Progenitor Cells to Sites of Arterial Injury. Circ. Res. 2007, 100, 590–597. [Google Scholar] [CrossRef] [Green Version]
- Li, M.; Yu, J.; Li, Y.; Li, D.; Yan, D.; Qu, Z.; Ruan, Q. CXCR4 positive bone mesenchymal stem cells migrate to human endothelial cell stimulated by ox-LDL via SDF-1α/CXCR4 signaling axis. Exp. Mol. Pathol. 2010, 88, 250–255. [Google Scholar] [CrossRef] [PubMed]
- Abi-Younes, S.; Sauty, A.; Mach, F.; Sukhova, G.K.; Libby, P.; Luster, A.D. The Stromal Cell–Derived Factor-1 Chemokine Is a Potent Platelet Agonist Highly Expressed in Atherosclerotic Plaques. Circ. Res. 2000, 86, 131–138. [Google Scholar] [CrossRef] [PubMed]
- Brenner, C.; Franz, W.M.; Kühlenthal, S.; Kuschnerus, K.; Remm, F.; Gross, L.; Theiss, H.D.; Landmesser, U.; Kränkel, N. DPP-4 inhibition ameliorates atherosclerosis by priming monocytes into M2 macrophages. Int. J. Cardiol. 2015, 199, 163–169. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, S.; Liu, C.; Aviv, A.; Ho, J.E.; Courchesne, P.; Muntendam, P.; Larson, M.G.; Cheng, S.; Wang, T.J.; Mehta, N.N.; et al. Stromal Cell–Derived Factor 1 as a Biomarker of Heart Failure and Mortality Risk. Arter. Thromb. Vasc. Biol. 2014, 34, 2100–2105. [Google Scholar] [CrossRef] [Green Version]
- Donath, M.Y. Multiple benefits of targeting inflammation in the treatment of type 2 diabetes. Diabetologia 2016, 59, 679–682. [Google Scholar] [CrossRef]
- Assar, M.E.; Angulo, J.; Rodríguez-Mañas, L. Diabetes and ageing-induced vascular inflammation. J. Physiol. 2016, 594, 2125–2146. [Google Scholar] [CrossRef] [Green Version]
- Teague, H.L.; Ahlman, M.A.; Alavi, A.; Wagner, D.D.; Lichtman, A.H.; Nahrendorf, M.; Swirski, F.K.; Nestle, F.; Gelfand, J.M.; Kaplan, M.J.; et al. Unraveling Vascular Inflammation. J. Am. Coll. Cardiol. 2017, 70, 1403–1412. [Google Scholar] [CrossRef]
- Gordon, S.; Plüddemann, A. Tissue macrophages: heterogeneity and functions. BMC Biol. 2017, 15, 53. [Google Scholar] [CrossRef]
- Chen, L.; Deng, H.; Cui, H.; Fang, J.; Zuo, Z.; Deng, J.; Li, Y.; Wang, X.; Zhao, L. Inflammatory responses and inflammation-associated diseases in organs. Oncotarget 2018, 9. [Google Scholar] [CrossRef] [Green Version]
- Virdis, A.; Schiffrin, E.L. Vascular inflammation: A role in vascular disease in hypertension? Curr. Opin. Nephrol. Hypertens. 2003, 12, 181–187. [Google Scholar] [CrossRef] [PubMed]
- Hamuro, M.; Polan, J.; Natarajan, M.; Mohan, S. High glucose induced nuclear factor kappa B mediated inhibition of endothelial cell migration. Atherosclerosis 2002, 162, 277–287. [Google Scholar] [CrossRef]
- Bennett, M.R.; Sinha, S.; Owens, G.K. Vascular Smooth Muscle Cells in Atherosclerosis. Circ Res. 2016, 118, 692–702. [Google Scholar] [CrossRef] [PubMed]
- Aoki, T.; Frȍsen, J.; Fukuda, M.; Bando, K.; Shioi, G.; Tsuji, K.; Ollikainen, E.; Nozaki, K.; Laakkonen, J.; Narumiya, S. Prostaglandin E 2 –EP2–NF-κB signaling in macrophages as a potential therapeutic target for intracranial aneurysms. Sci. Signal. 2017, 10, eaah6037. [Google Scholar] [CrossRef] [Green Version]
