1. Introduction
Nanomaterials have become increasingly popular in the production of a wide range of products and processes including, but not limited to, cosmetics [
1], pharmaceuticals [
2], medical research [
3], semiconductor fabrication [
4], food [
5], and electronic manufacturing [
6]. The global market for nanomaterial-based products is estimated to reach
$55 billion by 2022, with about a 20% compound annual growth rate [
7]. Increased use of nanoparticles (NPs) heightens the potential for human exposure, especially from airborne particles or the consumption of products containing them. Workers in various industries are at higher risk of exposure to NPs used in manufacturing via inhalation [
8]. While some NPs are relatively harmless, others produce moderate to severe toxic effects. In vitro studies have demonstrated that NPs are cellularly internalized where they can cause injuries [
9,
10,
11,
12,
13]. This is observed as increased reactive oxidative stress, mitochondrial dysfunction, severe DNA damage, cell cycle arrest, induction of apoptosis, and increased necrosis [
14].
Toxicity depends on the physicochemical properties of NPs [
15]. For instance, the crystal structure and morphology of NPs affect cytotoxicity. The amorphous form of TiO
2 generated more reactive oxygen species (ROS) than the anatase and rutile forms [
16]. Rod-shaped CeO
2 produced toxic responses in RAW 264.7 cells while octahedron and cubic CeO
2 elicited little change [
17]. Surface charge may also influence toxicity, with positively charged ZnO producing a higher degree of toxicity than negatively charged particles in A549 cells [
18]. Three iron NPs (Fe
3O
4, oleic acid-coated Fe
3O
4, and carbon-coated Fe) with different positive charges also produced toxic responses in BEL-7402 cells, with higher positive charge correlating with worse toxic responses [
19]. Dissolution rate, relative available binding sites on particle surfaces, and particle surface charge of various transition metal oxides correlated with toxicity in A549 cells [
20]. It is important to note that the toxicity mechanisms of NPs are not always, but can be, cell line-dependent [
9,
21]. For instance, NiO NPs arrested BEAS-2B cells in the G
0/G
1 phase of cell cycle while arrest of A549 cells occurred in the G
2/M phase [
21]. Furthermore, NiO NPs induced a higher rate of apoptosis in BEAS-2B cells than in A549 cells [
21]. ZnO exposure also induced cell cycle alterations in A549 cells but not in BEAS-2B cells [
9]. Collectively, morphology, surface charge, dissolution rate, and relative surface binding sites influence NP toxicity.
NiO NPs are used in coloring agents for enamels, nanowires, automotive rear-view mirrors, and more products [
22]. Ni(OH)
2 NPs are used in rechargeable battery electrodes, nickel cadmium batteries, and nickel metal hydride batteries [
23]. Nickel can be released to the environment via various anthropogenic processes. It exists in various oxidation states, though Ni(II), nickel in the +2 valence state, is its prevalent form [
24]. The environmental levels of Ni-associated compounds and their effective toxic concentrations are influenced by nickel’s oxidation state, agglomeration, and media (i.e., water, soil, foods, anaerobic, aerobic), as well as by interactions with other organic and inorganic matrixes. Although the environmental concentrations of nano-sized nickel are still unknown, their existence in the environment may impose risk to human health. The mechanism of toxicity is an essential element of and forms the base of risk assessment. Toxic responses upon exposure to NiO and Ni(OH)
2 NPs have been characterized to a limited extent in in vivo and in vitro settings. Treatment with NiO or Ni(OH)
2 NPs induced inflammation in the lungs of rodents [
25,
26]. NiO induced ROS and lipid peroxidation in A549 cells [
27]. Oxidative stress, apoptosis, and reduced viability in the breast cancer cell line MCF-7 and the human airway epithelial cell line HEp-2 were also caused by NiO NP exposure [
28]. Particulate and soluble nickel compound treatments (including NiO and Ni(OH)
2) led to a varying degree of toxicity in modified Chinese hamster ovary CHO-K1 (AS52) cells [
29]. However, Ni(OH)
2 cytotoxicity has not been studied in human cell lines. Adverse responses to nickel NPs observed in animals and cells indicate human health could be threatened by exposure.
