Disorders of Sex Development—Novel Regulators, Impacts on Fertility, and Options for Fertility Preservation


Abstract
:1. Gonadal and Germ Cell Development
2. Disorders of Sex Development
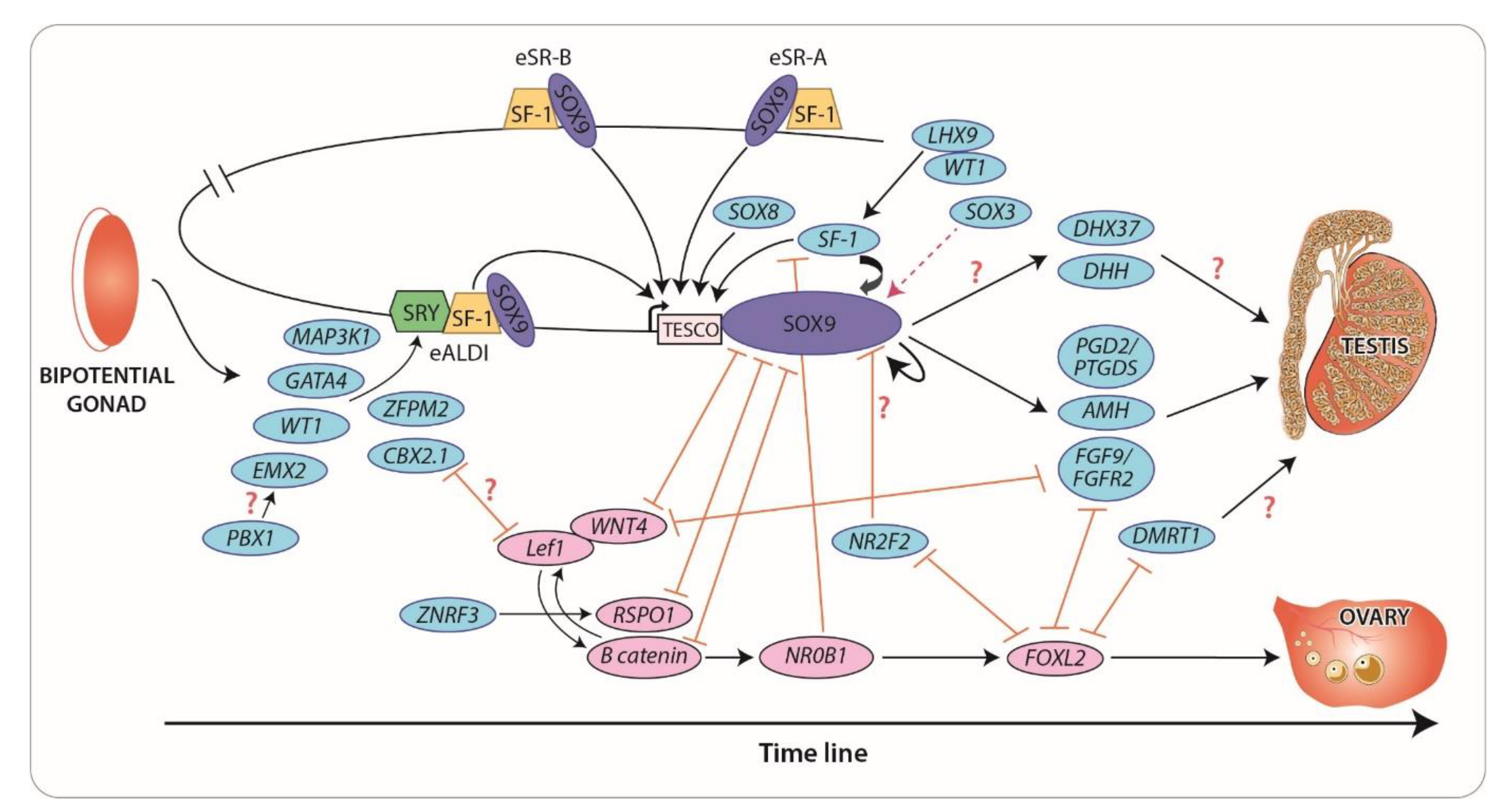
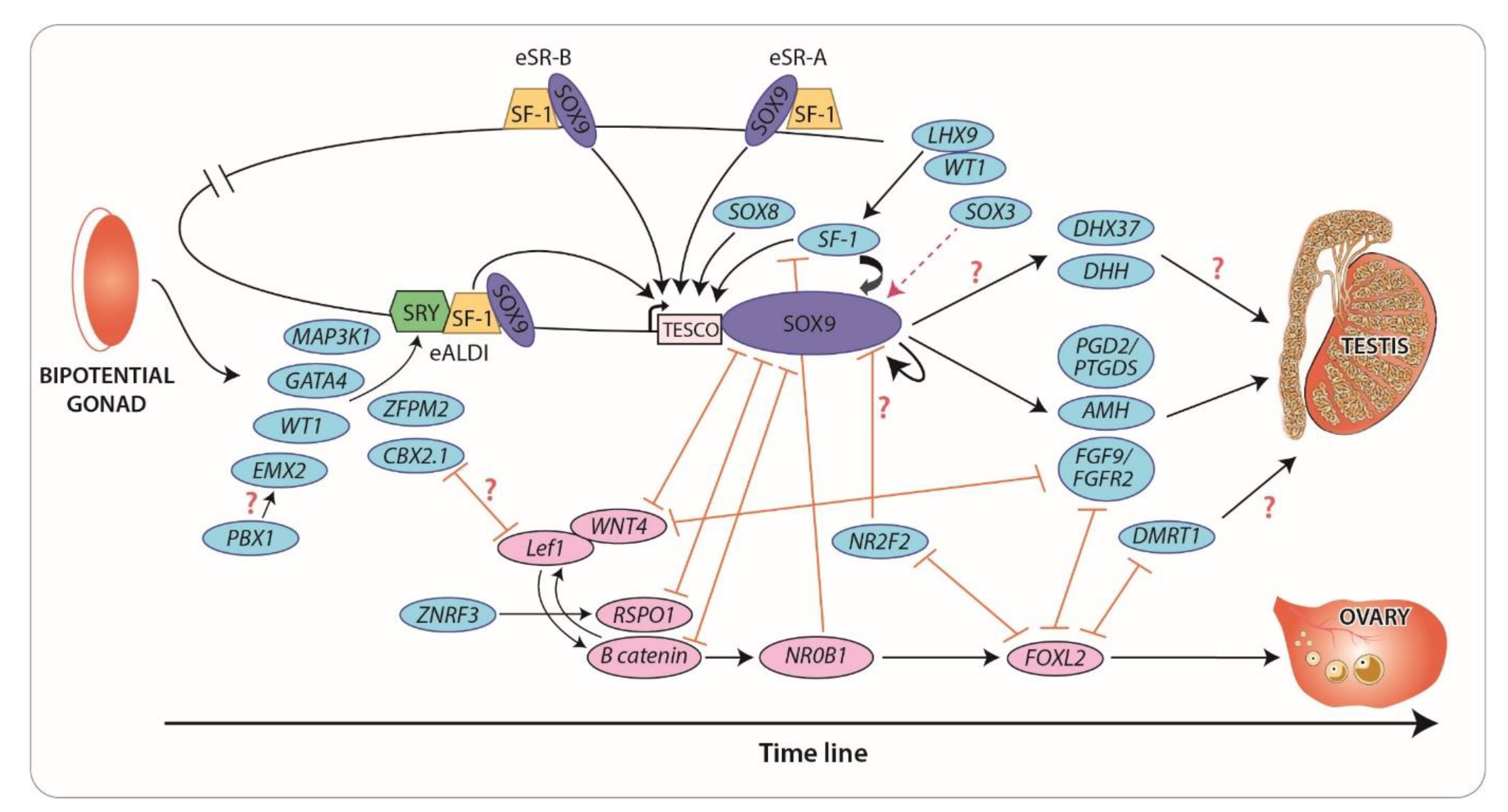
3. Novel Regulators and Mechanisms of Gonadal Development
3.1. The SRY/SOX Family: New Concepts in Gene Dosage and Regulation
3.2. NR5A1 (SF1) and WT1: Old Genes, New Mechanisms
3.3. PBX1 and CBX2: Gene Interactions that Promote Testis Development
3.4. NR2F2: A “Pro-Ovary and Anti-Testis” Gene
3.5. FGF and WNT Signaling: Antagonistic Pathways in Gonadal Sex Differentiation
4. Maintenance of Sex-Specific Somatic Cell Lineages
4.1. FOXL2: Maintenance of Female Fate
4.2. DMRT1: Maintenance of Male Fate
4.3. DHX37: A Novel Participant of Gonadal Development and Maintenance
5. Impact of DSD on Fertility and Options for Fertility Preservation
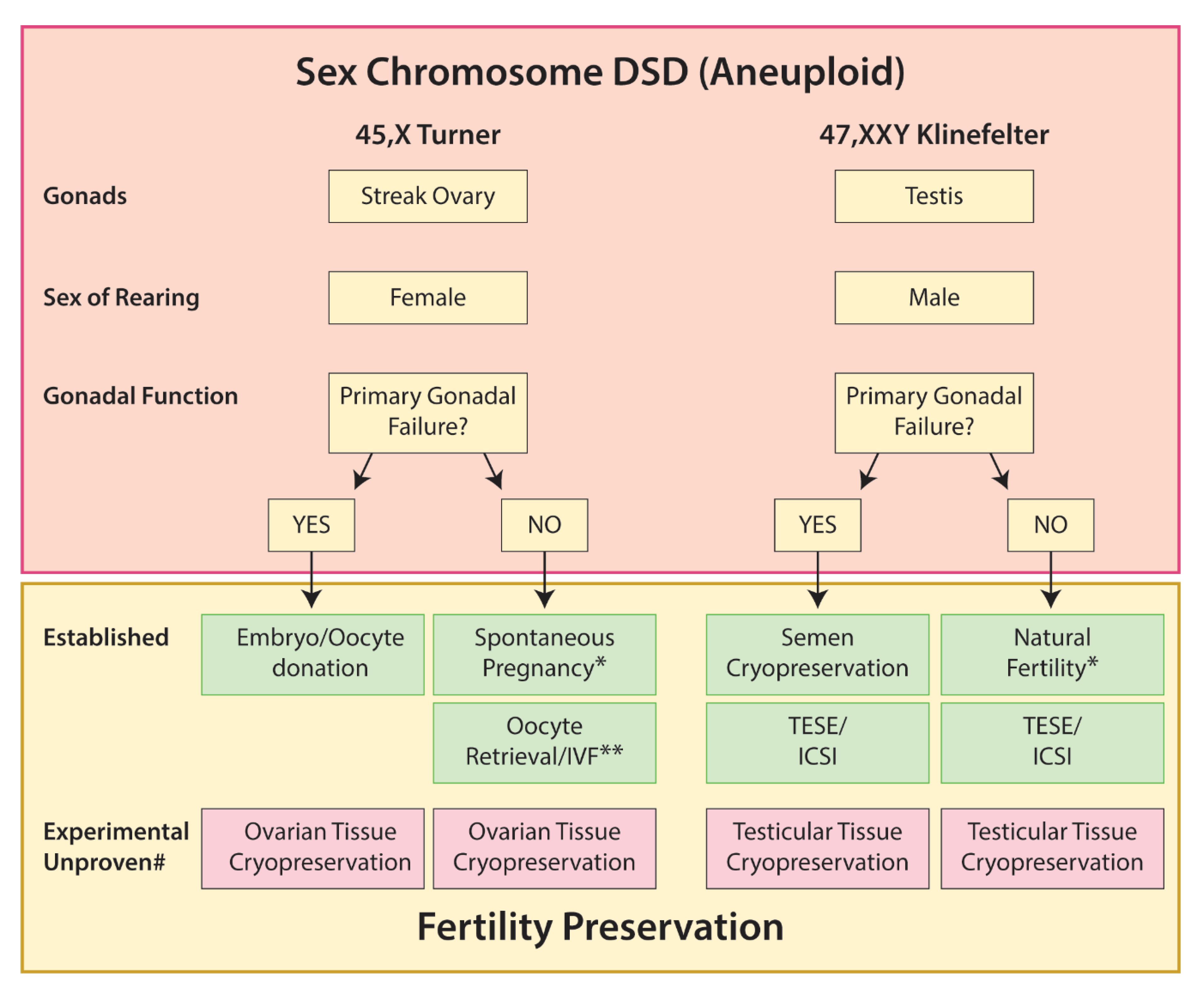
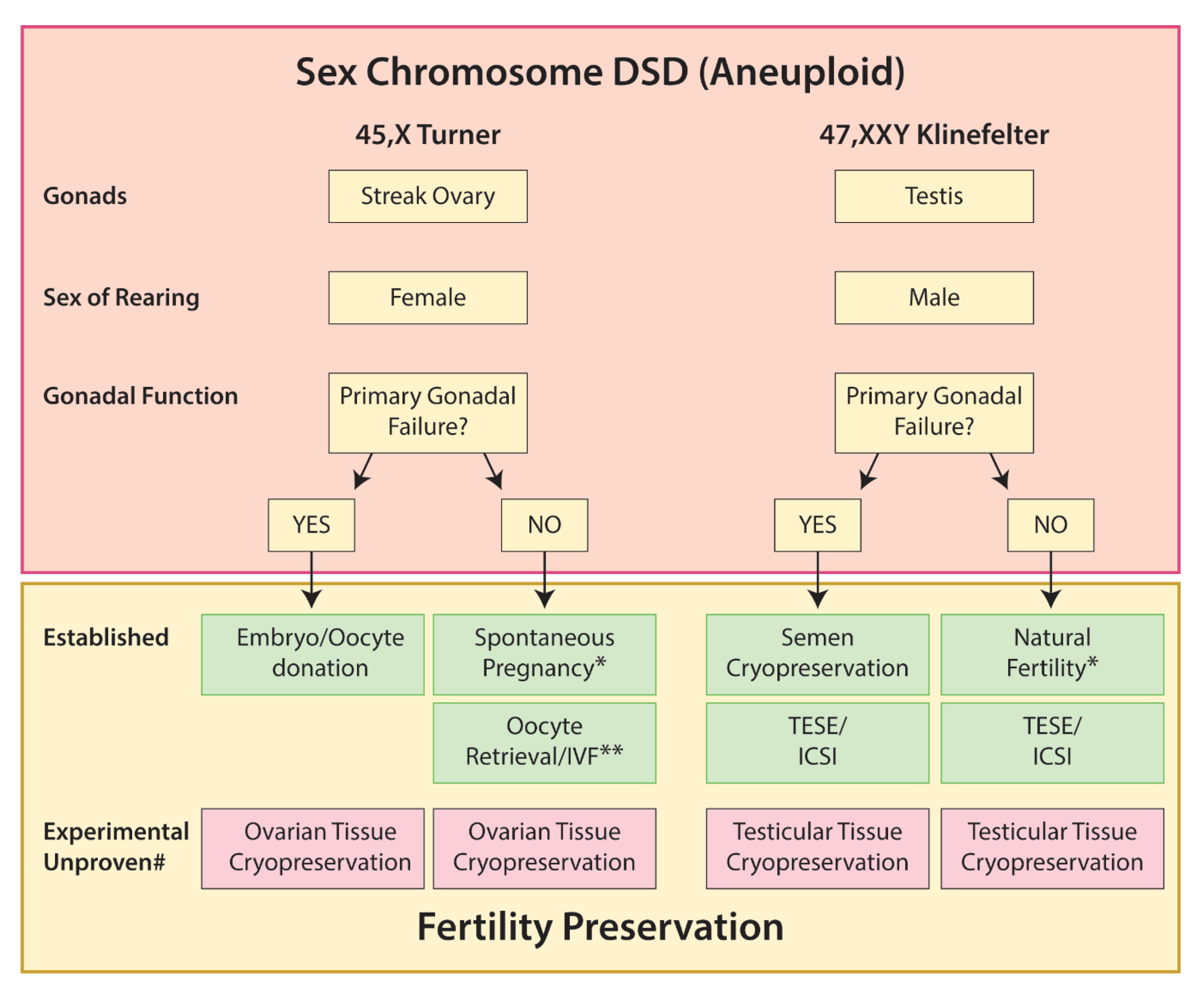
5.1. Sex Chromosome DSD
5.1.1. Turner Syndrome
5.1.2. Klinefelter Syndrome
5.1.3. 45,X/46,XY
5.1.4. 45,XX/46,XY
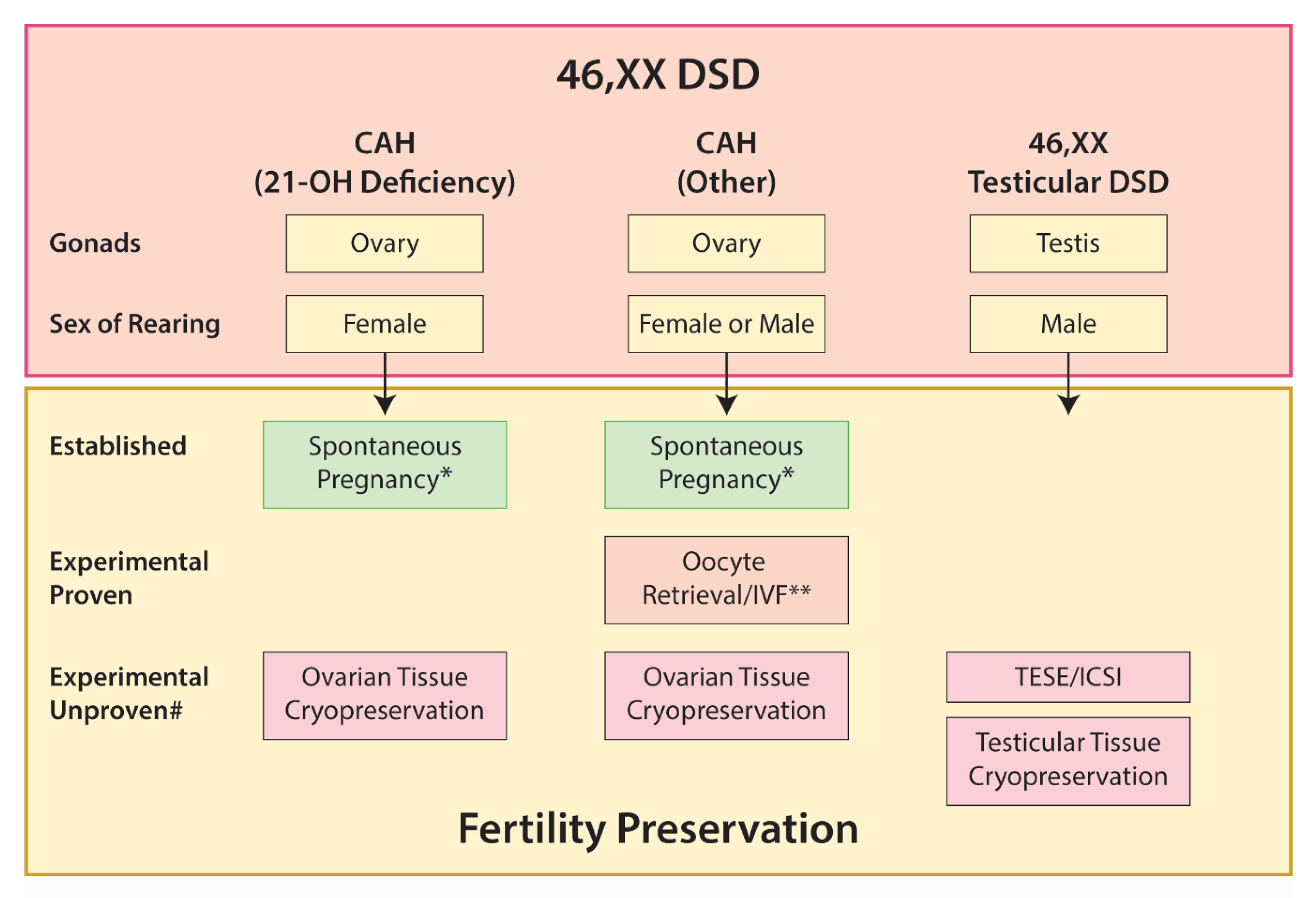
5.2. 46,XX DSD
5.2.1. Disorders of Androgen Production or Action
5.2.2. 46,XX Testicular DSD
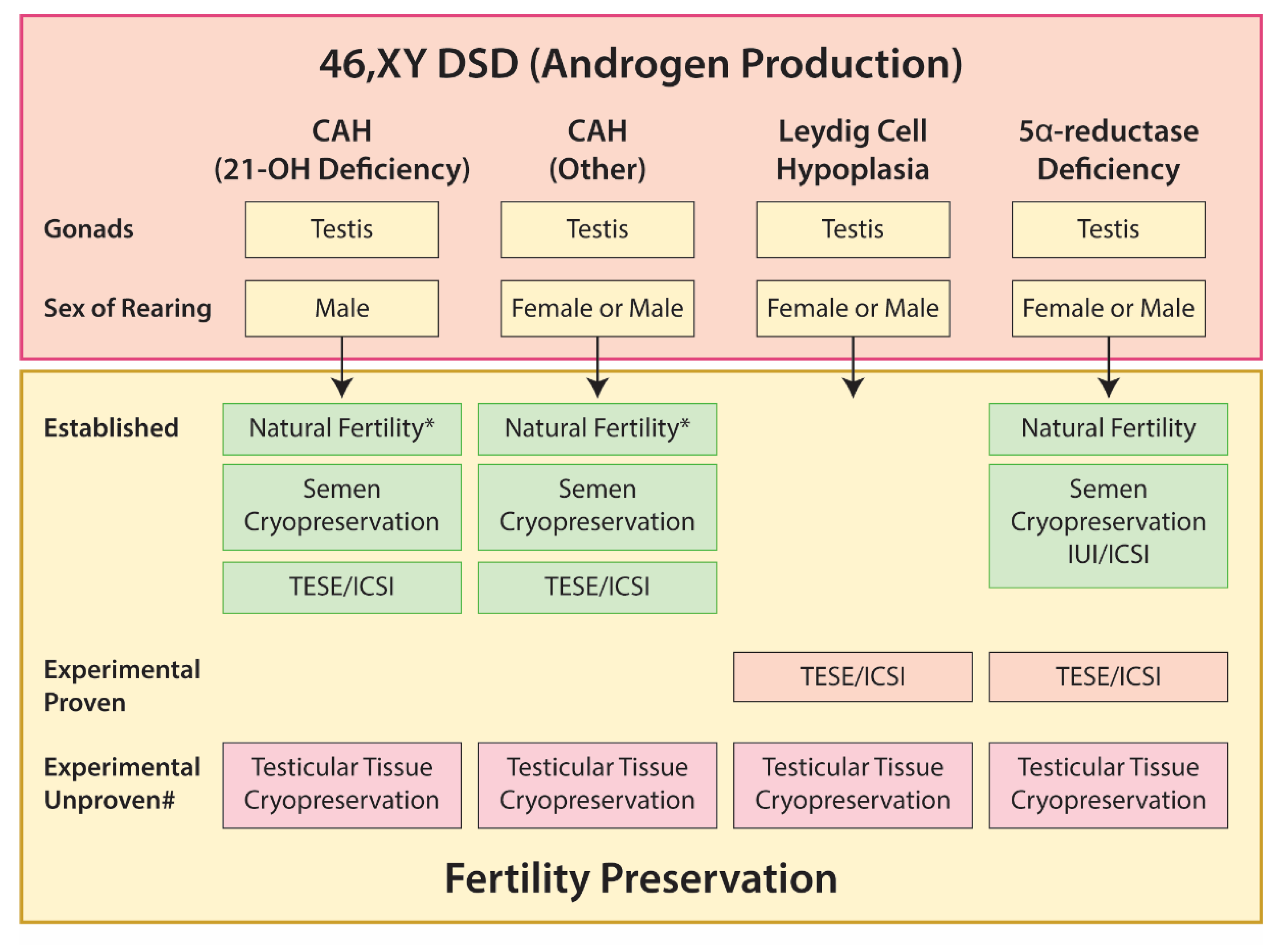
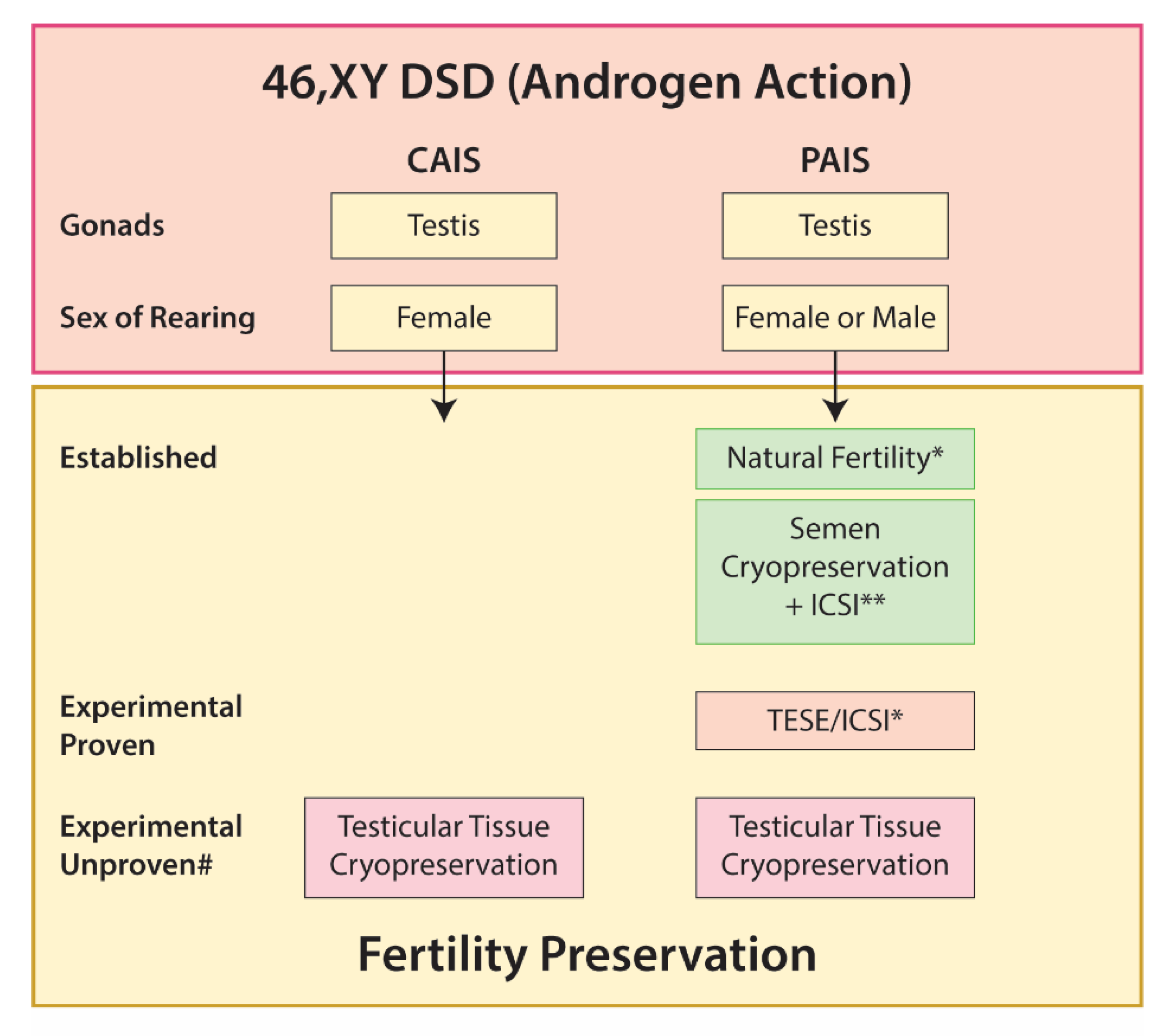
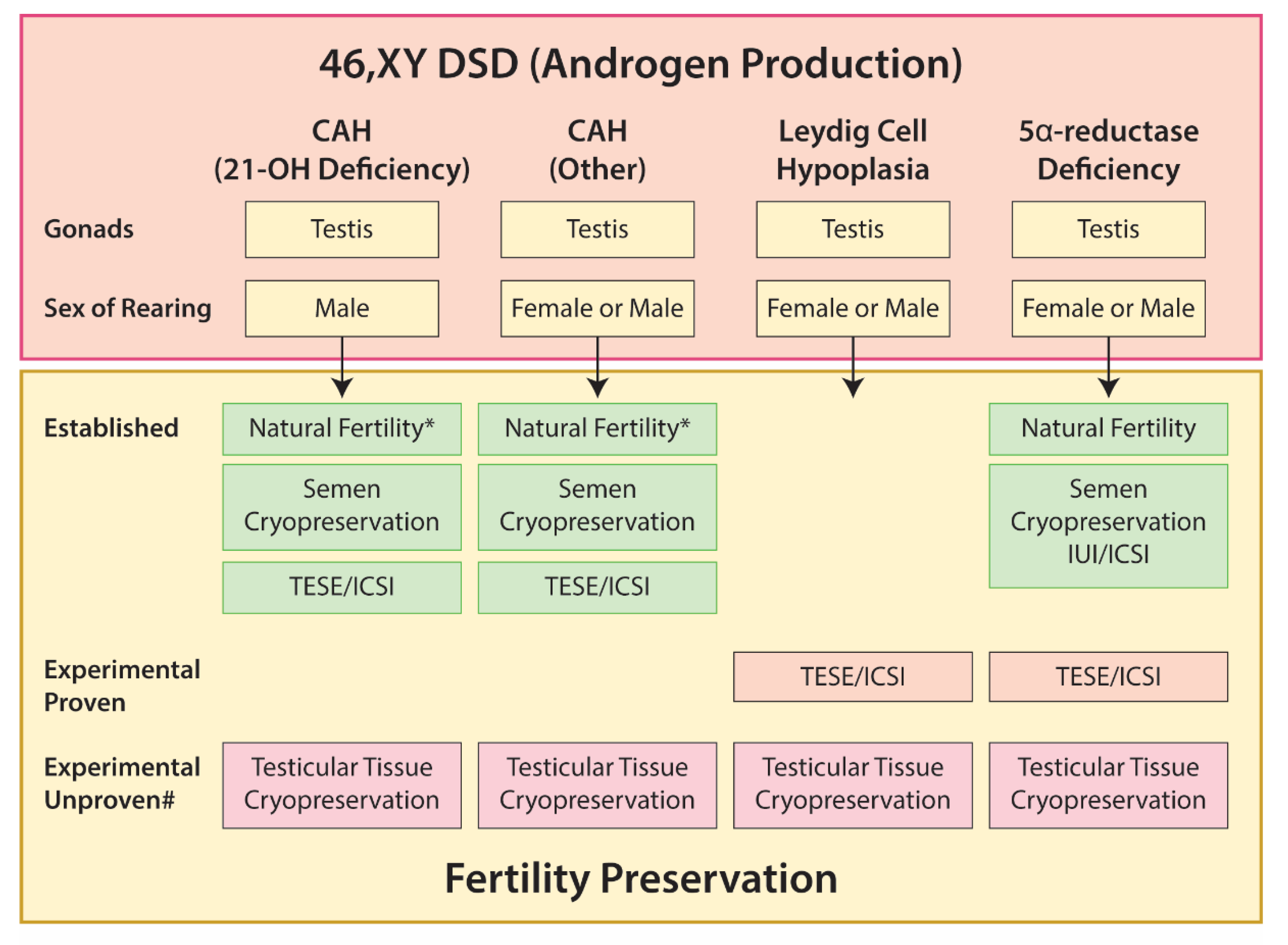
5.3. 46,XY DSD
5.3.1. Gonadal Dysgenesis
5.3.2. Disorders of Androgen Production
5.3.3. Disorders of Androgen Action
Androgen Insensitivity Syndrome
5.4. Ovotesticular DSD
6. Fertility Preservation in Prepubertal Individuals with DSD—Ovarian or Testicular Tissue Cryopreservation
7. Conclusions
Funding
Acknowledgments
Conflicts of Interest
References
- Bashamboo, A.; Donohoue, P.A.; Vilain, E.; Rojo, S.; Calvel, P.; Seneviratne, S.N.; Buonocore, F.; Barseghyan, H.; Bingham, N.; Rosenfeld, J.A.; et al. A recurrent p.Arg92Trp variant in steroidogenic factor-1 (NR5A1) can act as a molecular switch in human sex development. Hum. Mol. Genet. 2016, 25, 3446–3453. [Google Scholar] [CrossRef] [Green Version]
- Morohashi, K. The ontogenesis of the steroidogenic tissues. Genes Cells Devoted Mol. Cell. Mech. 1997, 2, 95–106. [Google Scholar] [CrossRef] [PubMed]
- Hannema, S.E.; Hughes, I.A. Regulation of Wolffian duct development. Horm. Res. 2007, 67, 142–151. [Google Scholar] [CrossRef] [PubMed]
- Acien, P. Embryological observations on the female genital tract. Hum. Reprod. 1992, 7, 437–445. [Google Scholar] [CrossRef] [PubMed]
- Wilhelm, D.; Palmer, S.; Koopman, P. Sex determination and gonadal development in mammals. Physiol. Rev. 2007, 87, 1–28. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Workman, S.; Wilson, M. The molecular pathways underlying early gonadal development. J. Mol. Endocrinol. 2018, 62, R47–R64. [Google Scholar] [CrossRef] [Green Version]
- Koopman, P.; Münsterberg, A.; Capel, B.; Vivian, N.; Lovell-Badge, R. Expression of a candidate sex-determining gene during mouse testis differentiation. Nature 1990, 348, 450–452. [Google Scholar] [CrossRef]
- Sekido, R.; Lovell-Badge, R. Sex determination and SRY: Down to a wink and a nudge? Trends Genet. TIG 2009, 25, 19–29. [Google Scholar] [CrossRef]
- Bashamboo, A.; Eozenou, C.; Rojo, S.; McElreavey, K. Anomalies in human sex determination provide unique insights into the complex genetic interactions of early gonad development. Clin. Genet. 2017, 91, 143–156. [Google Scholar] [CrossRef] [Green Version]
- Lindeman, R.E.; Gearhart, M.D.; Minkina, A.; Krentz, A.D.; Bardwell, V.J.; Zarkower, D. Sexual cell-fate reprogramming in the ovary by DMRT1. Curr. Biol. CB 2015, 25, 764–771. [Google Scholar] [CrossRef] [Green Version]
- Clarke, T.R.; Hoshiya, Y.; Yi, S.E.; Liu, X.; Lyons, K.M.; Donahoe, P.K. Mullerian inhibiting substance signaling uses a bone morphogenetic protein (BMP)-like pathway mediated by ALK2 and induces SMAD6 expression. Mol. Endocrinol. 2001, 15, 946–959. [Google Scholar] [CrossRef] [PubMed]
- Barsoum, I.B.; Bingham, N.C.; Parker, K.L.; Jorgensen, J.S.; Yao, H.H. Activation of the Hedgehog pathway in the mouse fetal ovary leads to ectopic appearance of fetal Leydig cells and female pseudohermaphroditism. Dev. Biol. 2009, 329, 96–103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahmed, S.F.; Bashamboo, A.; Lucas-Herald, A.; McElreavey, K. Understanding the genetic aetiology in patients with XY DSD. Br. Med. Bull. 2013, 106, 67–89. [Google Scholar] [CrossRef] [PubMed]
- Auchus, R.J. The backdoor pathway to dihydrotestosterone. Trends Endocrinol. Metab. TEM 2004, 15, 432–438. [Google Scholar] [CrossRef]
- Culty, M. Gonocytes, the forgotten cells of the germ cell lineage. Birth Defects Res. C Embryo Today 2009, 87, 1–26. [Google Scholar] [CrossRef]
- Mitchell, R.T.; Cowan, G.; Morris, K.D.; Anderson, R.A.; Fraser, H.M.; McKenzie, K.J.; Wallace, W.H.; Kelnar, C.J.; Saunders, P.T.; Sharpe, R.M. Germ cell differentiation in the marmoset (Callithrix jacchus) during fetal and neonatal life closely parallels that in the human. Hum. Reprod. 2008, 23, 2755–2765. [Google Scholar] [CrossRef] [Green Version]
- Skakkebaek, N.E.; Berthelsen, J.G.; Giwercman, A.; Muller, J. Carcinoma-in-situ of the testis: Possible origin from gonocytes and precursor of all types of germ cell tumours except spermatocytoma. Int. J. Androl. 1987, 10, 19–28. [Google Scholar] [CrossRef]
- Rabinovici, J.; Jaffe, R.B. Development and regulation of growth and differentiated function in human and subhuman primate fetal gonads. Endocr. Rev. 1990, 11, 532–557. [Google Scholar] [CrossRef]
- Hughes, I.A.; Houk, C.; Ahmed, S.F.; Lee, P.A. Consensus statement on management of intersex disorders. J. Pediatr. Urol. 2006, 2, 148–162. [Google Scholar] [CrossRef]
- Deebel, N.A.; Galdon, G.; Zarandi, N.P.; Stogner-Underwood, K.; Howards, S.; Lovato, J.; Kogan, S.; Atala, A.; Lue, Y.; Sadri-Ardekani, H. Age-related presence of spermatogonia in patients with Klinefelter syndrome: A systematic review and meta-analysis. Hum. Reprod. Update 2020, 26, 58–72. [Google Scholar] [CrossRef]