- Venuraju, S.M.; Yerramasu, A.; Corder, R.; Lahiri, A. Osteoprotegerin as a Predictor of Coronary Artery Disease and Cardiovascular Mortality and Morbidity. J. Am. Coll. Cardiol. 2010, 55, 2049–2061. [Google Scholar] [CrossRef] [Green Version]
- Sprague, A.H.; Khalil, R.A. Inflammatory cytokines in vascular dysfunction and vascular disease. Biochem. Pharmacol. 2009, 78, 539–552. [Google Scholar] [CrossRef] [Green Version]
- Yoshida, S.; Ono, M.; Shono, T.; Izumi, H.; Ishibashi, T.; Suzuki, H.; Kuwano, M. Involvement of interleukin-8, vascular endothelial growth factor, and basic fibroblast growth factor in tumor necrosis factor alpha-dependent angiogenesis. Mol. Cell. Biol. 1997, 17, 4015–4023. [Google Scholar] [CrossRef] [Green Version]
- Vlahopoulos, S.; Boldogh, I.; Casola, A.; Brasier, A.R. Nuclear factor-kappaB-dependent induction of interleukin-8 gene expression by tumor necrosis factor alpha: evidence for an antioxidant sensitive activating pathway distinct from nuclear translocation. Blood 1999, 94, 1878–1889. [Google Scholar] [CrossRef]
- Michel, J.-B.; Martin-Ventura, J.L.; Nicoletti, A.; Ho-Tin-Noé, B. Pathology of human plaque vulnerability: Mechanisms and consequences of intraplaque haemorrhages. Atherosclerosis 2014, 234, 311–319. [Google Scholar] [CrossRef]
- Zaman, A.G.; Helft, G.; Worthley, S.G.; Badimon, J.J. The role of plaque rupture and thrombosis in coronary artery disease. Atherosclerosis 2000, 149, 251–266. [Google Scholar] [CrossRef]
- Stratmann, B.; Tschoepe, D. Pathobiology and cell interactions of platelets in diabetes. Diabetes Vasc. Dis. Res. 2005, 2, 16–23. [Google Scholar] [CrossRef] [Green Version]
- Haznedaroglu, I.C.; Malkan, U.Y. Lipotoxicity-Related Hematological Disorders in Obesity. In Obesity and Lipotoxicity; Engin, A.B., Engin, A., Eds.; Springer International Publishing: Cham, Switzerland, 2017; Volume 960, pp. 469–487. ISBN 978-3-319-48380-1. [Google Scholar]
- Wang, L.; Chen, Y.; Li, X.; Zhang, Y.; Gulbins, E.; Zhang, Y. Enhancement of endothelial permeability by free fatty acid through lysosomal cathepsin B-mediated Nlrp3 inflammasome activation. Oncotarget 2016, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, F.; Chen, J.; Zhu, H. A potential strategy for treating atherosclerosis: improving endothelial function via AMP-activated protein kinase. Sci. China Life Sci. 2018, 61, 1024–1029. [Google Scholar] [CrossRef] [PubMed]
- Cacicedo, J.M.; Yagihashi, N.; Keaney, J.F.; Ruderman, N.B.; Ido, Y. AMPK inhibits fatty acid-induced increases in NF-κB transactivation in cultured human umbilical vein endothelial cells. Biochem. Biophys. Res. Commun. 2004, 324, 1204–1209. [Google Scholar] [CrossRef] [PubMed]
- Dinarello, C.A. Interleukin-1 in the pathogenesis and treatment of inflammatory diseases. Blood 2011, 117, 3720–3732. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dai, Y.; Dai, D.; Wang, X.; Ding, Z.; Mehta, J.L. DPP-4 Inhibitors Repress NLRP3 Inflammasome and Interleukin-1beta via GLP-1 Receptor in Macrophages Through Protein Kinase C Pathway. Cardiovasc. Drugs 2014, 28, 425–432. [Google Scholar] [CrossRef] [PubMed]