To date, there are no studies comparing the difference in cellular toxicity and toxicological mechanism upon exposure of HepG2 (a human hepatocellular carcinoma cell line) and A549 (a human bronchoalveolar carcinoma cell line) to NiO and Ni(OH)2 NPs. Further, there have been no studies on the role of suppression of cell proliferation induced by these NPs on overall toxicity. Our preliminary data suggest that Ni(OH)2 NPs decrease viability more significantly than NiO NPs in A549 cells. We thus hypothesized that (1) differential cytotoxicity of NiO and Ni(OH)2 NPs is cell line-, particle-, time-, and dose-dependent, (2) cytotoxicity is mediated by oxidative stress and subsequent cellular events including modulation of mitochondrial membrane potential and caspase-3 enzyme activity, and (3) exposure to NiO and Ni(OH)2 NPs alters cell cycle and suppresses cell proliferation. Our specific aims were to (1) demonstrate that cytotoxicity is cell line-, particle-, time-, and dose-dependent, (2) measure the differences in various biochemical responses upon NiO or Ni(OH)2 exposure, and (3) investigate whether cell viability is a function of cell killing and inhibition of cell proliferation. To achieve our goals, we measured cell viability in HepG2 and A549 cells upon NiO or Ni(OH)2 exposure. We then delineated the toxicological mechanism of action in the context of the physical and chemical properties of NPs and oxidative stress-mediated cellular injuries, including changes in mitochondrial membrane potential, caspase-3 activity, apoptosis, cell proliferation, and cell cycle, in A549 cells.
3. Discussion
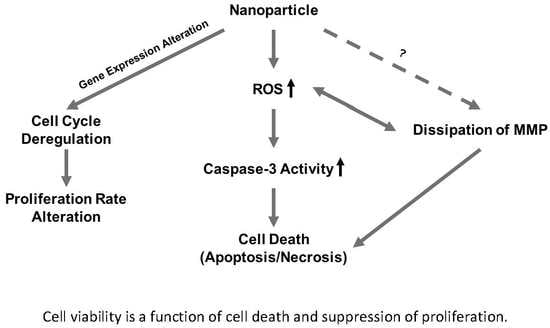
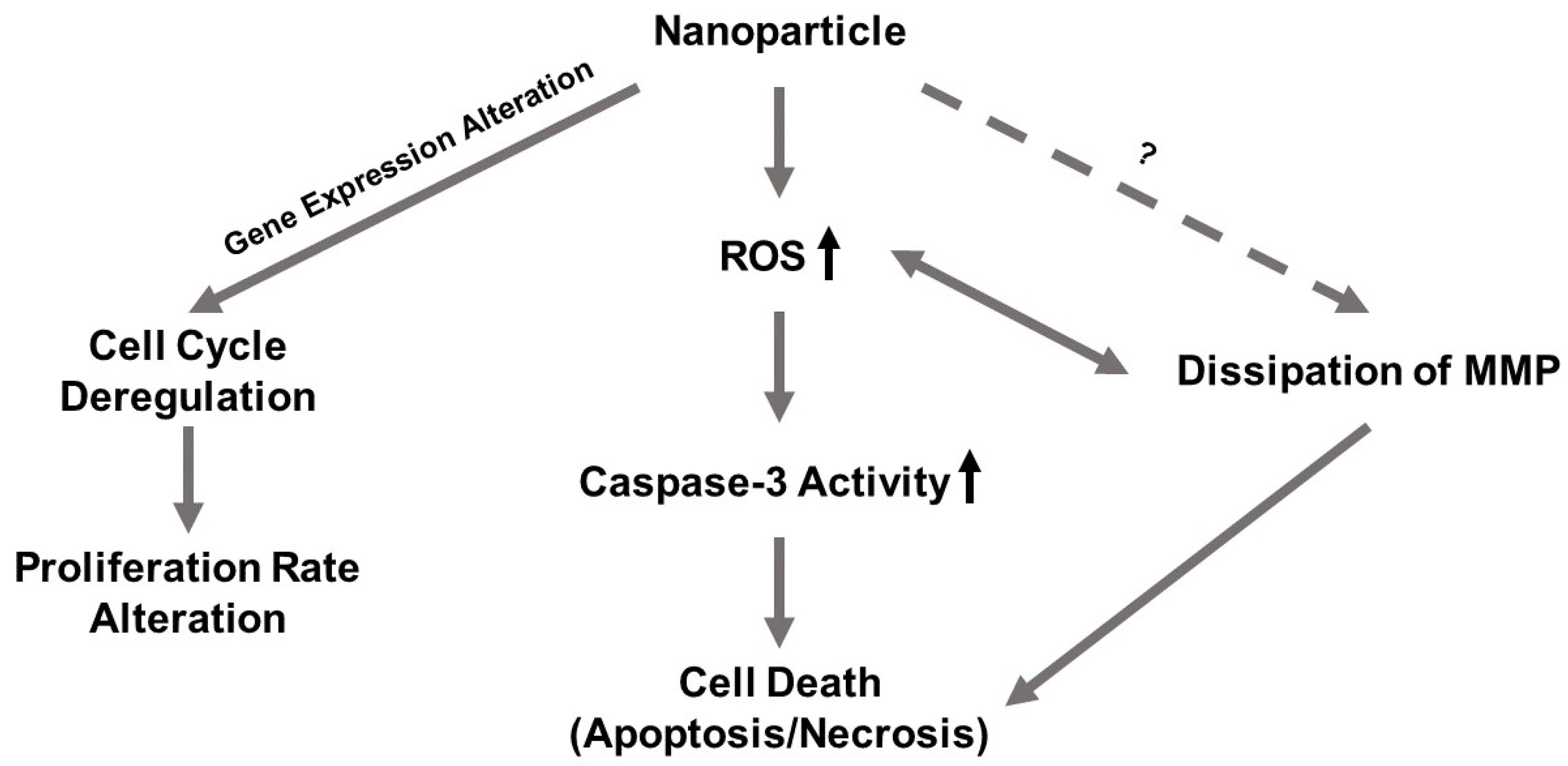
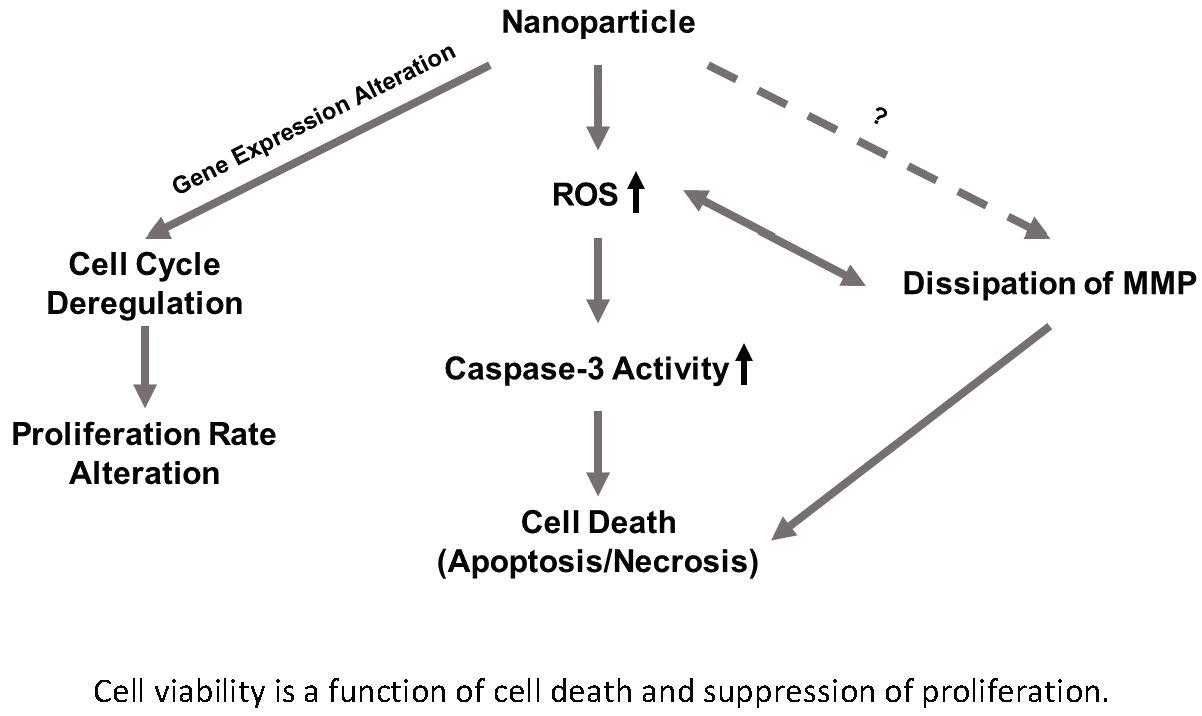
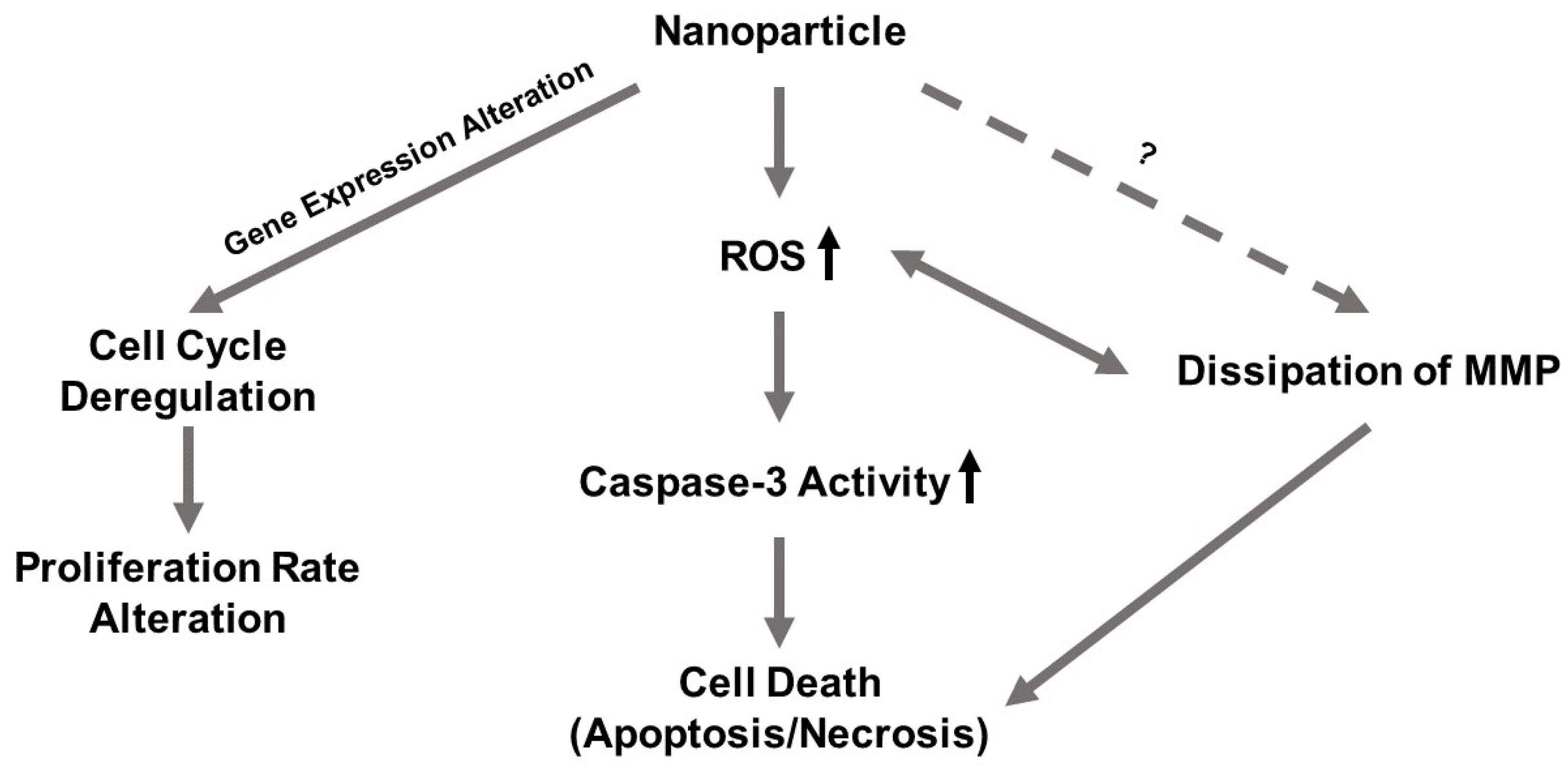
In this study, we investigated and compared the cytotoxicity of two nickel NPs. Several cellular responses were explored as components of this cytotoxicity. We hypothesized that (1) the differential cytotoxicity of NiO and Ni(OH)2 NPs is cell line-, particle-, time-, and dose-dependent, (2) cytotoxicity is mediated by oxidative stress and subsequent cellular events including modulation of mitochondrial membrane potential and caspase-3 enzyme activity, and (3) exposure to NiO and Ni(OH)2 NPs alters cell cycle and suppresses cell proliferation.
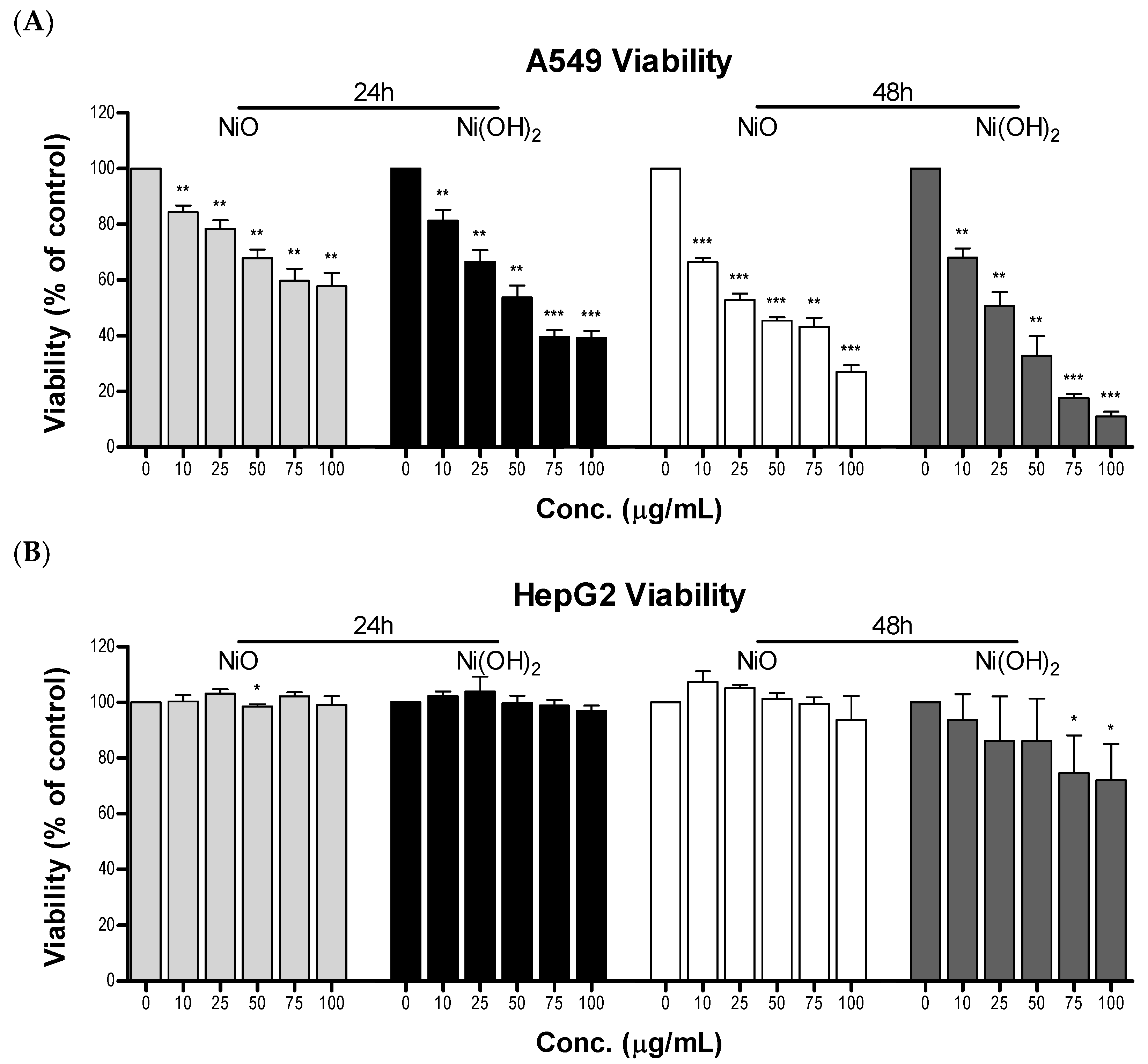
Cell viability is cell line-dependent. A549 cells (a lung cell line) are much more sensitive to NPs than HepG2 cells (a liver cell line). As A549 cells are epithelial cells in a respiratory organ, it is reasonable that they would be more sensitive to particle exposure than hepatic cells, which are suited to interact with toxic compounds. Other studies are in agreement with this notion. For instance, A549 cells experienced greater induction of OS, lactate dehydrogenase leakage, reduction in glutathione levels, dissipation of MMP, elevation of apoptotic gene expression, and decline in cellular viability compared to HepG2 cells upon exposure to CuFe
2O
4 and ZnFe
2O
4 NPs [
30,
31]. Upon exposure to a variety of sizes and concentrations of silica NPs, HepG2 cells were less susceptible than A549 cells and exhibited a lower degree of toxic responses, including decreased ROS induction, lower decline in glutathione (GSH), and less reduction of cell viability [
32]. A549 cells also experienced a greater reduction in MMP and reduction of viability than HepG2 cells upon treatment with silver NPs [
33]. One possible explanation regarding the discrepancy between the in vitro toxic responses of the two cell types may be due to the fact that the liver is primarily responsible for removing toxic compounds from the body and has a higher capacity for detoxification (i.e., phase I & II enzymes) than the lung. On a different note, previously we conducted a study on comparative cytotoxicity between two lung cell lines using seven transition metal oxide nanoparticles [
20]. BEAS-2B is an immortalized, but not cancerous, human bronchial epithelial cell line, whereas A549 is a human bronchoalveolar carcinoma-derived cell line. Both cell types showed similar trends of toxicity. The issue of organ-specific and cell type-specific cytotoxicity is still unsettled and deserves further attention.
We found that NiO- and Ni(OH)
2-induced cytotoxicity is concentration-, time-, and particle-specific in A549 cells. A549 cells experienced concentration-dependent viability at all tested dosages. Ni(OH)
2 consistently produced more severe outcomes compared to NiO and increased treatment time led to increased cytotoxic effects for both particles. Previously, the toxicity of Ni(OH)
2 had not been examined in human cells. However, our viability results have a similar trend to a study conducted in AS52 cells, where viability was found to be concentration-dependent and the median lethal concentration (LC
50) of Ni(OH)
2 was found to be six times greater than that of NiO [
29].