- James, P.A.; Rose, K.; Francis, D.; Norris, F. High-level 46XX/46XY chimerism without clinical effect in a healthy multiparous female. Am. J. Med. Genet. A 2011, 155A, 2484–2488. [Google Scholar] [CrossRef] [PubMed]
- Looijenga, L.H.; Hersmus, R.; Oosterhuis, J.W.; Cools, M.; Drop, S.L.; Wolffenbuttel, K.P. Tumor risk in disorders of sex development (DSD). Best Pract. Res. Clin. Endocrinol. Metab. 2007, 21, 480–495. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- King, T.F.; Conway, G.S. Swyer syndrome. Curr. Opin. Endocrinol. Diabetes Obes. 2014, 21, 504–510. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Claahsen-van der Grinten, H.L.; Stikkelbroeck, N.M.; Sweep, C.G.; Hermus, A.R.; Otten, B.J. Fertility in patients with congenital adrenal hyperplasia. J. Pediatr. Endocrinol. Metab. 2006, 19, 677–685. [Google Scholar] [CrossRef]
- Miller, W.L.; Auchus, R.J. The molecular biology, biochemistry, and physiology of human steroidogenesis and its disorders. Endocr. Rev. 2011, 32, 81–151. [Google Scholar] [CrossRef] [Green Version]
- Kang, H.J.; Imperato-McGinley, J.; Zhu, Y.S.; Cai, L.Q.; Schlegel, P.; Palermo, G.; Rosenwaks, Z. The first successful paternity through in vitro fertilization-intracytoplasmic sperm injection with a man homozygous for the 5alpha-reductase-2 gene mutation. Fertil. Steril. 2011, 95, e2125–e2128. [Google Scholar] [CrossRef]
- Hughes, I.A.; Davies, J.D.; Bunch, T.I.; Pasterski, V.; Mastroyannopoulou, K.; MacDougall, J. Androgen insensitivity syndrome. Lancet (Lond. Engl.) 2012, 380, 1419–1428. [Google Scholar] [CrossRef] [Green Version]
- Hannema, S.E.; Scott, I.S.; Rajpert-De Meyts, E.; Skakkebaek, N.E.; Coleman, N.; Hughes, I.A. Testicular development in the complete androgen insensitivity syndrome. J. Pathol. 2006, 208, 518–527. [Google Scholar] [CrossRef]
- Vorona, E.; Zitzmann, M.; Gromoll, J.; Schuring, A.N.; Nieschlag, E. Clinical, endocrinological, and epigenetic features of the 46,XX male syndrome, compared with 47,XXY Klinefelter patients. J. Clin. Endocrinol. Metab. 2007, 92, 3458–3465. [Google Scholar] [CrossRef] [Green Version]
- Hacker, A.; Capel, B.; Goodfellow, P.; Lovell-Badge, R. Expression of Sry, the mouse sex determining gene. Development 1995, 121, 1603–1614. [Google Scholar]
- Morais da Silva, S.; Hacker, A.; Harley, V.; Goodfellow, P.; Swain, A.; Lovell-Badge, R. Sox9 expression during gonadal development implies a conserved role for the gene in testis differentiation in mammals and birds. Nat. Genet. 1996, 14, 62–68. [Google Scholar] [CrossRef]
- Hanley, N.A.; Hagan, D.M.; Clement-Jones, M.; Ball, S.G.; Strachan, T.; Salas-Cortes, L.; McElreavey, K.; Lindsay, S.; Robson, S.; Bullen, P.; et al. SRY, SOX9, and DAX1 expression patterns during human sex determination and gonadal development. Mech. Dev. 2000, 91, 403–407. [Google Scholar] [CrossRef]
- Sekido, R.; Lovell-Badge, R. Sex determination involves synergistic action of SRY and SF1 on a specific Sox9 enhancer. Nature 2008, 453, 930–934. [Google Scholar] [CrossRef]
- Gonen, N.; Quinn, A.; O’Neill, H.C.; Koopman, P.; Lovell-Badge, R. Normal Levels of Sox9 Expression in the Developing Mouse Testis Depend on the TES/TESCO Enhancer, but This Does Not Act Alone. PLoS Genet. 2017, 13, e1006520. [Google Scholar] [CrossRef] [Green Version]
- Georg, I.; Bagheri-Fam, S.; Knower, K.C.; Wieacker, P.; Scherer, G.; Harley, V.R. Mutations of the SRY-responsive enhancer of SOX9 are uncommon in XY gonadal dysgenesis. Sex. Dev. 2010, 4, 321–325. [Google Scholar] [CrossRef] [PubMed]
- Gonen, N.; Futtner, C.R.; Wood, S.; Garcia-Moreno, S.A.; Salamone, I.M.; Samson, S.C.; Sekido, R.; Poulat, F.; Maatouk, D.M.; Lovell-Badge, R. Sex reversal following deletion of a single distal enhancer of Sox9. Science 2018, 360, 1469–1473. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Croft, B.; Ohnesorg, T.; Sinclair, A.H. The Role of Copy Number Variants in Disorders of Sex Development. Sex. Dev. 2018, 12, 19–29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Croft, B.; Ohnesorg, T.; Hewitt, J.; Bowles, J.; Quinn, A.; Tan, J.; Corbin, V.; Pelosi, E.; van den Bergen, J.; Sreenivasan, R.; et al. Human sex reversal is caused by duplication or deletion of core enhancers upstream of SOX9. Nat. Commun. 2018, 9, 5319. [Google Scholar] [CrossRef] [Green Version]
- Weiss, J.; Meeks, J.J.; Hurley, L.; Raverot, G.; Frassetto, A.; Jameson, J.L. Sox3 is required for gonadal function, but not sex determination, in males and females. Mol. Cell. Biol. 2003, 23, 8084–8091. [Google Scholar] [CrossRef] [Green Version]
- Rizzoti, K.; Brunelli, S.; Carmignac, D.; Thomas, P.Q.; Robinson, I.C.; Lovell-Badge, R. SOX3 is required during the formation of the hypothalamo-pituitary axis. Nat. Genet. 2004, 36, 247–255. [Google Scholar] [CrossRef]
- Haines, B.; Hughes, J.; Corbett, M.; Shaw, M.; Innes, J.; Patel, L.; Gecz, J.; Clayton-Smith, J.; Thomas, P. Interchromosomal insertional translocation at Xq26.3 alters SOX3 expression in an individual with XX male sex reversal. J. Clin. Endocrinol. Metab. 2015, 100, E815–E820. [Google Scholar] [CrossRef] [Green Version]
- Sutton, E.; Hughes, J.; White, S.; Sekido, R.; Tan, J.; Arboleda, V.; Rogers, N.; Knower, K.; Rowley, L.; Eyre, H.; et al. Identification of SOX3 as an XX male sex reversal gene in mice and humans. J. Clin. Investig. 2011, 121, 328–341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chaboissier, M.C.; Kobayashi, A.; Vidal, V.I.; Lützkendorf, S.; van de Kant, H.J.; Wegner, M.; de Rooij, D.G.; Behringer, R.R.; Schedl, A. Functional analysis of Sox8 and Sox9 during sex determination in the mouse. Development 2004, 131, 1891–1901. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barrionuevo, F.; Georg, I.; Scherthan, H.; Lécureuil, C.; Guillou, F.; Wegner, M.; Scherer, G. Testis cord differentiation after the sex determination stage is independent of Sox9 but fails in the combined absence of Sox9 and Sox8. Dev. Biol. 2009, 327, 301–312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Portnoi, M.F.; Dumargne, M.C.; Rojo, S.; Witchel, S.F.; Duncan, A.J.; Eozenou, C.; Bignon-Topalovic, J.; Yatsenko, S.A.; Rajkovic, A.; Reyes-Mugica, M.; et al. Mutations involving the SRY-related gene SOX8 are associated with a spectrum of human reproductive anomalies. Hum. Mol. Genet. 2018, 27, 1228–1240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guran, T.; Yesil, G.; Turan, S.; Atay, Z.; Bozkurtlar, E.; Aghayev, A.; Gul, S.; Tinay, I.; Aru, B.; Arslan, S.; et al. PPP2R3C gene variants cause syndromic 46,XY gonadal dysgenesis and impaired spermatogenesis in humans. Eur. J. Endocrinol. 2019, 180, 291–309. [Google Scholar] [CrossRef]