- Ridker, P.M.; Everett, B.M.; Thuren, T.; MacFadyen, J.G.; Chang, W.H.; Ballantyne, C.; Fonseca, F.; Nicolau, J.; Koenig, W.; Anker, S.D.; et al. Antiinflammatory Therapy with Canakinumab for Atherosclerotic Disease. N. Engl. J. Med. 2017, 377, 1119–1131. [Google Scholar] [CrossRef]
- Libby, P.; Ridker, P.M.; Hansson, G.K. Inflammation in Atherosclerosis. J. Am. Coll. Cardiol. 2009, 54, 2129–2138. [Google Scholar] [CrossRef] [Green Version]
- Choy, P.C.; Siow, Y.L.; Mymin, D. Lipids and atherosclerosis. Biochem. Cell Biol. 2004, 82, 212–224. [Google Scholar] [CrossRef]
- Rafieian-Kopaei, M.; Setorki, M.; Doudi, M.; Baradaran, A.; Nasri, H. Atherosclerosis: process, indicators, risk factors and new hopes. Int. J. Prev. Med. 2014, 5, 927–946. [Google Scholar]
- Sharma, G.; She, Z.-G.; Valenta, D.T.; Stallcup, W.B.; Smith, J.W. Scavenger Receptor-mediated Targeting of Macrophage Foam Cells In Atherosclerotic Plaque Using Oligonucleotide-functionalized Nanoparticles. Nano Life 2010, 1, 207–214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vincent, J. Lipid Lowering Therapy for Atherosclerotic Cardiovascular Disease: It Is Not So Simple. Clin. Pharm. 2018, 104, 220–224. [Google Scholar] [CrossRef]
- Bhatt, D.L.; Steg, P.G.; Miller, M.; Brinton, E.A.; Jacobson, T.A.; Ketchum, S.B.; Doyle, R.T.; Juliano, R.A.; Jiao, L.; Granowitz, C.; et al. Cardiovascular Risk Reduction with Icosapent Ethyl for Hypertriglyceridemia. N. Engl. J. Med. 2019, 380, 11–22. [Google Scholar] [CrossRef] [PubMed]
- Kersten, S. Physiological regulation of lipoprotein lipase. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2014, 1841, 919–933. [Google Scholar] [CrossRef] [PubMed]
- Minicocci, I.; Montali, A.; Robciuc, M.R.; Quagliarini, F.; Censi, V.; Labbadia, G.; Gabiati, C.; Pigna, G.; Sepe, M.L.; Pannozzo, F.; et al. Mutations in the ANGPTL3 Gene and Familial Combined Hypolipidemia: A Clinical and Biochemical Characterization. J. Clin. Endocrinol. Metab. 2012, 97, E1266–E1275. [Google Scholar] [CrossRef] [Green Version]
- Abu-Farha, M.; Al-Khairi, I.; Cherian, P.; Chandy, B.; Sriraman, D.; Alhubail, A.; Al-Refaei, F.; AlTerki, A.; Abubaker, J. Increased ANGPTL3, 4 and ANGPTL8/betatrophin expression levels in obesity and T2D. Lipids Health Dis. 2016, 15, 181. [Google Scholar] [CrossRef] [Green Version]
- Gusarova, V.; Alexa, C.A.; Wang, Y.; Rafique, A.; Kim, J.H.; Buckler, D.; Mintah, I.J.; Shihanian, L.M.; Cohen, J.C.; Hobbs, H.H.; et al. ANGPTL3 blockade with a human monoclonal antibody reduces plasma lipids in dyslipidemic mice and monkeys. J. Lipid Res. 2015, 56, 1308–1317. [Google Scholar] [CrossRef] [Green Version]
- Weisberg, I.S.; Park, E.; Ballman, K.V.; Berger, P.; Nunn, M.; Suh, D.S.; Breksa, A.P.; Garrow, T.A.; Rozen, R. Investigations of a common genetic variant in betaine–homocysteine methyltransferase (BHMT) in coronary artery disease. Atherosclerosis 2003, 167, 205–214. [Google Scholar] [CrossRef]