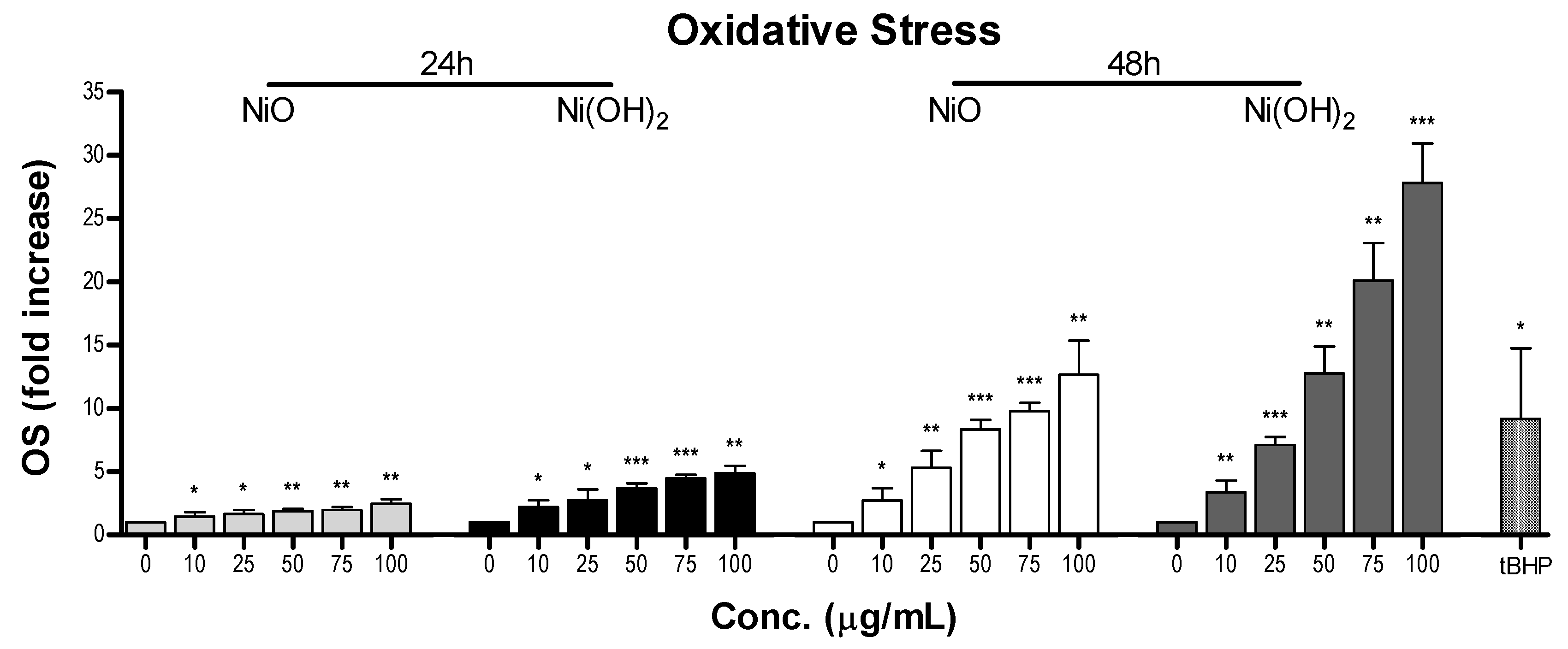
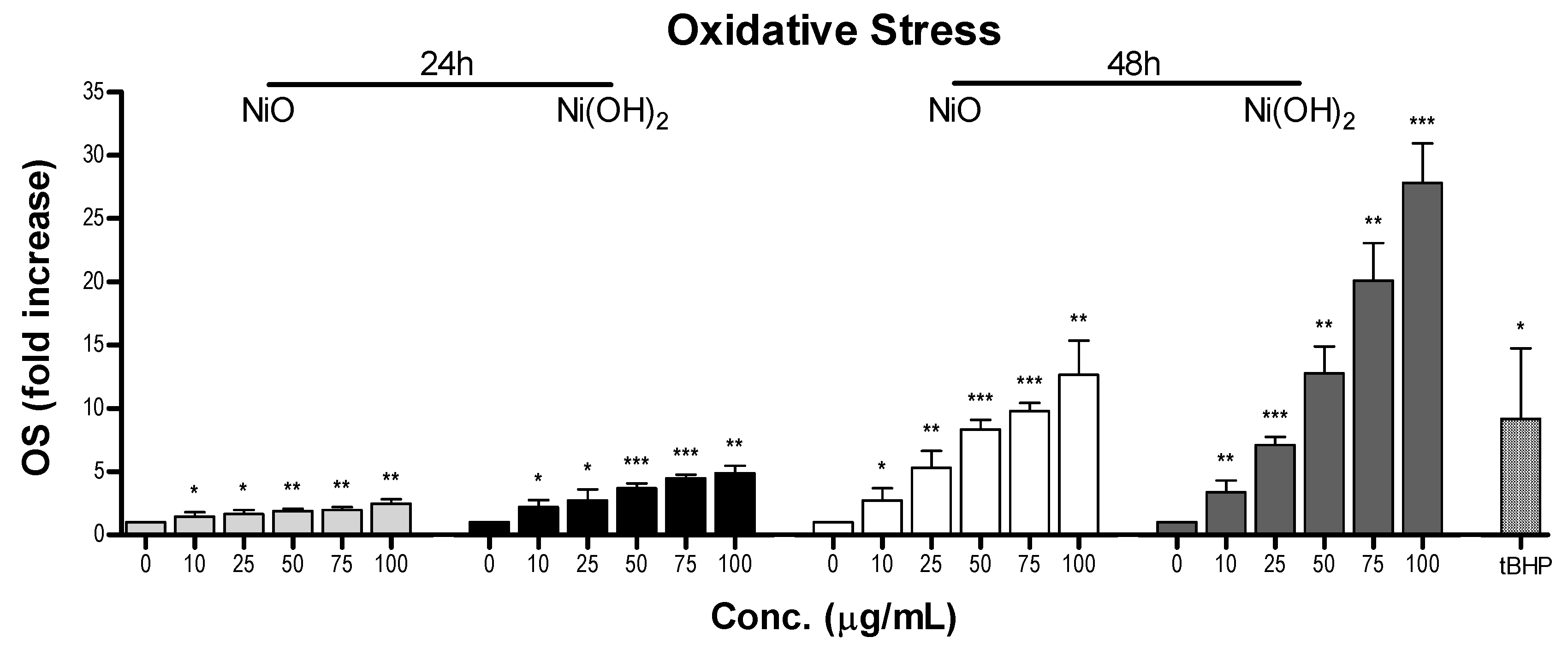
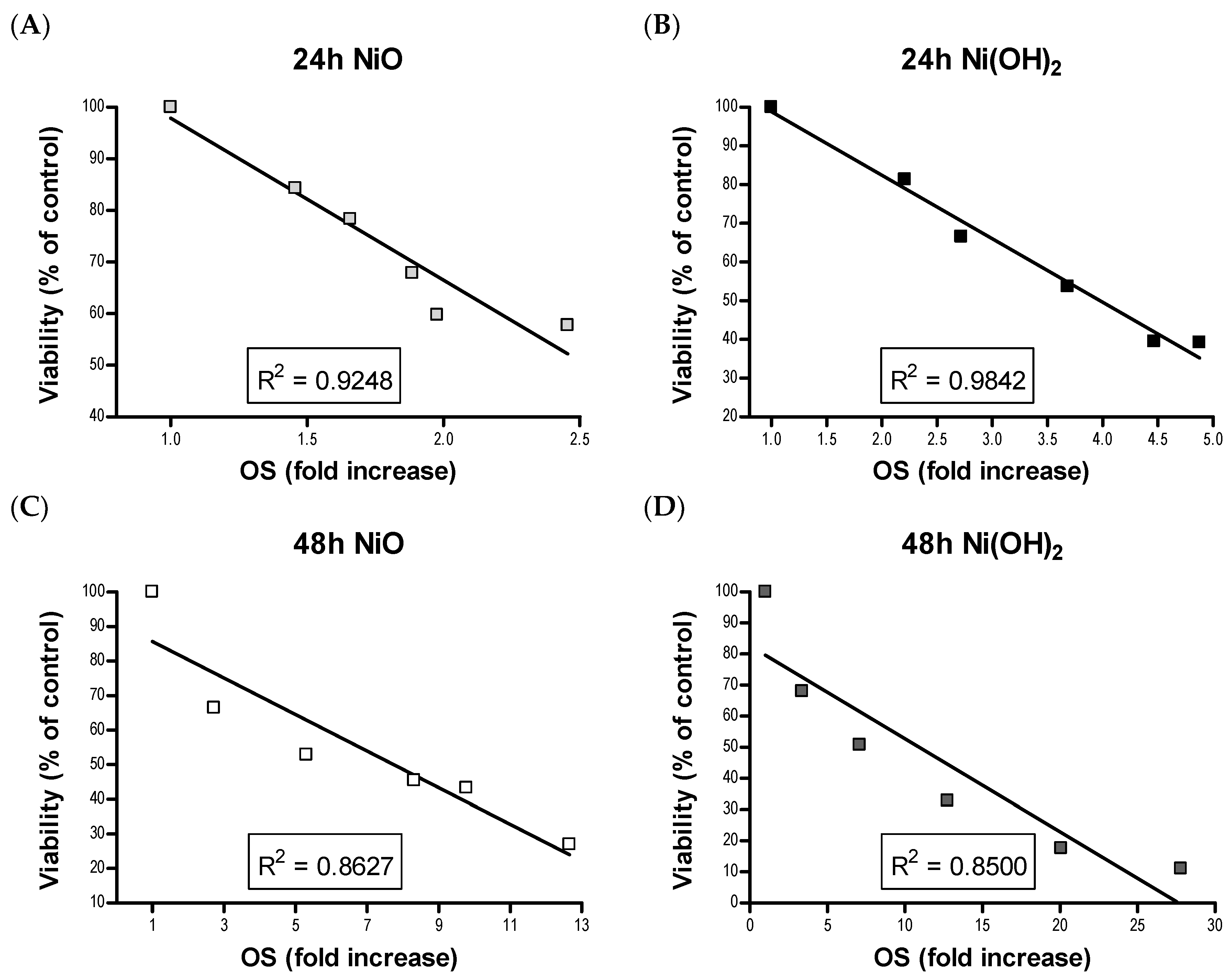
OS was elevated upon exposure to NiO and Ni(OH)
2 and had a strong correlation with cell viability at both 24 and 48 h (
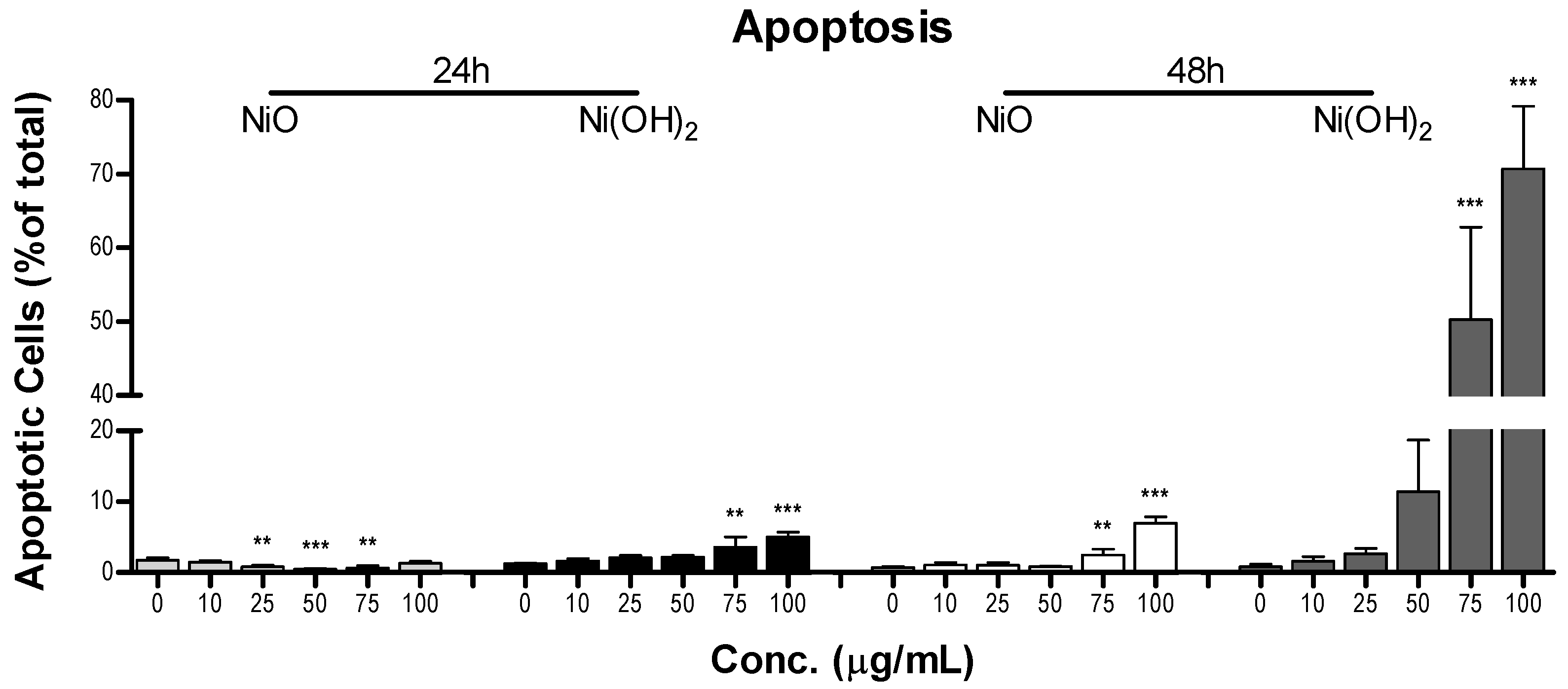
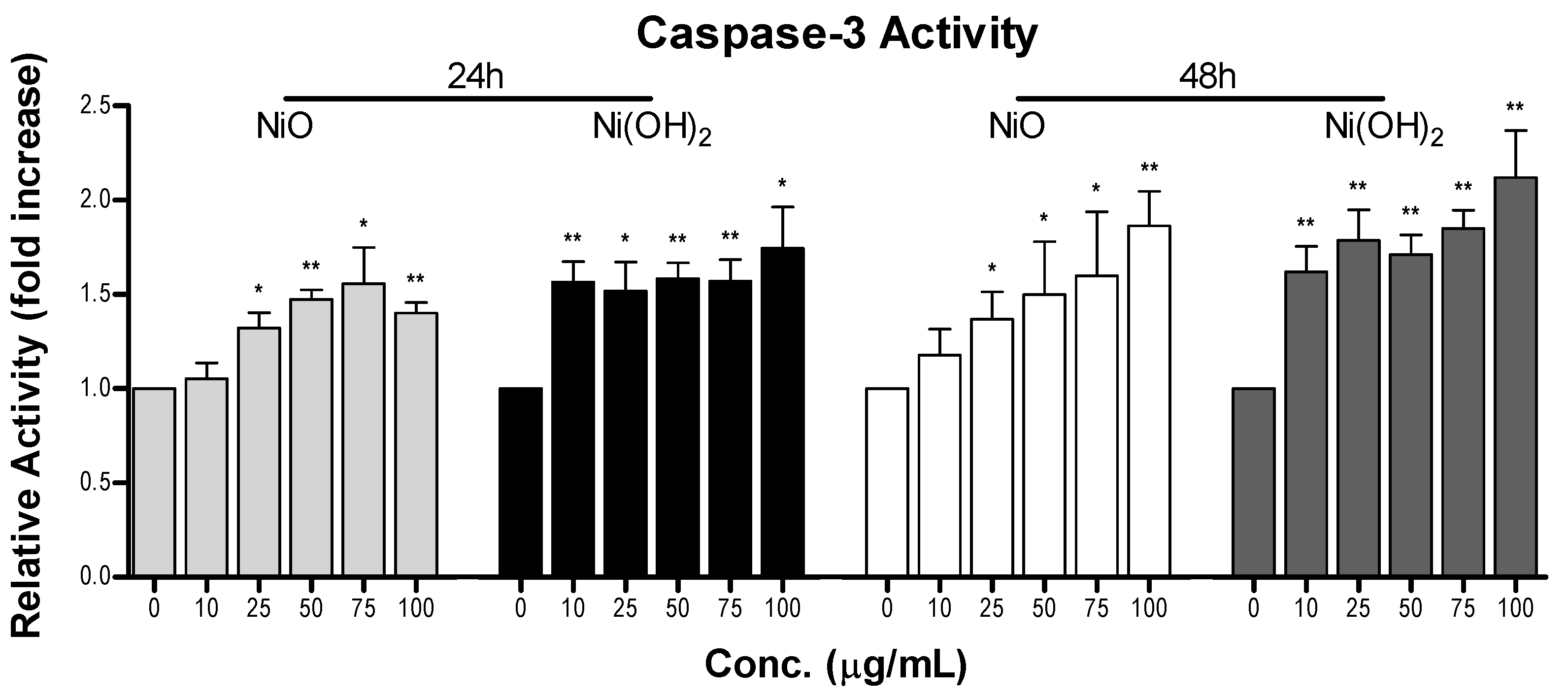
Figure 11). This indicates that the generation of free radicals and oxidants is a hallmark of NP toxicity and stress from these oxidative species triggers consequential molecular events leading to cell death. OS-mediated dissipation of MMP due to the presence of both NPs was supported by the apparent reduction in influx of cationic JC-1 into mitochondria. Reduction in the number of healthy mitochondria, or their general functionality, in a cell plays a substantial role in perturbing the homeostasis of bioenergetics and multiple signaling pathways pertaining to cell survival. One such signaling alteration is the increase of caspase-3 enzymatic activity and subsequent apoptosis. Our data demonstrate that both NiO and Ni(OH)