- van den Ham, R.; van Dissel-Emiliani, F.M.; van Pelt, A.M. Expression of the scaffolding subunit A of protein phosphatase 2A during rat testicular development. Biol. Reprod. 2003, 68, 1369–1375. [Google Scholar] [CrossRef] [Green Version]
- de Boer, P.; Giele, M.; Lock, M.T.; de Rooij, D.G.; Giltay, J.; Hochstenbach, R.; te Velde, E.R. Kinetics of meiosis in azoospermic males: A joint histological and cytological approach. Cytogenet. Genome Res. 2004, 105, 36–46. [Google Scholar] [CrossRef] [Green Version]
- Dudiki, T.; Kadunganattil, S.; Ferrara, J.K.; Kline, D.W.; Vijayaraghavan, S. Changes in Carboxy Methylation and Tyrosine Phosphorylation of Protein Phosphatase PP2A Are Associated with Epididymal Sperm Maturation and Motility. PLoS ONE 2015, 10, e0141961. [Google Scholar] [CrossRef] [Green Version]
- Achermann, J.C.; Ito, M.; Hindmarsh, P.C.; Jameson, J.L. A mutation in the gene encoding steroidogenic factor-1 causes XY sex reversal and adrenal failure in humans. Nat. Genet. 1999, 22, 125–126. [Google Scholar] [CrossRef]
- Achermann, J.C.; Ozisik, G.; Ito, M.; Orun, U.A.; Harmanci, K.; Gurakan, B.; Jameson, J.L. Gonadal determination and adrenal development are regulated by the orphan nuclear receptor steroidogenic factor-1, in a dose-dependent manner. J. Clin. Endocrinol. Metab. 2002, 87, 1829–1833. [Google Scholar] [CrossRef] [PubMed]
- Correa, R.V.; Domenice, S.; Bingham, N.C.; Billerbeck, A.E.; Rainey, W.E.; Parker, K.L.; Mendonca, B.B. A microdeletion in the ligand binding domain of human steroidogenic factor 1 causes XY sex reversal without adrenal insufficiency. J. Clin. Endocrinol. Metab. 2004, 89, 1767–1772. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferraz-de-Souza, B.; Lin, L.; Achermann, J.C. Steroidogenic factor-1 (SF-1, NR5A1) and human disease. Mol. Cell. Endocrinol. 2011, 336, 198–205. [Google Scholar] [CrossRef] [PubMed]
- Domenice, S.; Zamboni Machado, A.; Moraes Ferreira, F.; Ferraz-de-Souza, B.; Marcondes Lerario, A.; Lin, L.; Yumie Nishi, M.; Lisboa Gomes, N.; Evelin da Silva, T.; Barbosa Silva, R.; et al. Wide spectrum of NR5A1-related phenotypes in 46,XY and 46,XX individuals. Birth Defects Res. C Embryo Today 2016, 108, 309–320. [Google Scholar] [CrossRef]
- Eggers, S.; Sadedin, S.; van den Bergen, J.A.; Robevska, G.; Ohnesorg, T.; Hewitt, J.; Lambeth, L.; Bouty, A.; Knarston, I.M.; Tan, T.Y.; et al. Disorders of sex development: Insights from targeted gene sequencing of a large international patient cohort. Genome Biol. 2016, 17, 243. [Google Scholar] [CrossRef] [Green Version]
- Ferlin, A.; Rocca, M.S.; Vinanzi, C.; Ghezzi, M.; Di Nisio, A.; Foresta, C. Mutational screening of NR5A1 gene encoding steroidogenic factor 1 in cryptorchidism and male factor infertility and functional analysis of seven undescribed mutations. Fertil. Steril. 2015, 104, 163–169 e161. [Google Scholar] [CrossRef]
- Fabbri-Scallet, H.; de Sousa, L.M.; Maciel-Guerra, A.T.; Guerra-Júnior, G.; de Mello, M.P. Mutation update for the NR5A1 gene involved in DSD and infertility. Hum. Mutat. 2020, 41, 58–68. [Google Scholar] [CrossRef]
- Sreenivasan, R.; Ludbrook, L.; Fisher, B.; Declosmenil, F.; Knower, K.C.; Croft, B.; Bird, A.D.; Ryan, J.; Bashamboo, A.; Sinclair, A.H.; et al. Mutant NR5A1/SF-1 in patients with disorders of sex development shows defective activation of the SOX9 TESCO enhancer. Hum. Mutat. 2018, 39, 1861–1874. [Google Scholar] [CrossRef]
- Mazen, I.; Abdel-Hamid, M.; Mekkawy, M.; Bignon-Topalovic, J.; Boudjenah, R.; El Gammal, M.; Essawi, M.; Bashamboo, A.; McElreavey, K. Identification of NR5A1 Mutations and Possible Digenic Inheritance in 46,XY Gonadal Dysgenesis. Sex. Dev. 2016, 10, 147–151. [Google Scholar] [CrossRef]
- Camats, N.; Fernández-Cancio, M.; Audí, L.; Schaller, A.; Flück, C.E. Broad phenotypes in heterozygous NR5A1 46,XY patients with a disorder of sex development: An oligogenic origin? Eur. J. Hum. Genet. 2018, 26, 1329–1338. [Google Scholar] [CrossRef] [Green Version]
- Robevska, G.; van den Bergen, J.A.; Ohnesorg, T.; Eggers, S.; Hanna, C.; Hersmus, R.; Thompson, E.M.; Baxendale, A.; Verge, C.F.; Lafferty, A.R.; et al. Functional characterization of novel NR5A1 variants reveals multiple complex roles in disorders of sex development. Hum. Mutat. 2018, 39, 124–139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Werner, R.; Mönig, I.; Lünstedt, R.; Wünsch, L.; Thorns, C.; Reiz, B.; Krause, A.; Schwab, K.O.; Binder, G.; Holterhus, P.M.; et al. New NR5A1 mutations and phenotypic variations of gonadal dysgenesis. PLoS ONE 2017, 12, e0176720. [Google Scholar] [CrossRef] [PubMed]
- Baetens, D.; Stoop, H.; Peelman, F.; Todeschini, A.L.; Rosseel, T.; Coppieters, F.; Veitia, R.A.; Looijenga, L.H.; De Baere, E.; Cools, M. NR5A1 is a novel disease gene for 46,XX testicular and ovotesticular disorders of sex development. Genet. Med. Off. J. Am. Coll. Med. Genet. 2016, 19, 367–376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Igarashi, M.; Takasawa, K.; Hakoda, A.; Kanno, J.; Takada, S.; Miyado, M.; Baba, T.; Morohashi, K.I.; Tajima, T.; Hata, K.; et al. Identical NR5A1 Missense Mutations in Two Unrelated 46,XX Individuals with Testicular Tissues. Hum. Mutat. 2016, 38, 39–42. [Google Scholar] [CrossRef]
- Guran, T.; Buonocore, F.; Saka, N.; Ozbek, M.N.; Aycan, Z.; Bereket, A.; Bas, F.; Darcan, S.; Bideci, A.; Guven, A.; et al. Rare Causes of Primary Adrenal Insufficiency: Genetic and Clinical Characterization of a Large Nationwide Cohort. J. Clin. Endocrinol. Metab. 2016, 101, 284–292. [Google Scholar] [CrossRef]