- Aviram, M.; Rosenblat, M.; Bisgaier, C.L.; Newton, R.S.; Primo-Parmo, S.L.; La Du, B.N. Paraoxonase inhibits high-density lipoprotein oxidation and preserves its functions. A possible peroxidative role for paraoxonase. J. Clin. Investig. 1998, 101, 1581–1590. [Google Scholar] [CrossRef] [Green Version]
- Mackness, B.; Durrington, P.; McElduff, P.; Yarnell, J.; Azam, N.; Watt, M.; Mackness, M. Low Paraoxonase Activity Predicts Coronary Events in the Caerphilly Prospective Study. Circulation 2003, 107, 2775–2779. [Google Scholar] [CrossRef] [Green Version]
- Tward, A.; Xia, Y.-R.; Wang, X.-P.; Shi, Y.-S.; Park, C.; Castellani, L.W.; Lusis, A.J.; Shih, D.M. Decreased Atherosclerotic Lesion Formation in Human Serum Paraoxonase Transgenic Mice. Circulation 2002, 106, 484–490. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shih, D.M.; Xia, Y.-R.; Wang, X.-P.; Miller, E.; Castellani, L.W.; Subbanagounder, G.; Cheroutre, H.; Faull, K.F.; Berliner, J.A.; Witztum, J.L.; et al. Combined Serum Paraoxonase Knockout/Apolipoprotein E Knockout Mice Exhibit Increased Lipoprotein Oxidation and Atherosclerosis. J. Biol. Chem. 2000, 275, 17527–17535. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.-Q.; Wan, H.-Q.; Wei, X.-J.; Zhang, Y.; Qu, P. CLI-095 decreases atherosclerosis by modulating foam cell formation in apolipoprotein E-deficient mice. Mol. Med. Rep. 2016, 14, 49–56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weber, C.; Noels, H. Atherosclerosis: current pathogenesis and therapeutic options. Nat Med. 2011, 17, 1410–1422. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Palucci, D.; Law, K.; Yanagawa, B.; Yam, J.; Butany, J. Atherosclerosis: pathogenesis and pathology. Diagn. Histopathol. 2012, 18, 461–467. [Google Scholar] [CrossRef]
- Raines, E.W. PDGF and cardiovascular disease. Cytokine Growth Factor Rev. 2004, 15, 237–254. [Google Scholar] [CrossRef]
- Johnson, J.L. Matrix metalloproteinases: influence on smooth muscle cells and atherosclerotic plaque stability. Expert Rev. Cardiovasc. Ther. 2007, 5, 265–282. [Google Scholar] [CrossRef]
- Linton, M.F.; Yancey, P.G.; Davies, S.S.; Jerome, W.G.; Linton, E.F.; Song, W.L.; Doran, A.C.; Vickers, K.C. The Role of Lipids and Lipoproteins in Atherosclerosis. In Endotext; Feingold, K.R., Anawalt, B., Boyce, A., Chrousos, G., Dungan, K., Grossman, A., Hershman, J.M., Kaltsas, G., Koch, C., Kopp, P., et al., Eds.; MDText.com, Inc.: South Dartmouth, MA, USA, 2000. [Google Scholar]
- Tao, H.; Yancey, P.G.; Babaev, V.R.; Blakemore, J.L.; Zhang, Y.; Ding, L.; Fazio, S.; Linton, M.F. Macrophage SR-BI mediates efferocytosis via Src/PI3K/Rac1 signaling and reduces atherosclerotic lesion necrosis. J. Lipid Res. 2015, 56, 1449–1460. [Google Scholar] [CrossRef] [Green Version]
- Khera, A.V.; Cuchel, M.; de la Llera-Moya, M.; Rodrigues, A.; Burke, M.F.; Jafri, K.; French, B.C.; Phillips, J.A.; Mucksavage, M.L.; Wilensky, R.L.; et al. Cholesterol Efflux Capacity, High-Density Lipoprotein Function, and Atherosclerosis. N. Engl. J. Med. 2011, 364, 127–135. [Google Scholar] [CrossRef] [Green Version]
- Tabas, I. Macrophage Apoptosis in Atherosclerosis: Consequences on Plaque Progression and the Role of Endoplasmic Reticulum Stress. Antioxid. Redox Signal. 2009, 11, 2333–2339. [Google Scholar] [CrossRef]