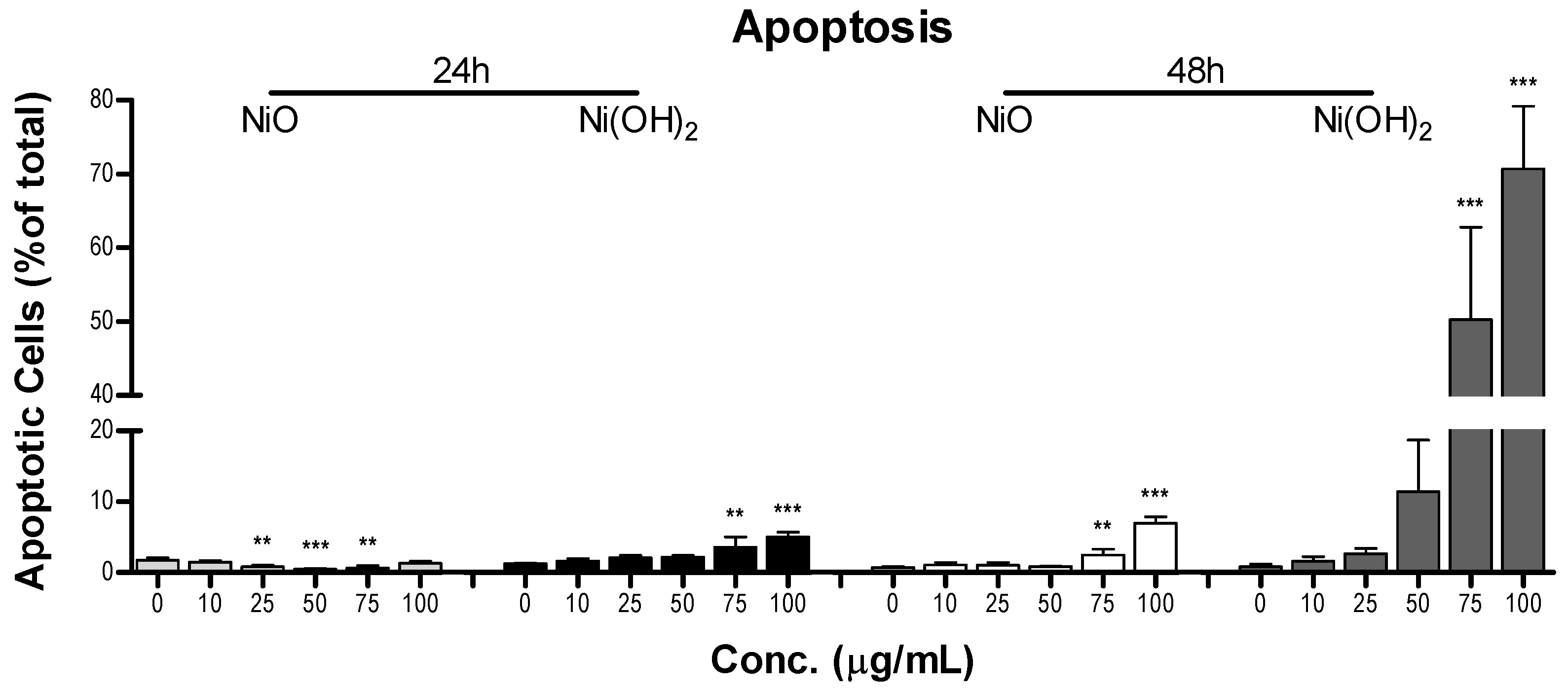
2 elevate caspase-3 enzymatic activity and apoptosis in a time- and concentration-dependent manner. NP-induced cell death is complex. In the present study, Ni(OH)
2 NPs imposed a much more significant elevation of apoptosis than NiO NPs, particularly towards 48-h exposure. Besides apoptosis, necrosis is also involved in NP-induced cell death. Capasso et al. found a concentration-dependent increase of necrosis mediated by NiO NPs [
21]. Another study found that CuO, ZnO, and Mn
2O
3 induced apoptosis in A549 cells but apoptotic cell populations increased in various increments, with the apoptotic rate staying relatively the same between two concentrations before drastically increasing in subsequent concentrations [
20]. Additionally, poly vinyl pyrrolidone-coated Ag and Ag
+ NPs induced both apoptosis and necrosis in time- and particle-dependent manners in THP-1 monocyte cells [
34]. Collectively, the roles of apoptosis and necrosis seem to be dynamic in the context of acute response and prolonged exposure.