- Swartz, J.M.; Ciarlo, R.; Guo, M.H.; Abrha, A.; Weaver, B.; Diamond, D.A.; Chan, Y.M.; Hirschhorn, J.N. A 46,XX Ovotesticular Disorder of Sex Development Likely Caused by a Steroidogenic Factor-1 (NR5A1) Variant. Horm. Res. Paediatr. 2017, 87, 191–195. [Google Scholar] [CrossRef] [Green Version]
- Knarston, I.M.; Robevska, G.; van den Bergen, J.A.; Eggers, S.; Croft, B.; Yates, J.; Hersmus, R.; Looijenga, L.H.J.; Cameron, F.J.; Monhike, K.; et al. NR5A1 gene variants repress the ovarian-specific WNT signaling pathway in 46,XX disorders of sex development patients. Hum. Mutat. 2019, 40, 207–216. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Li, G.; Zhang, J.; Gao, F.; Li, W.; Qin, Y.; Chen, Z.J. Novel WT1 Missense Mutations in Han Chinese Women with Premature Ovarian Failure. Sci. Rep. 2015, 5, 13983. [Google Scholar] [CrossRef] [Green Version]
- Hoefele, J.; Kemper, M.J.; Schoenermarck, U.; Mueller, S.; Klein, H.G.; Lemke, A. Truncating Wilms Tumor Suppressor Gene 1 Mutation in an XX Female with Adult-Onset Focal Segmental Glomerulosclerosis and Streak Ovaries: A Case Report. Nephron 2017, 135, 72–76. [Google Scholar] [CrossRef]
- Gomes, N.L.; de Paula, L.C.P.; Silva, J.M.; Silva, T.E.; Lerário, A.M.; Nishi, M.Y.; Batista, R.L.; Faria Júnior, J.A.D.; Moraes, D.; Costa, E.M.F.; et al. A 46,XX testicular disorder of sex development caused by a Wilms’ tumour Factor-1 (WT1) pathogenic variant. Clin. Genet. 2019, 95, 172–176. [Google Scholar] [CrossRef] [Green Version]
- Schnabel, C.A.; Selleri, L.; Cleary, M.L. Pbx1 is essential for adrenal development and urogenital differentiation. Genesis 2003, 37, 123–130. [Google Scholar] [CrossRef] [PubMed]
- Le Tanno, P.; Breton, J.; Bidart, M.; Satre, V.; Harbuz, R.; Ray, P.F.; Bosson, C.; Dieterich, K.; Jaillard, S.; Odent, S.; et al. PBX1 haploinsufficiency leads to syndromic congenital anomalies of the kidney and urinary tract (CAKUT) in humans. J. Med. Genet. 2017, 54, 502–510. [Google Scholar] [CrossRef] [PubMed]
- Heidet, L.; Moriniere, V.; Henry, C.; De Tomasi, L.; Reilly, M.L.; Humbert, C.; Alibeu, O.; Fourrage, C.; Bole-Feysot, C.; Nitschke, P.; et al. Targeted Exome Sequencing Identifies PBX1 as Involved in Monogenic Congenital Anomalies of the Kidney and Urinary Tract. J. Am. Soc. Nephrol. 2017, 28, 2901–2914. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eozenou, C.; Bashamboo, A.; Bignon-Topalovic, J.; Merel, T.; Zwermann, O.; Lourenco, D.; Lottmann, H.; Lichtenauer, U.; Rojo, S.; Beuschlein, F.; et al. The TALE homeodomain of PBX1 is involved in human primary testis-determination. Hum. Mutat. 2019, 40, 1071–1076. [Google Scholar] [CrossRef]
- Katoh-Fukui, Y.; Miyabayashi, K.; Komatsu, T.; Owaki, A.; Baba, T.; Shima, Y.; Kidokoro, T.; Kanai, Y.; Schedl, A.; Wilhelm, D.; et al. Cbx2, a polycomb group gene, is required for Sry gene expression in mice. Endocrinology 2012, 153, 913–924. [Google Scholar] [CrossRef] [Green Version]
- Katoh-Fukui, Y.; Tsuchiya, R.; Shiroishi, T.; Nakahara, Y.; Hashimoto, N.; Noguchi, K.; Higashinakagawa, T. Male-to-female sex reversal in M33 mutant mice. Nature 1998, 393, 688–692. [Google Scholar] [CrossRef]
- Biason-Lauber, A.; Konrad, D.; Meyer, M.; DeBeaufort, C.; Schoenle, E.J. Ovaries and female phenotype in a girl with 46,XY karyotype and mutations in the CBX2 gene. Am. J. Hum. Genet. 2009, 84, 658–663. [Google Scholar] [CrossRef] [Green Version]
- Garcia-Moreno, S.A.; Lin, Y.T.; Futtner, C.R.; Salamone, I.M.; Capel, B.; Maatouk, D.M. CBX2 is required to stabilize the testis pathway by repressing Wnt signaling. PLoS Genet. 2019, 15, e1007895. [Google Scholar] [CrossRef] [Green Version]
- Bouazzi, L.; Sproll, P.; Eid, W.; Biason-Lauber, A. The transcriptional regulator CBX2 and ovarian function: A whole genome and whole transcriptome approach. Sci. Rep. 2019, 9, 17033. [Google Scholar] [CrossRef]
- Sproll, P.; Eid, W.; Gomes, C.R.; Mendonca, B.B.; Gomes, N.L.; Costa, E.M.; Biason-Lauber, A. Assembling the jigsaw puzzle: CBX2 isoform 2 and its targets in disorders/differences of sex development. Mol. Genet. Genom. Med. 2018, 6, 785–795. [Google Scholar] [CrossRef] [Green Version]
- Bashamboo, A.; Eozenou, C.; Jorgensen, A.; Bignon-Topalovic, J.; Siffroi, J.P.; Hyon, C.; Tar, A.; Nagy, P.; Sólyom, J.; Halász, Z.; et al. Loss of Function of the Nuclear Receptor NR2F2, Encoding COUP-TF2, Causes Testis Development and Cardiac Defects in 46,XX Children. Am. J. Hum. Genet. 2018, 102, 487–493. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carvalheira, G.; Malinverni, A.M.; Moysés-Oliveira, M.; Ueta, R.; Cardili, L.; Monteagudo, P.; Mathez, A.L.G.; Verreschi, I.T.; Maluf, M.A.; Shida, M.E.F.; et al. The Natural History of a Man With Ovotesticular 46,XX DSD Caused by a Novel 3-Mb 15q26.2 Deletion Containing. J. Endocr. Soc. 2019, 3, 2107–2113. [Google Scholar] [CrossRef] [PubMed]
- Zhao, F.; Franco, H.L.; Rodriguez, K.F.; Brown, P.R.; Tsai, M.J.; Tsai, S.Y.; Yao, H.H. Elimination of the male reproductive tract in the female embryo is promoted by COUP-TFII in mice. Science 2017, 357, 717–720. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mendoza-Villarroel, R.E.; Robert, N.M.; Martin, L.J.; Brousseau, C.; Tremblay, J.J. The nuclear receptor NR2F2 activates star expression and steroidogenesis in mouse MA-10 and MLTC-1 Leydig cells. Biol. Reprod. 2014, 91, 26. [Google Scholar] [CrossRef] [Green Version]
- Mendoza-Villarroel, R.E.; Di-Luoffo, M.; Camiré, E.; Giner, X.C.; Brousseau, C.; Tremblay, J.J. The INSL3 gene is a direct target for the orphan nuclear receptor, COUP-TFII, in Leydig cells. J. Mol. Endocrinol. 2014, 53, 43–55. [Google Scholar] [CrossRef] [Green Version]
- Mehanovic, S.; Mendoza-Villarroel, R.E.; Viger, R.S.; Tremblay, J.J. The Nuclear Receptor COUP-TFII Regulates. J. Endocr. Soc. 2019, 3, 2236–2257. [Google Scholar] [CrossRef]
- Rotgers, E.; Jorgensen, A.; Yao, H.H. At the Crossroads of Fate-Somatic Cell Lineage Specification in the Fetal Gonad. Endocr. Rev. 2018, 39, 739–759. [Google Scholar] [CrossRef]