- Vaughan, D.E. PAI-1 and atherothrombosis. J. Thromb. Haemost. 2005, 3, 1879–1883. [Google Scholar] [CrossRef] [PubMed]
- Cesari, M.; Pahor, M.; Incalzi, R.A. REVIEW: Plasminogen Activator Inhibitor-1 (PAI-1): A Key Factor Linking Fibrinolysis and Age-Related Subclinical and Clinical Conditions: Plasminogen Activator Inhibitor-1 (PAI-1). Cardiovasc. Ther. 2010, 28, e72–e91. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mehta, J.; Lawson, D.; Saldeen, T. Reduction in plasminogen activator inhibitor-1 (PAI-1) with omega-3 polyunsaturated fatty acid (PUFA) intake. Am. Heart J. 1988, 116, 1201–1206. [Google Scholar] [CrossRef]
- Eitzman, D.T.; Westrick, R.J.; Xu, Z.; Tyson, J.; Ginsburg, D. Plasminogen activator inhibitor-1 deficiency protects against atherosclerosis progression in the mouse carotid artery. Blood 2000, 96, 4212–4215. [Google Scholar] [CrossRef]
- Jung, R.G.; Motazedian, P.; Ramirez, F.D.; Simard, T.; Di Santo, P.; Visintini, S.; Faraz, M.A.; Labinaz, A.; Jung, Y.; Hibbert, B. Association between plasminogen activator inhibitor-1 and cardiovascular events: A systematic review and meta-analysis. Thromb. J. 2018, 16, 12. [Google Scholar] [CrossRef] [Green Version]
- Bolívar, J.J. Essential Hypertension: An Approach to Its Etiology and Neurogenic Pathophysiology. Int. J. Hypertens. 2013, 2013, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Oparil, S.; Acelajado, M.C.; Bakris, G.L.; Berlowitz, D.R.; Cífková, R.; Dominiczak, A.F.; Grassi, G.; Jordan, J.; Poulter, N.R.; Rodgers, A.; et al. Hypertension. Nat. Rev. Dis. Primers 2018, 4, 18014. [Google Scholar] [CrossRef] [Green Version]
- Lastra, G.; Syed, S.; Kurukulasuriya, L.R.; Manrique, C.; Sowers, J.R. Type 2 Diabetes Mellitus and Hypertension. Endocrinol. Metab. Clin. North Am. 2014, 43, 103–122. [Google Scholar] [CrossRef] [Green Version]
- Drozdz, D.; Kawecka-Jaszcz, K. Cardiovascular changes during chronic hypertensive states. Pediatr. Nephrol. 2014, 29, 1507–1516. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Kuang, Z.-M.; Feng, S.-J.; Jiang, L.; Chen, Q.-X.; Ji, X.-Y.; Cheng, W.-L.; Hu, H.-J. Combined antihypertensive and statin therapy for the prevention of cardiovascular events in patients with hypertension without complications: protocol for a systematic review and meta-analysis. BMJ Open 2018, 8, e019719. [Google Scholar] [CrossRef] [Green Version]
- Ott, C.; Raff, U.; Schmidt, S.; Kistner, I.; Friedrich, S.; Bramlage, P.; Harazny, J.M.; Schmieder, R.E. Effects of saxagliptin on early microvascular changes in patients with type 2 diabetes. Cardiovasc. Diabetol. 2014, 13, 19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zografou, I.; Sampanis, C.; Gkaliagkousi, E.; Iliadis, F.; Papageorgiou, A.; Doukelis, P.; Vogiatzis, K.; Douma, S. Effect of vildagliptin on hsCRP and arterial stiffness in patients with type 2 diabetes mellitus. HJ 2014. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shirwany, N.A.; Zou, M. Arterial stiffness: A brief review. Acta Pharm. Sin. 2010, 31, 1267–1276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ecobici, M.; Voiculescu, M. Importance of arterial stiffness in predicting cardiovascular events. Rom. J. Intern. Med. 2017, 55, 8–13. [Google Scholar] [CrossRef] [Green Version]