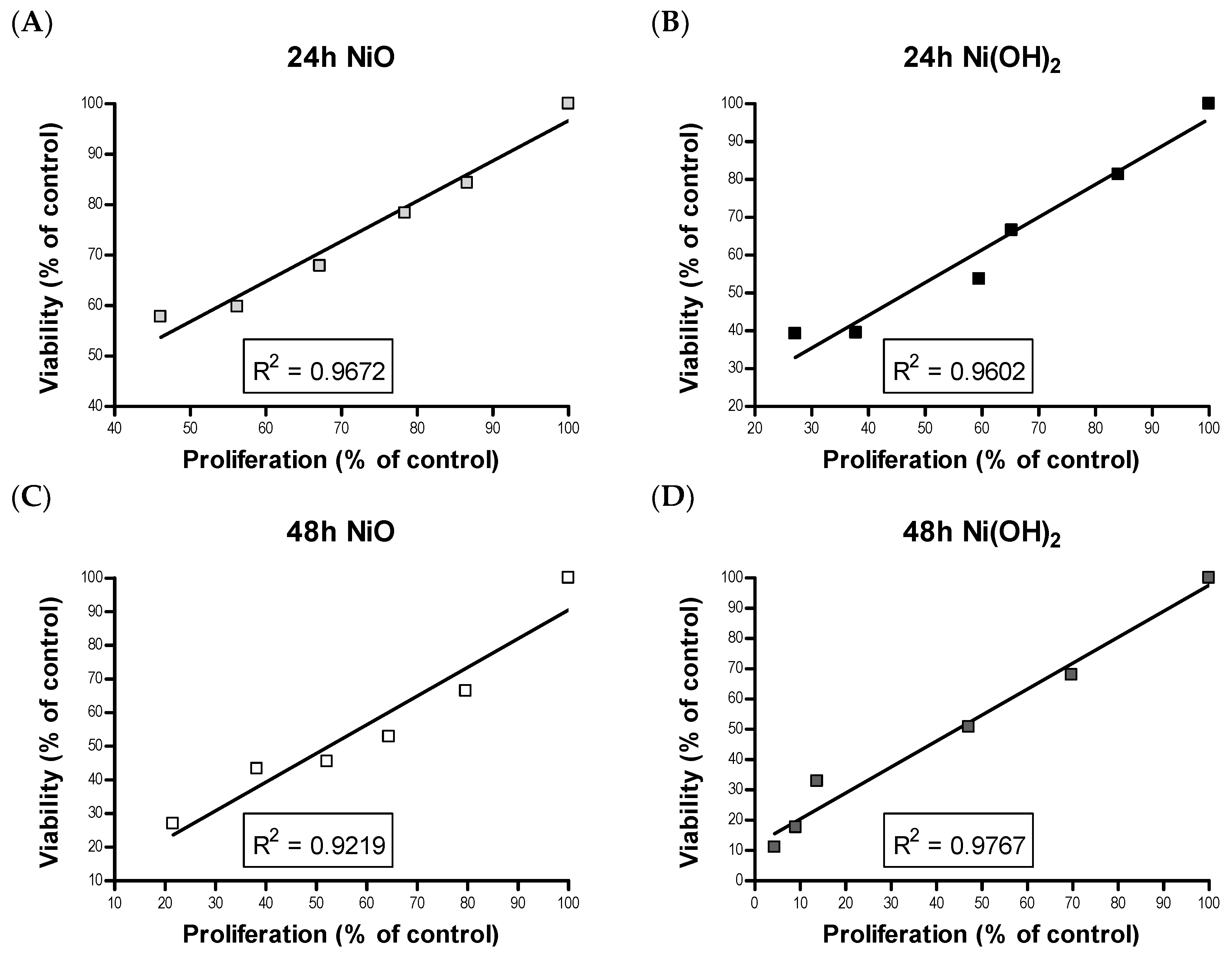
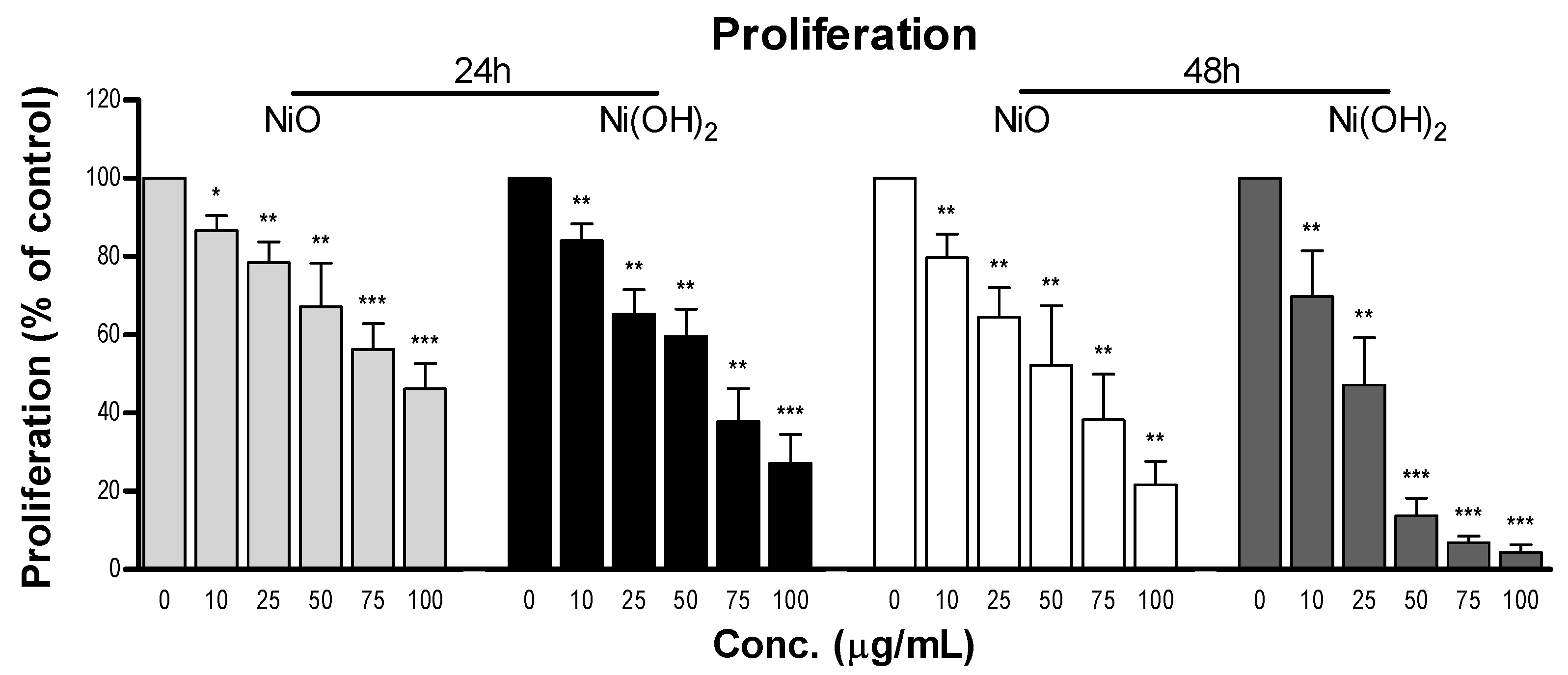
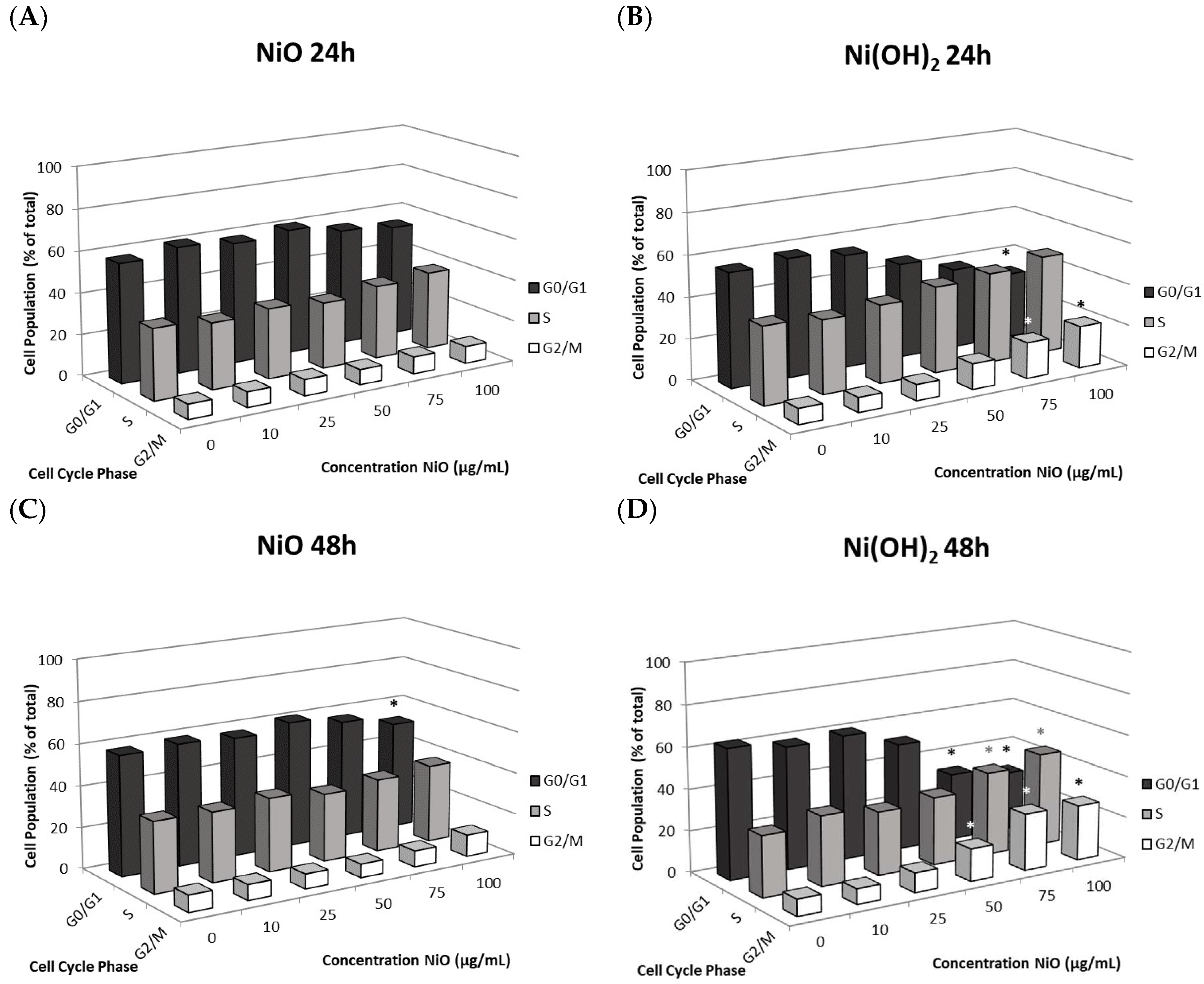
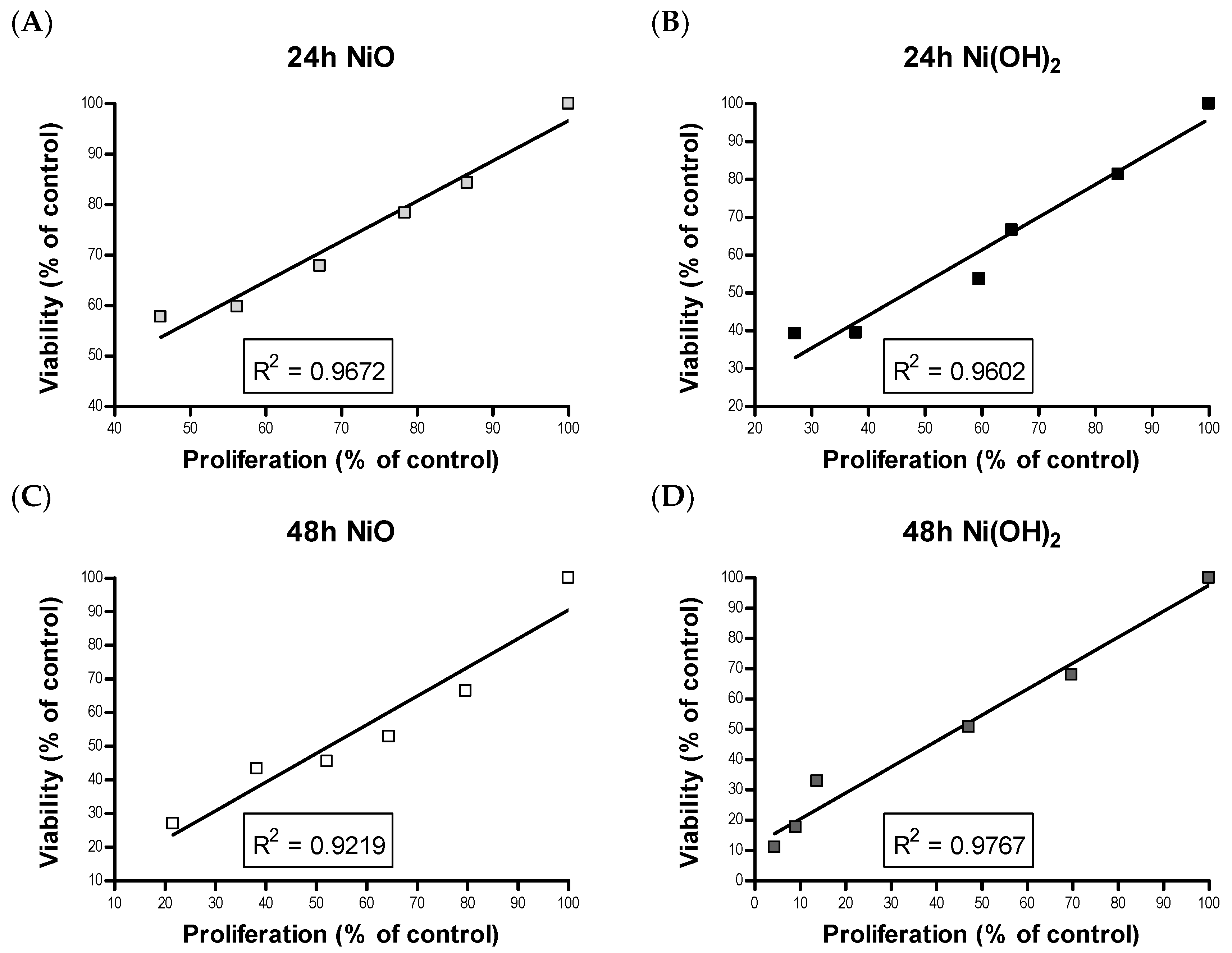
Although cell death induced by NPs has been demonstrated by a wealth of literature, suppression of cell proliferation is relatively under-studied. We hypothesized that the degree of cell viability imposed by exposure to NPs would be a function of cell death and cell proliferation. Our tritiated thymidine incorporation assays provided proliferation data which possessed a very strong linear correlation with cell viability for NiO and Ni(OH)
2 over a period of 24 and 48 h (
Figure 12). These correlations indicate that suppression of proliferation is a key factor in determining the reduction of cell viability. Modulation of cell proliferation has multiple causations, alteration of cell cycle being one. Our results showed that Ni(OH)
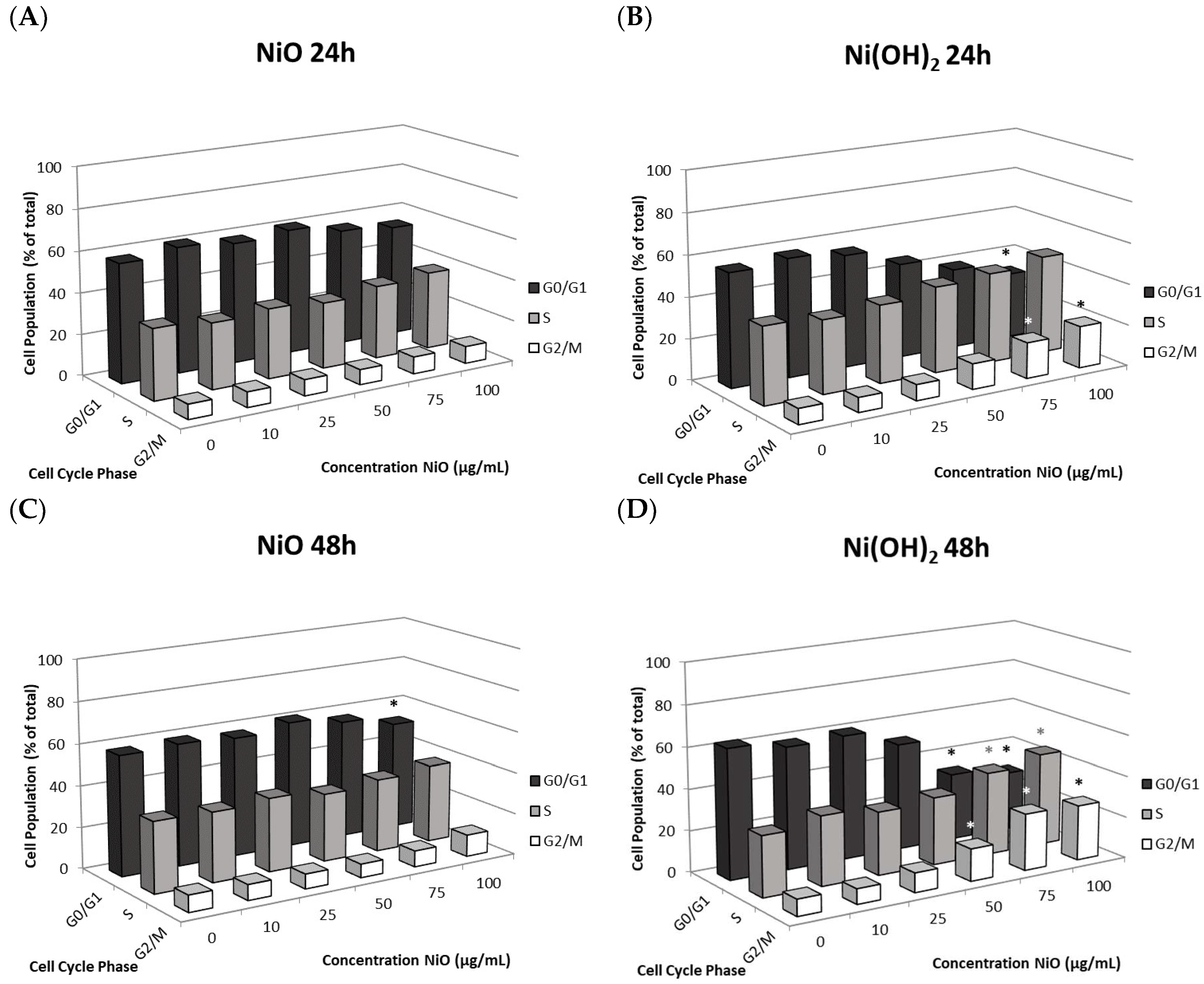
2 arrests cells in the S and G
2/M phases while NiO did not influence the cycle significantly. This also indicated that more factors play into proliferation rate. Although studies have found NP-mediated, phase-specific alterations of cell cycle, the effects are not all the same. For instance, exposure to TiO
2 caused HaCat cells to arrest in the S phase while ZnO and CuO exposure caused the same cell type to arrest in G
2/M [
35,
36,
37]. NPs may be composed of the same elements but have different properties based on their structure, which can also influence phase-specific arrest. Spherical TiO
2 NPs arrested A549 cells in the G
0/G
1 phase while needle-like TiO
2 NPs arrested A549 cells in G
2/M [
38,