- Kim, Y.; Kobayashi, A.; Sekido, R.; DiNapoli, L.; Brennan, J.; Chaboissier, M.C.; Poulat, F.; Behringer, R.R.; Lovell-Badge, R.; Capel, B. Fgf9 and Wnt4 act as antagonistic signals to regulate mammalian sex determination. PLoS Biol. 2006, 4, e187. [Google Scholar] [CrossRef] [Green Version]
- Jameson, S.A.; Lin, Y.T.; Capel, B. Testis development requires the repression of Wnt4 by Fgf signaling. Dev. Biol. 2012, 370, 24–32. [Google Scholar] [CrossRef] [Green Version]
- Bagheri-Fam, S.; Ono, M.; Li, L.; Zhao, L.; Ryan, J.; Lai, R.; Katsura, Y.; Rossello, F.J.; Koopman, P.; Scherer, G.; et al. FGFR2 mutation in 46,XY sex reversal with craniosynostosis. Hum. Mol. Genet. 2015, 24, 6699–6710. [Google Scholar] [CrossRef] [Green Version]
- Lavery, R.; Chassot, A.A.; Pauper, E.; Gregoire, E.P.; Klopfenstein, M.; de Rooij, D.G.; Mark, M.; Schedl, A.; Ghyselinck, N.B.; Chaboissier, M.C. Testicular differentiation occurs in absence of R-spondin1 and Sox9 in mouse sex reversals. PLoS Genet. 2012, 8, e1003170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nicol, B.; Yao, H.H. Gonadal Identity in the Absence of Pro-Testis Factor SOX9 and Pro-Ovary Factor Beta-Catenin in Mice. Biol. Reprod. 2015, 93, 35. [Google Scholar] [CrossRef] [PubMed]
- Mamsen, L.S.; Ernst, E.H.; Borup, R.; Larsen, A.; Olesen, R.H.; Ernst, E.; Anderson, R.A.; Kristensen, S.G.; Andersen, C.Y. Temporal expression pattern of genes during the period of sex differentiation in human embryonic gonads. Sci. Rep. 2017, 7, 15961. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tomaselli, S.; Megiorni, F.; Lin, L.; Mazzilli, M.C.; Gerrelli, D.; Majore, S.; Grammatico, P.; Achermann, J.C. Human RSPO1/R-spondin1 is expressed during early ovary development and augments beta-catenin signaling. PLoS ONE 2011, 6, e16366. [Google Scholar] [CrossRef] [PubMed]
- Biason-Lauber, A.; Konrad, D.; Navratil, F.; Schoenle, E.J. A WNT4 mutation associated with Mullerian-duct regression and virilization in a 46,XX woman. N. Engl. J. Med. 2004, 351, 792–798. [Google Scholar] [CrossRef] [Green Version]
- Philibert, P.; Biason-Lauber, A.; Gueorguieva, I.; Stuckens, C.; Pienkowski, C.; Lebon-Labich, B.; Paris, F.; Sultan, C. Molecular analysis of WNT4 gene in four adolescent girls with mullerian duct abnormality and hyperandrogenism (atypical Mayer-Rokitansky-Kuster-Hauser syndrome). Fertil. Steril. 2011, 95, 2683–2686. [Google Scholar] [CrossRef]
- Parma, P.; Radi, O.; Vidal, V.; Chaboissier, M.C.; Dellambra, E.; Valentini, S.; Guerra, L.; Schedl, A.; Camerino, G. R-spondin1 is essential in sex determination, skin differentiation and malignancy. Nat. Genet. 2006, 38, 1304–1309. [Google Scholar] [CrossRef]
- Tomaselli, S.; Megiorni, F.; De Bernardo, C.; Felici, A.; Marrocco, G.; Maggiulli, G.; Grammatico, B.; Remotti, D.; Saccucci, P.; Valentini, F.; et al. Syndromic true hermaphroditism due to an R-spondin1 (RSPO1) homozygous mutation. Hum. Mutat. 2008, 29, 220–226. [Google Scholar] [CrossRef]
- Jordan, B.K.; Mohammed, M.; Ching, S.T.; Delot, E.; Chen, X.N.; Dewing, P.; Swain, A.; Rao, P.N.; Elejalde, B.R.; Vilain, E. Up-regulation of WNT-4 signaling and dosage-sensitive sex reversal in humans. Am. J. Hum. Genet. 2001, 68, 1102–1109. [Google Scholar] [CrossRef] [Green Version]
- Wieacker, P.; Volleth, M. WNT4 and RSPO1 are not involved in a case of male-to-female sex reversal with partial duplication of 1p. Sex. Dev. 2007, 1, 111–113. [Google Scholar] [CrossRef]
- Harris, A.; Siggers, P.; Corrochano, S.; Warr, N.; Sagar, D.; Grimes, D.T.; Suzuki, M.; Burdine, R.D.; Cong, F.; Koo, B.K.; et al. ZNRF3 functions in mammalian sex determination by inhibiting canonical WNT signaling. Proc. Natl. Acad. Sci. USA 2018, 115, 5474–5479. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gustin, S.E.; Hogg, K.; Stringer, J.M.; Rastetter, R.H.; Pelosi, E.; Miles, D.C.; Sinclair, A.H.; Wilhelm, D.; Western, P.S. WNT/beta-catenin and p27/FOXL2 differentially regulate supporting cell proliferation in the developing ovary. Dev. Biol. 2016, 412, 250–260. [Google Scholar] [CrossRef] [PubMed]
- Ottolenghi, C.; Omari, S.; Garcia-Ortiz, J.E.; Uda, M.; Crisponi, L.; Forabosco, A.; Pilia, G.; Schlessinger, D. Foxl2 is required for commitment to ovary differentiation. Hum. Mol. Genet. 2005, 14, 2053–2062. [Google Scholar] [CrossRef] [PubMed]
- Uda, M.; Ottolenghi, C.; Crisponi, L.; Garcia, J.E.; Deiana, M.; Kimber, W.; Forabosco, A.; Cao, A.; Schlessinger, D.; Pilia, G. Foxl2 disruption causes mouse ovarian failure by pervasive blockage of follicle development. Hum. Mol. Genet. 2004, 13, 1171–1181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uhlenhaut, N.H.; Jakob, S.; Anlag, K.; Eisenberger, T.; Sekido, R.; Kress, J.; Treier, A.C.; Klugmann, C.; Klasen, C.; Holter, N.I.; et al. Somatic sex reprogramming of adult ovaries to testes by FOXL2 ablation. Cell 2009, 139, 1130–1142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmidt, D.; Ovitt, C.E.; Anlag, K.; Fehsenfeld, S.; Gredsted, L.; Treier, A.C.; Treier, M. The murine winged-helix transcription factor Foxl2 is required for granulosa cell differentiation and ovary maintenance. Development 2004, 131, 933–942. [Google Scholar] [CrossRef] [Green Version]
- Jorgensen, A.; Nielsen, J.E.; Perlman, S.; Lundvall, L.; Mitchell, R.T.; Juul, A.; Rajpert-De Meyts, E. Ex vivo culture of human fetal gonads: Manipulation of meiosis signalling by retinoic acid treatment disrupts testis development. Hum. Reprod. 2015, 30, 2351–2363. [Google Scholar] [CrossRef] [Green Version]
- Crisponi, L.; Deiana, M.; Loi, A.; Chiappe, F.; Uda, M.; Amati, P.; Bisceglia, L.; Zelante, L.; Nagaraja, R.; Porcu, S.; et al. The putative forkhead transcription factor FOXL2 is mutated in blepharophimosis/ptosis/epicanthus inversus syndrome. Nat. Genet. 2001, 27, 159–166. [Google Scholar] [CrossRef]
- Harris, S.E.; Chand, A.L.; Winship, I.M.; Gersak, K.; Aittomaki, K.; Shelling, A.N. Identification of novel mutations in FOXL2 associated with premature ovarian failure. Mol. Hum. Reprod. 2002, 8, 729–733. [Google Scholar] [CrossRef]
- Georges, A.; L’Hote, D.; Todeschini, A.L.; Auguste, A.; Legois, B.; Zider, A.; Veitia, R.A. The transcription factor FOXL2 mobilizes estrogen signaling to maintain the identity of ovarian granulosa cells. Elife 2014, 3, e04207. [Google Scholar] [CrossRef] [Green Version]