- Favero, G.; Paganelli, C.; Buffoli, B.; Rodella, L.F.; Rezzani, R. Endothelium and Its Alterations in Cardiovascular Diseases: Life Style Intervention. BioMed Res. Int. 2014, 2014, 1–28. [Google Scholar] [CrossRef] [Green Version]
- Joyner, M.J.; Dietz, N.M. Nitric oxide and vasodilation in human limbs. J. Appl. Physiol. 1997, 83, 1785–1796. [Google Scholar] [CrossRef]
- Bonaventura, D.; Lunardi, C.N.; Rodrigues, G.J.; Neto, M.A.; Bendhack, L.M. A novel mechanism of vascular relaxation induced by sodium nitroprusside in the isolated rat aorta. Nitric Oxide 2008, 18, 287–295. [Google Scholar] [CrossRef]
- Collins, M.J.; Li, X.; Lv, W.; Yang, C.; Protack, C.D.; Muto, A.; Jadlowiec, C.C.; Shu, C.; Dardik, A. Therapeutic strategies to combat neointimal hyperplasia in vascular grafts. Expert Rev. Cardiovasc. Ther. 2012, 10, 635–647. [Google Scholar] [CrossRef]
- Pandey, A.K.; Singhi, E.K.; Arroyo, J.P.; Ikizler, T.A.; Gould, E.R.; Brown, J.; Beckman, J.A.; Harrison, D.G.; Moslehi, J. Mechanisms of VEGF (Vascular Endothelial Growth Factor) Inhibitor-Associated Hypertension and Vascular Disease. Hypertension 2018, 71, e1–e8. [Google Scholar] [CrossRef]
- Han, X.; Liu, L.; Niu, J.; Yang, J.; Zhang, Z.; Zhang, Z. Serum VEGF Predicts Worse Clinical Outcome of Patients with Coronary Heart Disease After Percutaneous Coronary Intervention Therapy. Med. Sci. Monit. 2015, 21, 3247–3251. [Google Scholar] [CrossRef] [Green Version]
- Kannel, W.B. Diabetes and cardiovascular disease. The Framingham study. JAMA J. Am. Med. Assoc. 1979, 241, 2035–2038. [Google Scholar] [CrossRef]
- Effect of intensive blood-glucose control with metformin on complications in overweight patients with type 2 diabetes (UKPDS 34). UK Prospective Diabetes Study (UKPDS) Group. Lancet 1998, 352, 854–865. [CrossRef]
- Zelniker, T.A.; Wiviott, S.D.; Raz, I.; Im, K.; Goodrich, E.L.; Bonaca, M.P.; Mosenzon, O.; Kato, E.T.; Cahn, A.; Furtado, R.H.M.; et al. SGLT2 inhibitors for primary and secondary prevention of cardiovascular and renal outcomes in type 2 diabetes: A systematic review and meta-analysis of cardiovascular outcome trials. Lancet 2019, 393, 31–39. [Google Scholar] [CrossRef]
- Bonora, B.M.; Avogaro, A.; Fadini, G.P. Extraglycemic Effects of SGLT2 Inhibitors: A Review of the Evidence. DMSO 2020, 13, 161–174. [Google Scholar] [CrossRef] [Green Version]
- Kristensen, S.L.; Rørth, R.; Jhund, P.S.; Docherty, K.F.; Sattar, N.; Preiss, D.; Køber, L.; Petrie, M.C.; McMurray, J.J.V. Cardiovascular, mortality, and kidney outcomes with GLP-1 receptor agonists in patients with type 2 diabetes: A systematic review and meta-analysis of cardiovascular outcome trials. Lancet Diabetes Endocrinol. 2019, 7, 776–785. [Google Scholar] [CrossRef]
- Standl, E. GLP-1 receptor agonists and cardiovascular outcomes: An updated synthesis. Lancet Diabetes Endocrinol. 2019, 7, 741–743. [Google Scholar] [CrossRef]