39]. The mechanism of how cell cycle deregulation eventually influences cell proliferation remains to be elucidated.
Among the measured physical and chemical properties, specific surface area, metal dissolution, and surface charge are different between NiO and Ni(OH)
2 NPs. Ni(OH)
2 NPs have a higher specific surface area, indicative of more available binding sites (
Table 2) to interact with biomolecules such as protein, lipids, and nucleic acids. Consequentially, more interactions can lead to a higher degree of observed cellular injuries. One remaining issue is identification of chemical mechanism(s) (e.g., oxidation-reduction potential between NPs and biomolecules) that may damage biomolecules. In our previous study [
20], ions dissolved from transition metal oxides correlated with the differential toxicity of seven NPs. Therefore, we suspect that dissolution and the effects of ions may play a role in the observed differential toxicity between NiO and Ni(OH)
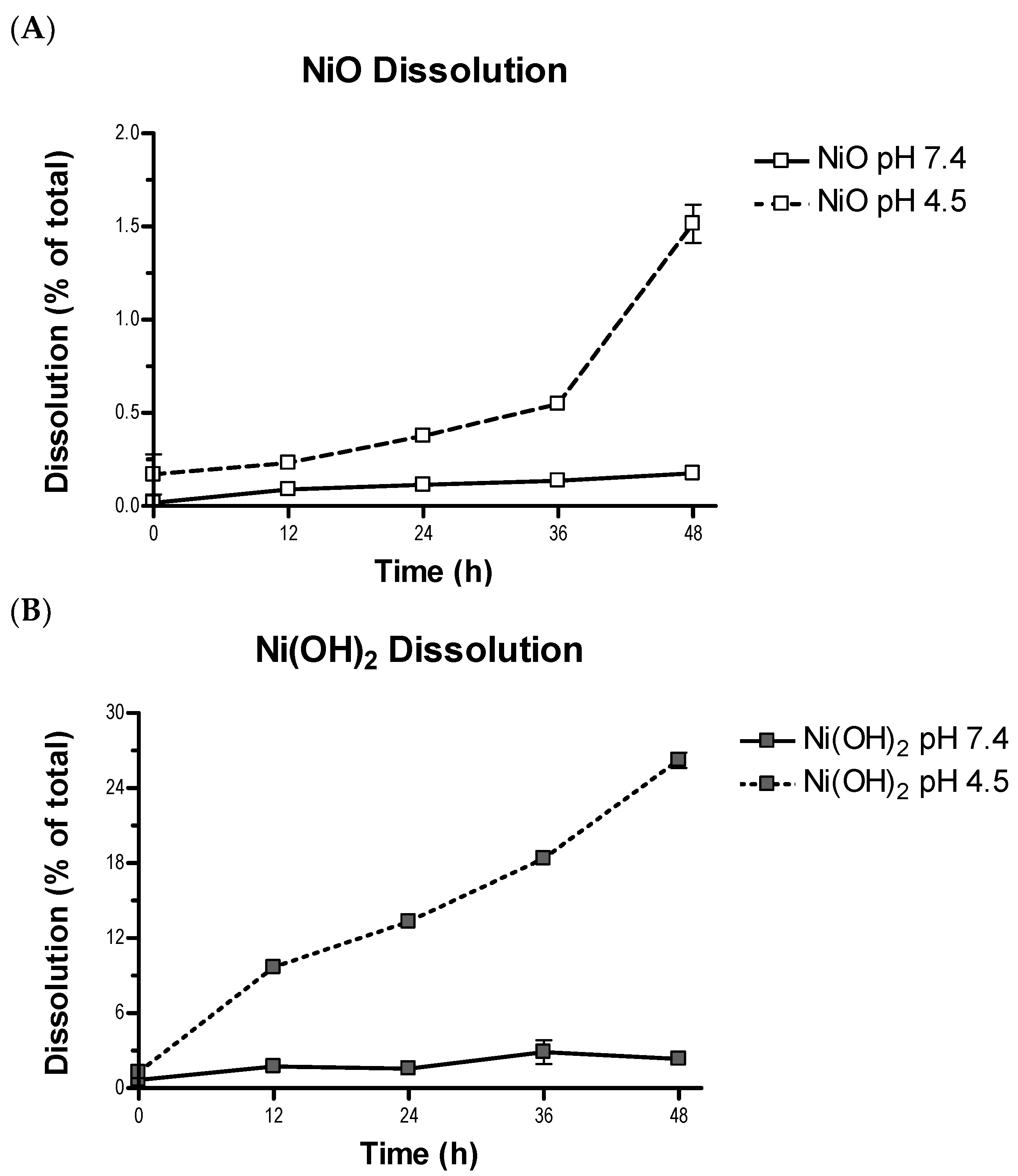
2. Compared with NiO, a higher degree of metal dissolution from Ni(OH)
2 might lead to Ni
2+-mediated toxicity. It is likely that once these particles are internalized and reside in the acidic lysosomal environment, the generation and intracellular action of Ni
2+ would be more exacerbated for Ni(OH)
2 than NiO as Ni(OH)
2 has been shown to have a much higher dissolution at lysosomal pH as compared to NiO (
Figure 3). Both NPs possess positive surface charge, which allows for electrostatic interactions with negative molecules, such as glycosaminoglycans, leading to endocytosis [
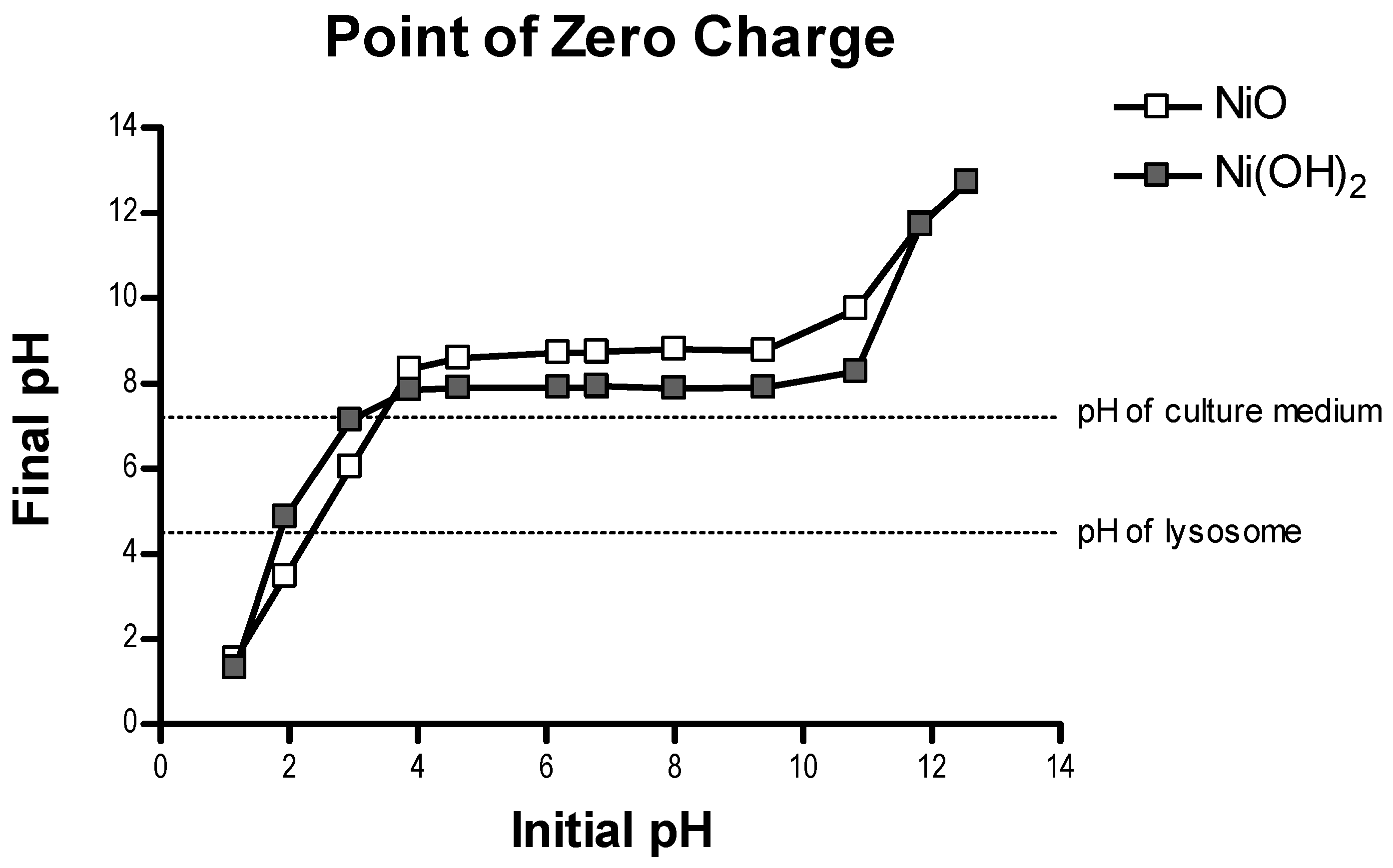
40]. The slight surface charge difference between these two NPs is not likely a key factor of cellular availability.