- Ottolenghi, C.; Pelosi, E.; Tran, J.; Colombino, M.; Douglass, E.; Nedorezov, T.; Cao, A.; Forabosco, A.; Schlessinger, D. Loss of Wnt4 and Foxl2 leads to female-to-male sex reversal extending to germ cells. Hum. Mol. Genet. 2007, 16, 2795–2804. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bagheri-Fam, S.; Bird, A.D.; Zhao, L.; Ryan, J.M.; Yong, M.; Wilhelm, D.; Koopman, P.; Eswarakumar, V.P.; Harley, V.R. Testis Determination Requires a Specific FGFR2 Isoform to Repress FOXL2. Endocrinology 2017, 158, 3832–3843. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Auguste, A.; Chassot, A.A.; Gregoire, E.P.; Renault, L.; Pannetier, M.; Treier, M.; Pailhoux, E.; Chaboissier, M.C. Loss of R-spondin1 and Foxl2 amplifies female-to-male sex reversal in XX mice. Sex. Dev. 2011, 5, 304–317. [Google Scholar] [CrossRef]
- Raymond, C.S.; Murphy, M.W.; O’Sullivan, M.G.; Bardwell, V.J.; Zarkower, D. Dmrt1, a gene related to worm and fly sexual regulators, is required for mammalian testis differentiation. Genes Dev. 2000, 14, 2587–2595. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matson, C.K.; Murphy, M.W.; Sarver, A.L.; Griswold, M.D.; Bardwell, V.J.; Zarkower, D. DMRT1 prevents female reprogramming in the postnatal mammalian testis. Nature 2011, 476, 101–104. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Svingen, T.; Ng, E.T.; Koopman, P. Female-to-male sex reversal in mice caused by transgenic overexpression of Dmrt1. Development 2015, 142, 1083–1088. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raymond, C.S.; Shamu, C.E.; Shen, M.M.; Seifert, K.J.; Hirsch, B.; Hodgkin, J.; Zarkower, D. Evidence for evolutionary conservation of sex-determining genes. Nature 1998, 391, 691–695. [Google Scholar] [CrossRef]
- Veitia, R.A.; Nunes, M.; Quintana-Murci, L.; Rappaport, R.; Thibaud, E.; Jaubert, F.; Fellous, M.; McElreavey, K.; Goncalves, J.; Silva, M.; et al. Swyer syndrome and 46,XY partial gonadal dysgenesis associated with 9p deletions in the absence of monosomy-9p syndrome. Am. J. Hum. Genet. 1998, 63, 901–905. [Google Scholar] [CrossRef] [Green Version]
- McDonald, M.T.; Flejter, W.; Sheldon, S.; Putzi, M.J.; Gorski, J.L. XY sex reversal and gonadal dysgenesis due to 9p24 monosomy. Am. J. Med. Genet. 1997, 73, 321–326. [Google Scholar] [CrossRef]
- Eggers, S.; Sinclair, A. Mammalian sex determination-insights from humans and mice. Chromosome Res. 2012, 20, 215–238. [Google Scholar] [CrossRef] [Green Version]
- Barbaro, M.; Balsamo, A.; Anderlid, B.M.; Myhre, A.G.; Gennari, M.; Nicoletti, A.; Pittalis, M.C.; Oscarson, M.; Wedell, A. Characterization of deletions at 9p affecting the candidate regions for sex reversal and deletion 9p syndrome by MLPA. Eur. J. Hum. Genet. 2009, 17, 1439–1447. [Google Scholar] [CrossRef] [Green Version]
- Raymond, C.S.; Parker, E.D.; Kettlewell, J.R.; Brown, L.G.; Page, D.C.; Kusz, K.; Jaruzelska, J.; Reinberg, Y.; Flejter, W.L.; Bardwell, V.J.; et al. A region of human chromosome 9p required for testis development contains two genes related to known sexual regulators. Hum. Mol. Genet. 1999, 8, 989–996. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Calvari, V.; Bertini, V.; De Grandi, A.; Peverali, G.; Zuffardi, O.; Ferguson-Smith, M.; Knudtzon, J.; Camerino, G.; Borsani, G.; Guioli, S. A new submicroscopic deletion that refines the 9p region for sex reversal. Genomics 2000, 65, 203–212. [Google Scholar] [CrossRef] [PubMed]
- Macdonald, J.; Kilcoyne, K.R.; Sharpe, R.M.; Kavanagh, A.; Anderson, R.A.; Brown, P.; Smith, L.B.; Jorgensen, A.; Mitchell, R.T. DMRT1 repression using a novel approach to genetic manipulation induces testicular dysgenesis in human fetal gonads. Hum. Reprod. 2018, 33, 2107–2121. [Google Scholar] [CrossRef] [Green Version]
- Choudhury, P.; Hackert, P.; Memet, I.; Sloan, K.E.; Bohnsack, M.T. The human RNA helicase DHX37 is required for release of the U3 snoRNP from pre-ribosomal particles. RNA Biol. 2019, 16, 54–68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McElreavey, K.; Jorgensen, A.; Eozenou, C.; Merel, T.; Bignon-Topalovic, J.; Tan, D.S.; Houzelstein, D.; Buonocore, F.; Warr, N.; Kay, R.G.G.; et al. Pathogenic variants in the DEAH-box RNA helicase DHX37 are a frequent cause of 46,XY gonadal dysgenesis and 46,XY testicular regression syndrome. Genet. Med. Off. J. Am. Coll. Med. Genet. 2019. [Google Scholar] [CrossRef] [Green Version]
- Evilen da Silva, T.; Gomes, N.L.; Lerário, A.M.; Keegan, C.E.; Nishi, M.Y.; Carvalho, F.M.; Vilain, E.; Barseghyanm, H.; Martinez-Aguayo, A.; Forclaz, M.V.; et al. Genetic evidence of the association of DEAH-box helicase 37 defects with 46,XY gonadal dysgenesis spectrum. J. Clin. Endocrinol. Metab. 2019, 104, 5923–5934. [Google Scholar] [CrossRef]
- Buonocore, F.; Clifford-Mobley, O.; King, T.F.J.; Striglioni, N.; Man, E.; Suntharalingham, J.P.; Del Valle, I.; Lin, L.; Lagos, C.F.; Rumsby, G.; et al. Next-Generation Sequencing Reveals Novel Genetic Variants (SRY, DMRT1, NR5A1, DHH, DHX37) in Adults With 46,XY DSD. J. Endocr. Soc. 2019, 3, 2341–2360. [Google Scholar] [CrossRef]
- Anderson, R.A.; Mitchell, R.T.; Kelsey, T.W.; Spears, N.; Telfer, E.E.; Wallace, W.H. Cancer treatment and gonadal function: Experimental and established strategies for fertility preservation in children and young adults. Lancet Diabetes Endocrinol. 2015, 3, 556–567. [Google Scholar] [CrossRef] [Green Version]
- Abir, R.; Fisch, B.; Nahum, R.; Orvieto, R.; Nitke, S.; Ben Rafael, Z. Turner’s syndrome and fertility: Current status and possible putative prospects. Hum. Reprod. Update 2001, 7, 603–610. [Google Scholar] [CrossRef] [Green Version]
- Bernard, V.; Donadille, B.; Zenaty, D.; Courtillot, C.; Salenave, S.; Brac de la Perriere, A.; Albarel, F.; Fevre, A.; Kerlan, V.; Brue, T.; et al. Spontaneous fertility and pregnancy outcomes amongst 480 women with Turner syndrome. Hum. Reprod. 2016, 31, 782–788. [Google Scholar] [CrossRef] [Green Version]