- Hirshberg, B.; Raz, I. Impact of the U.S. Food and Drug Administration Cardiovascular Assessment Requirements on the Development of Novel Antidiabetes Drugs. Diabetes Care 2011, 34, S101–S106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Green, J.B.; Bethel, M.A.; Armstrong, P.W.; Buse, J.B.; Engel, S.S.; Garg, J.; Josse, R.; Kaufman, K.D.; Koglin, J.; Korn, S.; et al. Effect of Sitagliptin on Cardiovascular Outcomes in Type 2 Diabetes. N. Engl. J. Med. 2015, 373, 232–242. [Google Scholar] [CrossRef] [Green Version]
- Scirica, B.M.; Bhatt, D.L.; Braunwald, E.; Steg, P.G.; Davidson, J.; Hirshberg, B.; Ohman, P.; Frederich, R.; Wiviott, S.D.; Hoffman, E.B.; et al. Saxagliptin and Cardiovascular Outcomes in Patients with Type 2 Diabetes Mellitus. N. Engl. J. Med. 2013, 369, 1317–1326. [Google Scholar] [CrossRef] [Green Version]
- Rosenstock, J.; Kahn, S.E.; Johansen, O.E.; Zinman, B.; Espeland, M.A.; Woerle, H.J.; Pfarr, E.; Keller, A.; Mattheus, M.; Baanstra, D.; et al. Effect of Linagliptin vs Glimepiride on Major Adverse Cardiovascular Outcomes in Patients With Type 2 Diabetes: The CAROLINA Randomized Clinical Trial. JAMA 2019, 322, 1155. [Google Scholar] [CrossRef] [Green Version]
- White, W.B.; Cannon, C.P.; Heller, S.R.; Nissen, S.E.; Bergenstal, R.M.; Bakris, G.L.; Perez, A.T.; Fleck, P.R.; Mehta, C.R.; Kupfer, S.; et al. Alogliptin after Acute Coronary Syndrome in Patients with Type 2 Diabetes. N. Engl. J. Med. 2013, 369, 1327–1335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosenstock, J.; Perkovic, V.; Johansen, O.E.; Cooper, M.E.; Kahn, S.E.; Marx, N.; Alexander, J.H.; Pencina, M.; Toto, R.D.; Wanner, C.; et al. Effect of Linagliptin vs Placebo on Major Cardiovascular Events in Adults With Type 2 Diabetes and High Cardiovascular and Renal Risk: The CARMELINA Randomized Clinical Trial. JAMA 2019, 321, 69. [Google Scholar] [CrossRef] [PubMed]
- Doggrell, S.A.; Dimmitt, S.B. Gliptins – do they increase cardiovascular risk or benefit? Expert Opin. Drug Saf. 2014, 13, 675–680. [Google Scholar] [CrossRef] [Green Version]
- McInnes, G.; Evans, M.; Del Prato, S.; Stumvoll, M.; Schweizer, A.; Lukashevich, V.; Shao, Q.; Kothny, W. Cardiovascular and heart failure safety profile of vildagliptin: A meta-analysis of 17 000 patients. Diabetes Obes. Metab. 2015, 17, 1085–1092. [Google Scholar] [CrossRef]
- Patil, H.R.; Al Badarin, F.J.; Al Shami, H.A.; Bhatti, S.K.; Lavie, C.J.; Bell, D.S.H.; O’Keefe, J.H. Meta-Analysis of Effect of Dipeptidyl Peptidase-4 Inhibitors on Cardiovascular Risk in Type 2 Diabetes Mellitus. Am. J. Cardiol. 2012, 110, 826–833. [Google Scholar] [CrossRef]
- Monami, M.; Ahrén, B.; Dicembrini, I.; Mannucci, E. Dipeptidyl peptidase-4 inhibitors and cardiovascular risk: A meta-analysis of randomized clinical trials. Diabetes Obes. Metab. 2013, 15, 112–120. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Authors | Subject of Study | Dose of Vildagliptin | Results |
|---|---|---|---|
| Lee et al. (2016) [13] | LPS stimulated RAW264.7 cells | varied at every stage of the experiment | ↓iNOS, ↓ NF-κB, ↓pJNK, ↓TLR-2, ↓TLR-4, |