4. Materials and Methods
4.1. Material Sources
NiO NPs were purchased from Nanostructured and Amorphous Materials (Los Alamos, Houston, TX, USA) and Ni(OH)2 NPs were purchased from US Research Nanomaterials (Houston, TX, USA). A549 cells and HepG2 cells were acquired from the American Tissue Culture Collection (Manassas, VA, USA). 2′,7′-dichlorodihydrofluorescein diacetate (H2DCFDA) and propidium iodide (PI) were obtained from Fisher Scientific (St. Peters, MO, USA). The JC-1 Mitochondrial Membrane Potential Detection Kit and sulforhodamine B were purchased from Biotium (Freemont, CA, USA). Ac-DEVD-pNA was obtained from Anaspec (Fremont, CA, USA). Annexin V-FITC and 7-aminoactinomycin D (7-AAD) were acquired from BD Biosciences (Franklin Lakes, NJ, USA). Tritiated thymidine was purchased from Perkin-Elmer (Waltham, MA, USA). Other chemicals used for experiments were of the highest purity that could be obtained.
4.2. Storage and Characterization of Nanoparticles
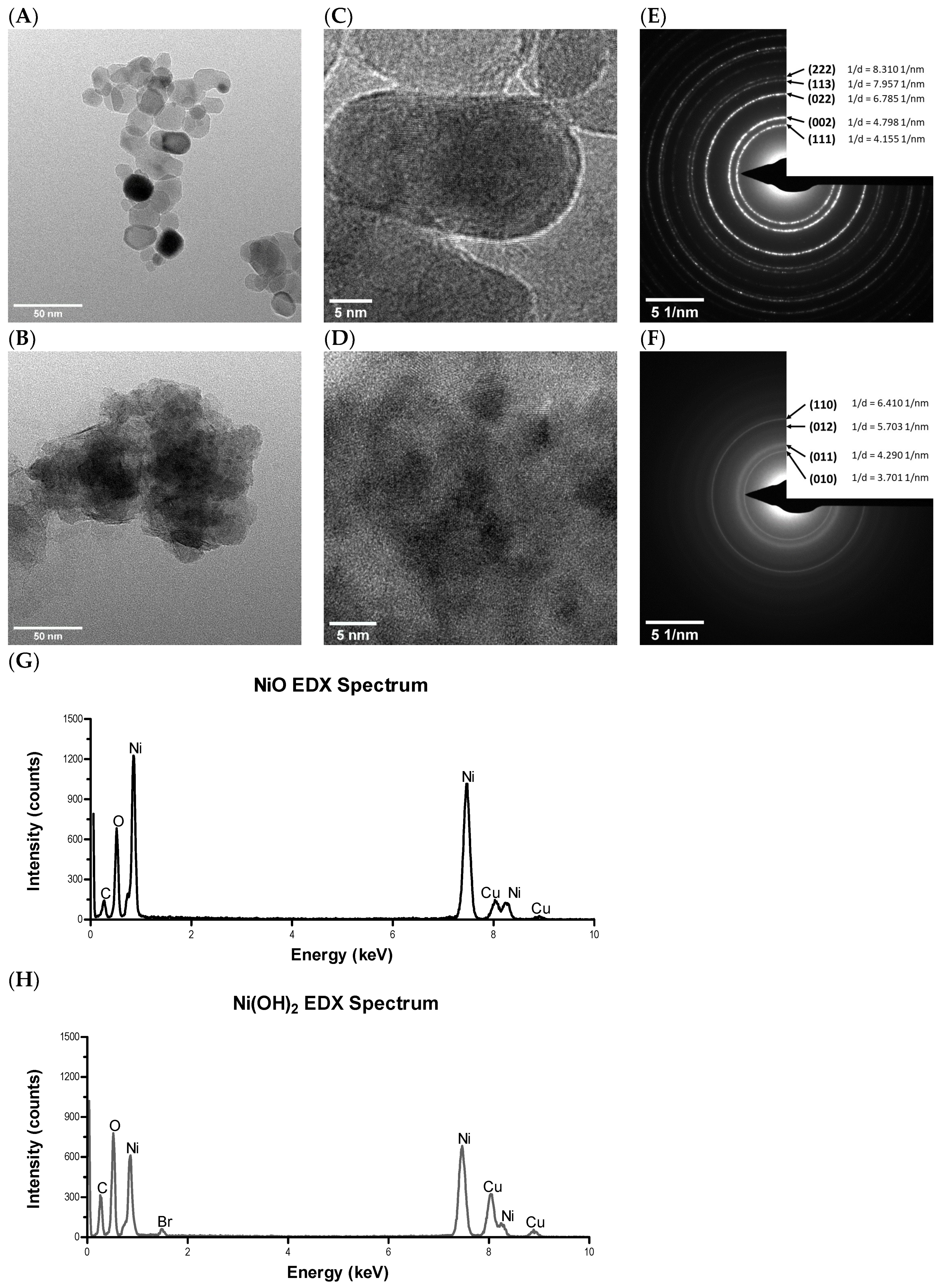
NPs were stored in an amber desiccator under a pure nitrogen atmosphere to protect them from moisture, oxidation, and UV damage. The instrumentation and protocols used to characterize NPs followed our previous publication [
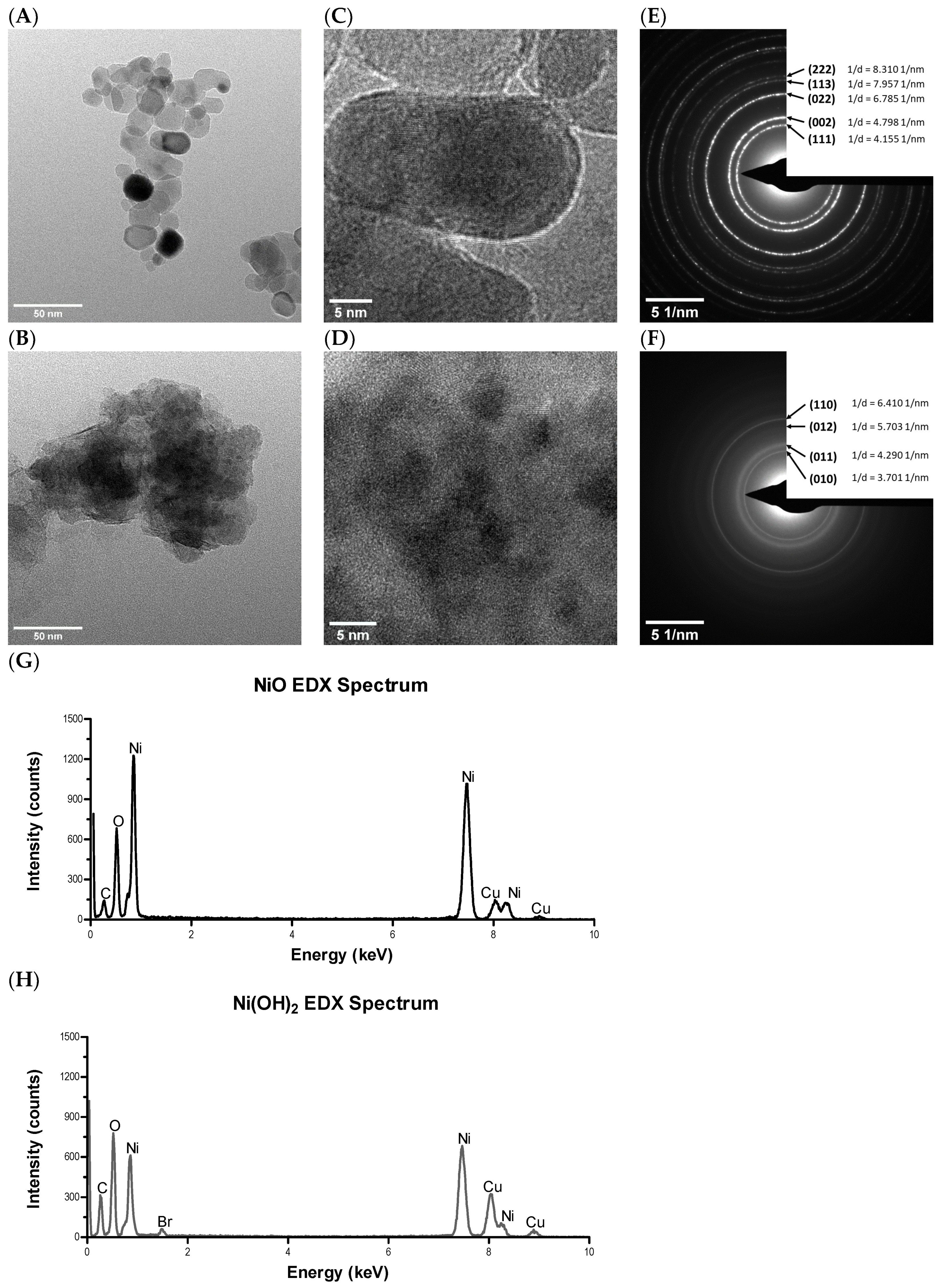
20]. SSA and APS of NPs in non-aqueous conditions were measured by Brunauer–Emmett–Teller (BET) and TEM, respectively. Morphology and crystal structure, surface charge, metal dissolution, and relative available surface binding sites of NPs in aqueous conditions were measured by high resolution transition electron microscopy (HRTEM), PZC, inductively coupled plasma-optical emission spectrometry (ICP-OES), and XPS.
4.3. TEM and HRTEM
NiO and Ni(OH)2 NPs were suspended in ethanol and dropped onto copper grids with amorphous carbon film. Grids were allowed to dry overnight before insertion into a Tecnai F20 TEM (Thermo Fisher Scientific, Hillsboro, OR, USA) equipped with an energy-dispersive detector (EDS) detector. TEM and HRTEM images were captured, as well as the SAED patterns for both samples.
4.4. Quantification of Available Binding Sites
XPS integrated peak areas of the O 1s binding energy core level were used to quantify the relative number of available binding sites for material in the extracellular matrix to interact with on the NiO and Ni(OH)
2 nanoparticle surfaces. Quantification was achieved by noting the physisorbed versus chemisorbed oxygen on the NP surfaces as described in our previous study [
20]. In the case of NiO NPs, the deconvoluted O 1s peak denoting the metal oxide represents the underlying substrate while those peak areas of those oxidation states not from the metal oxide denote physisorbed O. The number of available binding sites were quantified by dividing the XPS peak area of the physisorbed O by that of chemisorbed O.
The O 1s core levels of the NiO NPs were observed at 529.0 eV and 531.0 eV, denoting the metal oxide [
41,
42,
43] and physisorbed O on the NP surfaces, respectively. For the Ni(OH)
2 NP surfaces, the binding energy centers of the O 1s orbitals appear at 531.2 and 532.9 eV, denoting chemisorbed OH groups and physisorbed OH/H
2O [
41,
44] on the NP surfaces, respectively. While binding energies for adsorbed OH/H
2O are well established, they are not for chemisorbed OH on Ni(OH)
2; this information was obtained via XPS scans on the dried Ni(OH)
2 NPs performed in our laboratory as a reference standard. The binding energy peak center for this oxidation state was found to be 531.2 eV, having a full width at half maximum (FWHM) of 3.4 km/sec, the parameters of which were used in peak area deconvolution of the O 1s envelopes.