- Kalra, R.; Cameron, M.; Stern, C. Female fertility preservation in DSD. Best Pract. Res. Clin. Endocrinol. Metab. 2019, 33, 101289. [Google Scholar] [CrossRef] [PubMed]
- Mamsen, L.S.; Charkiewicz, K.; Anderson, R.A.; Telfer, E.E.; McLaughlin, M.; Kelsey, T.W.; Kristensen, S.G.; Gook, D.A.; Ernst, E.; Andersen, C.Y. Characterization of follicles in girls and young women with Turner syndrome who underwent ovarian tissue cryopreservation. Fertil. Steril. 2019, 111, 1217–1225. [Google Scholar] [CrossRef] [Green Version]
- Corona, G.; Pizzocaro, A.; Lanfranco, F.; Garolla, A.; Pelliccione, F.; Vignozzi, L.; Ferlin, A.; Foresta, C.; Jannini, E.A.; Maggi, M.; et al. Sperm recovery and ICSI outcomes in Klinefelter syndrome: A systematic review and meta-analysis. Hum. Reprod. Update 2017, 23, 265–275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Saen, D.; Vloeberghs, V.; Gies, I.; Mateizel, I.; Sermon, K.; De Schepper, J.; Tournaye, H.; Goossens, E. When does germ cell loss and fibrosis occur in patients with Klinefelter syndrome? Hum. Reprod. 2018, 33, 1009–1022. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stukenborg, J.B.; Jahnukainen, K.; Hutka, M.; Mitchell, R.T. Cancer treatment in childhood and testicular function: The importance of the somatic environment. Endocr. Connect. 2018, 7, R69–R87. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Batavia, J.P.; Kolon, T.F. Fertility in disorders of sex development: A review. J. Pediatr. Urol. 2016, 12, 418–425. [Google Scholar] [CrossRef] [PubMed]
- Flannigan, R.K.; Chow, V.; Ma, S.; Yuzpe, A. 45,X/46,XY mixed gonadal dysgenesis: A case of successful sperm extraction. Can. Urol. Assoc. J. 2014, 8, E108–E110. [Google Scholar] [CrossRef] [Green Version]
- Ljubicic, M.L.; Jorgensen, A.; Acerini, C.; Andrade, J.; Balsamo, A.; Bertelloni, S.; Cools, M.; Cuccaro, R.T.; Darendeliler, F.; Fluck, C.E.; et al. Clinical but Not Histological Outcomes in Males With 45,X/46,XY Mosaicism Vary Depending on Reason for Diagnosis. J. Clin. Endocrinol. Metab. 2019, 104, 4366–4381. [Google Scholar] [CrossRef]
- Landin-Wilhelmsen, K.; Bryman, I.; Hanson, C.; Hanson, L. Spontaneous pregnancies in a Turner syndrome woman with Y-chromosome mosaicism. J. Assist. Reprod. Genet. 2004, 21, 229–230. [Google Scholar] [CrossRef] [Green Version]
- Dumic, M.; Lin-Su, K.; Leibel, N.I.; Ciglar, S.; Vinci, G.; Lasan, R.; Nimkarn, S.; Wilson, J.D.; McElreavey, K.; New, M.I. Report of fertility in a woman with a predominantly 46,XY karyotype in a family with multiple disorders of sexual development. J. Clin. Endocrinol. Metab. 2008, 93, 182–189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dabrowski, E.; Johnson, E.K.; Patel, V.; Hsu, Y.; Davis, S.; Goetsch, A.L.; Habiby, R.; Brickman, W.J.; Finlayson, C. Turner Syndrome with Y Chromosome: Spontaneous Thelarche, Menarche, and Risk of Malignancy. J. Pediatr. Adolesc. Gynecol. 2020, 33, 10–14. [Google Scholar] [CrossRef] [PubMed]
- Abd Wahab, A.V.; Lim, L.M.; Mohamed Tarmizi, M.H. Ovotesticular Disorders of Sex Development: Improvement in Spermatogonia after Removal of Ovary and Mullerian Structures. J. Pediatr. Adolesc. Gynecol. 2019, 32, 74–77. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, Y.; Fujiwara, K.; Yamauchi, H.; Mikami, Y.; Kohno, I. Pregnancy in a woman with a Y chromosome after removal of an ovarian dysgerminoma. Gynecol. Oncol. 2000, 79, 519–521. [Google Scholar] [CrossRef]
- Sugawara, N.; Kimura, Y.; Araki, Y. Successful second delivery outcome using refrozen thawed testicular sperm from an infertile male true hermaphrodite with a 46, XX/46, XY karyotype: Case report. Hum. Cell. 2012, 25, 96–99. [Google Scholar] [CrossRef]
- Laursen, R.J.; Alsbjerg, B.; Vogel, I.; Gravholt, C.H.; Elbaek, H.; Lildballe, D.L.; Humaidan, P.; Vestergaard, E.M. Case of successful IVF treatment of an oligospermic male with 46,XX/46,XY chimerism. J. Assist. Reprod. Genet. 2018, 35, 1325–1328. [Google Scholar] [CrossRef]
- Hagenfeldt, K.; Janson, P.O.; Holmdahl, G.; Falhammar, H.; Filipsson, H.; Frisen, L.; Thoren, M.; Nordenskjold, A. Fertility and pregnancy outcome in women with congenital adrenal hyperplasia due to 21-hydroxylase deficiency. Hum. Reprod. 2008, 23, 1607–1613. [Google Scholar] [CrossRef]
- Simm, P.J.; Zacharin, M.R. Successful pregnancy in a patient with severe 11-beta-hydroxylase deficiency and novel mutations in CYP11B1 gene. Horm. Res. 2007, 68, 294–297. [Google Scholar] [CrossRef]
- Levran, D.; Ben-Shlomo, I.; Pariente, C.; Dor, J.; Mashiach, S.; Weissman, A. Familial partial 17,20-desmolase and 17alpha-hydroxylase deficiency presenting as infertility. J. Assist. Reprod. Genet. 2003, 20, 21–28. [Google Scholar] [CrossRef]
- Bianchi, P.H.; Gouveia, G.R.; Costa, E.M.; Domenice, S.; Martin, R.M.; de Carvalho, L.C.; Pelaes, T.; Inacio, M.; Codarin, R.R.; Sator de Faria, M.B.; et al. Successful Live Birth in a Woman With 17alpha-Hydroxylase Deficiency Through IVF Frozen-Thawed Embryo Transfer. J. Clin. Endocrinol. Metab. 2016, 101, 345–348. [Google Scholar] [CrossRef] [Green Version]
- Ben-Nun, I.; Siegal, A.; Shulman, A.; Ghetler, Y.; Kaneti, H.; Lunenfeld, B.; Beyth, Y.; Fejgin, M. Induction of artificial endometrial cycles with oestradiol implants and injectable progesterone: Establishment of a viable pregnancy in a woman with 17-alpha-hydroxylase deficiency. Hum. Reprod. 1995, 10, 2456–2458. [Google Scholar] [CrossRef] [PubMed]
- Albarel, F.; Perrin, J.; Jegaden, M.; Roucher-Boulez, F.; Reynaud, R.; Brue, T.; Courbiere, B. Successful IVF pregnancy despite inadequate ovarian steroidogenesis due to congenital lipoid adrenal hyperplasia (CLAH): A case report. Hum. Reprod. 2016, 31, 2609–2612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Delot, E.C.; Vilain, E.J. Nonsyndromic 46,XX Testicular Disorders of Sex Development. In GeneReviews®; Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Stephens, K., Amemiya, A., Eds.; Univisity of Washington: Seattle, WA, USA, 1993. [Google Scholar]
- Zhu, Y.; Hu, L.; Cao, D.; Ou, X.; Jiang, M. Chromosomal microarray analysis of infertile men with azoospermia factor microdeletions. Gene 2020, 735, 144389. [Google Scholar] [CrossRef] [PubMed]