| Dei et al. (2017) [14] | rBMVECs | 2.5–5 mg/day for 4–12 months | ↓ SDF-1α ↓ EPC |
| Zhang et al. (2018) [15] | HAECs | 5 and 10 μM for 24–72 h. | ↓ LDH, ↓ ROS, ↓TNF-α, ↓IL-8, ↓ICAM-1, ↓MCP-1, ↓TLR-4, ↓NF-κB |
| Qi et al. (2019) [16] | HUVECs | 2.5 and 5 μM for 24h | ↓ LDH, ↓ NAPHD, ↓AMPK, ↓IL-1β, ↓IL-18, ↑eNOS, ↑GSH |
| Liu et al. (2019) [17] | HUVECs and diabetic mice | varied at every stage of the experiment | ↓mtROS, ↑ATP, ↓Drp1 |
| Seo al. (2019) [18] | rabbit aortic rings | varied at every stage of the experiment | ↑VD |
| Oeseburg et al. (2010) [19] | HUVECs and diabetic fatty rats | varied at every stage of the experiment | ↑cAMP, ↑PKA, ↑CREB, ↑HO-1 |
| Terasaki et al. (2012 and 2013) [20,21] | Diabetic Apoe (–/–) mice | varied at every stage of the experiment | ↓MFCF |
| Khan et al. (2015) [22] | STZ-induced diabetic rats | 10 or 20 mg/kg/day for 3 weeks | ↓ TLC, ↓ TGL,↓ CRP ↓TNF-α, ↑aPTT, ↑NO |
| Jain et al. (2015) [23] | diabetic rats | varied at every stage of the experiment | ↑NO, ↑EDR, ↓ROS, ↓MPO, ↑GSH |
| Koyama et al. (2016) [24] | rabbits | 10 mg/kg/day for 5 weeks | ↑eNOS, ↑VGIH |
| Zhang et al. (2018) [25] | diabetic rats | 10 or 20 mg/kg/day for 12 weeks | ↓ TCL, ↓ ED, ↓Angptl3, ↓Bhmt,↓ Pon1 |
| Ji et al. (2019) [26] | diabetic mice | 35 mg/kg/day for 4 weeks | ↓ ERS, ↓ NF-κB, |
| van Poppel et al. (2011) [27] | DM2 patients | 50 mg /day for 4 weeks | ↑EDR |
| Noguchi et al. (2015) [28] | normoglycemic patients | 50 mg once | ↓ TGL, ↓EDs |
| Tani et al. (2015) [29] | DM2 patients | 50 mg/day for 8 weeks | ↓ PAI-1 |
| Evans et al. (2016) [30] | DM2 patients | 50 mg once or twice/day for 24 weeks | ↓SBP, ↓ DBP, ↓TGL, ↓VLDL,↓LDL, ↑HDL |
| Duvnjak et al. (2016) [31] | DM2 patients | 100 mg/day for 12 weeks | ↓TLC,↓LDL, ↓hsCRP, ↓AS, ↓CBP |
| Park et al. (2017) [32] | DM2 patients | 1 mg/twice a day for 12 weeks | ↓SDF-1α |
| Younis et al. (2017) [33] | patients with DM2 and CAD | Metformin + vildagliptin 25 or 50 mg/day | ↓ IL-1β, ↓hsCRP |
| El-Naggar et al. (2019) [34] | DM2 patients with hypertension | 50 mg/twice a day + 25 mg/day captopril for 24 weeks | ↓BP,↓VEGF |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wiciński, M.; Górski, K.; Wódkiewicz, E.; Walczak, M.; Nowaczewska, M.; Malinowski, B. Vasculoprotective Effects of Vildagliptin. Focus on Atherogenesis. Int. J. Mol. Sci. 2020, 21, 2275. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21072275
Wiciński M, Górski K, Wódkiewicz E, Walczak M, Nowaczewska M, Malinowski B. Vasculoprotective Effects of Vildagliptin. Focus on Atherogenesis. International Journal of Molecular Sciences. 2020; 21(7):2275. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21072275
Chicago/Turabian StyleWiciński, Michał, Karol Górski, Eryk Wódkiewicz, Maciej Walczak, Magdalena Nowaczewska, and Bartosz Malinowski. 2020. "Vasculoprotective Effects of Vildagliptin. Focus on Atherogenesis" International Journal of Molecular Sciences 21, no. 7: 2275. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21072275