4.5. Cell Culture and Nanoparticle Treatment
A549 cells were maintained in Ham’s F-12 modified medium supplemented with 10% HyClone FetalClone serum (GE Healthcare Life Sciences, Marlborough, MA, USA) and 1% penicillin/streptomycin. HepG2 cells were maintained in Eagle’s minimum essential medium supplemented with 10% FetalClone serum and 1% penicillin/streptomycin. Both cell lines were grown in 10-cm tissue culture dishes at 37 °C in a 5% CO2 humidified incubator. Upon reaching a confluence of ca. 70–80%, cells were trypsinized, and appropriate numbers of cells were seeded into tissue culture dishes or plates for various experiments. NPs were suspended in cell culture media to create a working concentration of 1 mg NP per 1 mL media. The working suspension of NPs was sealed with parafilm and sonicated for 3 min to break up aggregates. The suspension was vortexed to achieve a homogenous mixture before being added to cells and was diluted in cellular media to achieve a series of desired concentrations. Each treatment group was completed in at least triplicate and appropriate controls were included, with the most common controls being untreated cells.
4.6. Cell Viability
Cell viability was measured using the sulforhodamine B (SRB) assay. A549 cells and HepG2 cells were seeded into 24-well tissue culture plates and allowed to grow for 24 h before compound exposure. For A549 cells, 45,000 and 22,000 cells were seeded per well for 24- and 48-h exposure, respectively. For HepG2 cells, 120,000 cells were seeded per well for both 24 and 48 h. Cells were treated with nickel NPs (0, 10, 25, 50, 75, or 100 µg/mL) for 24 or 48 h. Upon termination of experiments, cell medium was discarded from the cells. The cells were fixed with cold 10% trichloroacetic acid (TCA) for 1 h at 4 °C. The fixed cells were then washed three times with distilled water and then allowed to dry completely. Cells were incubated with 0.5 mL of SRB staining solution (0.2% SRB in 1% acetic acid) for 30 min at room temperature. The cells were then washed three times with 1 mL of 1% acetic acid for 20 min on a rocker to eliminate excess dye. A Q-tip was used to remove excess solution stuck to the sides of the wells. Acetic acid removal was followed by addition of 400 µL of cold 10 mM Tris-HCl solution to each well for 20 min. Aliquots of 250 μL each were transferred onto a 96-well plate and absorbance was measured at 510 nm using a microplate reader (FLUOstar Omega, BMG Labtechnologies, Cary, NC, USA). Cell viability of treatment groups was calculated based on the percent absorbance relative to the control group with appropriate blanks subtracted.
4.7. Oxidative Stress (OS)
Reactive oxidative species were measured with H2DCFDA. Upon entry of cells, H2DCFDA was deacetylated by esterases to a non-fluorescent compound. When H2DCFDA is oxidized by reactive oxidative species, it is converted to the highly fluorescent compound 2′,7′-dichlorofluorescein (DCF) and can be detected by fluorescence spectroscopy. A549 cells were exposed to a series of concentrations of NPs (0, 10, 25, 50, 75, or 100 µg/mL) for 24 or 48 h. Cells were seeded into 96-well plates at 1500 or 750 cells per well for 24- and 48-h treatments, respectively, and grown for 24 h before exposure. As a positive control, cells were incubated with 400 µM tert-butyl hydroperoxide (tBHP) at 37 °C for 1 h before termination of the experiment. Upon termination, the media was removed from the cells followed by a wash with phosphate-buffered saline (PBS). Eighty µL of 0.87 mM H2DCFDA in ethanol was added to each well and the plate was incubated for 1 h. Cells were then washed with PBS three times followed by addition of 100 µL of PBS. Fluorescence was measured at 510 nm using a microplate reader. The florescence intensity of cells in experimental plates was divided by the fluorescence intensity of the control group to determine the percent increase in ROS with appropriate blank intensities considered.
4.8. Mitochondrial Membrane Potential
Mitochondrial membrane potential was determined with fluorescence microscopy using the JC-1 MMP Detection Kit (Cayman Chemical Company, Ann Arbor, MI, USA). JC-1 (5,5′,6,6′-Tetrachloro-1,1′,3,3′-tetraethylbenzimidazolylcarbocyanine iodide, CAS#: 3520-43-2) monomers fluoresce green when in the cytosol of cells. Accumulation of JC-1 in mitochondria allows the compound to form aggregates that exhibit red fluorescence in a concentration-dependent manner. Normal mitochondrial membrane potential, in healthy cells, allows JC-1 to influx and form aggregates in the mitochondria. In unhealthy cells, mitochondrial membrane potential is decreased, JC-1 concentration cannot increase high enough to form aggregates, and thus the compound remains green [
45]. The comparison of red and lack of red fluorescence allows the loss of mitochondrial membrane potential to be observed.
A549 cells were grown for 24 h in 35-mm glass-bottom culture dishes at 15,000 cells per dish. Cells were exposed to several concentrations of nickel NPs (0, 10, or 100 µg/mL) for 12 or 24 h. Following NP treatment, the plates were incubated with 100 µL of JC-1 staining solution per mL of medium at 37 °C for 15 min. Each plate was then washed with 1 mL of PBS followed by addition of 1 mL of PBS before fluorescence detection under an epifluorescence microscope (Olympus Corporation, Tokyo, Japan). Red fluorescence was observed with a Texas Red filter (excitation/emission: 590/610 nm) while green fluorescence was with a FITC filter (excitation/emission: 490/520 nm).
4.9. Caspase-3 Activity
Caspase-3 enzymatic activity was measured using Ac-DEVD-pNA as a substrate. A549 cells were grown for 24 h in 24-well plates at seeding densities of 45,000 or 22,000 cells per well for 24- or 48-h exposure, respectively. Cells were treated with a series of concentrations of NPs (0, 10, 25, 50, 75, or 100 µg/mL) for 24 or 48 h. After incubation with NPs, cells were washed once with 0.5 mL of PBS. Two hundred µL of cold lysis buffer (50 mM Tris-HCl, 1.5 mL of 5 M NaCl, 0.25 g of sodium deoxycholate, 1 mM ethylenediaminetetraacetic acid (EDTA), 0.5 mL of Triton-100, and 50 mL of distilled water) was added to each well, cells were scrapped off the well bottoms, resuspended in the lysis buffer, and incubated at 4 °C for 10 min. Samples were centrifuged at 15,000× g for 20 min at 4 °C. Total protein in each sample was measured using a Pierce BCA protein assay (Thermo Scientific, Rockford, IL, USA). Cell lysate and reaction buffer (20% glycerol, 0.5 mM EDTA, 5 mM dithiothreitol, and 100 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES), pH 7.5) were combined in a 96-well plate to have 20 µg of cellular protein and a total volume of 198 µL in each well. Following, 2 µL of 0.5 mg/mL Ac-DEVD-pNA substrate was added to each well. Samples were incubated at 37 °C for 6 h. Absorbance of enzyme-catalyzed release of p-nitroanilide was measured at 405 nm with a microplate reader.
4.10. Apoptosis
Apoptosis was measured with flow cytometry using annexin V-FITC and 7-AAD. A549 cells were seeded into 6-cm tissue culture dishes at densities of 250,000 and 120,000 for 24- and 48-h exposure, respectively, and allowed to grow for 24 h before treatment. Cells were treated with a series of NP concentrations (0, 10, 25, 50, 75, or 100 µg/mL) for 24 or 48 h. Upon termination of the NP exposure period, cells were washed with PBS and harvested with trypsinization. NP treatment medium, PBS washes, and trypsinized cells were all collected in the same centrifuge tube for each concentration of NP in order to avoid loss of floating cells undergoing apoptosis. The samples were centrifuged and the supernatant was discarded. The pellet was washed with 1 mL of ice-cold PBS, centrifuged again, and the supernatant was removed. The cells were resuspended in 100 µL of 1x annexin V binding buffer, 2 µL of annexin V-FITC, and 2 µL of 7-AAD. The cells were then incubated for 20 min in the dark. The stained cell solutions were transferred to the wells of a 96-well plate for flow cytometry analysis on a CytoFLEX flow cytometer (Beckman-Coulter, Brea, CA, USA). Cells in different stages of apoptosis were quantified using FCS Express 6 (DeNovo software, Pasadena, CA, USA). Early and late apoptotic cells were added to represent the total percentage of apoptotic cells.
4.11. Cell Proliferation
Proliferation was determined with a tritiated thymidine ([5′-3H]-thymidine) incorporation assay. A549 cells were seeded in 24-well plates with 45,000 and 22,000 cells per well for 24- and 48-h exposure, respectively. Cells were grown for 24 h before being dosed with nickel compounds. Cells were exposed to a series of concentrations of NPs (0, 10, 25, 50, 75, or 100 µg/mL) and treated with [5′-3H]-thymidine (Perkin-Elmer) simultaneously for 24 or 48 h. A working solution of [5′-3H]-thymidine was prepared with 20 µL of [5′-3H]-thymidine (1 µCi/µL) in 500 µL of PBS. Each well of the 24-well plate was treated with 20 µL of the [5′-3H]-thymidine working solution. Upon termination of each experiment, cells were washed twice with ice-cold PBS. The cells were then fixed in 0.5 mL of ice-cold 10% TCA for 5 min on ice. TCA fixation was repeated once. Cells were brought to room temperature and lysed using 0.5 mL of room temperature 1 N NaOH for 5 min. The solution was neutralized by adding an equal amount of 1 N HCl. The lysed cell solution was thoroughly mixed by pipetting up and down and then transferred to liquid scintillation counting vials with 4 mL of Econo-Safe scintillation counting fluid (Research Products International, Mt. Prospect, IL, USA). Sample vials were then subjected to scintillation counting using a Beckman liquid scintillation counter LS6500 (Beckman-Coulter). The total count of radioactivity was divided by the radioactivity from the 0 µg/mL control cells to determine the percentage of proliferating cells compared to cells not exposed to nickel compounds. All radioactive waste was disposed of following Missouri S&T’s Department of Environmental Health and Safety procedures.
4.12. Cell Cycle
Alteration of cell cycle due to nickel NP exposure was measured with flow cytometry and PI staining. A549 cells were grown in 6-cm tissue culture dishes for 24 h before treatment. The seeding density of cells per dish was 250,000 cells for 24-h treatment and 120,000 cells for 48-h treatment. Cells were exposed to a series of concentrations of NPs (0, 10, 25, 50, 75, or 100 µg/mL) for 24 or 48 h. Following incubation with NP treatments, cells were washed with PBS, harvested using trypsinization, and centrifuged. The cell pellet was then resuspended in 1 mL of PBS and 3 mL of ice-cold absolute methanol was added drop-wise to the solution while vortexing. The cells were stored at 4 °C for at least 24 h to fix. After fixation, the cells were centrifuged and washed once with PBS. The cells were then suspended in PI staining solution (50 µg/mL PI, 0.1% RNase A, and 0.05% Triton X-100 in PBS) for 20 min in the dark. One mL of PBS was added to each sample before centrifuging, the supernatant was removed, and cells were resuspended in 250 µL of PBS. The stained samples were then pipetted into a 96-well plate and analyzed with a flow cytometer. FCS Express 6 was used to determine the distribution of cells in different cell cycle phases. The number of cells in each phase of the cell cycle (G0/G1, S, and G2/M) was totaled and the percentage in each phase was calculated.
4.13. Statistical Analysis
Each experiment was repeated at least three times independently with each treatment group having at least triplicate samples. Data are presented as mean ± standard deviation. Statistical analysis was performed in Minitab 19. One-tailed unpaired t-tests were used to compare experimental groups to the control group in normalized data sets, with µ > control or µ < control depending on the experimental hypothesis. Analysis of variance (ANOVA) with Dunnett comparison was used to determine significant differences against the control group. Significance was set at p < 0.05. P-values less than 0.05, 0.01, and 0.001 are noted in figure legends. Linear regression to analyze correlations between data was completed using GraphPad Prism 4. All figures were produced using GraphPad Prism 4 except for the cell cycle distribution graphs, which were produced by Microsoft Excel 2016.

 ,
, 








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}