- Gupta, A.; Bajaj, R.; Jindal, U.N. A Rare Case of Swyer Syndrome in Two Sisters with Successful Pregnancy Outcome in Both. J. Hum. Reprod. Sci. 2019, 12, 267–269. [Google Scholar] [CrossRef] [PubMed]
- Gabriel Ribeiro de Andrade, J.; Marques-de-Faria, A.P.; Fabbri, H.C.; de Mello, M.P.; Guerra-Junior, G.; Maciel-Guerra, A.T. Long-Term Follow-Up of Patients with 46,XY Partial Gonadal Dysgenesis Reared as Males. Int. J. Endocrinol. 2014, 2014, 480724. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reisch, N.; Flade, L.; Scherr, M.; Rottenkolber, M.; Pedrosa Gil, F.; Bidlingmaier, M.; Wolff, H.; Schwarz, H.P.; Quinkler, M.; Beuschlein, F.; et al. High prevalence of reduced fecundity in men with congenital adrenal hyperplasia. J. Clin. Endocrinol. Metab. 2009, 94, 1665–1670. [Google Scholar] [CrossRef] [PubMed]
- Jha, S.; El-Maouche, D.; Marko, J.; Mallappa, A.; Veeraraghavan, P.; Merke, D.P. Individualizing Management of Infertility in Classic Congenital Adrenal Hyperplasia and Testicular Adrenal Rest Tumors. J. Endocr. Soc. 2019, 3, 2290–2294. [Google Scholar] [CrossRef] [Green Version]
- Burckhardt, M.A.; Udhane, S.S.; Marti, N.; Schnyder, I.; Tapia, C.; Nielsen, J.E.; Mullis, P.E.; Rajpert-De Meyts, E.; Fluck, C.E. Human 3beta-hydroxysteroid dehydrogenase deficiency seems to affect fertility but may not harbor a tumor risk: Lesson from an experiment of nature. Eur. J. Endocrinol. 2015, 173, K1–K12. [Google Scholar] [CrossRef] [Green Version]
- Bakircioglu, M.E.; Tulay, P.; Findikli, N.; Erzik, B.; Gultomruk, M.; Bahceci, M. Successful testicular sperm recovery and IVF treatment in a man with Leydig cell hypoplasia. J. Assist. Reprod. Genet. 2014, 31, 817–821. [Google Scholar] [CrossRef] [Green Version]
- Katz, M.D.; Kligman, I.; Cai, L.Q.; Zhu, Y.S.; Fratianni, C.M.; Zervoudakis, I.; Rosenwaks, Z.; Imperato-McGinley, J. Paternity by intrauterine insemination with sperm from a man with 5alpha-reductase-2 deficiency. N. Engl. J. Med. 1997, 336, 994–997. [Google Scholar] [CrossRef]
- Lucas-Herald, A.; Bertelloni, S.; Juul, A.; Bryce, J.; Jiang, J.; Rodie, M.; Sinnott, R.; Boroujerdi, M.; Lindhardt Johansen, M.; Hiort, O.; et al. The Long-Term Outcome of Boys With Partial Androgen Insensitivity Syndrome and a Mutation in the Androgen Receptor Gene. J. Clin. Endocrinol. Metab. 2016, 101, 3959–3967. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tordjman, K.M.; Yaron, M.; Berkovitz, A.; Botchan, A.; Sultan, C.; Lumbroso, S. Fertility after high-dose testosterone and intracytoplasmic sperm injection in a patient with androgen insensitivity syndrome with a previously unreported androgen receptor mutation. Andrologia 2014, 46, 703–706. [Google Scholar] [CrossRef] [PubMed]
- Rutgers, J.L.; Scully, R.E. The androgen insensitivity syndrome (testicular feminization): A clinicopathologic study of 43 cases. Int. J. Gynecol. Pathol. 1991, 10, 126–144. [Google Scholar] [CrossRef] [PubMed]
- Cools, M.; Wolffenbuttel, K.P.; Hersmus, R.; Mendonca, B.B.; Kaprova, J.; Drop, S.L.S.; Stoop, H.; Gillis, A.J.M.; Oosterhuis, J.W.; Costa, E.M.F.; et al. Malignant testicular germ cell tumors in postpubertal individuals with androgen insensitivity: Prevalence, pathology and relevance of single nucleotide polymorphism-based susceptibility profiling. Hum. Reprod. 2017, 32, 2561–2573. [Google Scholar] [CrossRef] [PubMed]
- Schultz, B.A.; Roberts, S.; Rodgers, A.; Ataya, K. Pregnancy in true hermaphrodites and all male offspring to date. Obstet. Gynecol. 2009, 113, 534–536. [Google Scholar] [CrossRef]
- Georgopapadakos, N.; Manoli, A.; Passia, G.; Skandalakis, P.N.; Filippou, D. Uterus Transplantation as a Therapy Method in Mayer-Rokitansky-Kuster-Hauser Syndrome. Cureus 2019, 11, e6333. [Google Scholar] [CrossRef] [Green Version]
- Picton, H.M.; Wyns, C.; Anderson, R.A.; Goossens, E.; Jahnukainen, K.; Kliesch, S.; Mitchell, R.T.; Pennings, G.; Rives, N.; Tournaye, H.; et al. A European perspective on testicular tissue cryopreservation for fertility preservation in prepubertal and adolescent boys. Hum. Reprod. 2015, 30, 2463–2475. [Google Scholar] [CrossRef] [PubMed]
- Islam, R.; Lane, S.; Williams, S.A.; Becker, C.M.; Conway, G.S.; Creighton, S.M. Establishing reproductive potential and advances in fertility preservation techniques for XY individuals with differences in sex development. Clin. Endocrinol. (Oxf.) 2019, 91, 237–244. [Google Scholar] [CrossRef]
- Johnson, E.K.; Finlayson, C.; Finney, E.L.; Harris, C.J.; Tan, S.Y.; Laronda, M.M.; Lockart, B.A.; Chen, D.; Rowell, E.E.; Cheng, E.Y.; et al. Gonadal Tissue Cryopreservation for Children with Differences of Sex Development. Horm. Res. Paediatr. 2019, 92, 84–91. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Consideration | Management | Implications for Fertility |
|---|---|---|
| Malignancy risk | Gonadectomy |
|
| Conservative |
| |
| Progressive germ cell loss | Fertility preservation |
|
| Gonads/gametes incongruent to sex of rearing | Fertility counseling |
|
| Transmission of genetic abnormality to subsequent generations | Genetic/fertility counseling |
|
| Presence/absence of Mullerian structures | Radiology/surgical assessment |
|
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gomes, N.L.; Chetty, T.; Jorgensen, A.; Mitchell, R.T. Disorders of Sex Development—Novel Regulators, Impacts on Fertility, and Options for Fertility Preservation. Int. J. Mol. Sci. 2020, 21, 2282. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21072282
Gomes NL, Chetty T, Jorgensen A, Mitchell RT. Disorders of Sex Development—Novel Regulators, Impacts on Fertility, and Options for Fertility Preservation. International Journal of Molecular Sciences. 2020; 21(7):2282. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21072282
Chicago/Turabian StyleGomes, Nathalia Lisboa, Tarini Chetty, Anne Jorgensen, and Rod T Mitchell. 2020. "Disorders of Sex Development—Novel Regulators, Impacts on Fertility, and Options for Fertility Preservation" International Journal of Molecular Sciences 21, no. 7: 2282. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21072282